

ABSTRACT
Hantaviruses successfully replicate in primary human endothelial cells by restricting the early induction of beta interferon (IFN-β) and interferon-stimulated genes (ISGs). Gn proteins from NY-1V, ANDV, and TULV, but not PHV, harbor elements in their 142-residue cytoplasmic tails (GnTs) that inhibit RIG-I/MAVS/TBK1-TRAF3-directed IFN-β induction. Here, we define GnT interactions and residues required to inhibit TRAF3-TBK1-directed IFN-β induction and IRF3 phosphorylation. We observed that GnTs bind TRAF3 via residues within the TRAF-N domain (residues 392 to 415) and that binding is independent of the MAVS-interactive TRAF-C domain (residues 415 to 568). We determined that GnT binding to TRAF3 is mediated by C-terminal degrons within NY-1V or ANDV GnTs and that mutations that add degrons to TULV or PHV GnTs confer TRAF3 binding. Further analysis of GnT domains revealed that TRAF3 binding is a discrete GnT function, independent of IFN regulation, and that residues 15 to 42 from the NY-1V GnT C terminus are required for inhibiting TBK1-directed IFN-β transcription. Mutagenesis of the NY-1V GnT revealed that altering tyrosine 627 (Y627A/S/F) abolished GnT regulation of RIG-I/TBK1-directed IRF3 phosphorylation and transcriptional responses of ISRE, κB, and IFN-β promoters. Moreover, GnTs from NY-1V, ANDV, and TULV, but not PHV, inhibited RIG-I-directed IRF3 phosphorylation. Collectively, these findings suggest a novel role for GnTs in regulating RIG-I/TBK1 pathway-directed IRF3 phosphorylation and IFN-β induction and define virulence determinants within GnTs that may permit the attenuation of pathogenic hantaviruses.
IMPORTANCE
INTRODUCTION
Hantaviruses primarily infect human endothelial cells (ECs) and nonlytically cause hemorrhagic fever with renal syndrome (HFRS) or hantavirus pulmonary syndrome (HPS) (1–7). HFRS results from infection by Eurasian hantaviruses (Hantaan virus [HTNV], Dobrava virus [DOBV], and Puumala virus[PUUV]) (8–11), while hantaviruses identified throughout the Americas (i.e., Andes virus [ANDV], Sin Nombre virus [SNV], and New York 1 virus [NY-1V]) are associated with HPS (1, 4, 5, 12–14). In contrast, Tula virus (TULV) and Prospect Hill virus (PHV) are hantaviruses that have not been associated with any human disease (15, 16). TULV and PHV differ from pathogenic hantaviruses by their use of discrete integrin receptors (17, 18), and in addition, PHV fails to regulate early interferon responses or replicate productively in human endothelial cells (19–21).
Hantaviruses are enveloped viruses with a trisegmented (segments S, M, and L) negative-sense RNA genome and constitute a unique genus within the Bunyaviridae family (11). The hantavirus M segment encodes a polyprotein precursor that is cotranslationally cleaved into two integral membrane surface glycoproteins, Gn and Gc, that are trafficked to the endoplasmic reticulum (ER)/cis-Golgi (22). GnGc hetero-oligomers are acquired on the virion's surface by viral budding into the lumen of the ER/cis-Golgi (5). Gc contains a short (<10-residue) cytoplasmic tail, while Gn contains a 142-residue cytoplasmic tail (GnT) with several potential functions in the viral life cycle (5). The GnT directs binding of viral nucleocapsid complexes, has matrix protein functions that nucleate viral assembly and budding (23), and with the exception of the PHV GnT, regulates RIG-I-directed beta interferon (IFN-β) induction (19, 20, 24–26).
Type I IFN is induced by the stimulation of innate cellular sensors that signal the activation of IFN response factors (IRFs) 3, 5, and 7 and NF-κB through cytoplasmic pathways (27, 28). Replicating RNA viruses generate small amounts of cytoplasmic double-stranded RNA (dsRNA), or 5′-triphosphate-containing RNAs, that are detected by MDA5 or RIG-I sensors and result in the mitochondrial assembly of MAVS complexes (28–33). MAVS binds tumor necrosis factor receptor-associated factor 3 (TRAF3), which recruits TBK1/IKKε kinases that activate NF-κB and direct the phosphorylation of constitutively expressed IRF3 (27, 34–40). Activated IRFs and NF-κB translocate to the nucleus and form transcriptional complexes with CBP/p300 and ATF2/cJUN on the IFN-β enhanceosome that transcriptionally induce IFN-β (28, 29, 32, 41, 42). IRFs additionally direct transcription from promoters containing IFN-stimulated responses elements (ISREs). IFN-β binds in an autocrine or paracrine fashion to type I IFN receptors, directing the induction of a variety of antiviral interferon-stimulated genes (ISGs) (43).
TRAF3 is required for IFN induction from virtually all inducers and a central point of regulation of IFN responses (27, 34–39). TRAF3 is an E3 ligase that normally directs the constitutive degradation of the NF-κB inducer NIK and forms complexes with TRAF2-cIAP or ORUD7B that regulate NF-κB and TBK1/IKKε activation (27, 34–36, 38, 39, 44). TRAF3 and TBK1 are further regulated by degradative K48-linked and nondegradative K63-linked ubiquitination, and deubiquitinating TRAF3 inhibits IRF3/7-directed IFN-β induction (36, 45, 46).
Hantavirus replication is highly sensitive to IFN pretreatment, and hantaviruses are grown in IFN locus-defective Vero E6 cells (19). However, the effects of IFN addition are nearly absent when IFN is added 1 day postinfection (19), and at late times after infection of human endothelial cells, hantaviruses induce high-level ISG responses (21). In order to productively replicate in their human endothelial cell targets, hantaviruses have evolved mechanisms for regulating the early production of IFN and antiviral ISGs (19–21, 24, 25, 47). In fact, PHV rapidly induces IFN-β and ISG responses that restrict its replication in human endothelial cells, and this potentially explains the absence of PHV-associated human disease (21, 47). These findings suggest that permissive hantavirus replication in human endothelial cells results from the selective restriction of early, but not late, IFN responses (11, 19–21).
Hantavirus GnGc and GnT proteins have been shown to regulate IFN induction in response to the expression of pathway-specific RIG-I and TBK1 inducers, suggesting a mechanism for early IFN regulation within hantavirus-infected endothelial cells (19, 20, 24, 25). Expression of GnGc or GnT proteins from NY-1V, ANDV, TULV, and HTNV, but not PHV, results in the regulation of RIG-I/TBK1-directed ISRE, κB, and IFN-β transcriptional responses (19–21). GnTs fail to regulate transcription directed by constitutively active IRF3-5D, suggesting that GnTs prevent IRF3 and NF-κΒ activation by regulating TBK1-TRAF3 complexes (20). This regulatory mechanism is supported by findings indicating that the NY-1V GnT binds TRAF3 and that GnT expression or NY-1V infection disrupts the formation of TRAF3-TBK1 complexes (20). Degrons at the C termini of NY-1V, HTNV, and ANDV GnTs direct ubiquitination and degradation of GnTs and further suggest a means by which GnTs might regulate ubiquitination-sensitive TRAF3-TBK1 complexes (26). However, the TULV GnT regulates RIG-I/TBK1-directed IFN-β induction yet lacks a degron domain (24, 25). These findings question whether degrons are required for TRAF3 binding and whether TBK1 inhibition is a function of discrete elements within hantavirus GnTs.
Here, we investigate interactive GnT and TRAF3 domains and demonstrate that degrons within hantavirus GnTs confer TRAF3 binding. We found that the TRAF3 residues required for binding to MAVS (residues 415 to 568) are dispensable for binding GnTs but that deleting residues 392 to 415 of TRAF3 abolished GnT binding to TRAF3. Our studies demonstrate that IFN regulation by the NY-1V GnT requires a domain 15 to 42 residues from the GnT C terminus that is upstream of the C-terminal degron. Site-directed mutagenesis of GnT residues within this domain resulted in the discovery of a single tyrosine residue (Y627) in the NY-1V GnT that abolished regulation of RIG-I- and TBK1-directed IFN-β transcription and IRF3 phosphorylation. Consistent with GnT regulation of IRF3 responses, expression of GnTs from NY-1V, ANDV, and TULV, but not PHV, inhibited RIG-I-directed phosphorylation of IRF3. These findings define potential virulence determinants within the GnT C terminus that are required for NY-1V GnT regulation of IFN signaling pathway activation.
MATERIALS AND METHODS
Cells and antibodies.
Cos7 and HEK293T cells (ATCC CRL-1586 and CRL-1573) were grown in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), as previously described (18). Antibodies were procured as follows: monoclonal anti-Gal4 (RK5C1, sc-510; Stratagene); monoclonal anti-myc (9E10; Santa Cruz, Cell Signaling); anti-actin (A5441; Sigma); monoclonal anti-β-actin (12CA5; Roche); monoclonal antibody (MAb) and polyclonal anti-Gal4 (sc-510 and sc-577; Santa Cruz); ANDV Gn MAb (H1808-50; US Biologicals); antibodies to TBK1 (3504), phospho-TBK1 (Ser172) (5483), IRF3 (4302), phospho-IRF3 (pSer396; 4947), anti-IKKε (2905) and anti-IRF3, anti-RIG-I, anti-MAVS, anti-MDA5, or anti-pIRF3-S396 (all from Cell Signaling); and monoclonal anti-Flag M2 (Agilent). Antinucleocapsid rabbit polyclonal serum directed at the NY-1V nucleocapsid protein was used to detect nucleocapsid protein as previously described (18, 19, 21). Horseradish peroxidase (HRP)-conjugated anti-mouse and goat anti-rabbit-HRP conjugates were purchased from GE Healthcare.
