

Abstract
We analyzed the mechanism of developmental failure in implanted β1 integrin-null blastocysts and found that primitive endoderm cells are present but segregate away from, instead of forming an epithelial layer covering, the inner cell mass. This cell segregation phenotype was also reproduced in β1 integrin-null embryoid bodies, in which primitive endoderm cells segregated and appeared as miniature aggregates detached from the core spheroids, and a primitive endoderm layer failed to form on the surface. Restricted β1 integrin gene deletion in embryos using Ttr-Cre or Sox2-Cre indicated that the loss of integrin function in the cells of the inner core rather than the outer layer is responsible for the failure to form a primitive endoderm layer. We conclude that β1 integrin is essential for the attachment of the primitive endoderm layer to the epiblast during the formation of a basement membrane, a process concurrent with the transition from cadherin- to integrin-mediated cell adhesion.
INTRODUCTION
The mechanisms by which cells spontaneously differentiate and organize in early embryos have long presented an enigma for developmental biologists. Starting from mammalian blastocysts, a small number of stem cells comprising the inner cell mass (ICM) multiply and differentiate, and the progeny self-organize into structures that eventually pattern the entire embryo (1–4). The ICM cells are pluripotent and can be derived and cultured in vitro, giving rise to embryonic stem (ES) cells (5, 6). One of the first structures to differentiate from the ICM is the primitive endoderm (PrE), a simple monolayer epithelium covering the surface, while the remainder of the ICM emerges into the epiblast at the embryonic day 4.5 (E4.5) stage of mouse embryogenesis (7–9). The receptor tyrosine kinase–Ras–mitogen-activated protein kinase (MAPK) signal pathway is critical in triggering the differentiation of the PrE lineage (10, 11), since PrE fails to form in embryos with genetic deletions in components of the pathway, including Fgf4 (12, 13), its cognate receptor Fgfr2 (14), and Grb2 (15, 16). Gata6 is the key transcription factor downstream of Grb2 (17) and is essential for PrE differentiation (18). The differentiation and morphogenesis of PrE are recognized to be a two-step process, and the mechanisms for the formation of the PrE epithelium have been updated recently (19, 20). Initial PrE differentiation is thought to occur randomly, both in the interior and at the periphery of the ICM (19–22). Subsequently, the differentiated PrE cells sort to the surface to form a coherent PrE epithelial layer (15, 20, 22, 23), although another idea is that the differentiation is not random but is affected by the timing of cell internalization (24, 25).
Aggregation of ES cells in suspension culture as embryoid bodies models some aspects of lineage differentiation and morphogenesis in early embryos (26, 27). Studies using embryoid bodies support the model that PrE differentiation often occurs in the interior of the cell aggregates and that subsequently the differentiated cells sort to and form an epithelium on the surface (27). The movement of the newly differentiated cells is dynamic and random, though the cells are retained and positioned on the surface upon establishing an apical polarity (27). Further analysis using E-cadherin-null ES cells indicates that the sorting and positioning of PrE cells at the surface are not in accord with differential adhesion (28); rather, they are underpinned by the ability of the PrE cells to establish and maintain an apical polarity (29). The intermixing of endoderm and epiblast cells in Dab2-knockout embryos supports this idea (30, 31). The endocytic adaptor protein Dab2 mediates directional vesicular trafficking required for the genesis of an apical polarity (32). Deletion of Dab2 leads to the loss of epithelial polarity and consequently the failure of PrE cells to position at the surface (29, 31–33).
β1 integrin is one of a few genes known to be required for the development of mouse embryos at the peri-implantation stage, since β1 integrin-knockout mouse embryos die shortly following implantation, at around E5.5 (34, 35). Heterodimers of β1 integrin with various α integrin subunits bind many components of the basement membrane and extracellular matrix, including collagens, laminins, vitronectin, and fibronectin (36). In addition to the mechanical function of cell attachment, engagement of integrins is known to modulate signaling pathways and thus influences cell behavior (37, 38).
In the early mouse embryos, β1 integrin is expressed in both epiblast and extraembryonic endoderm (35). Several explanations have been proposed to account for the requirement of β1 integrin in peri-implantation development. An early suggestion was that β1 integrin gene deletion leads to retarded ICM growth and embryonic failure (35). Using embryoid bodies as models, an initial analysis suggested that β1 integrin-deficient cells fail to express laminin α1 and other basement membrane components (39), though a subsequent study concluded that laminin and other components can be synthesized and secreted, but the proper assembly of laminin polymers requires β1 integrin (40). A more recent analysis of β1 integrin-deficient embryoid bodies stated that integrins are required for the differentiation of visceral endoderm by mediating the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and p38 MAPK (41).
Although extensively investigated in various systems (36) and developmental processes (42), the biology related to the defect of the β1 integrin-null embryos still lacks clarity. Here, we revisited the phenotype of β1 integrin-deficient embryos and sought further clarification and insights to ascertain the detail of PrE differentiation and morphogenesis in mice.
MATERIALS AND METHODS
Mutant mice and genotyping.
The experiments using mutant mouse models described in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Miami. Mutant mouse colonies were housed inside the barrier area of the institutional mouse facility. Transgenic and mutant lines used in this project are as follows: β1 integrinflox/flox (abbreviated as β1 integrinfl/fl) (Itgb1tm1Efu/J) (43), purchased from Jackson Lab; Ttr-Cre mice, a gift from Anna-Katerina Hadjantonakis (Sloan-Kettering Institute, New York, NY) (44); and Sox2-Cre [Tg(Sox2-cre)#Amc/J] (45), purchased from Jackson Lab. Genotyping of mutant mice was performed using PCR amplification of DNA extracted from tail biopsy specimens, according to protocols recommended by Jackson Lab. For the genotyping of the β1 integrin flox mice, new primers were designed for a tripartite PCR with oligonucleotides 5′ CGG CTC AAA GCA GAG TGT CAG TC 3′, 5′ CCA CAA CTT TCC CAG TTA GCT CTC 3′, and 5′ CGC AGA ACA ATA GGT GCT GAA ATT AC 3′ to genotype simultaneously for wild-type (160-bp), flox (240-bp), and deltaflox (280-bp) alleles. The constitutive β1 integrin-knockout allele was generated by first mating β1 integrinfl/fl with Sox2-Cre mice. β1 integrin-null embryos were produced by matings of the resulting β1 integrindf/+ parents.
The same PCR assay was also used for genotyping of embryonic tissues. For mosaic deletion (using Ttr-Cre and Sox2-Cre), embryonic and extraembryonic tissues from the same embryos were PCR amplified in separate reactions. In mosaic deletion embryos from earlier stages (E5.5), when clear separation of endoderm and epiblast was not possible, genotyping was performed by immunostaining to verify the β1 integrin-deficient embryos.
Derivation and culture of ES cells.
RW4 mouse ES cell lines have been described previously (29). The CFG37 ES cell line is a wild-type ES cell line that has stably inherited the βACT-GFP (green fluorescent protein) transgene (29). β1 integrin-heterozygous (G119) and -null (G201) ES cells were gifts from Reinhard Fässler via Lionel Larue (34). All ES cell lines were maintained in a pluripotent state by culturing upon irradiated murine embryonic fibroblasts in ES cell medium supplemented with 1,000 U/ml of recombinant leukemia inhibitory factor (LIF) (Esgro; Chemicon International). The ES cell culture medium was Dulbecco's modified Eagle medium (DMEM) with 15% (vol/vol) fetal bovine serum, 2 mM l-glutamine, 1× nonessential amino acids, 50 IU/ml penicillin, 50 mg/ml streptomycin, and 0.1 mM β-mercaptoethanol. The ES cells were routinely cultured on gelatin-coated tissue-culture-grade plastic. G201 β1 integrin–/– ES cells were cultured on the same type of plates without gelatin treatment since they adhered better without the coating. All cell cultures were propagated in a humidified incubator with 5% CO2. To differentiate into endoderm, the ES cells were exposed to 1 μM all-trans-retinoic acid (RA) dissolved in dimethyl sulfoxide (DMSO) for 4 to 7 days added as a supplement to the ES cell medium described above. More than 95% of the cell population differentiated into endoderm-like cells, in which Oct3/4 expression was lost and Dab2 and GATA4 expression was induced.
