

Abstract
Human cytomegalovirus (HCMV) genome replication requires host DNA damage responses (DDRs) and raises the possibility that DNA repair pathways may influence viral replication. We report here that a nucleotide excision repair (NER)-associated-factor is required for efficient HCMV DNA replication. Mutations in genes encoding NER factors are associated with xeroderma pigmentosum (XP). One of the XP complementation groups, XPE, involves mutation in ddb2, which encodes DNA damage binding protein 2 (DDB2). Infectious progeny virus production was reduced by >2 logs in XPE fibroblasts compared to levels in normal fibroblasts. The levels of immediate early (IE) (IE2), early (E) (pp65), and early/late (E/L) (gB55) proteins were decreased in XPE cells. These replication defects were rescued by infection with a retrovirus expressing DDB2 cDNA. Similar patterns of reduced viral gene expression and progeny virus production were also observed in normal fibroblasts that were depleted for DDB2 by RNA interference (RNAi). Mature replication compartments (RCs) were nearly absent in XPE cells, and there were 1.5- to 2.0-log reductions in viral DNA loads in infected XPE cells relative to those in normal fibroblasts. The expression of viral genes (UL122, UL44, UL54, UL55, and UL84) affected by DDB2 status was also sensitive to a viral DNA replication inhibitor, phosphonoacetic acid (PAA), suggesting that DDB2 affects gene expression upstream of or events associated with the initiation of DNA replication. Finally, a novel, infection-associated feedback loop between DDB2 and ataxia telangiectasia mutated (ATM) was observed in infected cells. Together, these results demonstrate that DDB2 and a DDB2-ATM feedback loop influence HCMV replication.
INTRODUCTION
Human cytomegalovirus (HCMV) is a large, complex DNA virus, which infects 40 to 90% of the population and can cause severe health problems in immunocompromised individuals. HCMV-associated pneumonitis and retinitis are the most prevalent problems detected following reactivation of latent virus (1). HCMV is the leading cause of birth defects upon congenital infection, as well as morbidity in immunosuppressed populations (2). Sequelae associated with congenital HCMV infections include deafness, blindness, mental disability, and bone defects (3). An association between HCMV and malignant gliomas has also been reported (4–8).
Living organisms are exposed to a variety of endogenous or exogenous DNA-damaging agents that generate a wide variety of DNA lesions. To maintain the integrity of their genomes, cells respond with a variety of defensive strategies to repair lesions caused by DNA-damaging insults. Recent studies demonstrate that infections by DNA viruses or viruses with a DNA genome stage during infection induce host DNA damage responses (DDRs) (9–15). Upon infection, DNA viruses create nuclear environments conducive to viral DNA replication through interactions with host proteins. As with other DNA viruses, ataxia telangiectasia mutated (ATM) is required for efficient replication of herpesviruses (10, 13, 16). HCMV was perhaps the first herpesvirus shown to induce a host DDR (17). ATM is activated by autophosphorylation and phosphorylation of p53 on Ser15, a downstream target of ATM signaling during infection or expression of IE1 or IE2 (10, 17). Subsequently, other ATM targets, including H2AX, CHK2, and p53, are phosphorylated, at least in part, in an ATM-dependent manner during infection or immediate early (IE) protein expression (10, 17). ATM is required for efficient HCMV replication (10).
DNA repair plays a critical role in the prevention of cell death, mutations, replication errors, persistent DNA damage, and genomic instability. In addition to ATM-mediated DNA damage signal transduction, numerous DNA repair systems have evolved to respond to DNA damage signals, including nucleotide excision repair (NER), which is a versatile DNA repair pathway that eliminates a wide variety of helix-distorting base lesions. Abnormalities of DNA repair have been found in cancer and aging, and it was recently discovered that NER functions in UV-irradiated HCMV-infected cells almost exclusively to repair the viral genome to the detriment of the host cell genome (18). NER removes UV-induced photolesions, including cyclobutane pyrimidine dimers (CPD) and pyrimidine-pyrimidone photoproducts (6-4PP), as well as other bulky DNA adducts induced by chemicals (19). Impaired NER activity is associated with several rare autosomal recessive disorders in humans, including xeroderma pigmentosum (XP) (20, 21) and Cockayne syndrome (22). XP is characterized by hypersensitivity to sunlight and an incidence of developing skin cancer that is ∼1,000 times that of the general population (23, 24). XP genotypes generally segregate into 8 major complementation groups (XPA through XPH), although there is an additional “variant” complementation group (XPV). Basic NER contains four distinguishable steps: recognition of damage within compact chromatin, incision and excision of the short damaged region (24 to 32 nucleotides [nt]), repair synthesis to fill the gap, and ligation (25).
NER can be divided into two subpathways: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). For GG-NER, detection of DNA lesions is dependent on specific DNA-binding proteins that have high affinity for damaged DNA. UV-damaged DNA binding (UV-DDB) complex is a crucial player in recognizing and processing CPD in the chromatin context (26, 27). UV-DDB is involved in global genomic repair (26–28). UV-DDB consists of two subunits, DDB1 and DDB2, which function in DNA repair and cell cycle regulation (29). The DDB complex has an inherently high binding affinity for DNA lesions, especially for 6-4PP (30–34). DDB can be considered an early damage recognition factor for UV-induced photolesions. Some reports suggest that DDB2 is specifically required for the repair of certain lesions within chromatin (35–39).