Plasmids.
Plasmids expressing NY-1V, ANDV, TULV, and PHV cytoplasmic tail domains (pBIND PHV GnT) were generated by C-terminally fusing the GnT coding region to a Gal4 tag as previously described (19, 48, 49). pBIND GnT and pBIND GnT-C42 constructs were generated by PCR amplification of GnGc coding regions of NY-1V, ANDV, TULV, and PHV as previously described (19, 20, 24, 26) using PCR primers containing BamHI and XbaI sites and directionally ligated into pBIND. Optimized ANDV-GnGc was synthesized by GenScript and cloned BamHI-XbaI into pcDNA3 (pcDNA3-ANDV-optiGnGc). The following constructs were purchased from Addgene: human pcDNA3-TBK-1-flag, human pEF-BOS-MAVS-flag, and human pEF-BOS-MDA5-flag (Addgene-27225) (50). RIG-I-flag (RIG-I CARD, residues 1 to 284) expression plasmid was from Michael Gale (51). pCMVBL-IRF3-T7 (52), pRK-TRAF3-wt (19, 20), and pRK-TRAF3-N415 or pRK-TRAF3-N392 truncated expression constructs were generously provided, purchased, or generated by site-directed mutagenesis as previously described (19, 20).
Transcriptional reporter assays.
For luciferase assays, transfections were performed using calcium phosphate or polyethyleneimine (PEI; 1 μg DNA/3 μg) and 60% confluent HEK293T cells in triplicate with a constant amount of total plasmid DNA as previously described (19, 24, 53). HEK293T were cotransfected with either ISRE-luciferase, κB-luciferase, or IFN-β-luciferase promoter constructs (Clontech) as indicated and an internal control renilla luciferase construct (PRL-null; Promega) as previously described (19, 20, 24). Cells were cotransfected with a plasmid expressing myc-tagged TBK1, Flag-N-RIG-I (helicase domain deficient) (33, 51), or Flag-MDA5 as pcDNA3 constructs (0.25 μg) or empty vector (19). Cells were cotransfected with constant amounts of total DNA using indicated amounts of NY-1V, ANDV, PHV, or TULV GnTs, GnT-C42, or deletion/mutated constructs as described or empty pBIND vector. Cells were lysed 48 h posttransfection with 1× passive lysis buffer (Promega) for 15 min at room temperature. For poly(I·C) stimulation, cells were transfected as above, and 1 day posttransfection cells were PEI transfected (53) with 3 μg/ml of Poly I·C (Sigma) and assayed 1 day later for luciferase promoter responses. Luciferase assays were performed using the Dual Luciferase Assay kit (Promega) according to the manufacturer's protocol, and detection was done using a luminometer. Assays were performed in triplicate at least three times with similar results (33, 46, 54–56). In the figures, error bars indicate the standard deviations from negative controls, with asterisks indicating statistical significance as determined by Student's t test (GraphPad Prism software), and P values are indicated in figure legends (33, 57–59).
Immunoprecipitation and Western blot analysis.
For coimmunoprecipitation experiments, Cos7 or HEK293T cells were transfected with 1 μg of pBIND-GnT constructs and 0.5 μg pRK-TRAF3 N415 using Fugene 6 (Roche) (20). Transfected cells were analyzed 48 h posttransfection in coimmunoprecipitation lysis buffer (20, 60). Where indicated, MG132 (50 μM) was added 6 h before cell lysis. Lysates were clarified by centrifugation, and the GnTs were immunoprecipitated with anti-Gal4 monoclonal antibodies (sc-2003) and protein A/G Plus agarose beads (24). Coimmunoprecipitated proteins were analyzed by Western blotting as previously described.
pBIND, pBIND-GnT, pBIND-GnT-C42, pcDNA3-TBK1, and pRK-TRAF3 or pRK-TRAF3-N415/N392 expression was analyzed by Western blotting of cotransfected HEK293 cells or Cos7 cells (24). Cells were lysed in Laemmli buffer 48 h posttransfection and subjected to Western blotting using anti-Gal4 (GnT) (1:1,000), anti-myc (1:1,000), or anti-Flag M2 (1:1,000) (20). Blots were washed, incubated with HRP-conjugated secondary antibodies, and developed by chemiluminescence with ECL reagent (Pierce) as previously described (24). Where indicated, blots were treated with stripping buffer (62.5 mM Tris-HCl [pH 6.8], 20% SDS, 100 mM β-mercaptoethanol), incubated with monoclonal anti-β-actin (1:5,000), and developed as described above.
TBK1 and IRF3 analysis.
HEK293T cells were lysed in 0.5% SDS lysis buffer (150 mM NaCl, 40 mM Tris, 2 mM EDTA, 5 mM NaF, 1 mM Na4P2O7, 1 mM Na3VO4, 0.5% SDS, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitor) and clarified by centrifugation at 14,000 rpm for 30 min at 4°C. Equivalent amounts of lysate were separated on SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose and incubated with a 1:1,000 dilution of antiactin, anti-Gal4, anti-ANDV Gn, anti-TBK1, anti-pTBK1-S172, anti-IRF3, or anti-pIRF3-S396 followed by horseradish peroxidase-conjugated sheep anti-mouse or goat anti-rabbit immunoglobulin G (GE Healthcare). Proteins were detected by fluorography using the Luminata Forte system (Millipore).
RESULTS
Gn regulation of RIG-I- and TBK1-directed transcriptional responses and IRF3 activation.
In contrast to ANDV, NY-1V, HTNV, and TULV, which transiently restrict the early induction of IFN, PHV fails to regulate early interferon induction or replicate within human endothelial cells (19, 20, 24). Previous studies demonstrated that expressing the cytoplasmic tail of Gn proteins from hantaviruses other than PHV inhibits RIG-I- and TBK1-directed activation of IRF3 and NF-κB as well as transcription from ISRE, κB, and IFN-β promoters (19, 20, 24). However, GnTs fail to inhibit ISRE transcription induced by expression of constitutively active IRF3-5D, and this pathway-specific analysis suggests that regulation occurs at the level of the TBK1 complex (19, 20, 24). Here, we comparatively evaluate roles for the GnT in regulating IFN-β transcriptional response and IRF3 phosphorylation. We expressed NY-1V, ANDV, TULV, and PHV GnTs and evaluated their ability to inhibit IFN-β promoter transcriptional responses directed by poly(I·C), RIG-I, MDA5, and TBK1. We found that GnTs from NY-1V, ANDV, and TULV, but not PHV, dramatically inhibited IFN-β promoter transcriptional responses directed by each pathway-specific activator (Fig. 1A to D).
FIG 1.
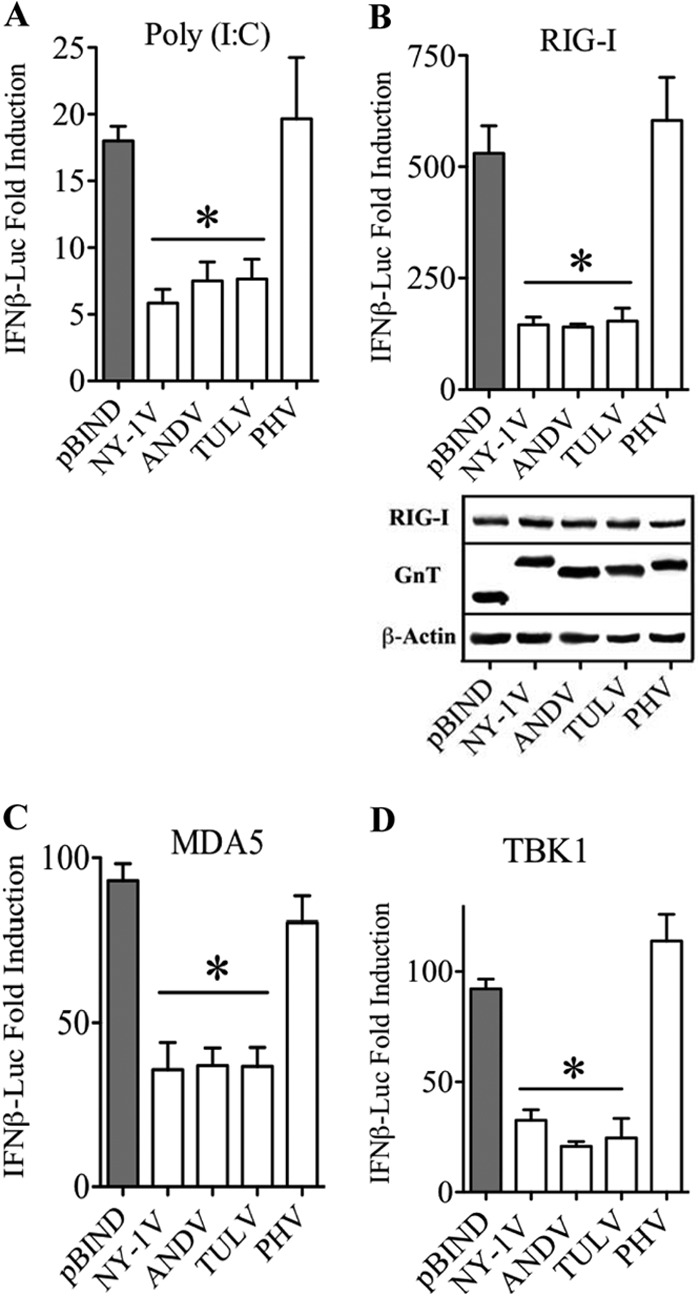
NY-1V, ANDV, and TULV GnTs inhibit IFN-β transcriptional responses. HEK293 cells were transfected with an IFN-β enhanceosome (42)-driven firefly luciferase reporter and a constitutively expressed Renilla luciferase plasmid in the presence or absence of poly(I·C) (A), RIG-I (B), MDA5 (C), or TBK1 (D) expression vectors. Cells were cotransfected with plasmids expressing NY-1V, ANDV, TULV, or PHV GnTs. Empty vector (pBIND) was used to maintain constant DNA transfection levels and as a positive control. Luciferase activity within lysates was determined 48 h posttransfection, normalized to Renilla luciferase activity, and reported as the fold increase compared to that of controls lacking inducer. Assays were performed in triplicate with similar results in at least 2 separate experiments. Asterisks indicate statistical significance (P < 0.05) as determined by Student's t test. Cell lysates were analyzed by Western analysis for Gn protein, RIG-I, MDA5, and TBK1 expression levels and compared to endogenous β-actin protein controls.