Culture and generation of embryoid bodies.
ES cells were cultured in suspension in nonadhesive plates to form cell aggregates/embryoid bodies. The details and time courses have been calibrated previously to form embryoid bodies with evenly distributed sizes that are comparable to cross sections of an E5.5 embryo (29, 33). Typically, 6 × 106 cells in suspension following dissociation with trypsin and EDTA in 10 ml of ES cell medium lacking LIF were used to initiate the embryoid body cultures. The medium was replenished after 2 days by collecting the spheroids with a brief centrifugation. Chimeric spheroids were prepared by mixing equal numbers (3 × 106 cells in 5 ml) of dissociated ES cells, where one population was either undifferentiated or previously differentiated. For histochemical analysis, cell aggregates were fixed in formalin, washed twice with phosphate-buffered saline (PBS), mixed with 3% low-melting-point agarose, and processed for embedding in paraffin. The samples were subsequently subjected to sectioning, staining, and analysis.
Cell attachment assay to Matrigel.
Attachment of β1 integrin-positive and -negative ES cells to a basement membrane was assayed based on a published protocol (37). Cells were harvested by trypsinization, the trypsin was inactivated by 10% fetal bovine serum (FBS), and the cells were plated in triplicate at equal densities (105 cells in 0.1 ml of DMEM) in 96-well plates coated with Matrigel. The cells were allowed to attach in a tissue culture incubator at 37°C and 5% CO2. At the indicated times, the wells were washed twice with cold PBS, fixed with ice-cold methanol for 10 min, and stained with 0.5% crystal violet (in 25% methanol) at room temperature for 10 min. The fixed cells were washed in double-distilled water (ddH2O) until the rinse contained no color. Fixed, stained cells were examined using a microscope; at least 4 representative fields were counted for each well. Results are expressed as the mean ± standard deviation.
Dispase treatment of embryoid bodies.
The β1 integrin-heterozygous G119 and -null G201 ES cells were grown in suspension cultures for 5 days to form embryoid bodies. For the last 2 days, dispase (BD Biosciences, Bedford, MA) or solvent carrier control was added into the culture. The dispase is a neutral protease that can degrade extracellular matrix (particularly basement membrane) efficiently and is often used for the gentle dissociation of epithelial tissues in cultures (46). Various dosages of dispase were tested to optimize the removal of basement membrane without damaging cells prior to deciding on using 1 U/ml for subsequent experiments.
Western blotting and antibodies.
Cells or spheroids were used for Western blotting. The secondary antibodies were horseradish peroxidase (HRP)-conjugated goat or mouse anti-rabbit or anti-mouse antibody (Bio-Rad; Jackson Immunolab; Zymed). SuperSignal West extended-duration substrate (Pierce) was used for chemiluminescence detection of protein bands.
Primary mouse monoclonal antibodies used included anti-E-cadherin (BD Transduction Labs; catalog no. 610181), mouse anti-Dab2 (BD Transduction Labs; catalog no. 610465), rabbit anti-Dab2 (in-house rabbit polyclonal antibody) (31), anti-N-cadherin (BD Transduction Labs; catalog no. 610920), anti-β-actin (Sigma; catalog no. A5441), pan-antilaminin (Sigma; catalog no. L9393), antifibronectin (Sigma; catalog no. F3648), anti-β1 integrin (BD Transduction Labs; catalog no. 610467), anti-GATA4 (Santa Cruz Biotechnology; catalog no. sc-1237 and sc-9053), anti-GATA6 (in-house rabbit polyclonal antibody) (18), anti-Oct3/4 (Santa Cruz Biotechnology; catalog no. sc-5279), anti-Nanog (Calbiochem; catalog no. SC1000), and antimegalin (Santa Cruz Biotechnology; catalog no. sc-16478). The above-mentioned antibodies were also used for immunofluorescence microscopy and immunohistochemistry.
Immunohistochemistry and immunofluorescence.
Embryos and spheroids were fixed with formalin, paraffin embedded, and sectioned at 5-μm thickness. Slides were deparaffinized in xylene, hydrated in a graded ethanol series, washed in water, and boiled in antigen retrieval solution (DakoCytomation). For frozen sections, embryoid bodies and embryos were fixed in 4% paraformaldehyde in PBS, preserved in 30% sucrose, embedded in OCT, and frozen. Sections were cut in 5-μm thicknesses using a cryostat. Following incubation with primary antibodies, corresponding species-specific secondary antibodies were applied. For immunohistochemistry, the secondary antibody was HRP conjugated (Vector Laboratories, CA) and was detected by a diaminobenzidine (DAB) peroxidase substrate kit (DakoCytomation). For immunofluorescence, multiple secondary antibodies conjugated with the appropriate Alexa fluorochrome were used for simultaneous imaging of multiple antigens. DAPI (4′,6-diamidino-2-phenylindole) solution was used as a generic nuclear counterstain and applied at the terminal stage of the procedure. Images were merged using Adobe Photoshop.
Phase-contrast, wide-field epifluorescence, and laser scanning confocal microscopy.
Conventional wide-field microscopy was performed with an inverted Zeiss AxioObserver Z1 microscope operated by Axio Vision 4.8 software. Objectives used were Plan-Apochromat 63× (oil immersion, numerical aperture [NA] of 1.4) and A-Plan ×10 (NA of 0.25). Images were acquired digitally with a monochrome Zeiss AxioCam MRm charge-coupled device (CCD) camera. Confocal imaging was performed with a Zeiss LSM510/uv Axiovert 200M inverted, laser scanning confocal microscope operated by Zeiss LSM software. Objectives used included Plan-Apochromat 63×, Plan-Neofluar 40× (oil immersion, NA of 1.3), and Plan-Neofluar 25× (water immersion, NA of 0.8).
For live-cell imaging, embryoid bodies were resuspended in media buffered with 10 mM HEPES, pH 7.4, and imaged in a glass-bottom microwell dish (MatTek Corporation, MA) with the 25× water-immersion lens.
Transmission electron microscopy.
Embryoid bodies were harvested from suspension culture and immediately processed by the institutional electron microscopy facility according to standard protocol. Briefly, the samples were initially immersed in 2% glutaraldehyde, 100 mM sucrose, 0.5 M phosphate buffer at pH 7.3 and subsequently in 2% osmium tetroxide in 1 M phosphate buffer. After a graded dehydration, the embryoid bodies were embedded by infiltration with Epon/Araldite resin (Electron Microscopy Sciences, Fort Washington, PA) and an overnight polymerization at 64°C. Gold/silver thin sections were cut with a Diatome diamond knife using a Leica Ultracut R ultramicrotome and then mounted on copper grids and contrasted with 4% uranyl acetate and 0.25% lead citrate. The sections were examined, and images were captured in vacuo with electrons accelerated at 60 kV and focused using the magnetic lens of a Philips CM10 transmission electron microscope.
RESULTS
Implanted β1 integrin-null blastocysts are competent in primitive endoderm differentiation but fail in morphogenesis.
β1 integrin is essential for early embryogenesis in mice, but the mechanism for the developmental failure of the knockout embryos is uncertain (34, 47). We analyzed the role and mechanism of β1 integrin in early embryogenesis in additional detail using both mutant embryos and embryoid bodies. Since previously it was reported that β1 integrin-null embryos show no particular phenotype prior to implantation but exhibit overt aberrant morphology at E5.5 (34, 35), we first analyzed newly implanted blastocysts at E4.5. The E4.5 embryos in utero from matings between β1 integrin-heterozygous mutant mice were sectioned and stained for markers, Dab2 for the PrE (30, 31) and Oct3/4 for the epiblast (48–50) (Fig. 1A). We were able to genotype the flushed-out blastocysts but could not perform PCR genotyping on the implanted blastocysts processed for histology. All implanted blastocysts harvested from 5 uteri comprise both Dab2- and Oct3/4-positive cells, and we estimated that 5 out of the 22 embryos were β1 integrin deficient based on their characteristic morphology. In these putative mutant embryos, the Dab2-positive PrE cells stagger in multiple layers adjacent to a cluster of Oct3/4-positive epiblast cells (Fig. 1A, upper right panel), whereas in wild-type embryos the PrE cells form a single epithelial layer covering a group of the Oct3/4-positive cells (Fig. 1A, upper left panel). Therefore, we conclude that the β1 integrin gene is not required for the lineage commitment to either PrE or epiblast in the implanted blastocysts.