HCMV infection induces a strong DDR, which is required for HCMV replication (10, 11, 17, 40). How this DDR affects viral replication is unclear. An important cellular response to DNA damage is to signal DNA repair programs that correct the damage. Based on this relationship, we hypothesize that DDR repair pathways will affect the fidelity and maturation of HCMV genomes. We found that the DDB2 NER factor was required for efficient viral genome replication, suggesting that the cellular NER machinery may also contribute to viral genome integrity during its replication. In the process of characterizing relationships between the DDR and DNA repair pathways, we discovered a novel feedback loop between ATM and DDB2 that became apparent during infection. Our data suggest a model wherein HCMV infection stimulates DNA damage and repair pathways that facilitate the replication or maturation of nascent virus.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic lung (HEL) fibroblasts were obtained from the Coriell Institute for Medical Research (Camden, NJ). HEK293T cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). These cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Telomerase-life-extended dermal fibroblasts from the XPE complementation group (TERT1389) and telomerase-life-extended normal dermal fibroblasts (TERTBJ) were generous gifts from Lisa D. McDaniel (Signature Genomic Laboratories, LLC, Spokane, WA) and cultured in DMEM supplemented with 15% FBS and 1% penicillin-streptomycin. All media, FBS, and antibiotics were from Gibco (Grand Island, NY). HCMV strain AD169 was obtained from the ATCC (Manassas, VA). HEL or XP fibroblasts were infected with HCMV AD169 at the indicated multiplicities of infection (MOI). Viral infections were performed in growth medium with 2% FBS for 2 h. The viral inoculum was removed and replaced with normal growth medium.
siRNA and transfections.
The ATM and control small interfering RNAs (siRNAs) used in this study were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). DDB2 and control siRNAs were ordered from Thermal Scientific (Pittsburgh, PA). Transfections were performed by electroporation in siPort transfection buffer (Ambion, Austin, TX). The nonspecific siRNA (siCON) was a nonsense sequence that had no effect on parameters tested relative to results with mock transfection. Transfection conditions for individual siRNAs were optimized and infections were performed 24 h posttransfection. The siRNAs used in this study are as follows: siCON, GATGCTGCATATAAGCAGC; siATM, CTATGTTGAGGAAGGAGGT; siGENOME SMARTpool DDB2, catalog no. M-011022-01; siGENOME RISK-Free control siRNA, catalog no. D-001220-01.
Immunoblot analysis.
Infected cells were harvested at the indicated times, and cells pellets were stored at −80°C. Thawed cell pellets were resuspended in radioimmunoprecipitation assay buffer (RIPA), which has been described (10), and incubated on ice for 1 h. Samples were sonicated for 15 s, and soluble proteins were collected by centrifugation for 10 min at 13,000 rpm in a microcentrifuge. Proteins were resolved by SDS-PAGE, and the proteins were transferred to a polyvinylidene difluoride membrane (PVDF) (Perkin-Elmer, Waltham, MA) by electroblotting. Detection of proteins was performed with antibodies specific for IE1-72 and IE2-86 (MAB810; Chemicon, Temecula, CA), pp65 (CA003-100; Virusys Corporation, Taneytown, MD), gB55 (Shan Lu, University of Massachusetts Medical School, Worcester, MA), ATM (Cell Signaling Technology, Danvers, MA), DDB2 (Abcam, Cambridge, MA), pUL44 and pUL84 (Virusys Corporation, Sykesville, MD), actin (A5316; Sigma, St. Louis, MO) and horseradish peroxidase (HRP)-conjugated secondary antibodies (GE Healthcare Life Sciences, Pittsburgh, PA). Protein bands were visualized by chemiluminescence using the ECL reagent (PerkinElmer, Waltham, MA).
Viral growth curves.
Fibroblasts were seeded and infected at the listed MOI in each experiment. At the indicated times postinfection (p.i.), a small aliquot (200 μl) of supernatant was harvested from each dish and stored at −80°C. Viral titers were then determined on HEL fibroblasts using standard plaque assay techniques (11). Plotted values represent the average for three independent experiments.
Immunofluorescence analysis.
Immunofluorescent detection of IE and UL44 proteins was performed as described previously (10). More than 200 cells were counted per sample when quantifying cell staining. Plotted values represent the average from three independent experiments. For immunofluorescence detection of DDB2, a construct expressing hemagglutinin (HA)-tagged DDB2 (HA-DDB2) (a kind gift from Yue Xiong, University of North Carolina, Chapel Hill, NC) was transiently transfected into HEL fibroblasts. Twenty-four hours after transfection, fibroblasts were infected with HCMV at an MOI of 1.0. HA-DDB2 and pUL44 were detected with antibodies against HA or pUL44 and fluorescent isothiocyanate (FITC)-conjugated goat anti-rabbit (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and Texas Red-conjugated goat anti-mouse IgG1 secondary antibodies (Southern Biotechnology Associates, Inc., Birmingham, AL). Images were captured on a Zeiss microscope (Zeiss AxioObserver Z1).
qRT-PCR.