The 1,138-residue hantavirus GnGc polyprotein is synthesized and cotranslationally processed in the ER to form Gn:Gc heterodimers, with a 142-residue GnT providing the only cytoplasmic element within Gn. Here, we determined whether the full-length GnGc regulates IFN transcriptional responses and IRF3 phosphorylation. Similar to GnT, expressing GnGc inhibits RIG-I-, MDA5-, and TBK1-directed, but not IRF3-5D-directed, transcription from an ISRE promoter (Fig. 2A) and the IFN-β enhanceosome (Fig. 2B). In addition, GnGc expression also inhibited RIG-I-directed IRF3 phosphorylation (Fig. 2C). Collectively, these findings demonstrate that NY-1V, ANDV, and TULV GnTs as well as the GnGc polyprotein inhibit RIG-I-induced transcriptional responses by impacting TBK1 phosphorylation of IRF3.
FIG 2.
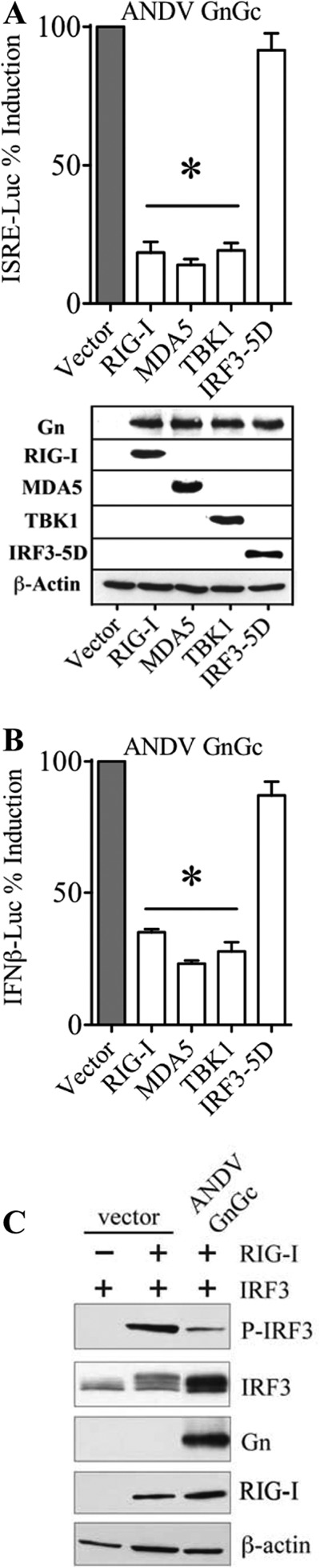
ANDV GnGc inhibits IRF3 phosphorylation and ISRE transcriptional responses. HEK293T cells were cotransfected with plasmid expressing ANDV GnGc, ISRE (A), or IFN-β (B) promoter-driven firefly luciferase reporter and a constitutively expressed Renilla luciferase plasmid in the absence (empty vector) or presence of RIG-I, MDA5, TBK1, or IRF3-5D expression plasmids. After 24 h, cells were harvested and measured for firefly luciferase activity, which was normalized to Renilla luciferase levels and presented as percent induction compared to control empty vector. Empty-vector positive-control induction was standardized to 100% for comparison. Asterisks indicate statistical significance (P < 0.05). Cell lysates were analyzed by Western analysis for Gn protein and RIG-I, MDA5, TBK1, IRF3-5D, and expression levels were compared to those of endogenous β-actin protein controls. (C) HEK293T cells were transfected with IRF3-T7 expression plasmid and empty vector or ANDV GnGc in the presence (+) or absence (−) of RIG-I expression plasmid (inducer). After 24 h, cells were harvested and analyzed by Western analysis for RIG-I, pIRF3 (α-pIRF3 S396), total IRF3, and ANDV GnGc (α-Gn) protein expression and compared to endogenous β-actin protein controls.
TRAF-N domains of TRAF3 mediate binding to NY-1V and ANDV GnTs.
We previously demonstrated that expression of the NY-1V GnT or NY-1V infection inhibits the formation of TRAF3-TBK1 complexes and that TRAF3 binds to the NY-1V GnT (20). TRAF3 recruits TBK1 to signaling complexes that are essential for the induction of type I IFN (38). Interactions that mediate TRAF3 binding to TBK1 are regulated by ubiquitination and impacted by a growing list of factors (34, 36, 40, 45). However, residues 440 to 442 within the TRAF-C domain of TRAF3 are required for binding to MAVS and UXT-V1 as well as for the downstream recruitment of TBK1 to MAVS complexes (Fig. 3A) (36, 45, 61). To investigate GnT regulatory interactions, we analyzed TRAF3 domains required for binding to the GnT using C-terminal truncations of TRAF3 (Fig. 3A). Immunoprecipitation of the ANDV GnT, but not the PHV GnT, resulted in the coprecipitation of TRAF3-N415, which lacks the MAVS interactive TRAF-C domain (Fig. 3A and B). The NY-1V GnT also coprecipitated TRAF3-N415 (Fig. 3C), but following deletion of an additional 24 residues within the TRAF-N domain (TRAF3-N391) neither NY-1V nor ANDV GnTs coprecipitated TRAF3-N391 (Fig. 3C). This indicates that TRAF3 binding to NY-1V and ANDV GnTs requires residues within the TRAF-N domain and that the MAVS-interactive TRAF-C domain of TRAF3 is dispensable for GnT binding.
FIG 3.
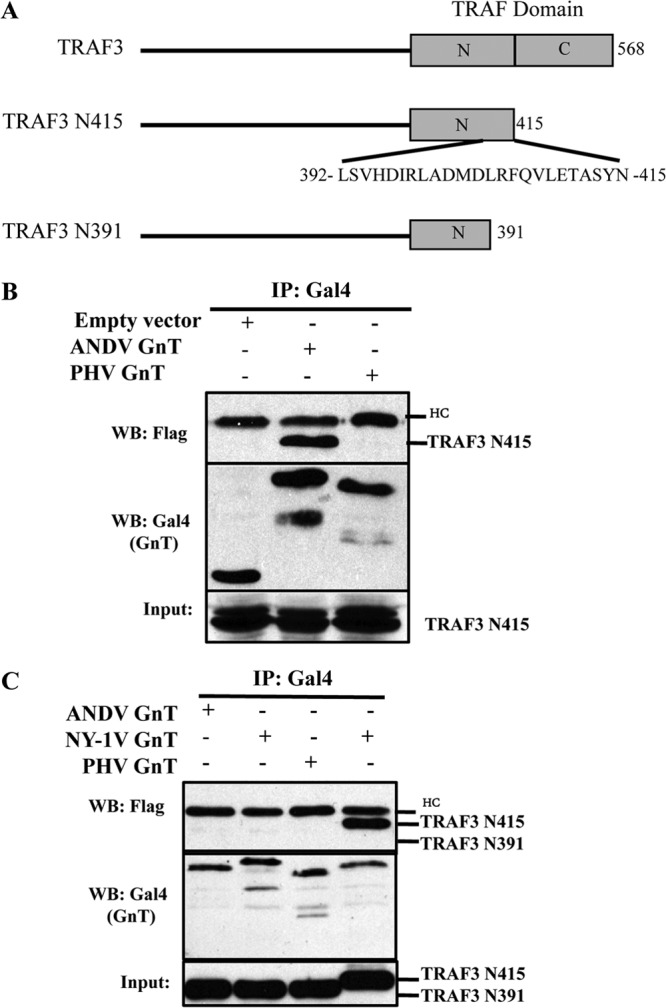
TRAF3 domains required for binding hantavirus GnTs. (A) Stick figure delineating TRAF-N and TRAF-C (MAVS binding) elements within the TRAF domain of full-length TRAF3 or truncated TRAF3 (TRAF3-415 and TRAF3-391) expression constructs. (B) Cos7 cells were transfected with pBIND empty vector, pBIND-ANDV GnT, or pBIND-PHV GnT as well as pRK-TRAF3-N415 as previously described (20). Briefly, 6 h prior to lysis, transfected cells were treated with MG132 and lysed 48 h posttransfection. Hantavirus GnTs were immunoprecipitated using anti-GAL4 antibody and protein A/G Plus agarose beads. Coprecipitated TRAF3-N415 was detected by Western blotting (WB) using the M2 anti-FLAG antibody (20). Input TRAF3-N415 and Gal4-GnT proteins were analyzed by anti-FLAG or anti-Gal4 WB as indicated. HC, IgG heavy chain. (C) Cells were transfected as described for Fig. 1B with NY1-V, ANDV, or PHV Gal4-GnT constructs and analyzed for their ability to coprecipitate TRAF3-N415 or TRAF3-N391 as indicated.
GnT degron is required for interactions with TRAF3.