FIG 1.

Segregation of primitive endoderm from inner cell mass in β1 integrin-deficient embryos. (A) E4.5 and E5.5 embryos in utero from timed matings between β1 integrin-heterozygous mutant parents were analyzed by immunofluorescence microscopy for the PrE marker Dab2 (red) and the epiblast marker Oct3/4 (green). Merged images of Dab2, Oct3/4, and nuclei labeled by DAPI (blue) are shown. Representative morphologically normal (WT) E4.5 and E5.5 embryos show a Dab2-positive PrE monolayer covering the Oct3/4-positive inner cell mass. A representative E4.5 mutant [Intb1 (−/−)] embryo shows that Dab2-positive PrE cells form multiple layers situated next to the Oct3/4-positive ICM. By E5.5, Dab2-positive extraembryonic endoderm cells segregate away from the Oct3/4-positive epiblast cells in the presumptive β1 integrin-deficient [Intb1 (−/−)] embryos. (B) Sequential sections from a pair of normal (WT, for β1 integrin wild-type or -heterozygous) and abnormal [β1 integrin-null, Intb1 (−/−)] E5.5 embryos were analyzed by immunofluorescence microscopy for the endoderm marker Dab2 (red) and epiblast marker Oct3/4 (green) and for nuclei labeled by DAPI (blue). The schematic illustration shows the tissue structure and the segregation of extraembryonic endoderm from embryonic ectoderm in the β1 integrin-null embryo. (C) E5.5 embryos in utero from matings between β1 integrin-heterozygous mutant parents were analyzed by immunofluorescence microscopy for laminin and Dab2. A representative morphologically normal (WT) E5.5 embryo shows a concentric monolayer of Dab2-positive (green) extraembryonic endoderm enveloping the central epiblast. The parietal endoderm cells are positive for laminin staining (red, arrow), and a thin ring of laminin-positive basement membrane is present (arrowhead) between the endoderm and epiblast. A representative morphologically abnormal [Intb1 (−/−)] embryo shows a patch of the Dab2-positive cells, some of which are also positive for laminin. No epithelial organization is recognizable in this presumptive β1 integrin-deficient embryo.
The defect in β1 integrin-deleted embryos becomes obvious 1 day later at E5.5, in which the PrE cells segregate from the epiblast cells (Fig. 1A, lower right panel). Although we were able to dissect both wild-type and mutant E5.5 embryos to confirm genotypes, the dissected mutant embryos no longer had a cohesive structure that was suitable for histology analysis. Thus, we resolved to analyze the undisturbed mutant embryos in situ following fixation and sectioning of the uterine horns. In wild-type E5.5 embryos, the Dab2-positive endoderm layer appears as a concentric rim surrounding the core of Oct3/4-positive epiblast cells. However, in the β1 integrin-null embryos, both Dab2- and Oct3/4-positive cells are present but segregate into clusters, as shown in a representative slide (Fig. 1A, lower right panel). An example comparing sequential sections of wild-type and mutant embryos stained with Dab2 and Oct3/4 is shown in Fig. 1B and illustrated schematically. Additionally, in the wild-type E5.5 embryos, intense laminin immunostaining shows a layer of Reichert's membrane with parietal endoderm cells embedded (arrow) and a weaker layer of basement membrane (arrowhead) lying under the visceral endoderm (Fig. 1C). In β1 integrin-null E5.5 embryos, laminin staining overlaps some of the Dab2-positive extraembryonic endoderm cells (Fig. 1B, lower panels); however, no distinct basement membrane layer can be observed in the mutant embryos. Therefore, β1 integrin is required for the attachment of the PrE to the epiblast and morphogenesis of PrE epithelial monolayer and associated basement membrane in early embryonic development.
β1 integrin-null ES cells undergo normal primitive endoderm differentiation in vitro but segregate in embryoid bodies.
To further analyze the defect in early embryogenesis of β1 integrin-knockout embryos, we investigated the properties of β1 integrin-null ES cells in culture. A β1 integrin-heterozygous ES line was used as a control; the heterozygous line is phenotypically indistinguishable from the wild type and was designated wild-type control (34). Upon treatment with all-trans-retinoic acid (RA), β1 integrin-null ES cells undergo endoderm differentiation to an extent comparable to that of wild-type controls, as judged by the induction of Dab2, laminin, Gata4, and Gata6 and the loss of Oct3/4 (Fig. 2A). Laminin proteins show no discrete size, likely because the secreted matrix proteins form polymers and are subjected to cross-linking and proteolytic processing. Binding to Matrigel was used as an assay to quantify cell binding affinity to basement membrane. As expected, β1 integrin-null ES cells have a reduced affinity for Matrigel (Fig. 2B). Attachment of undifferentiated wild-type ES cells to Matrigel reached maximum by 1 h, whereas minimal binding by β1 integrin-null cells occurred after 2 h of incubation. Differentiation slightly reduces the affinity of wild-type ES cells for Matrigel; in contrast, differentiation increases binding of β1 integrin-null ES cells. Interestingly, upon differentiation and a longer incubation, β1 integrin-null ES cells bind Matrigel to an extent similar to that of the wild-type cells (Fig. 2B). Thus, the assay suggests that β1 integrin contributes to the majority of the attachment of ES cells to Matrigel or basement membrane; however, upon differentiation, additional cell surface receptors can compensate and facilitate binding to Matrigel, though with delayed kinetics.
FIG 2.

Differentiation of β1 integrin-null ES cells in vitro. (A) β1 integrin-heterozygous G119 [Intb1 (+/−)] (designated WT) and the null G201 [Intb1 (−/−)] ES cells in monolayer culture were differentiated by treatment with 1 μM retinoic acid (RA) for 7 days. Cell lysates were assessed for PrE differentiation by Western blotting for β1 integrin, Dab2, GATA6, GATA4, Oct3/4, Nanog, laminin, and actin. (B) The differentiated and undifferentiated cells were assayed for their adhesive affinity to Matrigel. Upon incubation for 30, 60, and 120 min, the plates were washed and the numbers of remaining bound cells were determined. (C) Both the G119 (β1 integrin-heterozygous) and G201 (β1 integrin-null) cells were cultured in suspension for 4 days to produce embryoid bodies. Representative phase-contrast images of embryoid bodies are shown. (D) The embryoid bodies were harvested and subjected to histology analysis. Representative images at low and high magnification of the embryoid bodies stained for Dab2 are shown.
By maintaining the ES cells in suspension culture to form embryoid bodies, we were able to reproduce the endoderm-epiblast segregation phenotype in vitro (Fig. 2C and D). Unlike embryoid bodies derived from wild-type (or heterozygous) control ES cells, which typically have a smooth surface, embryoid bodies derived from β1 integrin-deficient ES cells often contain one or several clusters of cells located on the surface or scattered around the core of the embryoid bodies (Fig. 2C). By day 4 in suspension, a superficial endoderm layer forms in most of the cell aggregates derived from wild-type control ES cells, while embryoid bodies derived from β1 integrin-null ES cells lack an outer layer (Fig. 2C). By immunohistochemistry, the outer shell of wild-type embryoid bodies was confirmed to be the Dab2-positive extraembryonic endoderm (Fig. 2D). Small aggregates protruded from the surface or scattered around the cores of β1 integrin-null embryoid bodies are Dab2 positive (Fig. 2D, indicated by asterisks). Apparently, endoderm differentiation occurred in β1 integrin-null embryoid bodies, but rather than forming a concentric surface epithelial layer, the endoderm cells clustered and defoliated from the cores of the spheroids. Thus, the endoderm-epiblast segregation phenotype in embryos can be reproduced in vitro with β1 integrin-null embryoid bodies.
Segregation of primitive endoderm is caused by the defective basement membrane deposition in β1 integrin-null embryoid bodies.