Quantitative analyses of viral gene (Ul122, UL123, UL44, UL54, UL55, and UL84) and host gene (DDB2 and ATM) transcripts were performed using the SYBR GreenER quantitative PCR (qPCR) kit (Invitrogen, Carlsbad, CA) on a DNA Engine Opticon 3 continuous fluorescence detection system (MJ Research, Incorporated, Waltham, MA). Total RNA was extracted from the infected cells using TRIzol (Life Technologies), and 100 ng of purified total RNA was reverse transcribed to cDNA using a cDNA synthesis kit (Invitrogen, Carlsbad, CA). PCR conditions were set according to the manufacturer's recommendations. Briefly, after an initial 10 min of denaturation at 95°C, 30 cycles of amplification (45 cycles for DDB2) was performed at 95°C for 30 s and 60°C for 1 min, followed by melting-curve analyses. The amount of template RNA was normalized with the quantified GAPDH in each sample. All of the primer pairs are listed in Table 1. Quantitative experiments were performed at least three times, including a no-template control reaction. Relative expression was calculated using a modified comparative CT method (2−ΔΔCT) (41), in which CT was defined as the cycle numbers at which fluorescence reached a set threshold value. The differences in the CT value of the target genes from that of the corresponding internal control GAPDH gene, ΔCTgene (CTgene − CTGAPDH), were calculated. The changes in the ΔCT of the treated group from that of the control group, ΔΔCTgene (ΔCTtreated gene − ΔCTCON gene), were computed. The expression level of the treated group relative to that of the control group was described using the 2−ΔΔCT method.
TABLE 1.
Primers used for RT-qPCR
| Transcript | qPCR | Primer sequences |
|---|---|---|
| IE1 | SYBR green | 5′-TACCCGCGACTATCCCTCTG-3′ |
| 5′-GGCTCAGACTTGACAGACACA-3′ | ||
| IE2 | SYBR green | 5′-AGTCCGAGGAGATGAAATGCAGCA-3′ |
| 5′-CATGATATTGCGCACCTTCTC-3′ | ||
| UL44 | SYBR green | 5′-ACTGCCGTGCACGTTGCGTA-3′ |
| 5′-ACTTGCCGCTGTTCCCGACG-3′ | ||
| UL54 | SYBR green | 5′-CGTGCCGCGAGGTGTCATGT-3′ |
| 5′-CACCAGGGTCTCGCGCCAAG-3′ | ||
| UL84 | SYBR green | 5′-GCGGACGAGGGACTGGAGGT-3′ |
| 5′-AGCGCGGTAGCCAGACCGTA-3′ | ||
| UL55 | SYBR green | 5′-TCGACCCGCTACCGCCCTAC-3′ |
| 5′-GCAACGCCTTCGACCACGGA-3′ | ||
| GAPDH | SYBR green | 5′-GAAGGTGAAGGTCGGAGTC-3′ |
| 5′-GAAGATGGTGATGGGATTTC-3′ | ||
| DDB2 | SYBR green | 5′-GCCATCTGTCCAGCAGGGGC-3′ |
| 5′-GGGGTGAGTTGGGTGCCACG-3′ | ||
| ATM | SYBR green | 5′-AGCCCTGCGTGCACTGAAAGA-3′ |
| 5′-GCCAGAGGGAACAAAGCCGGA-3′ |
Real-time quantitative PCR analysis of viral DNA synthesis.
DNA was isolated from infected BJ and XPE cells or siRNA-treated HEL fibroblasts by using the Wizard SV genomic DNA purification system (Promega, Madison, WI) according to the manufacturer's instructions. Viral genomes were quantified with a primer pair (pp549s and pp812as) and a probe (pp770s) for UL83 (42), and the number of viral genomes was normalized to the number of cellular copies of β-ACTIN with a previously described set of primers and probe (43). Unknown sample values were determined on the basis of a standard curve of known copy numbers of UL83 (AD169-BAC) and β-ACTIN (pAB1-bactin-PCRscript) (kind gifts from Donald Coen, Harvard Medical School, Boston, MA). PCR mixtures contained 1 μl of 100 μl extracted DNA, 900 nM primers, 250 nM probe, 10 μl TaqMan Universal PCR master mix (Roche, Branchburg, NJ), and nuclease-free water (Ambion, Austin, TX) to 20 μl. Real-time PCR was run and analyzed by using a DNA Engine Opticon 3 continuous fluorescence detection system (MJ Research, Incorporated, Waltham, MA).
Retrovirus production and transduction.
The DDB2 cDNA was subcloned into the pQCXIP vector (Clontech, Mountain View, CA) from the HA-DDB2 construct and the sequence was verified. For retrovirus production and transduction, HEK293T cells were plated in 10-cm-diameter dishes (BD Biosciences, Franklin Lakes, NJ) and transfected with 27 μl of Mirus 293T lipid (Mirus, Madison, WI) together with the retroviral plasmids pQCXIP-HA-DDB2 (5 μg), pCG-GagPol (2 μg), and pCG-VSV-g (2 μg). After 48 h, the retrovirus-containing supernatant was filtered (0.45-um-pore-diameter low-protein-binding filter; Millipore, Billerica, MA), supplemented with 8 μg/ml Polybrene (Sigma, Milwaukeeè, WI), and then added to target cells which had been plated at 3 × 105 cells per 6-cm-diameter dish. This transduction was repeated with the second 72-h supernatant. Forty-eight hours after the second transduction, cells were replated, incubated overnight, and then selected with 200 μg/ml of Geneticin (Invitrogen, Carlsbad, CA) for 1 week.
RESULTS
HCMV replication is compromised in cells with reduced levels of or mutated DDB2.