The GnTs of NY-1V and ANDV contain a C-terminal degron domain that directs their ubiquitination and degradation (26), and TRAF3 is an E3 ubiquitin ligase that is required for RIG-I-directed activation of TBK1 (36, 38). In contrast, both PHV and TULV GnTs lack degron domains and are stably expressed (26). However, the TULV GnT regulates TBK1-directed ISRE and IFN transcriptional responses, similar to what is seen with degron-containing NY-1V and ANDV GnTs (19, 20, 24). These findings suggest that IFN regulation and TRAF3 binding may be conferred by discrete elements within specific hantavirus GnTs. To determine the role of the degron in TRAF3 binding, we mutated 3 to 5 residues within NY-1V, ANDV, PHV, and TULV GnTs in order to either abolish or add degron sequences (Fig. 4A). Figure 4A presents a summary of residue changes made to GnTs that ablate the NY-1V and ANDV degrons (−Deg) or add degron (+Deg) domains to TULV and PHV GnTs. We found that NY-1V or ANDV GnT degron-deficient mutants (GnT−Deg) are stably expressed (Fig. 4B). In contrast, wild-type (wt) PHV and TULV GnTs are normally stably expressed, but addition of degrons to PHV or TULV GnTs (GnT+Deg) results in their proteasomal degradation (Fig. 4C). Similar to what is shown in Fig. 3, we analyzed the ability of wt and mutant GnTs (+Deg and −Deg) to coprecipitate TRAF3 (Fig. 5A to C). Figure 5A demonstrates that the wt NY-1V GnT binds TRAF3 while a degron-deficient mutant failed to coprecipitate TRAF3. In contrast, the wt PHV GnT protein failed to bind TRAF3 while the PHV GnT+Deg mutant coprecipitated TRAF3 (Fig. 5A). Consistent with this, we observed that neither the stably expressed wt TULV GnT nor ANDV GnT−Deg mutant coprecipitated TRAF3 (Fig. 5B and C), while both the unstable TULV GnT+Deg mutant and the wt ANDV GnT coprecipitated TRAF3. These findings indicate that the presence of degron domains determines the ability of hantavirus GnT proteins to bind TRAF3.
FIG 4.

C-Terminal degron domains determine GnT stability. (A) C-terminal sequences of wt GnT of NY-1V, ANDV, PHV, and TULV proteins are contrasted with residue changes in GnT−Deg mutants. Residues mutated to add (+Deg) or abolish (−Deg) GnT degron domains are shown, and a summary of protein stability of the constructs is presented (26). (B, C) Cos7 cells were transfected with Gal4 GnT wt, +Deg, or −Deg constructs from NY-1V, ANDV, PHV, and TULV (20). Transfected cells were treated with MG132 6 h prior to lysis and lysed 48 h posttransfection (24). Gal4-GnT proteins were detected by anti-Gal4 Western analysis and compared to levels of β-actin (anti-β actin).
FIG 5.
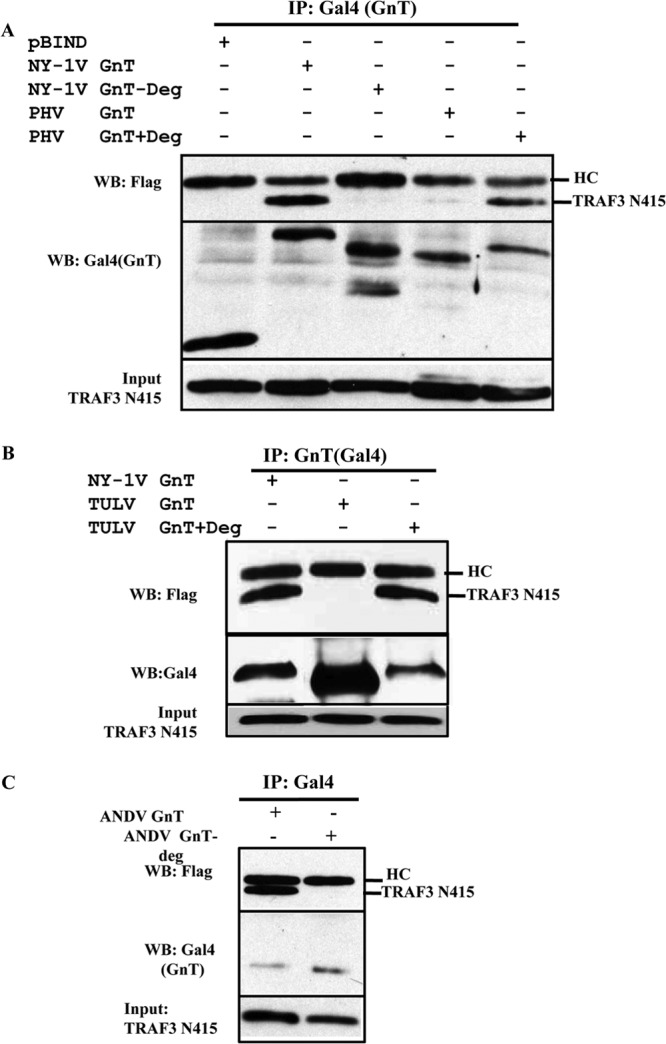
Hantavirus GnT degrons direct TRAF3 binding. Cells were transfected with pRK-TRAF3-N415 and wt Gal4-GnT or GnT+Deg or GnT−Deg mutants (Fig. 2A) of NY1-V or PHV (A), TULV (B), or ANDV (C) and analyzed for their ability to coimmunoprecipitate TRAF3-N415 (24). Hantavirus GnTs were immunoprecipitated using an anti-Gal4 antibody and protein A/G Plus agarose beads. Coprecipitated TRAF3-N415 was detected by Western blotting (WB) using the anti-FLAG antibody (24). Input TRAF3-N415 and Gal4-GnT proteins were analyzed by anti-FLAG or anti-Gal4 WB as indicated. HC, IgG heavy chain.
Role of the degron in GnT-regulated TBK1 signaling responses.
Our studies indicate that TRAF3 is recruited by GnT degrons, but this finding fails to explain GnT regulation of TBK1-directed ISRE responses by degron-deficient TULV GnTs (19, 20, 24, 26). To evaluate the effect of degrons on IFN pathway activation, we assayed wt and degron mutant GnT regulation of TBK1-directed ISRE and IFN-β promoter-directed transcriptional responses. Surprisingly, we found that deleting the degron from the NY-1V GnT had no effect on its ability to inhibit TBK1-directed ISRE transcription (Fig. 6A). Further, adding a degron to the PHV or TULV GnTs conferred TRAF3 binding to both mutants (Fig. 5A and B) but failed to cause the PHV GnT to inhibit TBK1-directed ISRE transcription (Fig. 6B). Curiously, degron addition to the TULV GnT partially reduced the ability of the TULV GnT+Deg mutant to inhibit TBK1-directed ISRE transcriptional responses (∼50%) (Fig. 6C). However, similar to the stably expressed NY-1V GnT−Deg mutant (Fig. 6A), the ANDV GnT−Deg mutant inhibited TBK1-directed ISRE transcription, similar to the wt ANDV GnT (Fig. 6D). Comparative analysis of wt and −Deg mutants of NY-1V and ANDV GnTs demonstrated that degron-containing and -deficient GnT proteins similarly inhibited TBK1-induced transcription from κB and IFN-β promoters (Fig. 6E and F). These findings demonstrate that degrons are not required for GnTs to inhibit TBK1-directed IFN-β transcriptional responses.
FIG 6.

Role of degrons in GnT regulation of TBK1 signaling. HEK293 cells were transfected with an ISRE (A to D), IFN-β promoter (E), or NF-κB promoter-driven firefly luciferase reporter and a constitutively expressed Renilla luciferase plasmid in the presence or absence of a TBK1 expression vector. Cells were cotransfected with plasmids expressing GnT wt or +Deg or −Deg mutants of NY-1V (A to C, E, and F), PHV (A and B), TULV (C), or ANDV (D to F) or with control pBIND empty vector to maintain constant DNA transfection levels. Luciferase activity within lysates was determined 48 h posttransfection, normalized to Renilla luciferase activity, and reported as the fold increase compared to that of controls lacking TBK1. Asterisks indicate statistical significance (P < 0.05). Cell lysates were analyzed for TBK1 levels by Western blotting and compared to endogenous β-actin protein controls. Gal4-GnT protein expression levels were determined by anti-Gal4 Western analysis of MG132-treated cells prior to lysis. Assays were performed in triplicate with similar results in at least 2 separate experiments.
Residues 15 to 42 from the NY-1V GnT C terminus inhibit TBK1 responses.
To define elements required for IFN regulation, we focused on NY-1V GnT domains and residues required for IFN regulation. We found that deleting 42 C-terminal residues (GnT-ΔC42) of the NY-1V GnT abolished TBK1 regulation (Fig. 7A) while deleting 14 C-terminal residues (GnT-ΔC14) had little effect on the ability of the protein to inhibit TBK1 responses (Fig. 7A). In fact, expressing only the C-terminal 42 residues of the NY-1V GnT inhibited TBK1 transcriptional responses in a concentration-dependent manner (Fig. 7B), and the GnT-C42 construct coprecipitated TRAF3 (Fig. 7C). These findings suggested that TBK1 regulation is primarily mediated by residues 15 to 42 from the GnT C terminus (NY-1V Gn residues 611 to 638).
FIG 7.