We next examined several molecular markers to decipher the potential mechanism responsible for the segregation of the outer endoderm from the inner epiblast cells. In wild-type embryoid bodies, the superficial Dab2-positive endoderm layer is polarized, as indicated by the apical distribution of the transmembrane receptor megalin and the complementary basolateral localization of the cell adhesion molecule E-cadherin (Fig. 3A). E-cadherin expression is relatively even throughout the spheroid, and expression levels are similar in both the endoderm and epiblast cells (Fig. 3A). On the Dab2-positive endoderm aggregates that protrude from the surface of β1 integrin-null embryoid bodies, megalin also is apical and polarized, and E-cadherin expression is contiguous and basolateral (Fig. 3A). The obvious difference appears to be the deposition of the laminin-containing basement membrane (Fig. 3B). Laminin staining marks endoderm cells and a thin layer of basement membrane partitioning the endoderm from the epiblast. However, in β1 integrin-deficient embryoid bodies, a diffuse laminin staining is present at the junction between endoderm and epiblast cells (Fig. 3B). Additionally, the delaminated clusters of Dab2-positive cells often exhibit laminin-positive matrix materials embedded at their center (Fig. 3C). An informative example of laminin deposition between segregating Dab2-positive endoderm cells and the core aggregate is shown in Fig. 3D.
FIG 3.

Characterization of embryoid bodies/spheroids. (A) Histological sections of 4-day embryoid bodies were analyzed by immunofluorescence microscopy for Dab2 (red), E-cadherin (green), megalin (white), and DAPI (blue). Representative examples are shown for embryoid bodies derived from β1 integrin-heterozygous (designated WT) or -null [Intb1 (−/−)] ES cells. (B) The embryoid bodies were analyzed by immunofluorescence microscopy for Dab2 (green) and laminin (red). Images from representative embryoid bodies are shown. (C) Two examples of small aggregates (presumably detached from the cores of embryoid bodies) among embryoid bodies derived from β1 integrin-null ES cells are stained for Dab2 (green) and laminin (red). (D) An example is shown for laminin deposition between a cluster of Dab2-positive endoderm cells in the process of detaching from the core aggregate of β1 integrin-deficient embryoid body. The sections were analyzed by immunofluorescence microscopy for Dab2 (green), laminin (red), and DAPI (blue).
Thus, the in vitro studies suggest that laminin synthesis occurs in the endoderm of both β1 integrin-positive and -deficient cells. Laminin is secreted and deposited; however, a continuous basement membrane layer forms only in the wild type, whereas the matrix materials appear irregularly distributed in β1 integrin-deficient embryoid bodies.
We next examined the presence of basement membrane in the embryoid bodies using transmission electron microscopy (Fig. 4). In mature (4- to 7-day-old) wild-type embryoid bodies, a basement membrane lies underneath the endoderm layer and separates the outer first layer of cells from the inner core (arrow, Fig. 4A). In β1 integrin-deficient embryoid bodies, usually no basement membrane is present between the first outer layer and the inner core of cells (Fig. 4B). This observation is similar to that reported previously in a study of β1 integrin-deficient embryoid bodies also using transmission electron microscopy (51). However, most of the surface cells of β1 integrin-deficient embryoid bodies lack microvilli, indicating that these cells, though positioned on the surface, are not differentiated endoderm. In rare cases, microvillus-decorated cells are present at the surface of β1 integrin-deficient embryoid bodies (Fig. 4C). These β1 integrin-deficient endoderm cells exhibit a dilated endoplasmic reticulum, indicating active synthesis and secretion. A basement membrane structure is also present (arrow, Fig. 4C), though the extracellular matrix materials appear to associate only loosely with either the endoderm or the inner cells.
FIG 4.
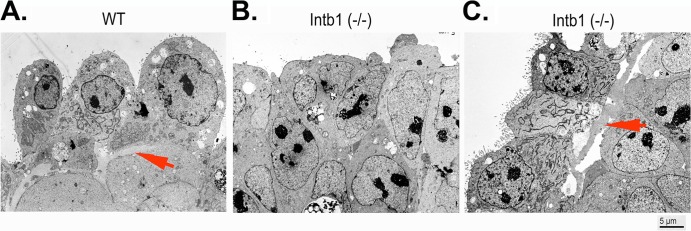
Analysis of embryoid bodies/spheroids using electron microscopy. Embryoid bodies (5 days in suspension culture) were analyzed by transmission electron microscopy. (A and B) Representative images are shown for surface layers of cells for wild-type (WT) (arrow indicates basement membrane) (A) and for β1 integrin-null [Intb1 (−/−)] (B) cells. (C) In some surface areas of the β1 integrin-null embryoid bodies, microvillus-containing endoderm cells are present, and loosely attached basement membranes are observable (arrow indicates basement membrane).
Thus, our results suggest that β1 integrin-deficient cells are able to secrete and assemble a basement membrane but that the cells have greatly reduced binding affinity and adhere poorly to the polymers.
Restricted deletion in chimeric embryoid bodies shows differential requirement of β1 integrin in epiblast and primitive endoderm for morphogenesis.
Further, we used chimeric embryoid bodies to determine the relative contribution of β1 integrin from the endoderm compartment compared to that from the epiblast. Previously, combinations of differentiated and undifferentiated ES cells in chimeric embryoid bodies have been implemented to analyze cell sorting and the genesis of an endoderm layer (29, 33). Here, we mixed monodispersed, differentiated and undifferentiated, and wild-type (or β1 integrin-heterozygous) or β1 integrin-deficient ES cells to form embryoid bodies and analyze the resulting cell sorting patterns. The mixed cells were cultured in suspension for 2 days, a time adequate for cells to sort but insufficient for spontaneous endoderm differentiation to occur, which requires a longer duration (5 to 7 days) (29, 33). A line of GFP-labeled ES cells (CFG37) was used as a wild-type control. In a combination of exclusively undifferentiated cells, i.e., “GFP + WT” (GFP-labeled wild type [GFP] plus β1 integrin heterozygous [designated WT here]) or “GFP + B(−/−)” (GFP plus β1 integrin null), the resulting embryoid bodies show a random amalgam of cells in a checkerboard fashion (Fig. 5A, left upper and lower panels). This indicates that loss of one or both copies of β1 integrin does not affect cell-cell interaction and that the cells are randomly distributed in the aggregates. As positive controls, the differentiated cells of either the β1 integrin-heterozygous line (designated WT) (as shown in Fig. 5A, “GFP + WT:RA”) or the wild type (in “GFP:RA + WT”) sort to the periphery and form a superficial endoderm layer (Fig. 5A, upper middle and right panels), consistent with our previous conclusion that differentiated endoderm cells polarize and position to the outer layer (29).
FIG 5.

Analysis of chimeric embryoid bodies derived from β1 integrin-positive and -deficient ES cells. Wild-type ES cells expressing GFP (GFP), with or without differentiation by retinoic acid (1 μM for 7 days), were mixed with the β1 integrin-heterozygous G119 (designated “WT”) and the null G201 [B1(−/−)] ES cells with or without differentiation. The chimeric cell mixtures were allowed to aggregate and sort in suspension culture for 2 days. The time period was adequate for cell sorting but insufficient to induce spontaneous differentiation (29, 33). (A) The live chimeric embryoid bodies formed were analyzed for cell sorting patterns. Confocal GFP images of embryoid bodies in the middle section are shown. “GFP + B1(−/−):RA” refers to GFP-labeled wild-type ES cells mixing with β1 integrin-null ES cells that have been differentiated with retinoic acid. “WT” refers to unlabeled G119 β1 integrin-heterozygous ES cells. (B) A representative image of a chimeric embryoid body consisting of GFP-labeled wild type (GFP) mixed with retinoic acid-differentiated β1 integrin-heterozygous (WT:RA) ES cells that was stained for laminin (red), GFP (green), and DAPI (blue). Overlaying GFP and DAPI produces a pale blue color. (C) The embryoid bodies were produced from a mixture of the undifferentiated GFP-labeled and the differentiated β1 integrin-null ES cells [GFP + B1(−/−):RA]. Two examples show confocal images of GFP (green), laminin (red), and DAPI (blue). (D) Embryoid bodies were produced from a chimeric mixture of the differentiated GFP-labeled wild-type and the undifferentiated β1 integrin-null ES cells [GFP:RA + B1(−/−)]. Two examples of confocal images of GFP (green), laminin (red), and DAPI (blue) are shown.
