

ABSTRACT
Macrophage infection is considered to play an important role in HIV-1 pathogenesis and persistence. Using a primary cell-based coculture model, we show that monocyte-derived macrophages (MDM) efficiently transmit a high-multiplicity HIV-1 infection to autologous CD4+ T cells through a viral envelope glycoprotein (Env) receptor- and actin-dependent virological synapse (VS), facilitated by interactions between ICAM-1 and LFA-1. Virological synapse (VS)-mediated transmission by MDM results in high levels of T cell HIV-1 integration and is 1 to 2 orders of magnitude more efficient than cell-free infection. This mode of cell-to-cell transmission is broadly susceptible to the activity of CD4 binding site (CD4bs) and glycan or glycopeptide epitope-specific broadly neutralizing monoclonal antibodies (bNMAbs) but shows resistance to bNMAbs targeting the Env gp41 subunit membrane-proximal external region (MPER). These data define for the first time the structure and function of the macrophage-to-T cell VS and have important implications for bNMAb activity in HIV-1 prophylaxis and therapy.
IMPORTANCEin vivo
INTRODUCTION
Human immunodeficiency virus-1 (HIV-1)-infected macrophages are found in infected patients (1) in antiretroviral sanctuaries and contain lower intracellular antiretroviral drug concentrations than CD4+ T cells (2). Long-lived HIV-1-infected macrophages resist viral cytopathic effects and shelter replication-competent virions in surface-accessible (3–6), but antibody-occluding (7, 8), virus-containing compartments (VCCs) (6) for several weeks (9), which strongly implicates macrophages in HIV-1 persistence (10, 11). Moreover, by efficiently spreading HIV-1 to uninfected CD4+ T cells through direct cell-to-cell contact (4, 12), macrophages may actively contribute to viral pathogenesis (10). Despite the potential importance of cell-to-cell transmission by macrophages to T cells (7, 13), the molecular organization and function of this intercellular interaction remains unknown (13).
Cell-to-cell transmission is the dominant mode of retroviral dissemination in vitro (14) and has been recently reported in vivo in murine models (15, 16). HIV-1 can spread directly from macrophages to CD4+ T cells in a contact-dependent manner (4, 12, 17–19), but the functional consequences of this for viral multiplicity of infection and for potential evasion of neutralizing antibody (NAb) attack are not known (20, 21). Over the past 4 years, a series of potent and broad-spectrum monoclonal antibodies, termed broadly neutralizing monoclonal antibodies (bNMAbs), have been isolated from infected individuals. These include MAbs against the CD4 binding site (CD4bs), a quaternary V1V2-dependent epitope and a high-mannose region on gp120, and the membrane-proximal external region (MPER) on gp41 (22–24). The ability of bNMAbs to inhibit HIV-1 replication is central to current HIV-1 prophylactic vaccine design and also may influence viral replication in an established infection (25). While NAb inhibit cell-free virus spread efficiently, there are conflicting reports regarding their relative activity in cell-to-cell transmission, in part due to heterogeneous experimental approaches (21, 26). Thus, cell-to-cell spread of HIV-1 between T cells at the virological synapse (VS) has been proposed to either have no significant effect on neutralization efficacy (20), selective effects on CD4bs-specific bNMAb (27), or a more generalized impact (28). No data are currently available on the activity of bNMAbs against macrophage-initiated cell-to-cell transmission of HIV-1 (21). Therefore, we sought to define the organization and function of the macrophage-T cell VS and to address the implications for bNMAb inhibition in a physiologically relevant model of primary monocyte-derived macrophages (MDM) and autologous CD4+ T cells (18).
MATERIALS AND METHODS
Cell isolation, infection, and HIV-1 preparation.
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood samples from healthy HIV-1-uninfected donors by density gradient centrifugation (Histopaque; Sigma), and monocytes were enriched to high purity (>95% CD14+) by untouched magnetic selection (MACS monocyte isolation kit II; Miltenyi Biotech) as previously described (18). Monocytes were seeded in 24- or 96-well plates at 2.5 × 105 to 5 × 105 cells/ml and differentiated to monocyte-derived macrophages (MDM) for 7 days in X-VIVO 10 (Lonza) medium supplemented with 1% heat-inactivated filtered human serum as previously described (4, 18). Autologous CD4+ T cells were enriched from PBMCs by untouched magnetic selection (MACS CD4+ T cell isolation kit; Miltenyi Biotech) to high purity (>95% CD3+ CD4+) and stimulated for 3 days with 1 μg/ml phytohemagglutinin (PHA) and 10 IU/ml interleukin-2 (IL-2; Centre for AIDS Reagents [CFAR], NIBSC, United Kingdom) in RPMI medium with 10% fetal bovine serum and 1% penicillin-streptomycin (RPMI-10) (4, 18).
MDM were infected for 7 days (4) at various multiplicities of infection (MOI; from 100 to 10−3) (18) with either the primary CCR5-using HIV-1 BaL (4) or one of several recombinant infectious molecular clones: pNL4.3-Gag-GFP (29) bearing the mac-tropic Env JRFL (kindly provided by B. Chen), Renilla luciferase expressing pNL4.3-LucR-T2A-BaL.ecto and pNL4.3-LucR-T2A-YU2.ecto (30), or diffusible green fluorescent protein (GFP) expressing pNL4.3-eGFP-BaL.ecto. Viral stocks were prepared by 293T transfection with polyethyleneimine, and titers were determined on TZM-bl (JC53) cells (20).
Antibodies and inhibitors.
Cytoskeleton inhibitors (Sigma) were diluted to nontoxic working concentration in serum-free RPMI: jasplakinolide (2 μM; F-actin), nocodazole (10 μM; microtubule), colchicine (10 μM; microtubule), paclitaxel (10 μM; microtubule), dynasore (80 μM; dynamin), dimethylamiloride (DMA; 100 μM; endocytosis), and blebbistatin (100 μM; myosin II). Toxicity in the presence of inhibitors was assessed on primary MDM using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) viability assay (Promega) as previously described (31). bNMAbs tested in neutralization assays were gp120-specific VRC01, b12, 2G12, and PGT121 and gp41 MPER-specific 2F5, 4E10, 10E8 (all human IgG1; obtained from the International AIDS Vaccine Initiative [IAVI] Neutralizing Antibody Consortium or transiently expressed in 293T cells under serum-free conditions essentially as previously described [32]), and anti-human IgG1 isotype control (kindly provided by H. Waldmann). 10E8 Fab was produced according to the manufacturer's instructions using the Pierce Fab preparation kit (Pierce). For functional assays, blocking antibodies all were purified mouse IgG1 used at 10 μg/ml unless otherwise stated: CD4 (13B.8.2; Beckman-Coulter), CCR5 (2D7; BD Biosciences), ICAM-1 (LB-2 IgG2b; BD Biosciences or Santa-Cruz), LFA-1 (L130; BD Biosciences), ICAM-2 (B-T1; AbD Serotec), and ICAM-3 (101-1D2; Santa-Cruz). For laser scanning confocal microscopy (LSCM), we used the following primary MAb: CD4 (nonblocking; clone L120; mouse IgG1; CFAR), CD81 (clone 454720; mouse IgG2b; R&D Systems), anti-gp120 (2G12-biotin), and mouse anti-p18 Gag IgG2a, together with the appropriate isotype controls. The following secondary antibodies were used: goat anti-mouse Alexa-488 IgG2b, Alexa-488, Alexa-647 IgG2a, Alexa-647 IgG1, goat anti-rabbit Alexa-488 IgG (Invitrogen), streptavidin-tetramethyl rhodamine isothiocyanate (TRITC) (Jackson), or directly conjugated phalloidin-TRITC (Sigma). For flow cytometry staining, we used directly conjugated mouse anti-CD3 (clone UCHT1; IgG1)-fluorescein isothiocyanate (FITC) or -allophycocyanin (APC) (BD Biosciences) and mouse anti-Gag (clone KC57; IgG1)-FITC or -phycoerythrin (PE; Beckmann) together with fluorochrome-conjugated isotype controls at the same concentration.