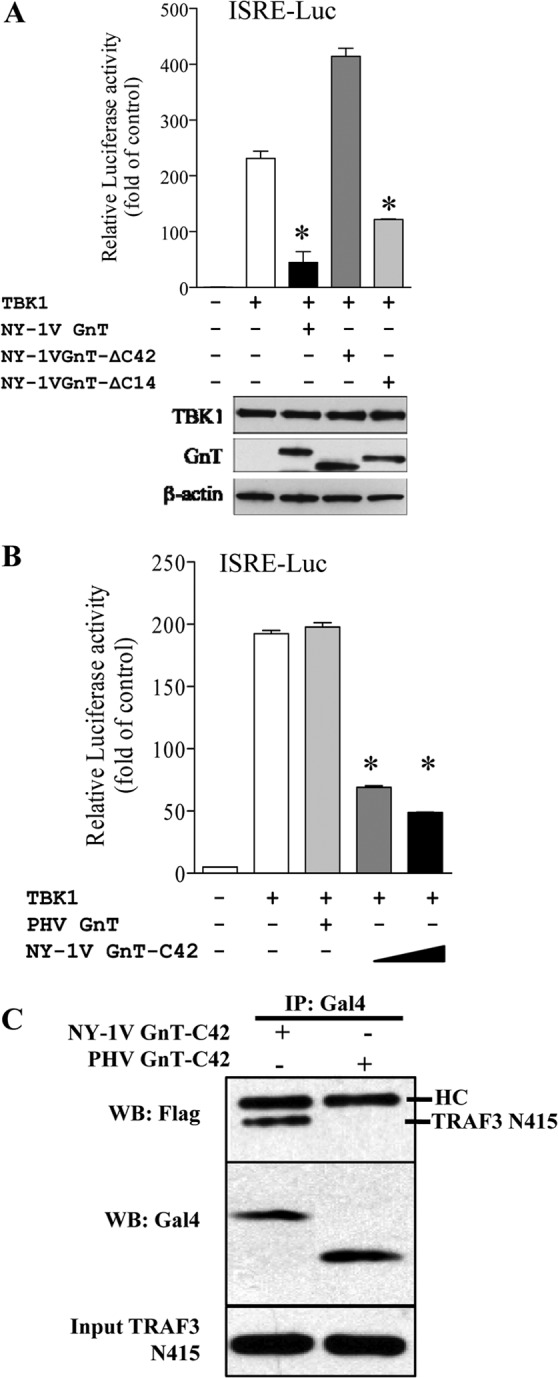
C-terminal GnT deletions regulate TBK1 and bind TRAF3. (A) Plasmids expressing full-length NY-1V GnT or GnT with C-terminal 42- or 14-residue deletions (GnT-ΔC42, GnT-ΔC14) were transfected into HEK293 cells along with ISRE-luciferase (ISRE-Luc) reporter and TBK1 expression plasmids as described for Fig. 6. Luciferase activity within lysates was determined 48 h posttransfection, normalized to Renilla luciferase activity, and reported as the fold increase compared to that of controls lacking TBK1 (19, 24). Protein expression levels were analyzed as indicated for Fig. 1. (B) Increasing concentrations of NY-1V GnTs (0.5 to 1 μg) expressing only the C-terminal 42 residues (GnT-C42) were assayed for their ability to regulate TBK1-directed ISRE transcription shown in panel A and compared to wt PHV GnT responses. Luciferase activity within lysates was determined 48 h posttransfection, normalized to Renilla luciferase activity, and reported as in panel A (19, 24). Assays were performed in triplicate with similar results in at least 2 separate experiments. Asterisks indicate statistical significance (P < 0.05). (C) The ability of NY-1V GnT-C42 and PHV GnT-C42 proteins to coimmunoprecipitate TRAF3-N415 was analyzed as described for Fig. 3.
Mutagenesis of conserved tyrosine residues within the NY-1V GnT.
The potential IFN pathway regulatory domain (residues 611 to 638) of the NY-1V GnT contains 3 conserved tyrosine residues (Y619, Y627, and Y632) (Fig. 8A). We mutated tyrosines to phenylalanine individually or in groups within wt GnT or GnT-ΔC14 constructs and evaluated their effect on TBK1 regulation. A summary of mutants and TBK1-regulated findings resulting in >60% TBK1-directed ISRE transcriptional inhibition (+) is provided in Fig. 8A. We found that only mutants containing the Y627F mutation were able to regulate TBK1-directed ISRE transcription (Fig. 8A and B). We further generated and analyzed the GnT Y627F mutation within a wt or degron mutant background (Fig. 4A) as presented in Fig. 8C and D. Within wt or degron-mutant NY-1V GnTs, the Y627F mutation prevented GnT regulation of TBK1-directed IFN-β and κB transcriptional responses (Fig. 8C and D). These findings indicate that the C-terminal degron and Y619F or Y632F mutations are irrelevant to the regulation of TBK1 responses (Fig. 8B) and suggest the importance of Y627 in NY-1V GnT regulation.
FIG 8.

Role of NY-1V GnT tyrosines in TBK1 regulation. (A) A summary of Y-to-F mutations made in 3 tyrosine residues within the NY-1V GnT-ΔC14 construct and the ability of the GnT-mutants to regulate TBK1 signaling responses is presented. (B to D) HEK293 cells were transfected with TBK1 expression plasmid or empty vector, ISRE (B), IFN-β (C), or NF-κB (D) transcriptional reporters and NY-1V GnT-ΔC14 constructs with or without mutations in Y627F, Y619F, or Y632F (+Deg or −Deg) as indicated. GnT effects on TBK1-directed ISRE responses were evaluated as described for Fig. 6, normalized to Renilla luciferase levels, and evaluated for comparable protein expression levels by Western analysis using antibodies to Gal4 and β-actin as in Fig. 1. Asterisks indicate statistical significance (P < 0.05).
Tyrosine 627 is required for NY-1V GnT inhibition of RIG-I/TBK1-directed signaling.
Alanine scanning mutagenesis was used to further evaluate residues required for IFN regulation within the NY-1V GnT. We changed 15 residues within the NY-1V GnT 611-to-638 domain, including 7 residues that differ from PHV GnTs (indicated by asterisks in Fig. 9A). Only mutations in tyrosine 627 (Y627 A/S) altered the ability of the NY-1V GnT to inhibit TBK1-directed ISRE transcriptional responses (>75%) (indicated by plus signs in Fig. 9A). Figure 9B provides representative GnT findings demonstrating that mutagenesis of conserved residues adjacent to residue 627 (S629A and S630A) in the NY-1V GnT failed to alter TBK1-directed ISRE transcriptional responses. In contrast, mutagenesis of Y627 to alanine (Y627A) or serine (Y627S) abrogated inhibitory effects of the NY-1V GnT on TBK1-directed ISRE transcription (Fig. 9C). Consistent with discrete NY-1V GnT domains mediating TRAF3 binding and IFN regulation, the GnT-Y627A mutant still coprecipitated TRAF3-N415 (Fig. 9D) but lacked the ability to regulate TBK1-directed transcriptional responses (Fig. 8C and D and 9C). These results suggest that TRAF3 binding and IFN regulation require discrete elements within the GnT and demonstrate that tyrosine 627 is required to direct NY-1V GnT regulation of TBK1 signaling responses.
FIG 9.

Residues required for NY-1V GnT-directed TBK1 regulation. (A) A summary of mutagenesis of NY-1V GnT residues (26) and their effects on TBK1-directed ISRE responses is presented. Representative NY-1V GnT residue changes at Y627, S629, and R630 are presented and evaluated in panels B and C for effects on TBK1-directed ISRE transcription as in Fig. 6. Asterisks (*) denote NY-1V residues that were changed to PHV residues and resulted in no change in the ability of GnT mutants to regulate TBK1-directed ISRE transcriptional responses. (B) Analysis of TBK1-directed ISRE transcriptional responses directed by GnT-R630A or GnT-S629A in comparison to wt NY-1V GnT regulation as described above. (C) Comparison of NY-1V wt, GnT-Y627A, and GnT-Y627S mutants (0.5 or 1 μg) for regulation of TBK1-directed ISRE responses was performed as described for Fig. 7. Lysates were evaluated for comparable protein expression levels by Western analysis using antibodies to TBK1, Gal4, and β-actin as in Fig. 1. *, statistical significance (P < 0.05). (D) Cells were transfected with NY-1V GnT wt or GnT-Y627A mutant and assayed for the ability of the mutant to coimmunoprecipitate TRAF3-N415 as described for Fig. 3.
RIG-I senses dsRNA, activating a signaling pathway that directs TBK1 phosphorylation of IRF3 and IFN-β induction (27–29). Here, we compared the ability of the NY-1V GnT Y627A mutant to alter TBK1 and IRF3 phosphorylation. We observed no difference in the total resulting TBK1 or phospho-TBK1 levels between wt and Y627A mutant NY-1V GnTs (Fig. 10A). In contrast, following activation of the RIG-I pathway, we observed that wt GnT, but not the GnT-Y627A mutant, dramatically reduced IRF3 phosphorylation (Fig. 10B and D). This finding suggests that the NY-1V GnT inhibits IRF3 phosphorylation via a Y627-requiring mechanism.
FIG 10.