It has been reported that ATM is activated by HCMV infection (10, 17, 44) and HCMV replication is affected by functional changes in ATM mediated by mutation, drug inhibitor, or RNAi (10). Given the requirement of DDR for replication, we obtained life-extended (“telomerized”) XPE dermal fibroblasts that do not express DDB2 and normal (control), life-extended dermal fibroblasts (BJ) to determine the contribution of DDB2, a DNA repair factor, to HCMV replication. As shown in Fig. 1A, there was a 2- to 3-log reduction in infectious progeny production in XPE (ddb2) cells. Early (pp65) and early/late (E/L) (gB55) gene expression was reduced, along with that of IE2, whose expression levels are linked to viral DNA replication (Fig. 1B). The levels of IE1, the first viral protein expressed during infection, were largely unaffected, as we have also observed when DDR signaling is activated (10).
FIG 1.

DDB2 is required for efficient HCMV replication. (A to D) Replication of HCMV in normal and XPE (ddb2) cells. Normal (BJ) and XPE telomerase-life-extended dermal fibroblasts were infected at an MOI of 0.1. (A) Cell supernatants were assayed for infectious virus production by plaque assay. (B) Viral protein expression is altered in XPE cells. Immunoblot analyses for viral IE (IE1/IE2), E (pp65), and E/L (gB55) protein expression in BJ and XPE (lanes E) dermal fibroblasts are shown. (C and D) DDB2 depletion by siRNA compromises HCMV replication. HEL fibroblasts were transfected with siRNAs specific for DDB2 (siDDB2) or with a control siRNA (siCON) 24 h prior to infection with HCMV at an MOI of 0.1. (C) Cell supernatants were assayed for infectious virus production by plaque assay. (D) Transient depletion of DDB2 alters viral expression. The levels of DDB2 and viral IE (IE1/IE2), E (pp65), and E/L (gB55) protein expression were assessed by immunoblot analysis. (E and F) Effect of rescuing ddb2 on HCMV replication. XPE cells were infected with a retrovirus containing a DDB2 cDNA, and BJ and XPE cells were infected with a retrovirus containing an empty vector. Stably transduced cells were infected with HCMV at an MOI of 0.1. (E) Cell supernatants were assayed for infectious virus production by plaque assay. (F) Protein levels of DDB2 and actin were measured by immunoblot analysis. (A, C, and E) Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. For certain data points, error bars may be too tight to be visible.
A concern with using XPE fibroblasts as a model is the possibility of secondary mutations accumulating given that the originating ddb2 mutation was a germ line event. To complement experiments using XPE fibroblasts, we employed siRNAs to transiently deplete DDB2 protein levels in HEL fibroblasts. Cells were transfected with siRNA specific for DDB2 (siDDB2) 24 h prior to HCMV infection. Viral replication (Fig. 1C) and gene expression (Fig. 1D) were then monitored during a 5-day infection time course. Immunoblotting for DDB2 confirmed the effectiveness of DDB2 depletion with siDDB2 (Fig. 1D). Progeny virus production was reduced ∼15-fold throughout the replication time course in cells depleted for DDB2 (Fig. 1C). Similar to what we observed in XPE fibroblasts (Fig. 1B), we found reduced levels of IE2, pp65, and gB55 but little or no change in IE1 expression in DDB2-depleted HEL fibroblasts (Fig. 1D). At the same time, we noted that the siDDB2 treated cells had a milder phenotype than the XPE cells. This could be due to the levels of DDB2 depletion, additional lesions in the XPE fibroblasts, or a combination of both. To address these possibilities, we introduced wild-type cDDB2 into XPE fibroblasts via retrovirus transduction in an attempt to rescue the wild-type phenotype for viral replication. DDB2 protein levels were restored to near-normal levels in the transduced XPE fibroblasts (Fig. 1F). Infection of the DDB2-tranduced, XPE fibroblasts with HCMV resulted in complete rescue of HCMV progeny production with viral yields that were similar to vector control BJ fibroblasts (Fig. 1E). In contrast, viral yields were 2 to 3 logs lower in XPE fibroblasts transduced with an empty vector. Taken together, these results demonstrate that DDB2 expression influences HCMV replication.
DDB2 is necessary for the formation of mature replication compartments.
We next determined whether cells deficient in DDB2 are compromised in the formation of viral replication compartments (RCs), which are sites of viral DNA replication and maturation. BJ and XPE fibroblasts were infected with HCMV and immunostained with anti-IE and anti-pUL44 antibodies to detect HCMV replication compartments (Fig. 2). IE protein expression was used to indicate infected cells. pUL44 is a virally encoded PCNA-like processivity factor of the viral DNA polymerase and was used to identify viral RCs. In XPE fibroblasts, “immature” or preRCs formed (Fig. 2A), but the percentage of merged, “mature” RCs was greatly reduced relative to BJ fibroblasts (Fig. 2B). In fact, nearly all of the pUL44 immunostaining was observed as nuclear diffuse, a combination of diffuse and pre-RC forms, or pre-RC with only a few cells displaying mature RCs. This difference in the percentages of mature RCs between BJ and XPE fibroblasts might explain the replication phenotype observed in XPE fibroblasts (Fig. 1).
FIG 2.