Somewhat unexpectedly, differentiated β1 integrin-deficient cells are able to sort and form an outer layer enveloping wild-type ES cells [Fig. 5A, “GFP + B1(−/−):RA,” middle lower panel], and to a lesser extent, differentiated wild-type ES cells can also sort and form a partial layer outside a core of β1 integrin-null cells [Fig. 5A, “GFP:RA + B1(−/−),” right lower panel]. In control chimeras composed of two wild types (or β1 integrin-heterozygous cells), of differentiated and undifferentiated constituents, typically a continuous laminin-positive basement membrane is sandwiched between the outer endoderm and the inner epiblast cells, as shown in a representative example (Fig. 5B). In embryoid bodies composed of a β1 integrin-deficient endoderm and a wild-type core [GFP + B1(−/−):RA], a basement membrane is also observed in some aggregates, but the attached β1 integrin-deficient endoderm cells aggregate into multiple layers instead of forming a monolayered epithelium (Fig. 5C, left panel). In other embryoid bodies, a well-formed basement membrane is present on the outer shell of the wild-type core, but no endoderm cells reside on top (Fig. 5C, right panel, arrow). Presumably, the β1 integrin-deficient endoderm cells detach due to compromised binding to the basement membrane. In another chimeric combination, differentiated GFP-labeled wild-type cells segregate from unlabeled β1 integrin-null cells without forming a concentric outer layer (Fig. 5A, lower right panel). However, some of these embryoid bodies also exhibit a partial outer endoderm layer (Fig. 5A, lower right panel). Upon closer examination, we found that the surface endoderm layer of these embryoid bodies either lacks a basement membrane between the endoderm and epiblast (Fig. 5D, left panels) or has a basement membrane where wild-type (GFP-labeled) cells reside on both sides (Fig. 5D, right panels). Probably, a small percentage of undifferentiated cells among the majority of differentiated wild-type ES cells participate in the formation of the basement membrane. These results suggest that the presence of β1 integrin in the core epiblast cells is essential for the formation of the primitive endoderm layer, since β1 integrin in the epiblast compartment is critical for the retention of a continuous basement membrane.
β1 integrin in epiblast has critical role for primitive endoderm morphogenesis shown by Ttr-Cre- or Sox2-Cre-mediated deletion.
To analyze the role and relative importance of β1 integrin in endoderm and epiblast in early embryonic germ layer patterning in vivo, we designed experiments to delete the β1 integrin gene in endoderm using Ttr-Cre (44) or in the epiblast using Sox2-Cre (45). Transthyretin (Ttr) is expressed in cells of the visceral yolk sac and fetal liver in developing embryos, and the expression pattern and timing of the promoter as well as the Ttr-Cre mice have been characterized in detail previously (44). Sox2-Cre can be used to efficiently delete a floxed gene in the epiblast without significantly affecting extraembryonic endoderm (32, 45). The cell-restricted β1 integrin gene deletion in embryos was achieved by crossing β1 integrinfl/fl females with β1 integrindf/+; Sox2-Cre or β1 integrindf/+; Ttr-Cre males. E5.5 and E9.5 embryos from timed matings were analyzed.
For deletion using Ttr-Cre, from 12 E5.5 embryos that we examined in detail, two embryos were found slightly abnormal and all 10 other embryos were indistinguishable from the wild-type control. One of these two abnormal embryos is shown (Fig. 6B, left panel), compared to a wild-type control (Fig. 6A, left panel). This β1 integrin-deleted (by Ttr-Cre) embryo shows a mislocalized, Dab2-positive endoderm cell in the interior of the embryo (Fig. 6B). However, the visceral endoderm appeared intact and polarized, as shown by normal megalin and E-cadherin staining (Fig. 6B, right panel), compared to wild-type control (Fig. 6A, middle panel). The abnormal morphology of mutant embryos was apparent by the E9.5 stage, with 4 out of 17 (29%) embryos found grossly defective (Fig. 6D, middle panel). These 4 deformed embryos were confirmed by PCR genotyping to be β1 integrinfl/df; Ttr-Cre, and the rest of the 10 morphologically normal embryos were either wild type or heterozygous for β1 integrin.
FIG 6.

Deletion of β1 integrin in the extraembryonic endoderm by Ttr-Cre and the epiblast by Sox2-Cre. E5.5 and E9.5 embryos were harvested from matings between male β1 integrin+/df; Ttr-Cre and female β1 integrinfl/fl mice. The E5.5 embryos were sectioned and stained for Dab2 (green), laminin (red), and DAPI (blue). The E9.5 embryos were dissected, photographed, and genotyped by PCR. Representative examples of β1 integrin-heterozygous (fl/df), Ttr-Cre-mediated deletion in extraembryonic endoderm and Sox2-Cre-mediated deletion in epiblast are shown. (A) Wild-type [Intb1 (fl/df)] E5.5 embryos were sectioned and stained with Dab2, laminin, and DAPI; E-cadherin, megalin, and DAPI; and Oct3/4, Dab2, and DAPI. (B) Mutant (β1 integrinfl/df; Ttr Cre) E5.5 embryos were sectioned and stained with Dab2, laminin, and DAPI and E-cadherin, megalin, and DAPI. (C) Mutant (β1 integrinfl/df; Sox2 Cre) E5.5 embryos were sectioned and stained with Dab2, laminin, and DAPI and Oct3/4, Dab2, and DAPI. (D) Representative E9.5 embryos of β1 integrinfl/df, β1 integrinfl/df; Ttr-Cre, and β1 integrinfl/df; Sox2-Cre types are shown.
For deletion using Sox2-Cre, in 16 E5.5 embryos that we examined in detail, 4 (25%) embryos were found defective and all 12 other embryos were indistinguishable from the wild-type control. The abnormal E5.5 embryos are very similar in the degree of deformation to those of constitutive β1 integrin-knockout embryos, showing segregation of the PrE from the epiblast (Fig. 6C, right panel) and the clustering of Dab2-positive PrE cells and associated laminin (Fig. 6C, right panel). Nevertheless, the conditional mutants still persisted and were genotyped by PCR as β1 integrinfl/df; Sox2-Cre at E9.5. Among the 28 embryos, the 6 (21%) deformed embryos were confirmed to have a β1 integrin deletion in the epiblast (Fig. 6D, right panel).
These results support the notion that β1 integrin in the epiblast plays a more crucial role than β1 integrin in the PrE cells for patterning of extraembryonic endoderm at the E4.5 to E5.5 stages. However, β1 integrin in both the extraembryonic endoderm and the epiblast is ultimately essential for further normal development and gastrulation.
Effacement of basement membrane by dispase prevents primitive endoderm detachment from inner cells in β1 integrin-deficient embryoid bodies.
The segregation of endoderm cells from the core of the β1 integrin-deficient embryoid bodies suggests that the formation of basement membrane prevents E-cadherin-mediated cell-cell adhesion and leads to the detachment of the endoderm cells. To test this model, we included dispase (43) in the cell culture medium during the formation of embryoid bodies to digest the secreted extracellular basement membrane materials. In β1 integrin wild-type (or heterozygous) embryoid bodies, the surface endoderm layer was only slightly less contiguous upon dispase treatment than were untreated controls (Fig. 7A, left panel). A typical basement membrane was observed in untreated embryoid bodies (Fig. 7C, left panels) but was absent in those with dispase treatment (Fig. 7C, right panels). In these cells, the laminin staining was cytoplasmic, which was inaccessible to dispase cleavage. In embryoid bodies derived from β1 integrin-deficient ES cells, typically small Dab2-positive endoderm cell aggregates detached or protruded from the surface of the cores (Fig. 7B, left panel). Upon dispase treatment during the formation of cell aggregates, the β1 integrin-deficient embryoid bodies showed no detached small endoderm aggregates, and patches of endodermal monolayers resided on the surface of the embryoid bodies (Fig. 7B, right panel). Dispase treatment eliminated most of the laminin-containing basement membrane materials (Fig. 7D).
FIG 7.