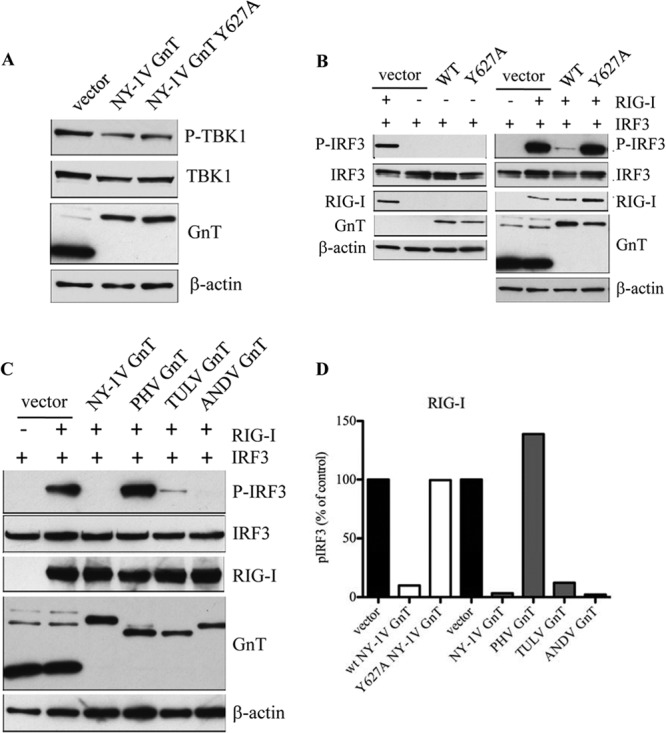
GnTs regulate total and phosphorylated IRF3 levels. (A, B) HEK293T cells were cotransfected with plasmid expressing empty vector, Gal4-NY-1V GnT, or Gal4-NY-1V GnT Y627A and IRF3-T7 or TBK1-Flag expression vectors as indicated. After 24 h, cells were harvested and analyzed by Western blotting for pTBK1 (α-pTBK1-S172), total TBK1, pIRF3 (α-pIRF3-S396), total IRF3, RIG-I, β-actin, and GnT protein expression (α-Gal4) as indicated. (C) HEK293T cells were cotransfected as in panel A to express wt Gal4-GnTs from NY-1V, ANDV, PHV, or TULV and IRF3-T7. Cells were harvested and analyzed by Western blotting as in panel A and Fig. 1. (D) RIG-I-directed phospho-IRF3 levels in panels B and C were quantitated using NIH image and standardized to endogenous β-actin protein controls.
To determine whether IRF3 phosphorylation is commonly inhibited by hantavirus GnTs, we comparatively evaluated RIG-I-induced phospho-IRF3 levels in cells expressing NY-1V, PHV, TULV, and ANDV GnT proteins. HEK293T cells were transfected with identical amounts of plasmids expressing RIG-I and IRF3 in the presence of cotransfected empty vector or GnT expression plasmids. We found that while PHV failed to reduce phospho-IRF3 levels in response to RIG-I, expression of wt NY-1V, ANDV, and TULV GnT proteins inhibited IRF3 phosphorylation (Fig. 10C and D). These findings demonstrate that wt NY-1V, ANDV, and TULV GnT proteins inhibit IRF3 phosphorylation and suggest a common mechanism for GnTs to inhibit RIG-I/MDA5 induction of IFN-β.
DISCUSSION
The successful replication of hantaviruses within human endothelial cells is at least in part due to their ability to regulate the induction of IFN-β (6, 19, 21, 47). Previous studies demonstrate that human endothelial cells elicit a biphasic IFN response following hantavirus infection as a result of hantavirus regulation of early, but not late, IFN and ISG responses (21). Hantaviruses express few proteins with the potential to regulate cytoplasmic RIG-I or TBK1 signaling effectors that are activated intracellularly by RNA viruses. The hantavirus nucleocapsid (N) protein is highly expressed during infection, although expressing the NY-1V N protein, including potentially encoded NSs ORF, fails to regulate RIG-I- or TBK1-directed transcriptional responses (19, 20, 24). In contrast, expressing the complete GnGc or the GnT proteins from NY-1V, ANDV, and TULV regulates RIG-I- and TBK1-directed ISRE, NF-κB, or IFN-β transcription (19, 20, 24). GnTs fail to inhibit constitutively active IRF3-5D, indicating that regulation occurs at the level of the MAVS-TRAF3-TBK1 complex (19, 20, 24).
GnTs of pathogenic NY-1V, ANDV, and HTNV contain degron domains that direct GnT ubiquitination and degradation (26, 48). Consistent with this, the NY-1V GnT binds the E3 ubiquitin ligase TRAF3 and disrupts TBK1-TRAF3 interactions required for TBK1 activation (20). However, degrons are absent from TULV and PHV GnTs, yet the TULV GnT inhibits RIG-I-, MDA5-, and TBK1-directed ISRE, κB, and IFN-β transcriptional responses (Fig. 1 and 6) (24). As a result, discrete GnTs are likely to block TBK1-directed IRF3 and NF-κB activation by virus-specific mechanisms.
TRAF3 is required for IFN induction from nearly all stimuli, and TRAF3 forms complexes with TBK1 and MAVS that are regulated by ubiquitination and deubiquitination (36, 38). Here we evaluated the role of GnT degrons, TRAF3 binding domains, and specific GnT residues for their role in IFN regulation. We found that the TRAF-N domain of TRAF3 was required to bind the NY-1V GnT (Fig. 3). This distinguishes GnT-TRAF3 complexes from TRAF-C domains required for TRAF3 recruitment of MAVS/UXT-V1 proteins (36) (Fig. 3A) and suggests that the GnT does not regulate IFN signaling responses by dissociating TRAF3 from MAVS.
The roles of the degrons and TRAF3 binding in IFN regulation remain to be defined. We mutated residues within C-terminal degrons of NY-1V and ANDV GnTs to disrupt degrons or add degrons to TULV and PHV GnTs by mutating residues to mimic NY-1V GnTs (Fig. 4A). Mutating 3 to 5 residues within the C-terminal degrons of the NY-1V and ANDV GnTs stabilized the proteins, while adding degrons to PHV and TULV GnTs resulted in their degradation in the absence of proteasomal inhibitors (Fig. 4B and C). Interestingly, we observed that only GnTs containing degrons coprecipitated TRAF3 (Fig. 5), suggesting that the degron mediates GnT binding to TRAF3 complexes.
However, the wt TULV GnT lacks a degron and still inhibits RIG-I/TBK1-directed ISRE/IFN-β transcriptional responses (Fig. 1) (24). Similarly, when we evaluated NY-1V and ANDV GnT−Deg mutants, we found that they still inhibited TBK1-directed ISRE, κB, and IFN-β transcription responses like wt GnTs (Fig. 6A, D, E, and F). In contrast, adding a degron to PHV or TULV GnTs failed to confer TBK1 regulation by the PHV GnT+Deg mutant (Fig. 6B), while partially reducing TBK1 inhibition by the TULV GnT+Deg mutant. Overall, these findings indicate that degron binding to TRAF3 is not required for IFN regulation by hantavirus GnT proteins. However, we cannot exclude the possibility that TRAF3 binding facilitates GnT regulation of TBK1, IRF3, or ubiquitin ligases or alternatively TRAF3 heterotrimer formation with additional TRAF2/5 adapter proteins (36, 40, 58, 62).
Analysis of the NY-1V GnT establishes that residues 15 to 42 from the C terminus are required for TBK1 regulation (Fig. 7 and 8). Mutagenesis of residues within this NY-1V GnT (residues 15 to 42) domain resulted in only 1 change, Y627 to A, S, or F, that prevented GnT regulation of TBK1-directed ISRE, κB, or IFN-β transcriptional responses (Fig. 8 and 9). In contrast, mutating NY-1V GnT-Y619F, -Y632F, -S629A, or -R630A had no effect on TBK1-directed transcriptional responses (Fig. 8 and 9). Mutating Y627F within the stably expressed NY-1V GnT−Deg mutant or degron-containing wt NY-1V GnT prevented regulation of IFN-β transcription but was degron independent (Fig. 8C and D). Moreover, the Y627 residue was required for the NY-1V GnT to inhibit RIG-I-directed IRF3 phosphorylation (Fig. 10). These findings define a single tyrosine residue within the NY-1V GnT (Y627) required for regulating RIG-I-directed IRF3 activation and ISRE, κB, and IFN-β transcriptional responses.
It remains to be determined whether GnT residues critical for IFN pathway regulation in NY-1V are conserved across hantaviruses. Y627 is conserved in ANDV and TULV but also present in the PHV GnT, which fails to regulate RIG-I/TBK1-directed responses. However, the PHV GnT uniquely contains F632 instead of Y632, lacks a degron motif like TULV, and has 15/42 residue differences from the NY-1V GnT. The residue changes required for the PHV GnT to gain IFN regulating function as well as residue requirements for ANDV and TULV GnT function remain to be determined. Collectively, these findings indicate that unique hantavirus GnT configurations determine their function in regulating human IFN responses.
In addition to regulating RIG-I-directed transcriptional responses, the Fig. 10C data demonstrate that GnTs from NY-1V, ANDV, and TULV inhibit IRF3 phosphorylation. The absence of direct coprecipitation of GnT-TBK1 complexes suggests several ancillary mechanisms for regulating IRF3 phosphorylation. However, the list of potential GnT targets that regulate MAVS-TRAF3-TBK1 signaling responses and impact IRF3/7 and NF-κB activation is large and includes OTUD7B, Fox01, TRIAD3a, CYLD, UXT-V1, RNF11/125, Mindbomb1/2, NLRX1, SOCS1, TRIM21/23/28/38, ISG56, optineurin, OTUB1/2, MIP-T3, TRAF2, NIK, NLRP4, RAUL, TRIP, NLRC5, DUBA, NAP1, SINTBAD, ITCH, A20, TAX1BP, AIBIN1 and NEMO (36, 39, 40, 44, 45, 54, 59, 61–63). Many of these inhibitors are E3 ligases or deubiquitinases that regulate IFN-β induction by altering K48- or K63-linked polyubiquitination of MAVS, TBK1, TRAF3, or IRF3 (27, 40, 58, 62–64). Interestingly, tyrosine phosphorylation of the E3 ubiquitin ligase TRIM21 regulates IRF3 activity and demonstrates a role for phosphotyrosine regulation of serine-phosphorylated IRF3 (62). The observation that a GnT tyrosine is critical for TBK1 regulation suggests a potential role for tyrosine regulation of IRF3 activation, although Y627 is reportedly not phosphorylated (48, 49). However, RAUL, OTUD7B, Fox01, and E3 ligase regulators may be impacted by GnTs and alter the ubiquitination of TBK1, NEMO, TRAF3, IRF3/7, or NF-κB required for IFN-β induction (34, 40, 44, 45, 54, 58, 61, 62, 65, 66). These factors provide an array of potential GnT targets IFN regulation; however, the mechanism by which GnTs inhibit TBK1-directed IRF3 and NF-κB activation remains to be revealed.