Reduced formation of “mature” viral replication compartments (RCs) in XPE fibroblasts. Normal (BJ) and XPE dermal fibroblasts were infected at an MOI of 0.1. Cells were fixed each day p.i., and IE1 and IE2 proteins and pUL44 were detected by immunostaining. (A) Localization of IE proteins and pUL44. Immunofluorescent images of normal (BJ) and XPE dermal fibroblasts infected with HCMV. Cells with “immature” RCs were defined as those with multiple, small pUL44 compartments (yellow arrows), and cells with “mature” RCs were identified as those composed of single, larger pUL44 compartments (white arrows). 4′,6-Diamidino-2-phenylindole (DAPI) staining was used to define nuclei. (B) The percentage of fibroblasts with mature RCs was plotted relative to those lacking or having immature RCs. More than 200 cells were scored per sample. Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. For certain data points, error bars may be too tight to be visible.
Given the impact of DDB2 on RC formation, we determined its cellular location. Immunodetection of endogenous DDB2 has not been possible in other systems, and we were not able to detect endogenous DDB2 by immunofluorescence in fibroblasts (not shown). Instead, we transfected a plasmid that expresses HA-DDB2 into HEL fibroblasts prior to infection and detected DDB2 by immunostaining for HA. In mock-infected cells, HA-DDB2 was found diffusely expressed in the nucleus (Fig. 3). This result is consistent with findings in published work (45, 46). During infection, HA-DDB2 staining was biased toward viral RCs as detected by costaining for pUL44. By later times p.i., most of the DDB2 protein was observed in mature RCs, similar to what has been observed for DDR proteins, including ATM, γH2AX, phospho-p53 (10, 40), and other NER proteins (18).
FIG 3.
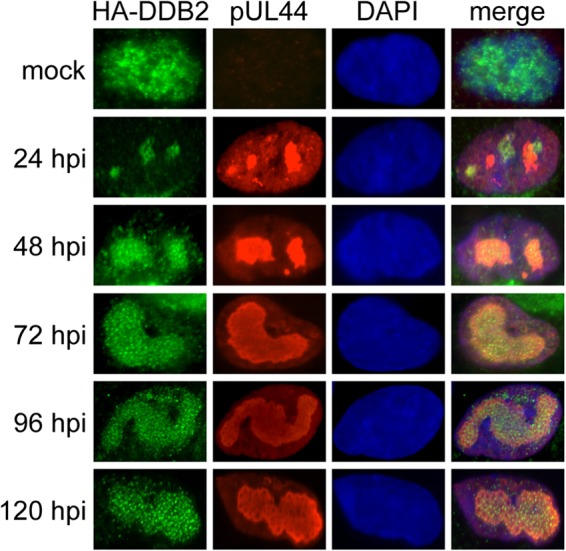
Localization of DDB2 to RCs during infection. HEL fibroblasts were transiently transfected with a plasmid that expresses HA-DDB2. Immunofluorescence detection of pUL44 and HA-DDB2 in HCMV-infected (MOI = 1.0) and mock-infected HEL fibroblasts is shown. DAPI staining was used to define nuclei.
DDB2 is required for efficient viral DNA synthesis.
Since RC formation is dependent upon viral DNA synthesis (47), we hypothesized that reduced mature RC formation might be a consequence of reduced viral DNA synthesis. To test this hypothesis, XPE and BJ fibroblasts were infected with HCMV and viral DNA quantified by qPCR. The amount of viral DNA present in each sample was estimated as the number of copies of HCMV UL83 relative to the copy number of a cellular locus (β-ACTIN). A >1- to 2-log reduction in the number of copies of UL83 was observed throughout a 14-day infection time course in XPE fibroblasts relative to results for BJ fibroblasts (Fig. 4A). This result was further confirmed with HEL fibroblasts. HEL fibroblasts were transfected with siDDB2 24 h prior to HCMV infection, and viral DNA was quantified by qPCR. An ∼5-fold reduction in HCMV UL83 copies was observed from days 2 to 5 p.i. in DDB2-depleted fibroblasts relative to results for control siRNA-treated fibroblasts (Fig. 4B). Of note, HCMV DNA was barely detectable at day 1 in siDDB2-treated fibroblasts. These results indicate that DDB2 may influence the events preceding or involved in initiating viral DNA synthesis.
FIG 4.
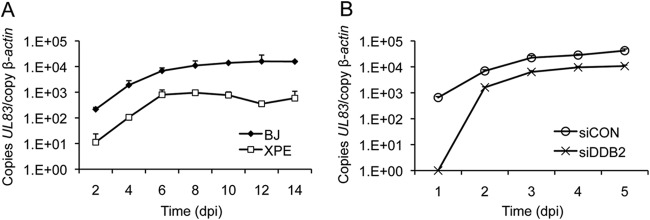
Viral DNA levels are reduced in XPE fibroblasts. (A) BJ and XPE fibroblasts were infected with HCMV at an MOI of 0.1. Viral DNA levels were determined using real-time quantitative PCR with samples collected at 2-day intervals p.i. (dpi, days p.i.). The amount of viral DNA assayed is represented as copies of the viral gene, UL83, per copy of the cellular gene, β-ACTIN. (B) Transient depletion of DDB2 alters DNA replication. HEL fibroblasts were transfected with the indicated siRNAs and infected with HCMV at an MOI of 0.1 24 h later. Viral DNA levels were determined using real-time quantitative PCR on samples collected each day p.i. for 5 days. Viral DNA levels were determined as described for panel A. In both panels, mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. For certain data points, error bars may be too tight to be visible.