Live-cell microscopy.
Live-cell time-lapse imaging of MDM and CD4+ T cell cocultures was performed in 35-mm glass-bottomed imaging dishes (MakTec) with a 40 × oil immersion objective Axiovert 200 microscope with an Axiovision MRm charge-coupled-device (CCD) camera and Colibri illumination. Image analysis was performed using AxioVision v 4.8.2. For short-term analysis of transfer, images were acquired every 2 min for up to 6 h. For longer-term conjugate analysis comparing GFP+ MDM to GFP− MDM, images were acquired every 5 min for up to 12 h. Image analysis of conjugate duration (min) and frequency (number of conjugates formed per MDM) were performed for 10 MDM for each condition (GFP+ and GFP−). Conjugates were scored if they persisted >5 min (i.e., were present in two sequential images).
LCSM.
CD4+ T cells were prelabeled with the nonblocking CD4 MAb L120 as previously described (4) and cocultured with HIV-1 BaL-infected MDM on glass coverslips for 2 to 3 h to allow VS formation. Nonadherent T cells were gently removed in warm RPMI, and MDM-CD4+ T cell conjugates were fixed and stained for LSCM as described previously (4), with minor modifications. Briefly, cells were fixed in 4% paraformaldehyde for 1 h at room temperature (RT) and washed in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA), and nuclei were stained with 1 μg/ml Hoechst in PBS (for 8 min), washed in PBS, and surface stained for 1 h at RT. They were then permeabilized using 0.1% saponin–0.5% BSA with 10% pooled human/goat serum to block Fc-receptor binding, and intracellular staining was performed for 1 h at RT. Coverslips were mounted in Mowiol 4-88 (Calbiochem) and analyzed using an Olympus FV1000 microscope. Images were acquired with 63× and 100× oil immersion objectives and processed in Olympus Fluoview 2.0b.
HIV-1 transfer assay.
CD4+ T cells were cocultured in duplicate with HIV-1-infected MDM for 3 h in RPMI-10. Nonadherent cells were gently aspirated in warm RPMI-10, washed, fixed, stained for CD3, permeabilized, stained for intracellular Gag, and analyzed by flow cytometry on a FACSCalibur (BD Biosciences) (4). Transfer was expressed relative to the percent CD3+ Gag+ cells in isotype or vehicle (dimethylsulfoxide [DMSO] or RPMI) control conditions: transfer (%) = 100 × [(transferinhibitor)/(transfercontrol)].
Luciferase infectivity assay.
Target CD4+ T cells were cocultured for 24 and 48 h at a 2:1 ratio with donor HIV-1+ MDM (7 days postinfection [dpi] with Renilla luciferase-expressing pNL4.3-LucR-T2A-BaL.ecto). In parallel conditions, cocultures were also gently shaken at 75 rpm at 37°C with 5% CO2 (20, 33), and CD4+ T cells were separated from HIV-1+ MDM by a 3.0-μm transwell membrane (Costar) or were incubated with virus-containing cell-free supernatants from HIV-1+ MDM (collected at 7 dpi). At the relevant time point, CD4+ T cells were gently removed (with <0.1% MDM contamination in coculture conditions determined by flow cytometry staining for CD3 and CD14), washed in PBS, and lysed with Glo-Lysis buffer (Promega). For luciferase quantification, cell lysates were mixed 1:1 with Ren-Glo assay solution (Promega) at room temperature, and luminescence was measured at 1,000 ms−1 integration on a SpectroMax M5 plate reader. Background values from uninfected wells were subtracted.
Neutralization assays.
Renilla luciferase-expressing HIV-1-infected MDM were untreated or preincubated with 10 μg/ml pooled purified human IgG to block Fc receptors for 1 h at 37°C before removal and preincubation with bNMAb or isotype control for 1 h at 37°C. In parallel, cell-free virus stocks were preincubated with bNMAbs or isotype control. We tested 5-fold dilutions of bNMAb from the starting concentration of 50 μg/ml (MPER, b12, and 2G12), 16.67 μg/ml (10E8 Fab), or 10 μg/ml (VRC01 and PGT121). Autologous CD4+ T cells were cultured with HIV-1-infected MDM or cell-free viral stocks (the MOI was titrated to achieve equivalent luciferase expression in both conditions) for 48 h in RPMI-10 with 10 IU/ml IL-2, followed by gentle aspiration, lysis, and luciferase analysis as described above. After subtracting background values, data were normalized to the isotype control as described above. Inhibitor titration data were fitted to a variable-slope sigmoidal dose-response curve in GraphPad Prism 5.0 (bottom constrained to 0), and the significance of differences in the 50% inhibitory concentration (IC50) was compared by an extra sum-of-squares F test.
qPCR for pol.
Target CD4+ T cells in duplicate wells were gently removed following coculture with HIV-1BaL-infected MDM as described above, washed in PBS, pelleted, and frozen at −80°C for later extraction with the DNeasy blood and tissue kit (Qiagen) according to the manufacturer's protocol. Primers and probes for the detection of HIV-1BaL pol and the cell reference human β-globin gene have been previously described (20). Singleplex PCRs were performed in triplicate in a 20-μl total volume, with 2× Agilent brilliant III fast master mix, 375 nM each primer, 125 nM probe, 0.3 μl ROX, and 2 μl of sample DNA. A standard curve for pol and β-globin was generated for each quantitative PCR (qPCR) by 5-fold titration of single vDNA copy/cell standards prepared from ACH2 cells (20). Reactions were performed on a StepOne plus (ABI) qPCR machine with the following steps: one cycle at 95°C for 3 min, followed by at least 40 cycles of 95°C for 5 s and then 60°C for 10 s. The results of triplicate technical replicates per sample for pol and β-globin were averaged, and the ratio of pol to β-globin was calculated to determine the relative abundance (expressed as vDNA copies/cell) (20).
qPCR for total gag.
Cell copy number was quantified in triplicate at two dilutions using an adapted albumin qPCR as described previously (34). The master mix contained 2× Roche LightCycler 480 probe MM, 200 nM probe (6-carboxyfluorescein [FAM]-CCTGTCATG CCCACACAAATCTCTCC-black hole quencher 1 [BHQ-1]), 250 nM Albumin_F (GCTGTCATCTCTTGTGGGCTGT), and 250 nM Albumin_R (AAACTCATGGGAGCTGCTGGTT) (Eurofins MWG Operon) with 10 μl DNA sample in a total volume of 25 μl. Input cells (2 × 104) in 10 μl were assayed in triplicate for total HIV using 500 nM probe (FAM-AGTRGTGTGTGCCCGTCTGTTG-BHQ-1), 500 nM LTR_OS (GRAACCCACTGCTTAASSCTCAA), 500 nM LTR_AS (TGTTCGGGCGCCACTGCTAGAGA) (Eurofins MWG Operon), and 2× Roche LightCycler 480 probe MM in a total volume of 25 μl. Both qPCR amplifications were performed with one cycle of 95°C for 10 min and then 45 cycles of 95°C for 15 s and 60°C for 1 min. Data were analyzed using Roche LightCycler software. ACH2 cells (8 × 105) containing one integrated copy of HIV-1 per cell were used in duplicate as standards with cell and HIV copy numbers ranging in serial 10-fold dilutions from 1 × 105 to 1 × 100 DNA copies/reaction.
qPCR for alu-gag integrated DNA.
Integrated HIV-1 provirus was quantified using an adapted alu-PCR assay (35). Briefly, 5 replicates of 7,500 cells underwent a first-round PCR amplification (95°C for 2 min; 20 cycles of 95°C for 15 s, 50°C for 15 s, and 72°C for 150 s) using 100 nM alu (GCCTCCCAAAGTGCTGGGATTACA) and 600 nM gag (GTTCCTGCTATGTCACTTCC) reverse primers (Eurofins MWG Operon), 1.5 U platinum Taq (Invitrogen), 1.5 mM MgCl2 (Invitrogen), 300 μM deoxynucleoside triphosphate (dNTP; Appleton Woods), and 10× master mix (Invitrogen) in a total volume of 50 μl. Ten μl of the first-round product was amplified in a nested protocol using the total gag assay described above. A first-round PCR with 3 replicates using only the gag reverse primer (gag only) acted as a background unintegrated control. Serially 3× diluted integration site standards (36) (ranging from 3,750 to 15 copies per 7,500 cells in duplicate) were used to construct a standard curve for each plate. Integration levels per cell were calculated only if the alu-gag and gag-only signals were statistically significantly different by Student's t test (P < 0.05).