Removal of basement membrane by dispase prevents detachment of primitive endoderm in β1 integrin-deficient embryoid bodies. The β1 integrin-heterozygous G119 (designated WT) and -null G201 [Intb1 (−/−)] ES cells were grown in suspension cultures for 5 days to form embryoid bodies. For the last 2 days, dispase (1 U/ml) or solvent carrier control was added into the culture. (A) Dab2 (green) immunofluorescence was overlaid on phase-contrast images shown for the β1 integrin-heterozygous embryoid bodies. (B) Dab2 (green) immunofluorescence was superimposed on phase-contrast images of the β1 integrin-null embryoid bodies. (C) Images of immunofluorescence microscopy for Dab2 (green), laminin (red), and DAPI (blue) of a representative β1 integrin-heterozygous (designated WT) embryoid body, either with or without dispase treatment, are shown. (D) Images of immunofluorescence microscopy for Dab2, laminin, and DAPI of a representative β1 integrin-null [Intb1 (−/−)] embryoid body, either with or without dispase treatment, are shown.
Thus, interference with basement membrane formation by dispase prevents the detachment of the differentiating endoderm from the epiblast in β1 integrin-deficient embryoid bodies. We conclude that the secretion and formation of basement membrane material displace E-cadherin-mediated interaction between the endoderm and the epiblast cells. As the deposited basement membrane material creates a physical gap between cells and prevents E-cadherin-mediated cell adhesion, the β1 integrin-deficient epiblast core fails to bind and retain a basement membrane, and the β1 integrin-deficient endoderm outer layer detaches and segregates.
DISCUSSION
In this study, we revisited the embryonic phenotype of β1 integrin-knockout mice and determined the mechanism for the developmental failure of the mutant embryos. By immunostaining implanted blastocysts, we found that abundant PrE differentiation occurs in E4.5 and E5.5 β1 integrin-deficient embryos. However, the PrE cells segregate from the epiblast in the mutant embryos, instead of forming a monolayer epithelium covering the inner epiblast. From additional in vitro analysis using embryoid bodies, we conclude that β1 integrin-deficient PrE cells form but fail to attach to and then segregate from the epiblast. A basement membrane forms, but the β1 integrin-deficient inner core cells fail to retain the extracellular matrix and the bound endoderm epithelium. Thus, β1 integrin is essential for the attachment of the PrE to the epiblast through binding to a basement membrane located between the PrE and epiblast. These studies of the β1 integrin-deficient embryonic phenotype provide new understanding of the development of the PrE.
Sorting and positioning of primitive endoderm cells on the surface.
Observations in both embryos and embryoid bodies lead to the conclusion that the initial differentiation of PrE occurs randomly in the ICM (15, 33). The cells of the ICM undergo active and dynamic movements and sort to the surface to form the PrE layer (19–21, 25). The force underlying the sorting of PrE cells is not differential adhesive affinity (29), which has been proposed to explain the segregation and sorting of embryonic germinal layers (28, 52, 53). Instead, the surface sorting and positioning of PrE cells are due to the ability of the endoderm cells to establish an apical polarity (29). Consistent with this idea, the glycoprotein megalin is induced early in differentiating endoderm cells and achieves an apical localization when the cells reach the surface to form an epithelium (54). Furthermore, loss of polarity in Dab2-deleted embryos leads to intermixing and failure of the PrE cells to anchor at the surface (30–32). Dab2-mediated directional trafficking of endocytic cargos is important in establishing the polarized distribution of megalin to and E-cadherin away from the apical surface of the endoderm epithelial cells, thus creating a nonadhesive apical edge (31).
One potential explanation for the failure of PrE surface positioning in β1 integrin-deficient embryos is a lack of polarization, since β1 integrin is known to participate in orienting epithelial polarity (55, 56). However, we observed that β1 integrin-deficient endoderm cells sort to the surface and are polarized as indicated by the apical distribution of megalin.
Accompanying differentiation, the PrE cells in the interior begin to express basement membrane components such as laminin and collagen IV prior to reaching the surface (27). The formation of a basement membrane appears to be a means of stabilizing the organization and surface positioning of PrE cells upon their sorting to the surface. Presumably, when the PrE cells reach the surface, the secreted laminin and collagen IV proteins assemble into an insoluble basement membrane polymer. This extracellular barrier prevents further E-cadherin-mediated binding and intermixing of PrE with epiblast cells and stabilizes the mature extraembryonic endoderm layer. Instead of cadherin-mediated cell adhesion, the association of the PrE epithelial layer and the epiblast is mediated by their mutual binding to a shared basement membrane between the two cell types.
Transition from cadherin- to integrin-mediated attachment of primitive endoderm.
The analysis of β1 integrin-deficient embryos and embryoid bodies reveals the time and mechanisms for the transition from cadherin-mediated cell-cell interactions to integrin-mediated cell-basement membrane binding during the process of PrE formation. In early-stage ES cell aggregates, cell-cell adhesion is mainly mediated by E-cadherin and N-cadherin, since E-cadherin-null ES cells have a greatly reduced mutual adhesion (29), and additional reduction of the N-cadherin gene copy further decreases ES cell adhesive affinity (R. Moore, unpublished observations). The current analysis suggests that a transition from cadherin-mediated adhesion to integrin-mediated anchoring between PrE and epiblast coincides with the formation of the PrE basement membrane at around the E4.5 stage.
The results suggest that the formation of basement membrane is the critical force that breaks cadherin-mediated cell-cell contact between the PrE and the epiblast beneath (Fig. 8). The demonstration that removal of secreted basement membrane components by dispase prevents the detachment of PrE cells from core aggregates in the β1 integrin-deficient embryoid bodies strongly supports the critical role of basement membrane formation in the switch from cadherin- to integrin-mediated cell adhesion. Normally, upon deposition and formation of basement membrane, the epithelial structure is then maintained by mutual binding of PrE and the epiblast core cells to the opposite side of the same basement membrane sheet. At this stage, β1 integrin becomes essential for maintaining the PrE monolayer epithelial structure.
FIG 8.
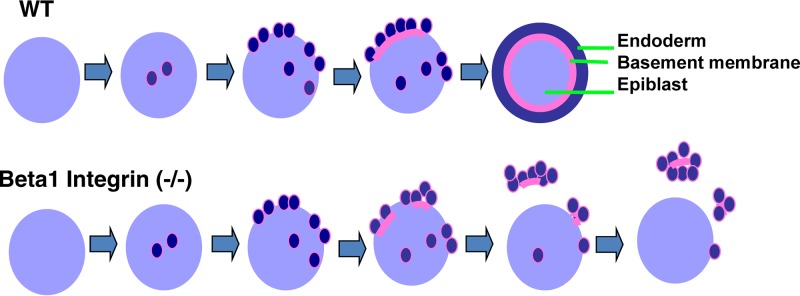
Model for the formation of primitive endoderm and the role of β1 integrin. PrE differentiation initiates randomly in both peripheral and central regions of the ICM, and subsequently, the differentiated cells sort and position on the surface. Laminin synthesis is induced upon differentiation. Basement membrane components, including laminin produced and secreted by the PrE cells, are deposited between the endoderm and epiblast compartment. The formation of a basement membrane drives the conversion of E-cadherin-mediated endoderm-epiblast adhesion to β1 integrin-mediated basement membrane anchorage. In β1 integrin-null embryos, strong cadherin-mediated adhesion between PrE cells and their weak binding to the basement membrane lead to the segregation and detachment of PrE cells from the core.
Mechanism of basement membrane formation and the critical requirement of β1 integrin in the epiblast compartment.
The current analysis also provides insights into the formation of basement membrane and the roles of integrins. In this study, we conclude that β1 integrin-deficient ES cells are capable of PrE differentiation and subsequent synthesis and deposition of basement membrane components. It has been postulated that the soluble extracellular matrix components can assemble spontaneously into insoluble polymers to form a basement membrane (57, 58). Consistent with this model, in β1 integrin-deficient embryoid bodies, the differentiated and detached PrE cell aggregates contain basement membrane polymers as seen by immunofluorescence and electron microscopy, suggesting that self-assembly does not require β1 integrin. Rather, β1 integrin is important for binding and retaining the deposited basement membrane fragments adjoining the cell basal surface. In accord with this notion, in chimeric embryoid bodies of undifferentiated wild-type and differentiated β1 integrin-deficient cells, an apparently normal basement membrane layer forms and covers the β1 integrin-positive core, but no PrE cells reside on the outer side of the basement membrane (Fig. 5C). Presumably, the β1 integrin-deficient endoderm cells secrete the protein components to form a basement membrane bound by the β1 integrin-positive inner core cells. However, the β1 integrin-deficient endoderm cells then detach from the basement membrane due to weak binding, leaving an unoccupied basement membrane surface on the outside.