DDB2 affects the expression of early genes involved in viral DNA replication.
Given the observations of reduced DNA replication and minimal RC formation in cells lacking DDB2, we next determined whether viral proteins involved in DNA replication were reduced in XPE cells. We targeted UL54 (encoding the HCMV DNA polymerase), UL44 (encoding the DNA polymerase accessory factor), and UL84, whose encoded protein is required for the initiation of viral DNA synthesis (48–50). We measured the expression of these genes by real-time PCR using the 2−ΔΔCT method (41, 51). All transcripts detected were specific and not the result of genomic DNA contamination, since mock-infected cells and qPCRs carried out in the absence of reverse transcriptase failed to produce any products (data not shown). As shown in the Fig. 5A, UL44 expression was reduced 2- to 7-fold in XPE fibroblasts relative to that in BJ fibroblasts during HCMV infection. UL54 gene expression displayed a 3- to 4-fold reduction and UL84 showed a 4- to 7-fold reduction in expression during XPE fibroblast infection relative to results for infected BJ fibroblasts. These data raise the possibility that DDB2 influences the expression of early genes that play important roles in viral DNA replication. In addition, the levels of pUL44 and pUL84 were barely detectable on immunoblots derived from infected XPE fibroblasts (Fig. 5B). We were not able to measure pUL54 levels due to a lack of antibody. Similar results were also observed in siDDB2-transfected HEL fibroblasts, where 2- to 5-fold reductions in transcript levels were detected for all three genes (Fig. 5C). Likewise, the levels of pUL44 and pUL84 were also reduced as observed on immunoblots (Fig. 5D). These results show that gene expression associated with lytic DNA replication is markedly reduced in cells lacking DDB2. All together, DDB2 is critical to early gene transcription and protein expression.
FIG 5.

DDB2 influences the expression of early genes involved in viral DNA replication. (A and B) BJ and XPE fibroblasts were infected with HCMV at an MOI of 0.1. (A) Measurement of HCMV UL44, UL54, and UL84 transcript levels by qRT-PCR. BJ and XPE fibroblasts were infected with HCMV at an MOI of 0.1. Levels of HCMV gene transcript levels in XPE fibroblasts relative to those in BJ fibroblasts are shown. (B) Accumulation of early proteins associated with viral DNA replication is altered in XPE fibroblasts. Cells infected in panel A were assayed for pUL44 and pUL84 protein expression by immunoblot analysis. (C and D) Transient depletion of DDB2 affects UL44, UL54, and UL84 gene expression. HEL fibroblasts were transfected with the indicated siRNAs and infected with HCMV at an MOI of 0.1 24 h later. (C) Measurement of HCMV UL44, UL54, and UL84 transcript levels by qRT-PCR. HCMV gene transcript levels in siDDB2-treated fibroblasts relative to those in siCON-treated fibroblasts are shown. (D) Early protein expression associated with viral DNA replication is altered in siDDB2-treated cells. Cells infected in panel C were assayed for pUL44 and pUL84 protein expression by immunoblot analysis. (A and C) Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments.
Viral genes affected by DDB2 status are also sensitive to a viral DNA replication inhibitor.
From Fig. 4 and 5, we know that DDB2 influences DNA replication and early gene expression. We next determined whether these changes in expression are dependent on DNA replication and whether the observed effects are limited to the early genes assayed, given that the expression of additional gene products, such as IE2 (IE), pp65 (E), and gB55 (E/L), was also affected by the absence of DDB2 (Fig. 1B and D). BJ and XPE fibroblasts were infected at an MOI of 0.1 and then treated with or without 100 μg/ml of the viral DNA replication inhibitor phosphonoacetic acid (PAA). As noted earlier, marked reductions in IE2, pp65, UL44, UL84, and gB55 protein levels were observed in XPE fibroblasts relative to levels in BJ fibroblasts with wild-type DDB2 (Fig. 6A). We found that all of the viral proteins examined were barely detectable in the presence of PAA in BJ and XPE fibroblasts, with the exception of IE1. Analysis of virus replication as assessed by plaque assay confirmed that HCMV replication was inhibited in the PAA-treated cells (Fig. 6B). Likewise, PAA treatment reduced the levels of all viral transcripts (IE, E, and E/L) examined in both XPE and BJ fibroblasts, although the effect was minimal for UL123, which codes for IE1 (Fig. 6C). While many of these genes are considered early (E) genes, DNA replication still influences their expression, as described previously (52, 53). The relative impact of PAA on transcripts was varied. PAA treatment had a greater influence on UL122, UL44, and UL54 transcript levels in BJ and XPE fibroblasts until very late in the infection time course (120 hours p.i. [hpi]) (Fig. 6D to F). For UL84, PAA treatment of XPE fibroblasts had the most dramatic impact on transcript levels, which were sustained throughout the infection time course (Fig. 6H). Like the transcript levels of viral early genes (UL44, UL54, and UL84) assayed for Fig. 5, which were reduced in XPE fibroblasts (Fig. 6E, F, and H), other gene transcript levels (UL122 and UL55) were also reduced in XPE fibroblasts (Fig. 6D and G). Given that UL123 transcripts and the encoded IE1 protein levels were not dramatically impacted by PAA or DDB2 status, we conclude that genes whose expression is influenced by viral DNA replication are the same as those affected by DDB2 status.
FIG 6.