Depending on the proteins, cells, and IFN assays used to evaluate regulation, several hantavirus proteins or complexes have been suggested to regulate IFN induction (19, 20, 24, 25, 47, 67, 68). Reports demonstrate that the NY-1V, TULV, or SNV GnGc proteins or GnTs inhibit IFN induction and that inhibition is blocked downstream of RIG-I and upstream of constitutively activated IRF3, at the level of the TBK1 complex (19, 20, 24, 25). Using pathway-specific inducers, ANDV GnGc and GnT proteins also inhibit RIG-I- and TBK1-directed ISRE, κB, and IFN-β transcription and IRF3 phosphorylation (Fig. 1, 2, and 10), while N proteins from NY-1V and PHV have no effect on RIG-I-induced ISRE or IFN induction (19, 20, 24, 25). One study indicates that the ANDV GnGc as well as SNV and ANDV N proteins fail to regulate Sendai virus (SeV)-induced IFN responses (68), but when coexpressed, the ANDV N and GnGc were partially inhibitory (68). However, SeV-induced IFN responses complicate analysis of hantavirus protein regulation since hantavirus proteins may interfere with SeV infection rather than IFN signaling responses. Further, it is unclear whether the slow transcription and transient IFN regulation following hantavirus infection is sufficient to, or needs to, regulate the robust induction of IFN by a rapid SeV transcriptional process.
Interestingly, human endothelial cells infected with HFRS- or HPS-causing hantaviruses elicit high-level ISG responses at late times postinfection, and hantavirus replication is sensitive to the effects of IFN at early times postinfection only (19, 21). In contrast to responses evaluated in human endothelial cells, a complete blockade of early and late IFN responses is suggested by one study (68), but this report uses A549 epithelial cells, which are poorly infected by hantaviruses and result in little or no viral RNA transcription (69). The combination of the multiplicity of infection (MOI) used (0.03) with a >100-fold reduction in hantavirus infection of A549 cells (68, 69) suggests that responses observed may result from limited infection of A549 cells rather than a late ISG blockade. Late ISG induction in hantavirus-infected endothelial cells (21, 47) also contrasts with findings suggesting that hantaviruses inhibit STAT signaling in response to type I IFN (47). Studies showed that hantavirus-infected endothelial cells elicit high-level IFN-directed ISG responses at late times postinfection (21), which may actually be an inherent plan for hantaviruses to establish persistence within host endothelial cells (70, 71). Although persistence is not accomplished in humans, IFN uniquely directs the proliferation, rather than apoptosis, of endothelial cells, and this may limit vascular damage during infection of the endothelium (72, 73).
Our studies demonstrate that unique hantavirus GnTs regulate RIG-I/MDA5 signaling pathways but leave open the specific regulatory mechanism by which GnTs inhibit TBK1-directed responses. Studies of additional regulatory or activating factors mentioned above have yet to be analyzed for their interactions with GnT proteins or used to assess components of GnT complexes that regulate IFN induction. Nonetheless, defining a residue within the GnT that abolishes RIG-I-TBK1 regulation and is required to inhibit IRF3 phosphorylation suggests a potential virulence determinant within NY-1V that may be used for viral attenuation. Collectively, our findings define GnT interactions that inhibit IFN-β transcriptional induction by regulating the activation of IRF3/NF-κB transcription factors. However, numerous potential effectors that impact TBK1-IRF3 signaling responses (36, 40, 62, 74) require further investigation to define regulatory interactions targeted by discrete hantavirus GnTs.
ACKNOWLEDGMENTS
We thank Eric Roth and Aleksandr Nasonov for technical support and Ken Marcu and Nancy Reich for insightful discussions.
This work was supported by grants AI75022, AI055621, AI1092191, and AI097951 from the National Institutes of Health.
Footnotes
Published ahead of print 2 January 2014
REFERENCES
- 1.Duchin JS, Koster FT, Peters CJ, Simpson GL, Tempest B, Zaki SR, Ksiazek TG, Rollin PE, Nichol S, Umland ET, Moolenaar RL, Reef SE, Nolte KB, Gallaher MM, Butler JC, Breiman RF, Hantavirus Study Group 1994. Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. N. Engl. J. Med. 330:949–955 [DOI] [PubMed] [Google Scholar]
- 2.Hjelle B, Jenison S, Torrez-Martinez N, Yamada T, Nolte K, Zumwalt R, MacInnes K, Myers G. 1994. A novel hantavirus associated with an outbreak of fatal respiratory disease in the southwestern United States: evolutionary relationships to known hantaviruses. J. Virol. 68:592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee HW. 1982. Hemorrhagic fever with renal syndrome (HFRS). Scand. J. Infect. Dis. Suppl. 36:82–85 [PubMed] [Google Scholar]
- 4.Nichol ST, Spiropoulou CF, Morzunov S, Rollin PE, Ksiazek TG, Feldmann H, Sanchez A, Childs J, Zaki S, Peters CJ. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262:914–917. 10.1126/science.8235615 [DOI] [PubMed] [Google Scholar]
- 5.Schmaljohn C. 2001. Bunyaviridae and their replication, p 1581–1602 In Knipe PM, Howley PM, Cohen JL, Griffin DL, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, vol 1 Lipppincott-Raven, Philadelphia, PA [Google Scholar]
- 6.Yanagihara R, Silverman DJ. 1990. Experimental infection of human vascular endothelial cells by pathogenic and nonpathogenic hantaviruses. Arch. Virol. 111:281–286. 10.1007/BF01311063 [DOI] [PubMed] [Google Scholar]
- 7.Zaki S, Greer P, Coffield L, Goldsmith C, Nolte K, Foucar K, Feddersen R, Zumwalt R, Miller G, Rollin P, Ksiazek T, Nichol S, Peters C. 1995. Hantavirus pulmonary syndrome: pathogenesis of an emerging infectious disease. Am. J. Pathol. 146:552–579 [PMC free article] [PubMed] [Google Scholar]
- 8.Lahdevirta J. 1982. Clinical features of HFRS in Scandinavia as compared with East Asia. Scand. J. Infect. Dis. Suppl. 36:93–95 [PubMed] [Google Scholar]
- 9.Lahdevirta J, Enger E, Hunderi OH, Traavik T, Lee HW. 1982. Hantaan virus is related to hemorrhagic fever with renal syndrome in Norway. Lancet ii:606. [DOI] [PubMed] [Google Scholar]
- 10.Lee HW, Baek LJ, Johnson KM. 1982. Isolation of Hantaan virus, the etiologic agent of Korean hemorrhagic fever, from wild urban rats. J. Infect. Dis. 146:638–644. 10.1093/infdis/146.5.638 [DOI] [PubMed] [Google Scholar]
- 11.Schmaljohn C, Hjelle B. 1997. Hantaviruses: a global disease problem. Emerg. Infect. Dis. 3:95–104. 10.3201/eid0302.970202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang B, Crowley M, Campen M, Koster F. 2007. Hantavirus cardiopulmonary syndrome. Semin. Respir. Crit. Care Med. 28:193–200. 10.1055/s-2007-976491 [DOI] [PubMed] [Google Scholar]
- 13.Enria D, Padula P, Segura EL, Pini N, Edelstein A, Posse CR, Weissenbacher MC. 1996. Hantavirus pulmonary syndrome in Argentina. Possibility of person to person transmission. Medicina 56:709–711 [PubMed] [Google Scholar]
- 14.Lopez N, Padula P, Rossi C, Lazaro ME, Franze-Fernandez MT. 1996. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology 220:223–226. 10.1006/viro.1996.0305 [DOI] [PubMed] [Google Scholar]
- 15.Plyusnin A, Vapalahti O, Lankinen H, Lehvaslaiho H, Apekina N, Myasnikov Y, Kallio-Kokko H, Henttonen H, Lundkvist A, Brummer-Korvenkontio M. 1994. Tula virus: a newly detected hantavirus carried by European common voles. J. Virol. 68:7833–7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanagihara R, Daum CA, Lee PW, Baek LJ, Amyx HL, Gajdusek DC, Gibbs CJ. 1987. Serological survey of Prospect Hill virus infection in indigenous wild rodents in the USA. Trans. R. Soc. Trop. Med. Hyg. 81:42–45. 10.1016/0035-9203(87)90275-6 [DOI] [PubMed] [Google Scholar]
- 17.Gavrilovskaya IN, Brown EJ, Ginsberg MH, Mackow ER. 1999. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 73:3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. 1998. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Natl. Acad. Sci. U. S. A. 95:7074–7079. 10.1073/pnas.95.12.7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alff PJ, Gavrilovskaya IN, Gorbunova E, Endriss K, Chong Y, Geimonen E, Sen N, Reich NC, Mackow ER. 2006. The pathogenic NY-1 hantavirus G1 cytoplasmic tail inhibits RIG-I- and TBK-1-directed interferon responses. J. Virol. 80:9676–9686. 10.1128/JVI.00508-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. 2008. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J. Virol. 82:9115–9122. 10.1128/JVI.00290-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geimonen E, Neff S, Raymond T, Kocer SS, Gavrilovskaya IN, Mackow ER. 2002. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. U. S. A. 99:13837–13842. 10.1073/pnas.192298899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pensiero MN, Hay J. 1992. The Hantaan virus M-segment glycoproteins G1 and G2 can be expressed independently. J. Virol. 66:1907–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hepojoki J, Strandin T, Wang H, Vapalahti O, Vaheri A, Lankinen H. 2010. Cytoplasmic tails of hantavirus glycoproteins interact with the nucleocapsid protein. J. Gen. Virol. 91:2341–2350. 10.1099/vir.0.021006-0 [DOI] [PubMed] [Google Scholar]