Statistical analysis.
Pairwise comparisons were done with Student's t test, and comparisons of more than two groups were done by analysis of variance (ANOVA) with Dunnett's posttest correction on log-transformed or normalized data. Comparison of IC50s was performed by an extra sum-of-squares F test. Analyses were performed in Prism version 5.0, and unless otherwise stated it was performed on experimental replicates from a minimum of 3 independent donors. An alpha value of <0.05 after correction for multiple comparisons was considered statistically significant.
RESULTS
Efficient contact-mediated HIV-1 infection from MDM to T cells.
To investigate HIV-1 spread from infected MDM to T cells under the most physiological in vitro conditions, we established a model in which infected MDM were cocultured with autologous activated primary CD4+ T cells for various times. We first wished to image in real time the contacts formed between infected MDM and CD4+ T cells to determine whether uninfected and HIV-1-infected MDM interacted with T cells with different kinetics. For this we used a GFP reporter virus (JRFL Env-bearing HIV-1NL-Gag-GFP-JRFL) to infect MDM over 7 days and then imaged the cocultures immediately after adding the T cells. We observed HIV-1 transfer from a strongly GFP-positive compartment with characteristics of the macrophage VCC to the interface with the T cell within 30 to 40 min. This rapid transfer resulted in a large quantity of Gag-GFP deposition on the T cell surface (Fig. 1A; also see Movie S1 in the supplemental material), much of which we suppose to be virions. Following transfer, T cells were observed to detach from MDM but retained the GFP signal (Fig. 1B; also see Movie S2). These data support our previous hypothesis regarding the transient nature of the macrophage-T cell VS (4). To quantify the frequency and duration of cell-cell interactions between infected MDM and CD4+ T cells, we performed live-cell coculture experiments with a mac-tropic GFP reporter infectious molecular clone (pNL4.3-GFP-BaL). We observed frequent transient cell-to-cell interactions (“scanning” of MDM by T cells [37]), and image analysis revealed that the frequency of interactions of CD4+ T cells with infected MDM was significantly higher than that with uninfected MDM (means, 5.5 and 1.5, respectively; P = 0.007 by t test) (Fig. 1C, upper). Average conjugate duration was statistically indistinguishable between infected and uninfected MDM (median, 35 min for each; P = 0.268 by t test) (Fig. 1C, lower), but there were significantly more long-lived (>200 min) interactions (8/54 versus 0/15; P < 0.001 by Fisher's exact test) (Fig. 1C, lower). We explored the kinetics of HIV-1 transfer to T cells using a flow cytometry-based Gag transfer assay. Transfer was rapid and significantly above background levels at all time points compared to t = 0 (Fig. 1D). The efficiency of transfer did not appear to be due to high-multiplicity cell-free virion diffusion from infected MDM, since cocultures separated by a transwell (TW) membrane, which permits cell-free virion diffusion but not direct cell-to-cell contact (20), did not result in a significant signal above background at 24 h (Fig. 1D). However, we reasoned that viral transfer measured by Gag staining of permeabilized cells did not necessarily imply productive transmission, since it could also correspond to uptake of Gag protein or virions into nonproductive compartments (14). Therefore, to assess whether efficient virion transfer led to productive T cell infection, we gently aspirated CD4+ T cells (yielding <0.1% MDM contamination [18]) at various time points postcoculture and measured the production of HIV-1BaL late reverse transcripts by quantitative PCR for HIV-1 pol or HIV-1NL-LucR-T2A.BaL luciferase expression. Both assays confirmed that the rapid kinetics of Gag transfer measured by flow cytometry corresponded to similar time-dependent increases in CD4+ T cell infection (Fig. 1E and F).
FIG 1.

Rapid HIV-1 transfer from the VCC results in productive T cell infection. (A) Live-cell time-lapse imaging of HIV-1NL-Gag-GFP-JRFL-infected MDM and autologous CD4+ T cell cocultures. Images were acquired every 2 min for up to 6 h, and minutes after initiation of coculture are shown above stills from time-lapse series. HIV-1 Gag transfer from an MDM (labeled “M”) to a T cell (labeled “T”) at the VS is arrowed. Gag-GFP, signal from Gag-GFP infecting virus; BF, bright-field illumination; Merge, merged Gag-GFP and BF signals. (B) Detachment of a Gag+ T cell following VS-mediated transfer at 54 min. Scale bar, 10 μm. (C) Image analysis of conjugate frequency (number of conjugates formed per MDM; left) and duration (min; right) was performed with long-term imaging (every 5 min for 12 h) of HIV-1NL-GFP-BaL-infected MDM (n = 10 MDM for each condition, MDM+ and MDM−). Green circles, infected MDM; open squares, uninfected MDM. Conjugates were scored if they persisted >5 min and were present in two sequential images. **, P < 0.01; ***, P < 0.001 (t test). (D) Kinetics of Gag transfer measured using a flow cytometry transfer assay. Bars, means ± standard errors of the means (SEM) for 10 independent donors (n = 4 for transwell). **, P < 0.01; ***, P < 0.001 (ANOVA with Dunnett's posttest compared to t = 0). TW, cell-free exposure through a virus-permeable 3.0-μm transwell membrane. (E and F) CD4+ T cells were gently aspirated after coculture with infected MDM at a 1:1 ratio and analyzed for productive infection by qPCR for HIV-1 pol (HIV-1BaL-infected MDM; means ± SEM from 4 independent donors) (E) or luciferase expression (HIV-1NL-LucR-T2A.BaL-infected MDM; means ± SEM from 3 independent donors) (F). *, P < 0.05; ***, P < 0.001 (ANOVA with Dunnett's posttest compared to t = 0). An arbitrary value of 0.1 (D) or 1 (E) was added to all data to permit log transformation prior to analysis. RLU, relative light units.
HIV-1 transfer to T cells requires actin remodeling in the infected MDM.
HIV-1 has been observed to colocalize with several tetraspanin molecules, such as CD81, within the VCC of infected MDM (reviewed in reference 38). To probe the localization of HIV-1 within infected MDM in our system, we carried out LSCM of permeabilized, infected MDM. HIV-1 Gag colocalized substantially with CD81 but was significantly reduced in regions labeled with lysosomal marker Lamp-1, consistent with the reported VCC-like location of virus (Fig. 2A and B) (3, 39, 40). Following coculture of infected MDM with CD4+ T cells for 3 h, CD81 and HIV-1 Gag were polarized to the VS, suggesting at least partial relocation/reorganization of components of the VCC to the intercellular interface (Fig. 2C). Since this process was relatively rapid, we suspected it to be active and cytoskeleton dependent. LSCM of permeabilized cells revealed F-actin proximal to the VCC in HIV-1BaL-infected MDM (Fig. 2D) that was polarized toward the interface with contacting CD4+ T cells (Fig. 2E). To investigate a functional role of actin in this process, we preincubated MDM for 1 h with nontoxic concentrations (Fig. 2F) of various inhibitors of cytoskeletal remodeling: the dynamin inhibitor dynasore, the amiloride endocytosis inhibitor DMA, the F-actin remodeling inhibitor jasplakinolide, the myosin II motor inhibitor blebbistatin, and inhibitors of microtubule remodeling, nocodazole, colchicine, and paclitaxel, alongside appropriate vehicle controls. Inhibitors were washed out prior to coculture with T cells to avoid confounding effects on the T cell cytoskeleton. We observed a significant (P < 0.05 by ANOVA) reduction in transfer to T cells over 3 h in jasplakinolide-treated MDM, whereas there was only a partial, nonsignificant reduction for microtubule or myosin inhibitors, implicating actin rearrangement in the reorganization of the VCC and subsequent viral transfer across the VS (Fig. 2G).