The effect of a viral DNA replication inhibitor on the expression of viral genes affected by DDB2 status. BJ and XPE fibroblasts were infected with HCMV at an MOI of 0.1 in the presence (+) or absence (−) of the viral DNA synthesis inhibitor phosphonoacetic acid (PAA) (100 μg/ml). Samples were harvested at the indicated times p.i. (A) Viral protein expression is altered in XPE and PAA-treated cells. The levels of IE1, IE2, pp65, pUL44, pUL84, and gB55 protein expression were assessed by immunoblot analysis. (B) Replication of HCMV in BJ and XPE fibroblasts with or without PAA. Cell supernatants were assayed for infectious virus production by plaque assay. (C to H) Measurement of UL123 (C), UL122 (D), UL44 (E), UL54 (F), UL55 (G), UL84 (H), and GAPDH transcript levels by qRT-PCR. Total RNA was isolated at 24 to 120 hpi, and the amounts of viral transcripts were measured by qRT-PCR and normalized to that of GAPDH. The fold change in transcript levels of each open reading frame (ORF) and for different cell types with or without PAA is plotted relative to the corresponding transcript levels measured in BJ fibroblasts. Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments.
DDB2 and ATM function in the same genetic pathway to affect HCMV replication.
We determined whether DDB2 and ATM functioned in the same genetic pathway given their similar biological phenotypes, including relocation to RCs and effects on HCMV replication. Depletion of ATM reduced HCMV titers by ∼10-fold in normal BJ fibroblasts (Fig. 7A), similar to results observed in HEL fibroblasts (10). If ATM and DDB2 affect HCMV replication through independent pathways, one would anticipate a cumulative effect on viral replication when ATM is depleted in XPE fibroblasts lacking DDB2. However, we find that siATM treatment did not reduce HCMV titers beyond those observed in control siRNA-transfected, XPE fibroblasts (Fig. 7A; ATM depletion is shown in Fig. 7B). These data are consistent with a model in which ATM and DDB2 function within the same genetic pathway to limit HCMV replication.
FIG 7.
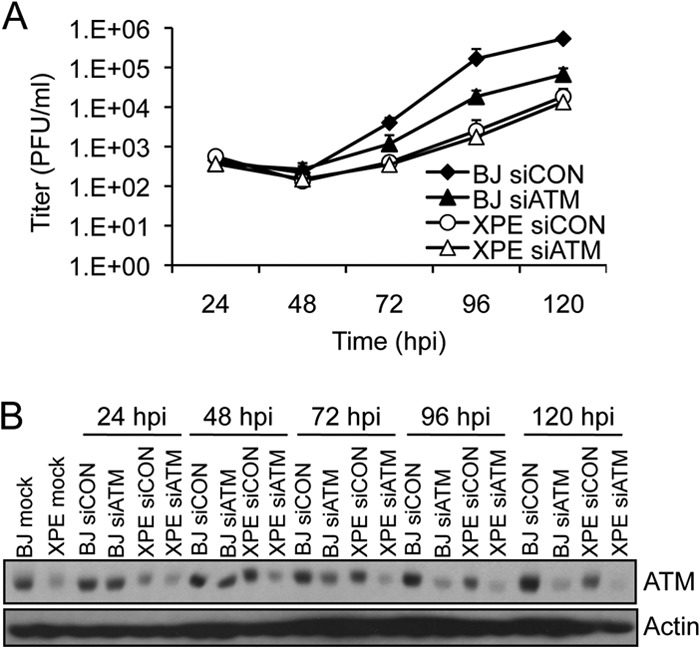
DDB2 and ATM function in the same genetic pathway to influence HCMV replication. (A) Viral replication when ATM is depleted in BJ and XPE fibroblasts. BJ and XPE fibroblasts were transfected with siRNAs specific for ATM (siATM) or with a control siRNA (siCON) 24 h prior to infection with HCMV at an MOI of 0.1. Infected cell supernatants were assayed for infectious virus production by plaque assay. The mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. For certain data points, error bars may be too tight to be visible. (B) Transient depletion of ATM in BJ and XPE fibroblasts. BJ and XPE fibroblasts were transfected with siATM or siCON and infected with HCMV at an MOI of 0.1. The levels of ATM protein expression were assessed by immunoblot analysis.
A novel ATM-DDB2 negative feedback loop exists in HCMV-infected cells.
We previously noted that ATM levels increase during HCMV infection (10). This is a novel observation, since ATM levels are unaffected in response to other infections, including infections by other herpesviruses (12, 13), nor are they affected by environmental insults that promote DDR signaling (54, 55). Here we observed that HCMV infection resulted in time-dependent increases in ATM levels that were delayed in cells depleted for DDB2 (Fig. 8A), even though ATM transcript levels remained largely unchanged (Fig. 8B). Therefore, ATM protein accumulation is independent of ATM RNA levels. In contrast, we found that DDB2 transcript levels were ∼2- to 4-fold higher in ATM-depleted cells relative to those in control siRNA-treated cells, at least through 96 hpi (Fig. 8C; ATM depletion is shown in Fig. 8D). To determine whether ATM accumulation is specific to DDB2 or secondary to viral DNA synthesis, we measured ATM levels in siDDB2- and siCON-treated HEL fibroblasts with or without PAA. As shown in Fig. 8E, ATM accumulation occurred in PAA-treated cells and remained DDB2 dependent. This observation demonstrates that ATM accumulation is a response to HCMV infection that is independent of viral DNA replication and that DDB2 regulates the kinetics of this process.