β1 integrin is essential for binding of the epiblast cells to the basement membrane. In the absence of β1 integrin, the basement membrane detaches from the inner epiblast core of embryoid bodies, carrying with it PrE cells that have acquired an increased affinity (over undifferentiated cells) to the matrix, presumably due to induction of additional integrin subtypes (42).
The critical importance of the β1 integrin requirement in the inner epiblast cells for the PrE organization is also supported by the phenotypes of the conditional deletion of β1 integrin. At the E5.5 stage, deletion of β1 integrin in the PrE cells (mediated by Ttr-Cre) produced nearly morphologically normal embryos; in contrast, deletion in the epiblast (mediated by Sox2-Cre) generated deformed embryos of severity similar to that of those from constitutive deletion of β1 integrin, showing segregation of PrE from epiblast. However, as a note of caution, Ttr-Cre is expressed at a slightly later stage (44), which likely contributes to a milder defective phenotype. Ttr-Cre would be effective only after the formation of the primitive endoderm epithelium, and thus, in these mosaic embryos, only the role of β1 integrin in the maintenance of the epithelium will be observed.
Model for the organization of primitive endoderm epithelium.
Based on this study, we propose a model for the development of PrE (illustrated in Fig. 8). As established previously, PrE differentiation occurs randomly within the ICM, where cell-cell adhesion is mediated by cadherins (19–21, 25). Upon differentiation, PrE cells produce laminin and other components of the basement membrane, sort to the surface, and deposit extracellular matrix components between the PrE and epiblast. The formation of basement membrane switches the adhesion between PrE and epiblast from cadherin- to integrin-mediated binding.
In β1 integrin-deficient embryos, the phenotype is shown here as physical segregation of PrE from the epiblast. Thus, β1 integrin is not essential for lineage differentiation; rather, its deletion leads to the segregation of PrE from epiblast in peri-implanted mouse embryos.
ACKNOWLEDGMENTS
We appreciate the gift of the β1 integrin-null and -heterozygous control ES cells from Reinhard Fässler via Lionel Larue (Institut Curie, France). We also appreciate the Ttr-Cre mice from Anna-Katerina Hadjantonakis (Memorial Sloan-Kettering Institute, New York, NY).We appreciate valuable comments from our colleagues, including Yue Meng, Santas Rosario, and James Hoy, for reading, suggestions, and comments on the manuscript. We are indebted to George T. McNamara (University of Miami Analytical Imaging Core Facility) for assistance with confocal microscopy and Margaret Bates (University of Miami Electron Microscope Core Facility) with transmission electron microscopy.
The work is supported by R01 CA095071, CA79716, and CA75389 to X.-X. Xu from NCI, NIH. R.M. is supported by the Sylvester Comprehensive Cancer Center and an American Cancer Society Institutional Research Grant (IRG-98-277-09).
Footnotes
Published ahead of print 25 November 2013
REFERENCES
- 1.Arnold SJ, Robertson EJ. 2009. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 10:91–103. 10.1038/nrm2618 [DOI] [PubMed] [Google Scholar]
- 2.Lu CC, Brennan J, Robertson EJ. 2001. From fertilization to gastrulation: axis formation in the mouse embryo. Curr. Opin. Genet. Dev. 11:384–392. 10.1016/S0959-437X(00)00208-2 [DOI] [PubMed] [Google Scholar]
- 3.Rossant J, Tam PP. 2009. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 136:701–713. 10.1242/dev.017178 [DOI] [PubMed] [Google Scholar]
- 4.Takaoka K, Hamada H. 2012. Cell fate decisions and axis determination in the early mouse embryo. Development 139:3–14. 10.1242/dev.060095 [DOI] [PubMed] [Google Scholar]
- 5.Martin GR. 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U. S. A. 78:7634–7638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans MJ, Kaufman MH. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292:154–156 [DOI] [PubMed] [Google Scholar]
- 7.Gardner RL. 1982. Investigation of cell lineage and differentiation in the extraembryonic endoderm of the mouse embryo. J. Embryol. Exp. Morphol. 68:175–198 [PubMed] [Google Scholar]
- 8.Gardner RL. 1983. Origin and differentiation of extraembryonic tissues in the mouse. Int. Rev. Exp. Pathol. 24:63–133 [PubMed] [Google Scholar]
- 9.Gardner RL. 1989. Cell allocation and lineage in the early mouse embryo. Ciba Found. Symp. 144:172–181 [PubMed] [Google Scholar]
- 10.Yamanaka Y, Lanner F, Rossant J. 2010. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development 137:715–724. 10.1242/dev.043471 [DOI] [PubMed] [Google Scholar]
- 11.Lanner F, Rossant J. 2010. The role of FGF/Erk signaling in pluripotent cells. Development 137:3351–6330. 10.1242/dev.050146 [DOI] [PubMed] [Google Scholar]
- 12.Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. 1995. Requirement of FGF-4 for postimplantation mouse development. Science 267:246–249. 10.1126/science.7809630 [DOI] [PubMed] [Google Scholar]
- 13.Goldin SN, Papaioannou VE. 2003. Paracrine action of FGF4 during periimplantation development maintains trophectoderm and primitive endoderm. Genesis 36:40–47. 10.1002/gene.10192 [DOI] [PubMed] [Google Scholar]
- 14.Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. 1998. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl. Acad. Sci. U. S. A. 95:5082–5087. 10.1073/pnas.95.9.5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chazaud C, Yamanaka Y, Pawson T, Rossant J. 2006. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev. Cell 10:615–624. 10.1016/j.devcel.2006.02.020 [DOI] [PubMed] [Google Scholar]
- 16.Cheng AM, Saxton TM, Sakai R, Kulkarni S, Mbamalu G, Vogel W, Tortorice CG, Cardiff RD, Cross JC, Muller WJ, Pawson T. 1998. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell 95:793–803. 10.1016/S0092-8674(00)81702-X [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Smedberg JL, Cai KQ, Capo-Chichi DC, Xu XX. 2011. Ectopic expression of GATA6 bypasses requirement for Grb2 in primitive endoderm formation. Dev. Dyn. 240:566–576. 10.1002/dvdy.22447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai KQ, Capo-Chichi CD, Rula ME, Yang DH, Xu XX. 2008. Dynamic GATA6 expression in primitive endoderm formation and maturation in early mouse embryogenesis. Dev. Dyn. 237:2820–2829. 10.1002/dvdy.21703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris SA, Teo RT, Li H, Robson P, Glover DM, Zernicka-Goetz M. 2010. Origin and formation of the first two distinct cell types of the inner cell mass in the mouse embryo. Proc. Natl. Acad. Sci. U. S. A. 107:6364–6369. 10.1073/pnas.0915063107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frankenberg S, Gerbe F, Bessonnard S, Belville C, Pouchin P, Bardot O, Chazaud C. 2011. Primitive endoderm differentiates via a three-step mechanism involving Nanog and RTK signaling. Dev. Cell 21:1005–1013. 10.1016/j.devcel.2011.10.019 [DOI] [PubMed] [Google Scholar]
- 21.Rossant J, Chazaud C, Yamanaka Y. 2003. Lineage allocation and asymmetries in the early mouse embryo. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358:1341–1348. 10.1098/rstb.2003.1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis AK. 2008. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 35:3081–3091. 10.1242/dev.021519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meilhac SM, Adams RJ, Morris SA, Danckaert A, Le Garrec JF, Zernicka-Goetz M. 2009. Active cell movements coupled to positional induction are involved in lineage segregation in the mouse blastocyst. Dev. Biol. 331:210–221. 10.1016/j.ydbio.2009.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris SA. 2011. Cell fate in the early mouse embryo: sorting out the influence of developmental history on lineage choice. Reprod. Biomed. Online 22:521–524. 10.1016/j.rbmo.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 25.Yamanaka Y. 2011. Response: cell fate in the early mouse embryo—sorting out the influence of developmental history on lineage choice. Reprod. Biomed. Online 22:525–527. 10.1016/j.rbmo.2011.03.011 [DOI] [PubMed] [Google Scholar]