FIG 2.

HIV-1 transfer from the macrophage VCC is actin dependent. (A) LSCM of permeabilized HIV-1BaL-infected MDM showing colocalization of HIV-1 p18 Gag with CD81 but not with the lysosomal marker LAMP-1. (B) Colocalization with p18 Gag was quantified in 10 positive MDM. ***, P < 0.001 by t test. (C) Gag p18 and CD81 colocalized at the synaptic junction with autologous CD4+ T cells 3 h postcoculture. T, T cell nuclei. Scale bar, 20 μm. (D) F-actin proximal to HIV-1 p18 Gag in the VCC of HIV-1BaL-infected MDM. Scale bar, 20 μm. (E) F-actin copolarized to the synaptic junction with autologous CD4+ T cells (nuclei labeled “T”) 3 h postcoculture. Scale bar, 10 μm. Nuclei were stained with Hoechst. (F) Toxicity of inhibitors on MDM was assessed after 1 h of treatment with the dynamin inhibitor dynasore (80 μM), the endocytosis inhibitor DMA (100 μM), the F-actin inhibitor jasplakinolide (2 μM), the microtubule inhibitors nocodazole (10 μM), paclitaxel (10 μM), and colchicine (10 μM), and the myosin II inhibitor blebbistatin (100 μM) in serum-free medium by MTS assay. Viability was expressed as a percentage of the DMSO/medium control. Data are means ± SEM from 3 donors. (G) HIV-1BaL-infected MDM were pretreated with these inhibitors, washed three times in warm medium, and incubated with autologous CD4+ T cells for 3 h, and HIV-1 transfer was measured in T cells stained for CD3 and HIV-1 Gag by flow cytometry and expressed as a percentage of the CD3+ Gag+ cells in DMSO control wells. Bars, means ± SEM from 3 independent donors. *, P < 0.05 by ANOVA with Dunnett's posttest compared to the DMSO control.
HIV-1 transmission requires Env receptor and adhesion molecule interactions.
The T cell-to-T cell HIV-1 VS has been previously reported to depend both on Env-CD4 and Env-coreceptor interactions and interactions between integrins and ICAMs (41, 42; reviewed in reference 37). To examine the molecular organization of the VS governing viral transfer, we quantified the impact of potential VS inhibitors in the flow cytometry-based functional assay of VS transfer. Preincubation of CD4+ T cells for 1 h with anti-CD4 or anti-CCR5 blocking MAb significantly inhibited transfer (P < 0.001 by ANOVA) (Fig. 3A), suggesting that transfer is dependent on Env binding to the target cell receptors on CD4+ T cells (4). This hypothesis was further tested by inclusion of the anti-gp120 NMAbs 2G12 and b12 with infected MDM both prior to and during T cell coculture. These bNMAbs, which block CD4-gp120 engagement and also prevent the conformational changes required for CCR5 engagement, strongly inhibited HIV-1 transfer (P < 0.001) (Fig. 3A). A functional role of ICAM-1–LFA-1 interactions was confirmed using a blocking ICAM-1 MAb that incompletely but significantly (50 to 60%) inhibited VS-mediated HIV-1 transfer (P < 0.001 by ANOVA) (Fig. 3B). The degree of inhibition by the ICAM-1 MAb was mirrored by the activity of a blocking MAb against LFA-1 (P < 0.001) (Fig. 3B). In contrast, neither anti-ICAM-2 nor anti-ICAM-3 blocking MAbs showed significant inhibition. These results are consistent with previous studies indicating that the ICAM-1–LFA-1 interaction dominates both physiological interactions between activated T cells and antigen-presenting cells and cell-to-cell transmission of HIV-1 (42, 43).
FIG 3.
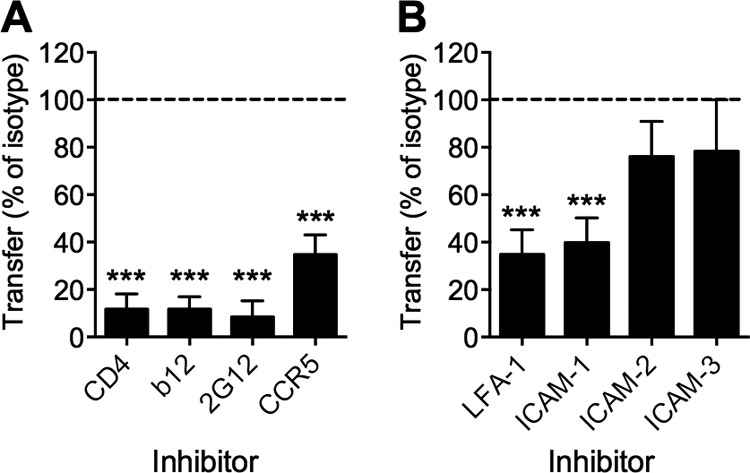
HIV-1 transmission requires Env receptor- and actin-dependent interactions. (A) Preincubation of CD4+ T cells with blocking anti-CD4 (13B8.2) or anti-CCR5 (2D7) antibody (10 μg/ml) or preincubation of HIV-1BaL-infected MDM with anti-gp120 (2G12 or b12; 10 μg/ml) after Fc block resulted in a significant reduction in HIV-1 transfer to CD4+ T cells during 3 h of coculture. All MAbs were maintained during coculture. Transfer was measured by flow cytometry (FACSCalibur) of aspirated CD4+ T cells stained for CD3 and HIV-1 Gag, expressed as a percentage of the CD3+ Gag+ cells in relevant isotype (mouse or human) control wells. (B) Preincubation of CD4+ T cells with blocking anti-LFA-1 (L130 n = 5 donors) or preincubation of HIV-1BaL-infected MDM with anti-ICAM-1 (LB-2; n = 5 donors), ICAM-2 (B-T1; 10 μg/ml), or ICAM-3 (101-1D2; 10 μg/ml; both n = 2 donors) after Fc block followed by assessment of HIV-1 transfer to CD4+ T cells during 3 h of coculture with HIV-1BaL-infected MDM. All MAbs were maintained during coculture. Transfer was measured as described for panel A and expressed as a percentage of the CD3+ Gag+ cells in isotype control wells. Bars, means ± SEM. ***, P < 0.001 by ANOVA with Dunnett's posttest.
MDM transmit a high-multiplicity infection to T cells across VS.
Although MDM cell-to-cell transmission of HIV-1 to CD4+ T cells appears considerably more efficient than cell-free transmission (4, 17, 19) (Fig. 1D), the relative efficiency of infection remains poorly defined. Various methods have been reported to isolate cell-free infection in coculture models to allow comparisons to cell-to-cell transmission, including the use of transwell membranes that permit viral diffusion without cell contact (4, 20), incubation with infected donor cell supernatant (20), or gentle shaking of cocultures to prevent sustained contacts (33). We compared all of these methods of cell-free infection to VS-mediated transmission by MDM infected 7 days previously with HIV-1NL-LucR-T2A.BaL, observing 20- to 250-fold higher CD4+ T cell infection by VS-mediated transmission at 24 and 48 h (Fig. 4A). The greater infection of T cells in the shaking system compared to the transwell or supernatant cell-free infection systems most probably reflects a proportion of contacts of sufficient duration in the former system to mediate some VS-mediated T cell infection and will overestimate the implied contribution of cell-free infection. The increased T cell infection across VS compared to cell-free infection may result from a high proportion of infectious virions with infection kinetics faster than those of cell-free infection, leading to a greater proportion of infected T cells, a higher multiplicity of individual T cell infections, or both. To directly quantify the levels of T cell integration following VS-mediated transmission, we used alu-gag qPCR to measure total vDNA and integrated proviral DNA levels in T cells infected by VS-mediated transmission from MDM compared to transwell or cell-free virus inoculation at an MOI of 0.1 at 48 h. VS-mediated transmission achieved a robust, high level of target cell integration with a mean of 1.98 integrated vDNA copies per cell (95% confidence intervals [CI], 0.12, 3.80) (Fig. 4B). In contrast, high-MOI supernatant infection resulted in 0.06 integrants per cell (95% CI, 0.04, 0.08) (Fig. 4B), and transmission across a transwell produced integration levels below the limit of detection of the assay. To confirm that this high level of VS-mediated integration was not the result of contamination by infected MDM, we pretreated T cells with the HIV-1 integrase inhibitor raltegravir (RAL; 2 μM), which significantly reduced the number of integrated vDNA copies per cell (by 40-fold), indicating de novo integration (Fig. 4B). When we compared the efficiency of integration for VS-mediated and cell-free infections expressed as the ratio of integrated to total vDNA (Fig. 4C), we found that this was equivalent for both transmission routes. These data indicate that the level of viral integration is governed primarily by the magnitude of the initial viral inoculum rather than any qualitative differences between cell-to-cell and cell-free viral infections. The incomplete effect of raltegravir against VS-mediated transmission is due to the high MOI of cell-cell transmission, as we have previously reported (18).