- 24.Matthys V, Gorbunova EE, Gavrilovskaya IN, Pepini T, Mackow ER. 2011. The C-terminal 42 residues of the TULV Gn protein regulate interferon induction. J. Virol. 85:4752–4760. 10.1128/JVI.01945-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthys V, Mackow ER. 2012. Hantavirus regulation of type I interferon responses. Adv. Virol. 2012:524024. 10.1155/2012/524024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sen N, Sen A, Mackow ER. 2007. Degrons at the C terminus of the pathogenic but not the nonpathogenic hantavirus G1 tail direct proteasomal degradation. J. Virol. 81:4323–4330. 10.1128/JVI.02279-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiscott J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 282:15325–15329. 10.1074/jbc.R700002200 [DOI] [PubMed] [Google Scholar]
- 28.Seth RB, Sun L, Chen ZJ. 2006. Antiviral innate immunity pathways. Cell Res. 16:141–147. 10.1038/sj.cr.7310019 [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 30.Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, Wilkinson P, Julkunen I, Vitour D, Meurs E, Hiscott J. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 80:6072–6083. 10.1128/JVI.02495-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 32.Yoneyama M, Fujita T. 2007. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18:545–551. 10.1016/j.cytogfr.2007.06.023 [DOI] [PubMed] [Google Scholar]
- 33.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- 34.Charoenthongtrakul S, Gao L, Parvatiyar K, Lee D, Harhaj EW. 2013. RING finger protein 11 targets TBK1/IKKi kinases to inhibit antiviral signaling. PLoS One 8:e53717. 10.1371/journal.pone.0053717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 36.Hacker H, Tseng PH, Karin M. 2011. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat. Rev. Immunol. 11:457–468. 10.1038/nri2998 [DOI] [PubMed] [Google Scholar]
- 37.Matsui K, Kumagai Y, Kato H, Sato S, Kawagoe T, Uematsu S, Takeuchi O, Akira S. 2006. Cutting edge: role of TANK-binding kinase 1 and inducible IkappaB kinase in IFN responses against viruses in innate immune cells. J. Immunol. 177:5785–5789 http://www.jimmunol.org/content/177/9/5785 [DOI] [PubMed] [Google Scholar]
- 38.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211. 10.1038/nature04374 [DOI] [PubMed] [Google Scholar]
- 39.Shen RR, Hahn WC. 2011. Emerging roles for the non-canonical IKKs in cancer. Oncogene 30:631–641. 10.1038/onc.2010.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, Li Y, Dunn GP, Chan E, Thai T, Yang S, Ficarro SB, Marto JA, Jeon H, Hahn WC, Barbie DA, Eck MJ. 2013. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 3:747–758. 10.1016/j.celrep.2013.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW, Nikolich-Zugich J, Moses AV, Gale M, Jr, Fruh K, Diamond MS. 2013. IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9:e1003118. 10.1371/journal.ppat.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panne D. 2008. The enhanceosome. Curr. Opin. Struct. Biol. 18:236–242. 10.1016/j.sbi.2007.12.002 [DOI] [PubMed] [Google Scholar]
- 43.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628. 10.1073/pnas.95.26.15623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, Xiao Y, Cheng X, Chang M, Fu YX, Zal T, Zhu C, Sun SC. 2013. OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature 494:371–374. 10.1038/nature11831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belgnaoui SM, Paz S, Samuel S, Goulet ML, Sun Q, Kikkert M, Iwai K, Dikic I, Hiscott J, Lin R. 2012. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 12:211–222. 10.1016/j.chom.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 46.Mao AP, Li S, Zhong B, Li Y, Yan J, Li Q, Teng C, Shu HB. 2010. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 285:9470–9476. 10.1074/jbc.M109.071043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spiropoulou CF, Albarino CG, Ksiazek TG, Rollin PE. 2007. Andes and Prospect Hill hantaviruses differ in early induction of interferon although both can downregulate interferon signaling. J. Virol. 81:2769–2776. 10.1128/JVI.02402-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geimonen E, Fernandez I, Gavrilovskaya IN, Mackow ER. 2003. Tyrosine residues direct the ubiquitination and degradation of the NY-1 hantavirus G1 cytoplasmic tail. J. Virol. 77:10760–10868. 10.1128/JVI.77.20.10760-10768.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geimonen E, LaMonica R, Springer K, Farooqui Y, Gavrilovskaya IN, Mackow ER. 2003. Hantavirus pulmonary syndrome-associated hantaviruses contain conserved and functional ITAM signaling elements. J. Virol. 77:1638–1643. 10.1128/JVI.77.2.1638-1643.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. 2005. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175:5260–5268 http://www.jimmunol.org/content/175/8/5260 [DOI] [PubMed] [Google Scholar]
- 51.Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699. 10.1128/JVI.79.5.2689-2699.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tom R, Bisson L, Durocher Y. 2008. Transfection of HEK293-EBNA1 cells in suspension with linear PEI for production of recombinant proteins. CSH Protoc. 2008:pdb.prot4977. 10.1101/pdb.prot4977 [DOI] [PubMed] [Google Scholar]
- 54.Lei CQ, Zhang Y, Xia T, Jiang LQ, Zhong B, Shu HB. 2013. FoxO1 negatively regulates cellular antiviral response by promoting degradation of IRF3. J. Biol. Chem. 288:12596–12604. 10.1074/jbc.M112.444794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang B, Wang J, Sun B. 2013. Trim21: a novel negative regulator in DNA sensor signaling. Cell. Mol. Immunol. 10:190–192. 10.1038/cmi.2013.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. 2013. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 14:172–178. 10.1038/ni.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. 2008. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J. Immunol. 181:1780–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Y, Hayward GS. 2010. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33:863–877. 10.1016/j.immuni.2010.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang M, Wang L, Zhao X, Zhao K, Meng H, Zhao W, Gao C. 2012. TRAF-interacting protein (TRIP) negatively regulates IFN-beta production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J. Exp. Med. 209:1703–1711. 10.1084/jem.20120024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207. 10.1038/nature04369 [DOI] [PubMed] [Google Scholar]
- 61.Huang Y, Liu H, Ge R, Zhou Y, Lou X, Wang C. 2012. UXT-V1 facilitates the formation of MAVS antiviral signalosome on mitochondria. J. Immunol. 188:358–366 [DOI] [PubMed] [Google Scholar]
- 62.Stacey KB, Breen E, Jefferies CA. 2012. Tyrosine phosphorylation of the E3 ubiquitin ligase TRIM21 positively regulates interaction with IRF3 and hence TRIM21 activity. PLoS One 7:e34041. 10.1371/journal.pone.0034041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oudshoorn D, Versteeg GA, Kikkert M. 2012. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev. 23:273–282. 10.1016/j.cytogfr.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6:644–658. 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- 65.Paz S, Vilasco M, Arguello M, Sun Q, Lacoste J, Nguyen TL, Zhao T, Shestakova EA, Zaari S, Bibeau-Poirier A, Servant MJ, Lin R, Meurs EF, Hiscott J. 2009. Ubiquitin-regulated recruitment of IkappaB kinase epsilon to the MAVS interferon signaling adapter. Mol. Cell. Biol. 29:3401–3412. 10.1128/MCB.00880-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou AY, Shen RR, Kim E, Lock YJ, Xu M, Chen ZJ, Hahn WC. 2013. IKKepsilon-mediated tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep. 3:724–733. 10.1016/j.celrep.2013.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaaskelainen KM, Kaukinen P, Minskaya ES, Plyusnina A, Vapalahti O, Elliott RM, Weber F, Vaheri A, Plyusnin A. 2007. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 79:1527–1536. 10.1002/jmv.20948 [DOI] [PubMed] [Google Scholar]
- 68.Levine JR, Prescott J, Brown KS, Best SM, Ebihara H, Feldmann H. 2010. Antagonism of type I interferon responses by New World hantaviruses. J. Virol. 84:11790–11801. 10.1128/JVI.00916-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khaiboullina SF, Rizvanov AA, Deyde VM, St Jeor SC. 2005. Andes virus stimulates interferon-inducible MxA protein expression in endothelial cells. J. Med. Virol. 75:267–275. 10.1002/jmv.20266 [DOI] [PubMed] [Google Scholar]
- 70.Odorizzi PM, Wherry EJ. 2013. Immunology. An interferon paradox. Science 340:155–156. 10.1126/science.1237568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. 10.1126/science.1235214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gomez D, Reich NC. 2003. Stimulation of primary human endothelial cell proliferation by IFN. J. Immunol. 170:5373–5381 http://www.jimmunol.org/content/170/11/5373.full.pdf [DOI] [PubMed] [Google Scholar]
- 73.Korherr C, Gille H, Schafer R, Koenig-Hoffmann K, Dixelius J, Egland KA, Pastan I, Brinkmann U. 2006. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc. Natl. Acad. Sci. U. S. A. 103:4240–4245. 10.1073/pnas.0511319103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng MH, Ho TH, Kok KH, Siu KL, Li J, Jin DY. 2011. MIP-T3 is a negative regulator of innate type I IFN response. J. Immunol. 187:6473–6482. 10.4049/jimmunol.1100719 [DOI] [PubMed] [Google Scholar]