- 26.Coucouvanis E, Martin GR. 1995. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 83:279–287. 10.1016/0092-8674(95)90169-8 [DOI] [PubMed] [Google Scholar]
- 27.Capo-Chichi CD, Rula ME, Smedberg JL, Vanderveer L, Parmacek MS, Morrisey EE, Godwin AK, Xu XX. 2005. Perception of differentiation cues by GATA factors in primitive endoderm lineage determination of mouse embryonic stem cells. Dev. Biol. 286:574–586. 10.1016/j.ydbio.2005.07.037 [DOI] [PubMed] [Google Scholar]
- 28.Steinberg MS. 1962. On the mechanism of tissue reconstruction by dissociated cells. I. Population kinetics, differential adhesiveness, and the absence of directed migration. Proc. Natl. Acad. Sci. U. S. A. 48:1577–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore R, Cai KQ, Escudero DO, Xu XX. 2009. Cell adhesive affinity does not dictate primitive endoderm segregation and positioning during murine embryoid body formation. Genesis 47:579–589. 10.1002/dvg.20536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang DH, Smith ER, Roland IH, Sheng Z, He J, Martin WD, Hamilton TC, Lambeth JD, Xu XX. 2002. Disabled-2 is essential for endodermal cell positioning and structure formation during mouse embryogenesis. Dev. Biol. 251:27–44. 10.1006/dbio.2002.0810 [DOI] [PubMed] [Google Scholar]
- 31.Yang DH, Cai KQ, Roland IH, Smith ER, Xu XX. 2007. Disabled-2 is an epithelial surface positioning gene. J. Biol. Chem. 282:13114–13122. 10.1074/jbc.M611356200 [DOI] [PubMed] [Google Scholar]
- 32.Moore R, Cai KQ, Tao W, Smith ER, Xu XX. 2013. Differential requirement for Dab2 in the development of embryonic and extra-embryonic tissues. BMC Dev. Biol. 13:39. 10.1186/1471-213X-13-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rula ME, Cai KQ, Moore R, Yang DH, Staub CM, Capo-Chichi CD, Jablonski SA, Howe PH, Smith ER, Xu XX. 2007. Cell autonomous sorting and surface positioning in the formation of primitive endoderm in embryoid bodies. Genesis 45:327–338. 10.1002/dvg.20298 [DOI] [PubMed] [Google Scholar]
- 34.Fässler R, Meyer M. 1995. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 9:1896–1908. 10.1101/gad.9.15.1896 [DOI] [PubMed] [Google Scholar]
- 35.Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. 1995. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 9:1883–1895. 10.1101/gad.9.15.1883 [DOI] [PubMed] [Google Scholar]
- 36.Brakebusch C, Hirsch E, Potocnik A, Fässler R. 1997. Genetic analysis of beta1 integrin function: confirmed, new and revised roles for a crucial family of cell adhesion molecules. J. Cell Sci. 110:2895–2904 [DOI] [PubMed] [Google Scholar]
- 37.Brakebusch C, Fässler R. 2005. Beta 1 integrin function in vivo: adhesion, migration and more. Cancer Metastasis Rev. 24:403–411. 10.1007/s10555-005-5132-5 [DOI] [PubMed] [Google Scholar]
- 38.Miranti CK, Brugge JS. 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4:E83–E90. 10.1038/ncb0402-e83 [DOI] [PubMed] [Google Scholar]
- 39.Aumailley M, Pesch M, Tunggal L, Gaill F, Fässler R. 2000. Altered synthesis of laminin 1 and absence of basement membrane component deposition in (beta)1 integrin-deficient embryoid bodies. J. Cell Sci. 113:259–268 [DOI] [PubMed] [Google Scholar]
- 40.Lohikangas L, Gullberg D, Johansson S. 2001. Assembly of laminin polymers is dependent on beta1-integrins. Exp. Cell Res. 265:135–144. 10.1006/excr.2001.5170 [DOI] [PubMed] [Google Scholar]
- 41.Liu J, He X, Corbett SA, Lowry SF, Graham AM, Fässler R, Li S. 2009. Integrins are required for the differentiation of visceral endoderm. J. Cell Sci. 122:233–242. 10.1242/jcs.037663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meighan CM, Schwarzbauer JE. 2008. Temporal and spatial regulation of integrins during development. Curr. Opin. Cell Biol. 20:520–524. 10.1016/j.ceb.2008.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E. 2000. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J. Cell Biol. 150:1149–1160. 10.1083/jcb.150.5.1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwon GS, Hadjantonakis AK. 2009. Transthyretin mouse transgenes direct RFP expression or Cre-mediated recombination throughout the visceral endoderm. Genesis 47:447–455. 10.1002/dvg.20522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayashi S, Lewis P, Pevny L, McMahon AP. 2002. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119(Suppl 1):S97–S101. 10.1016/S0925-4773(03)00099-6 [DOI] [PubMed] [Google Scholar]
- 46.Stenn KS, Link R, Moellmann G, Madri J, Kuklinska E. 1989. Dispase, a neutral protease from Bacillus polymyxa, is a powerful fibronectinase and type IV collagenase. J. Invest. Dermatol. 93:287–290. 10.1111/1523-1747.ep12277593 [DOI] [PubMed] [Google Scholar]
- 47.Fässler R, Georges-Labouesse E, Hirsch E. 1996. Genetic analyses of integrin function in mice. Curr. Opin. Cell Biol. 8:641–646. 10.1016/S0955-0674(96)80105-0 [DOI] [PubMed] [Google Scholar]
- 48.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, Smith A. 1998. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95:379–391. 10.1016/S0092-8674(00)81769-9 [DOI] [PubMed] [Google Scholar]
- 49.Niwa H, Miyazaki J, Smith AG. 2000. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24:372–376. 10.1038/74199 [DOI] [PubMed] [Google Scholar]
- 50.Pesce M, Schöler HR. 2001. Oct-4: gatekeeper in the beginnings of mammalian development. Stem Cells 19:271–278. 10.1634/stemcells.19-4-271 [DOI] [PubMed] [Google Scholar]
- 51.Li S, Harrison D, Carbonetto S, Fassler R, Smyth N, Edgar D, Yurchenco PD. 2002. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J. Cell Biol. 157:1279–1290. 10.1083/jcb.200203073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steinberg MS. 2007. Differential adhesion in morphogenesis: a modern view. Curr. Opin. Genet. Dev. 17:281–286. 10.1016/j.gde.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 53.Steinberg MS, Gilbert SF. 2004. Townes and Holtfreter (1955): directed movements and selective adhesion of embryonic amphibian cells. J. Exp. Zool. A Comp. Exp. Biol. 301:701–706. 10.1002/jez.a.114 [DOI] [PubMed] [Google Scholar]
- 54.Gerbe F, Cox B, Rossant J, Chazaud C. 2008. Dynamic expression of Lrp2 pathway members reveals progressive epithelial differentiation of primitive endoderm in mouse blastocyst. Dev. Biol. 313:594–602. 10.1016/j.ydbio.2007.10.048 [DOI] [PubMed] [Google Scholar]
- 55.Raghavan S, Vaezi A, Fuchs E. 2003. A role for alphabeta1 integrins in focal adhesion function and polarized cytoskeletal dynamics. Dev. Cell 5:415–427. 10.1016/S1534-5807(03)00261-2 [DOI] [PubMed] [Google Scholar]
- 56.Yu W, Datta A, Leroy P, O'Brien LE, Mak G, Jou TS, Matlin KS, Mostov KE, Zegers MM. 2005. Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol. Biol. Cell 16:433–445. 10.1091/mbc.E04-05-0435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yurchenco PD. 1990. Assembly of basement membranes. Ann. N. Y. Acad. Sci. 580:195–213. 10.1111/j.1749-6632.1990.tb17929.x [DOI] [PubMed] [Google Scholar]
- 58.Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H. 1986. Models for the self-assembly of basement membrane. J. Histochem. Cytochem. 34:93–102. 10.1177/34.1.3510247 [DOI] [PubMed] [Google Scholar]