FIG 4.
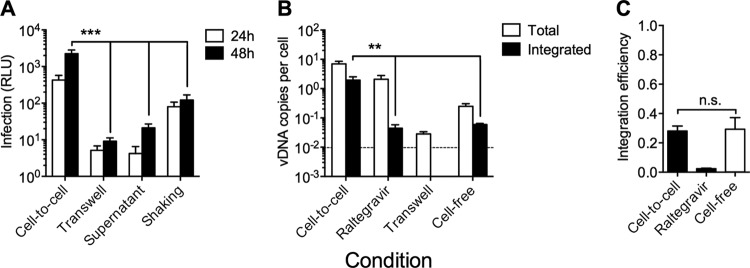
VS transmission is more efficient than cell-free infection. (A) Luciferase expression levels in aspirated CD4+ T cells after 24 and 48 h of coculture with HIV-1NL-LucR-T2A.BaL-infected MDM (cell-to-cell), coculture across a 3.0-μm transwell membrane (transwell), or exposure to cell-free supernatants collected from HIV-1NL-LucR-T2A.BaL-infected MDM 7 dpi HIV-1 (supernatant) or cocultured with HIV-1NL-LucR-T2A.BaL-infected MDM under gentle shaking conditions (75 rpm). Cell-to-cell infection levels were 250-, 100-, and 20-fold higher than those of the transwell, supernatant, and shaking controls, respectively, at 48 h. Means ± SEM are shown for 5 independent donors. ***, P < 0.001 by ANOVA with Dunnett's posttest compared to the cell-to-cell condition. (B) Total (white bars) and integrated (black bars) HIV-1 vDNA in CD4+ T cells was quantified by gag qPCR and alu-gag qPCR relative to human albumin, respectively. Conditions analyzed were coculture with HIV-1BaL-infected MDM for 48 h alone, in the presence of 2 μM raltegravir (RAL), or across a 3.0-μm transwell membrane or with cell-free infection of CD4+ T cells with HIV-1BaL (MOI, 0.1). Transwell integrated vDNA levels were below the limit of detection (represented by the dotted line). Means ± SEM are shown from 4 independent donors. **, P < 0.01 by ANOVA with Dunnett's posttest compared to the cell-to-cell condition. (C) Integration efficiency was expressed as the ratio of integrated to total HIV-1 Gag DNA (vDNAintegrated/vDNAtotal). *, P < 0.05; n.s., nonsignificant (ANOVA with Dunnett's posttest compared to the cell-to-cell condition). Means ± SEM from 4 independent donors are shown.
VS transmission selectively restricts bNMAb activity.
Although inhibitors of gp120-receptor interactions robustly inhibit VS-mediated transfer at saturating concentrations (Fig. 3A), bNMAbs may be differentially active in the cell-to-cell context compared to cell-free inhibition (27, 28). Therefore, we compared the efficiency of bNMAb inhibition over approximately one cycle of VS-mediated and cell-free infection of CD4+ T cells (18) using the clade B mac-tropic tier 1 (neutralization sensitive) and tier 2 (relatively neutralization resistant) Envs BaL and YU2, respectively (44). To avoid the confounding effects of differential MOI transmission between cell-to-cell and cell-free infection routes, we titrated down MDM infection levels and increased CD4+ T cell cell-free inocula to achieve equivalent T cell infection levels in both conditions at 48 h as previously described (18) (Fig. 5A and B). We compared bNMAb targeting three main epitope clusters on Env: the CD4bs (VRC01 and b12) and glycan (2G12) or glycopeptide (PGT121) epitope regions of gp120 and the MPER of gp41. The activity of 2G12, PGT121, and VRC01 was unaffected by route of transmission (Fig. 5C and D). In contrast, we observed a significant 16-fold reduction in the activity of the CD4bs NAb b12 against BaL (P = 0.002) (Fig. 5C) but not against YU2 (P = 0.612) (Fig. 5D). However, there was a striking reduction in the activity of MPER-directed bNMAbs 2F5, 4E10, and 10E8 against VS-mediated viral transmission, resulting in reductions in potency of >100-fold (Fig. 6). Indeed, the activity of 10E8 and 2F5 was negligible against YU2 Env even at the highest concentration used (50 μg/ml). The reduced activity of MPER bNMAbs against VS-mediated transmission was unchanged in experiments using a commercially available Fc blocking reagent (data not shown), suggesting that loss of function was not related to Fc receptor binding, in line with previous data (45). We hypothesized that steric inhibition reduces the on-rate of MPER bNMAbs for their epitopes, since they are sandwiched between the base of gp120 and the viral lipid envelope. If the virus is contained within an antibody-inaccessible compartment (the VCC) until the T cell contacts the MDM, then the time window available for antibody to bind gp41 will be limited, and slow on-rates might prevent most antibodies binding prior to virus entry. To investigate this, we tested inhibitors of gp41 fusion of smaller size than intact IgG molecules with the hypothesis that increased access to the MPER region reduces the differential. In agreement with this hypothesis, the Fab fragment of 10E8 showed a difference in inhibition of only 14-fold on BaL (compared to ∼100-fold for the IgG) and no significant difference on YU2 (Fig. 7A and B). Similarly, the activity of the 4.5-kDa peptide gp41 fusion inhibitor T20, although significantly reduced against VS compared to cell-free transmission on BaL (P = 0.001), was only 18-fold less effective and showed equivalent inhibitory activity against YU2 (P = 0.154) (Fig. 7C and D). Thus, the MPER region appears to be particularly susceptible to steric restriction at the macrophage-to-T cell VS, although Env sequence-dependent effects on neutralization efficacy also need to be taken into account (45).
FIG 5.

Neutralization of cell-free- and cell-to-cell-mediated infection. HIV-1-infected MDM and cell-free inocula were adjusted to produce equivalent infection levels at 48 h postinfection by the cell-to-cell (CTC; black bars) and cell-free (CF; open bars) routes for HIV-1NL-LucR-BaL (A) and HIV-1NL-LucR-YU2 (B). Means ± SEM are shown for 6 (BaL) and 4 (YU2) donors. The activity of bNAbs targeting the CD4bs (VRC01 and b12) and the glycan shield (PGT121 and 2G12) were tested against cell-to-cell (filled circles) and cell-free (open circles) infection of CD4+ T cells with HIV-1NL-LucR-T2A.BaL (C) and HIV-1NL-LucR-T2A.YU2 (D). Means ± SEM from 3 to 6 donors are shown. The significance of differences in IC50 was assessed by F test. A significant difference was observed for b12 (P = 0.002) against BaL. 2G12 was not tested against YU2.
FIG 6.
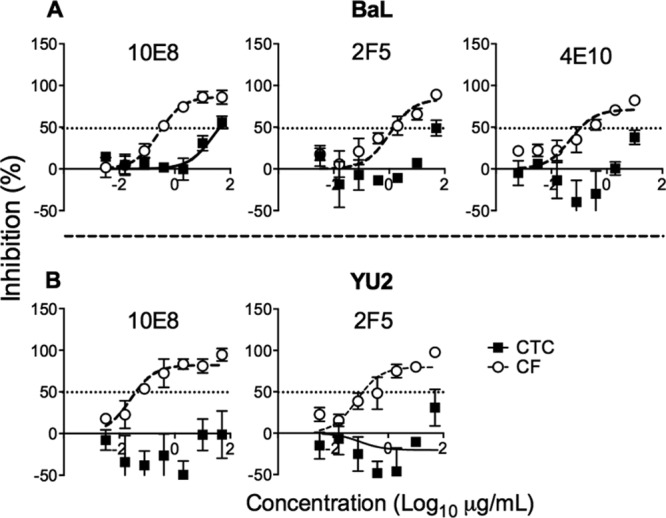
MDM to T cell VS-mediated transmission is resistant to MPER bNMAb neutralization. The activity of bNMAbs targeting the MPER region (10E8, 2F5, and 4E10) were tested against cell-to-cell (CTC; filled circles) and cell-free (CF; open circles) infection of CD4+ T cells with HIV-1NL-LucR-T2A.BaL (A) and HIV-1NL-LucR-T2A.YU2 (B). Means ± SEM from 3 to 6 donors are shown. The significance of differences in IC50 was assessed by F-test. A significant difference was observed for 10E8 (P < 0.001) against BaL. IC50s could not be fitted for 2F5 and 4E10 (BaL) or 10E8 and 2F5 (YU2). 4E10 was not tested against YU2.
FIG 7.
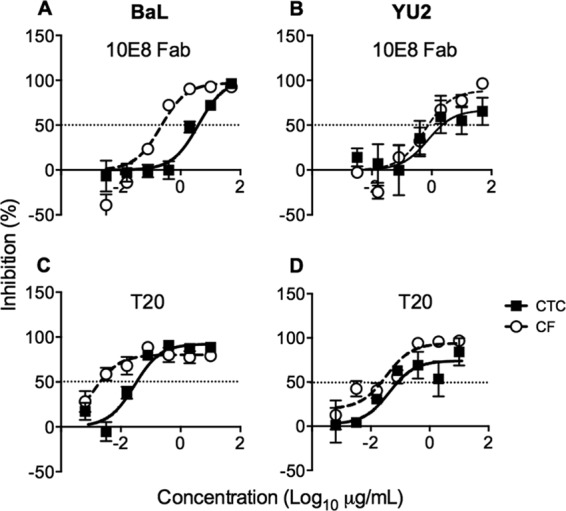
Inhibitor activity is dependent on size. Cell-to-cell (CTC; filled circles) and cell-free (CF; open circles) neutralization experiments were carried out with 10E8 Fab against HIV-1NL-LucR-T2A.BaL (A) and HIV-1NL-LucR-T2A.YU2 (B).There was a significant difference in 10E8 Fab IC50 for HIV-1NL-LucR-BaL (P = 0.013, F test, n = 3 donors) but not HIV-1NL-LucR-T2A.YU2 (P = 0.400, F test, n = 5 donors). The activity of T-20 was tested against cell-to-cell (CTC; filled circles) and cell-free (CF; open circles) infection of CD4+ T cells with HIV-1NL-LucR-T2A.BaL (C) and HIV-1NL-LucR-T2A.YU2 (D). Means ± SEM from 3 donors are shown. The significance of differences in IC50 was assessed by F test. A significant difference was observed for BaL (P = 0.001) but not YU2 (P = 0.154).
DISCUSSION
Here, we have defined for the first time the principal molecular components of the VS formed by contact between HIV-1-infected primary MDM and CD4+ T cells that results in infectious spread of HIV-1 to T cells that is 20- to 250-fold more efficient than cell-free infection. This mechanism of intercellular transfer results in high levels of T cell viral integration and provides an environment that substantially reduces the efficiency of neutralization by certain bNMAbs. These findings have implications for viral dissemination in vivo and may help to explain several facets of HIV-1 pathogenesis. High-multiplicity transmission of HIV-1 by directed cell-to-cell transfer has important implications for host-mediated and therapeutic antiviral strategies for the following reasons. (i) It overcomes antiretroviral inhibition in a probabilistic manner, potentially contributing to viral persistence by facilitating intermittent HIV-1 replication during antiretroviral therapy (46) and/or permitting ongoing dissemination in antiretroviral sanctuary sites (18, 47). (ii) It reduces the efficacy of NAb. In the current study, MPER bNMAbs were particularly affected, but a modest effect was also seen with the gp120 CD4bs bNMAb b12 on the neutralization-sensitive virus strain BaL, in accord with a previous study (27). (iii) Host restriction factors, such as tetherin/BST-2, may also be saturated by the magnitude of viral transmission at VS (48).
Efficient cell-to-cell transmission is likely to be the dominant mode of viral spread in densely packed primary and secondary lymphoid tissues (14) where intense HIV-1 replication occurs (49). Recent progress with intravital imaging using murine models shows that retrovirally infected cells frequently engage uninfected cells (15, 16) and suggests that T cells are important vectors for HIV-1 dissemination (15). Despite this, few studies have attempted to directly quantify the multiplicity of cell-to-cell transmission by measuring viral integration in target cells, and none have reported the multiplicity of macrophage-vectored transmission. Our finding of approximately 2 integrated copies of HIV-1 per target CD4+ T cell overall is comparable to studies of VS-mediated transmission between T cells using fluorescence in situ hybridization (FISH)-based measurements in highly permissive T cell lines (∼3 to 4 per infected cell) (50, 51). Interestingly, by examining the ratio of total DNA to integrated forms, we observed no difference compared to high-multiplicity cell-free infection of primary CD4+ T cells, suggesting that the enhanced efficiency of synaptic transmission derives from the quantity of infectious virions transferred rather than postentry effects on the target cell favoring nuclear trafficking and/or integration. This is an important insight in keeping with our previous observations (18) and supports efforts to target the entry pathway in cell-to-cell transmission.
The efficiency of this transmission route relates to the structure of the macrophage VS, which shares features of both the T cell VS and the dendritic cell (DC) infectious synapse (IS). In infected MDM, virions are principally sequestered in the VCC (38), and the modest effect of microtubule inhibitors, in contrast to the robust action of actin paralysis, suggests that transmission does not depend on polarized secretion of virions involving the microtubule organizing center (MTOC), as has been shown for T cells (52), but rather upon actin-mediated rearrangement of the VCC. However, these data do not exclude a role for the tubulin cytoskeleton in VCC regulation (53), and the molecular signaling involved in this active reorganization is an important area of future work. Unlike the DC IS, the macrophage VS is formed by productively infected cells, and as we show here, it remains dependent on gp120-CD4 interactions for viral transfer.
There are conflicting data in the literature regarding whether VS transmission represents a mechanism of dissemination that evades NAb attack (reviewed in reference 21). The conclusions of several studies appear to depend upon the cell-to-cell model system tested. We first demonstrated that transfer of HIV-1 from chronically HIV-1-infected Jurkat T cells to uninfected primary CD4+ target T cells was equivalently susceptible to NAb inhibition compared to cell-free infection (20). We concluded, based upon confocal and electron tomographic imaging and functional analyses, that this was because the VS formed between T cells was fully permissive for IgG entry; therefore, there was no kinetic barrier for NAb engagement with virus (20). However, another system based principally on cell-to-cell transmission of HIV-1 from T cells to the immortalized epithelial cell line HeLa expressing high levels of transgenic HIV-1 entry receptors (TZMbl [54]) showed loss of CD4bs-specific neutralization but preserved MPER antibody function (27). This result agrees with one recent report of DC-mediated transfer (45) but not another (55). Other studies, in which acutely transfected T cell lines transmit immature virions expressing Env in conformations that are unable to initiate immediate cell-membrane fusion (56), report more global resistance to bNMAb inhibition (28). Interestingly, deletion of the cytoplasmic tail of gp41 restores Env fusogenicity (28), resulting in enhanced neutralization by gp120-directed bNMAbs such as b12, but with considerably less impact on the MPER antibody 4E10 (28). Our data relating to macrophage VS-mediated transfer show that MPER-directed NAb suffer the most loss of activity, and results with 10E8 Fab and T20 suggest this is at least partially mediated by steric hindrance (28). This would be consistent with limited access of IgG to virus within the VCC (7, 8), to the rapid transfer of infectious HIV-1 from the VCC to contacting T cells (Fig. 1) (4), and with electron microscopic imaging which shows tight apposition of MDM and T cell membranes at the synaptic junction (4). MAb access to the VCC is likely to be limited by very low rates of diffusion through the narrow surface-connecting conduits (38). However, MAbs specific for cellular molecules may be actively trafficked to the VCC, as demonstrated by CD36-specific MAb binding to its cognate antigen on macrophages leading to internalization into the VCC and inhibition of virus release (39).
In contrast with MPER bNMAbs that most likely would act on virus transferring from MDM to T cells across an assembled functional VS, inhibition by CD4bs and 2G12 may occur largely by preventing VS assembly, similar to that observed for the T cell-T cell VS (20) and consistent with the significantly reduced number of Gag+ T cells recovered from MDM cocultures (Fig. 3A). Here, we have tested bNMAbs representing three major epitope clusters: the gp120 CD4bs, the glycan/glycopeptide supersite of vulnerability (57), and the MPER for comparative inhibition of cell-free and cell-to-cell infection. A fourth epitope cluster containing the V1V2 quaternary bNMAbs of which PG9 and PG16 are prototypes should also be evaluated in these systems.
As others have noted (45), we observed subtle but nevertheless potentially important Env strain-dependent effects on viral neutralization, which suggests that more than one Env should be tested in this type of study, as we have done here. Transmitted/founder virus Envs should also be investigated in this respect (58, 59). Such effects on neutralization are likely to relate both to global neutralization sensitivity (tier 1- versus tier 2-type viruses [44]) and to other features, such as glycan proximity to antibody binding sites. The impact of such glycans will be influenced not only by the env amino acid sequence but also by the producer cell type (60, 61). However, our data on the function of gp120-directed bNMAbs, particularly the glycan-dependent antibodies 2G12 and PGT121, which display equivalent activity against cell-cell and cell-free primary cell infection, are encouraging for vaccine developers. These data are consistent with reports that gp120-directed bNMAbs control established viral infection in a humanized mouse model of HIV-1 (62).
A number of recent studies, both in vitro and in vivo, are consistent with cell-to-cell spread of HIV-1 being the dominant mode of viral dissemination. Recent data, including those presented here, also highlight the potential importance of this mode of spread in allowing viral escape from antiretroviral therapy and some, but not all, NAb. The ability of the macrophage to survive for extended periods after infection and to store virus that can then be released across VS to efficiently infect T cells suggests that this cell type is more important to HIV-1 dissemination and pathogenesis than originally suspected.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Chen and U. O'Doherty for the generous gift of reagents and protocols, A. McMichael for support and guidance, and the Centre for AIDS Reagents, National Institute for Biological Standards and Control (NIBSC), and the Neutralizing Antibody Consortium for reagents.
C.J.A.D. was supported by the Wellcome Trust (094449/Z/10/Z), and Q.J.S. and R.A.R. are supported by the MRC (G0901732) and Dormeur Investment Services. J.C.K. and C.O. received funding through the NIH Center for HIV/AIDS Vaccine Immunology (CHAVI), UO1-AI067854, and the Bill & Melinda Gates Foundation's Collaboration for AIDS Vaccine Discovery (CAVD)/CA-VIMC, grant number 38619. Q.J.S. is a James Martin Senior Fellow and a Jenner Vaccine Institute Investigator.
C.J.A.D. and Q.J.S. devised the study. C.J.A.D., J.W., T.S., J.F., R.A.R., and Q.J.S. planned/conducted experiments. T.S., K.G., R.A.R., C.O., J.K., and J.F. contributed reagents/expertise. C.J.A.D. and Q.J.S. wrote and edited the paper.
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 4 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03245-13.
REFERENCES
- 1.Zalar A, Figueroa MI, Ruibal-Ares B, Bare P, Cahn P, de Bracco MM, Belmonte L. 2010. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antiviral Res. 87:269–271. 10.1016/j.antiviral.2010.05.005 [DOI] [PubMed] [Google Scholar]
- 2.Gavegnano C, Schinazi RF. 2009. Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir. Chem. Chemother. 20:63–78. 10.3851/IMP1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. 2007. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J. Cell Biol. 177:329–341. 10.1083/jcb.200609050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groot F, Welsch S, Sattentau QJ. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660–4663. 10.1182/blood-2007-12-130070 [DOI] [PubMed] [Google Scholar]
- 5.Bennett AE, Narayan K, Shi D, Hartnell LM, Gousset K, He H, Lowekamp BC, Yoo TS, Bliss D, Freed EO, Subramaniam S. 2009. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 5:e1000591. 10.1371/journal.ppat.1000591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welsch S, Groot F, Krausslich HG, Keppler OT, Sattentau QJ. 2011. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J. Virol. 85:7922–7927. 10.1128/JVI.00834-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koppensteiner H, Banning C, Schneider C, Hohenberg H, Schindler M. 2012. Macrophage internal HIV-1 is protected from neutralizing antibodies. J. Virol. 86:2826–2836. 10.1128/JVI.05915-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu H, Wang JJ, Qi M, Yoon JJ, Wen X, Chen X, Ding L, Spearman P. 2012. The intracellular virus-containing compartments in primary human macrophages are largely inaccessible to antibodies and small molecules. PLoS One 7:e35297. 10.1371/journal.pone.0035297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharova N, Swingler C, Sharkey M, Stevenson M. 2005. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J. 24:2481–2489. 10.1038/sj.emboj.7600707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aggarwal A, McAllery S, Turville SG. 2013. Revising the role of myeloid cells in HIV pathogenesis. Curr. HIV/AIDS Rep. 10:3–11. 10.1007/s11904-012-0149-1 [DOI] [PubMed] [Google Scholar]
- 11.Koppensteiner H, Brack-Werner R, Schindler M. 2012. Macrophages and their relevance in human immunodeficiency virus type I infection. Retrovirology 9:82. 10.1186/1742-4690-9-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. 2008. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 4:e1000015. 10.1371/journal.ppat.1000015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waki K, Freed EO. 2010. Macrophages and cell-cell spread of HIV-1. Viruses 2:1603–1620. 10.3390/v2081603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sattentau QJ. 2010. Cell-to-cell spread of retroviruses. Viruses 2:1306–1321. 10.3390/v2061306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. 2012. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490:283–287. 10.1038/nature11398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat. Commun. 3:1320. 10.1038/ncomms2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carr JM, Hocking H, Li P, Burrell CJ. 1999. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 265:319–329. 10.1006/viro.1999.0047 [DOI] [PubMed] [Google Scholar]
- 18.Duncan CJ, Russell RA, Sattentau QJ. 2013. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 27:2201–2206. 10.1097/QAD.0b013e3283632ec4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann DL, Gartner S, Le Sane F, Buchow H, Popovic M. 1990. HIV-1 transmission and function of virus-infected monocytes/macrophages. J. Immunol. 144:2152–2158 [PubMed] [Google Scholar]
- 20.Martin N, Welsch S, Jolly C, Briggs JA, Vaux D, Sattentau QJ. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 84:3516–3527. 10.1128/JVI.02651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schiffner T, Sattentau QJ, Duncan CJ. 2013. Cell-to-cell spread of HIV-1 and evasion of neutralizing antibodies. Vaccine 31:5789–5797. 10.1016/j.vaccine.2013.10.020 [DOI] [PubMed] [Google Scholar]
- 22.Burton DR, Poignard P, Stanfield RL, Wilson IA. 2012. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337:183–186. 10.1126/science.1225416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong PD, Mascola JR, Nabel GJ. 2013. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat. Rev. Immunol. 13:693–701. 10.1038/nri3516 [DOI] [PubMed] [Google Scholar]
- 24.Mouquet H, Nussenzweig MC. 2013. HIV: roadmaps to a vaccine. Nature 496:441–442. 10.1038/nature12091 [DOI] [PubMed] [Google Scholar]
- 25.Huang KH, Bonsall D, Katzourakis A, Thomson EC, Fidler SJ, Main J, Muir D, Weber JN, Frater AJ, Phillips RE, Pybus OG, Goulder PJ, McClure MO, Cooke GS, Klenerman P. 2010. B-cell depletion reveals a role for antibodies in the control of chronic HIV-1 infection. Nat. Commun. 1:102. 10.1038/ncomms1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin N, Sattentau Q. 2009. Cell-to-cell HIV-1 spread and its implications for immune evasion. Curr. Opin. HIV AIDS 4:143–149. 10.1097/COH.0b013e328322f94a [DOI] [PubMed] [Google Scholar]
- 27.Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 8:e1002634. 10.1371/journal.ppat.1002634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durham ND, Yewdall AW, Chen P, Lee R, Zony C, Robinson JE, Chen BK. 2012. Neutralization resistance of HIV-1 virological synapse-mediated infection is regulated by the gp41 cytoplasmic tail. J. Virol. 86:7484–7495. 10.1128/JVI.00230-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582–12595. 10.1128/JVI.00381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edmonds TG, Ding H, Yuan X, Wei Q, Smith KS, Conway JA, Wieczorek L, Brown B, Polonis V, West JT, Montefiori DC, Kappes JC, Ochsenbauer C. 2010. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology 408:1–13. 10.1016/j.virol.2010.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter GC, Bernstone L, Baskaran D, James W. 2011. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology 409:234–250. 10.1016/j.virol.2010.10.018 [DOI] [PubMed] [Google Scholar]
- 32.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. 2008. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329:112–124. 10.1016/j.jim.2007.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 81:1000–1012. 10.1128/JVI.01629-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mexas AM, Graf EH, Pace MJ, Yu JJ, Papasavvas E, Azzoni L, Busch MP, Di Mascio M, Foulkes AS, Migueles SA, Montaner LJ, O'Doherty U. 2012. Concurrent measures of total and integrated HIV DNA monitor reservoirs and ongoing replication in eradication trials. AIDS 26:2295–2306. 10.1097/QAD.0b013e32835a5c2f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agosto LM, Yu JJ, Dai J, Kaletsky R, Monie D, O'Doherty U. 2007. HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 368:60–72. 10.1016/j.virol.2007.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liszewski MK, Yu JJ, O'Doherty U. 2009. Detecting HIV-1 integration by repetitive-sampling Alu-gag PCR. Methods 47:254–260. 10.1016/j.ymeth.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasiliver-Shamis G, Dustin ML, Hioe CE. 2010. HIV-1 virological synapse is not simply a copycat of the immunological synapse. Viruses 2:1239–1260. 10.3390/v2051239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan J, Sattentau QJ. 2013. The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends Microbiol. 21:405–412. 10.1016/j.tim.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 39.Berre S, Gaudin R, Cunha de Alencar B, Desdouits M, Chabaud M, Naffakh N, Rabaza-Gairi M, Gobert FX, Jouve M, Benaroch P. 2013. CD36-specific antibodies block release of HIV-1 from infected primary macrophages and its transmission to T cells. J. Exp. Med. 210:2523–2538. 10.1084/jem.20130566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mlcochova P, Pelchen-Matthews A, Marsh M. 2013. Organization and regulation of intracellular plasma membrane-connected HIV-1 assembly compartments in macrophages. BMC Biol. 11:89. 10.1186/1741-7007-11-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293. 10.1084/jem.20030648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jolly C, Mitar I, Sattentau QJ. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 81:13916–13921. 10.1128/JVI.01585-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Plata MT, Puigdomenech I, Izquierdo-Useros N, Puertas MC, Carrillo J, Erkizia I, Clotet B, Blanco J, Martinez-Picado J. 2013. The infectious synapse formed between mature dendritic cells and CD4+ T cells is independent of the presence of the HIV-1 envelope glycoprotein. Retrovirology 10:42. 10.1186/1742-4690-10-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84:1439–1452. 10.1128/JVI.02108-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sagar M, Akiyama H, Etemad B, Ramirez N, Freitas I, Gummuluru S. 2012. Transmembrane domain membrane proximal external region but not surface unit-directed broadly neutralizing HIV-1 antibodies can restrict dendritic cell-mediated HIV-1 trans-infection. J. Infect. Dis. 205:1248–1257. 10.1093/infdis/jis183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98. 10.1038/nature10347 [DOI] [PubMed] [Google Scholar]
- 47.Sigal A, Baltimore D. 2012. As good as it gets? The problem of HIV persistence despite antiretroviral drugs. Cell Host Microbe 12:132–138. 10.1016/j.chom.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 48.Jolly C, Booth NJ, Neil SJ. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84:12185–12199. 10.1128/JVI.01447-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cory TJ, Schacker TW, Stevenson M, Fletcher CV. 2013. Overcoming pharmacologic sanctuaries. Curr. Opin. HIV AIDS 8:190–195. 10.1097/COH.0b013e32835fc68a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J. Virol. 85:7169–7176. 10.1128/JVI.00231-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russell RA, Martin N, Mitar I, Jones E, Sattentau QJ. 2013. Multiple proviral integration events after virological synapse-mediated HIV-1 spread. Virology 443:143–149. 10.1016/j.virol.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 52.Jolly C, Welsch S, Michor S, Sattentau QJ. 2011. The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog. 7:e1002226. 10.1371/journal.ppat.1002226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaudin R, Berre S, Cunha de Alencar B, Decalf J, Schindler M, Gobert FX, Jouve M, Benaroch P. 2013. Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PLoS One 8:e69450. 10.1371/journal.pone.0069450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choudhry V, Zhang MY, Harris I, Sidorov IA, Vu B, Dimitrov AS, Fouts T, Dimitrov DS. 2006. Increased efficacy of HIV-1 neutralization by antibodies at low CCR5 surface concentration. Biochem. Biophys. Res. Commun. 348:1107–1115. 10.1016/j.bbrc.2006.07.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su B, Xu K, Lederle A, Peressin M, Biedma ME, Laumond G, Schmidt S, Decoville T, Proust A, Lambotin M, Holl V, Moog C. 2012. Neutralizing antibodies inhibit HIV-1 transfer from primary dendritic cells to autologous CD4 T-lymphocytes. Blood 120:3708–3717. 10.1182/blood-2012-03-418913 [DOI] [PubMed] [Google Scholar]
- 56.Dale BM, McNerney GP, Thompson DL, Hubner W, de Los Reyes K, Chuang FY, Huser T, Chen BK. 2011. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10:551–562. 10.1016/j.chom.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong L, Lee JH, Doores KJ, Murin CD, Julien JP, McBride R, Liu Y, Marozsan A, Cupo A, Klasse PJ, Hoffenberg S, Caulfield M, King CR, Hua Y, Le KM, Khayat R, Deller MC, Clayton T, Tien H, Feizi T, Sanders RW, Paulson JC, Moore JP, Stanfield RL, Burton DR, Ward AB, Wilson IA. 2013. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 20:796–803. 10.1038/nsmb.2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 86:2715–2728. 10.1128/JVI.06157-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 206:1273–1289. 10.1084/jem.20090378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liedtke S, Adamski M, Geyer R, Pfutzner A, Rubsamen-Waigmann H, Geyer H. 1994. Oligosaccharide profiles of HIV-2 external envelope glycoprotein: dependence on host cells and virus isolates. Glycobiology 4:477–484. 10.1093/glycob/4.4.477 [DOI] [PubMed] [Google Scholar]
- 61.Willey RL, Shibata R, Freed EO, Cho MW, Martin MA. 1996. Differential glycosylation, virion incorporation, and sensitivity to neutralizing antibodies of human immunodeficiency virus type 1 envelope produced from infected primary T lymphocyte and macrophage cultures. J. Virol. 70:6431–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, Zang T, Dorner M, Billerbeck E, Labitt RN, Gaebler C, Marcovecchio PM, Incesu RB, Eisenreich TR, Bieniasz PD, Seaman MS, Bjorkman PJ, Ravetch JV, Ploss A, Nussenzweig MC. 2012. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 492:118–122. 10.1038/nature11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.