FIG 8.

An infection-associated DDB2-ATM negative feedback loop. (A and B) DDB2 contributes to the kinetics of ATM accumulation. HEL fibroblasts were transiently transfected with siCON or siDDB2 and subsequently infected with HCMV at an MOI of 0.1 24 h later. Samples were harvested at the indicated times p.i. (A) ATM and DDB2 levels were assessed by immunoblot analyses. (B) ATM transcript levels were measured by qRT-PCR. The fold change in ATM transcript levels in siDDB2-treated fibroblasts is shown relative to ATM transcript levels measured in siCON-treated fibroblasts. Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. (C and D) Influence of ATM on DDB2 transcript levels. HEL fibroblasts were treated with siCON or siATM and infected with HCMV at an MOI of 0.1 24 h later. Cells were harvested at the indicated times p.i. For panel C, DDB2 transcript levels were measured by qRT-PCR. The fold change in DDB2 transcript levels in siATM-treated fibroblasts is plotted relative to DDB2 transcript levels measured in siCON-treated fibroblasts. Mean values are shown, with bars denoting 1 standard deviation, for three independent experiments. For panel D, the levels of ATM protein expression in siCON- and siATM-treated fibroblasts were assessed by immunoblot analysis. (E) DDB2-dependent ATM accumulation in response to HCMV infection is independent of viral DNA replication. HEL fibroblasts were treated with siCON or siDDB2 and infected with HCMV at an MOI of 0.1 24 h later in the presence (+) or absence (−) of the viral DNA synthesis inhibitor phosphonoacetic acid (PAA) (100 μg/ml). The levels of ATM protein expression in siCON- and siDDB2-treated cells with or without PAA were assessed by immunoblot analyses.
In total, these results reveal an infection-associated negative feedback loop between ATM and DDB2 that regulates the HCMV replication cycle (Fig. 9).
FIG 9.
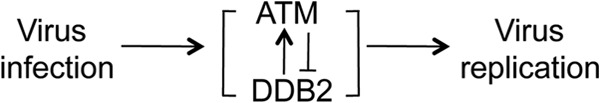
Model depicting the relationship among HCMV infection, DDB2, ATM, and virus replication.
DISCUSSION
Cells have many pathways with which to repair damaged DNA. With the exception of proofreading during viral DNA replication, DNA viruses generally lack repair programs. Given that DNA damage and errors occur during viral DNA replication (56–60), viruses likely rely on cellular machineries to provide repair activities. Complex interactions among viral and cellular DNA damage and repair systems have been reported in several studies of herpesviruses (13, 16, 57, 61). The protein kinase ATM is activated in response to DNA virus infections, including HCMV infection (10, 17, 40). We hypothesized that this infection-associated ATM activation stimulates cellular DNA repair pathways to facilitate successful replication of viral DNA. Here, evidence indicates that an essential NER DNA damage recognition factor, DDB2, contributes to HCMV lytic replication. While this is the first study to demonstrate such a role for an NER protein, this finding is consistent with a previous report that XPC, another NER factor, contributes to EBV DNA replication (61). In addition, we discovered a feedback loop between ATM and DDB2.
The described function of DDB2 is in recognizing and recruiting other factors to UV-damaged DNA for initiating NER (62–64). The observation of a functional link between ATM and DDB2 (Fig. 7A) was unexpected. However, a link between NER and ATM autophosphorylation has been observed, albeit in response to cross-links generated by cisplatin, which can be considered to be in a class similar to UV-induced adducts (65). This relationship between ATM and DDB2 during HCMV infection is further demonstrated by the novel infection-associated feedback loop wherein DDB2 promotes ATM accumulation and ATM modulates DDB2 expression (Fig. 8). ATM accumulation during infection correlates with increased ATM activation as measured by its autophosporylation (10), which is necessary for HCMV replication (44). Why an NER protein and ATM signaling are linked and relevant to HCMV replication awaits further study, but the reductions in viral RCs, E gene expression associated with lytic origin firing, gene expression influenced by DNA replication, and viral DNA loads in XPE cells suggest that the defect is linked to events preceding or associated with the initiation of DNA replication. One can imagine that the DDB2-associated amplification of ATM signaling during infection would generate a robust and broad DNA damage response (66) that could complement replication or repair activities not encoded by HCMV. That viruses tend to exploit preexisting cellular activities also raises the possibility that a DDB2-ATM relationship has been unappreciated in DDR research.
Why are NER proteins associated with RCs and affecting viral replication? It has been reported recently that NER takes place in HCMV-infected cells exposed to UV irradiation to selectively remove CPD from viral DNA in the absence of host DNA repair (18). It can be inferred from this study that all of the essential components of the NER pathway are available to preferentially repair viral DNA. The absence of host CPD repair in infected cells may be due to relocalization of NER proteins or condensation of host chromatin, since chromatin accessibility is an important step in the detection and efficient removal of DNA lesions by GG-NER (67, 68). Accessibility of damaged, viral DNA and localization of NER proteins to RCs (18; this study) may explain the bias in repair. What is left unanswered is the question of why NER proteins and NER capacity exist in HCMV RCs.
UV adduct formation is not likely in tissues and circulating monocytes where HCMV resides in vivo. Perhaps DDB2 functions independently of NER in infected cells. DDB2 is a component of a Cul4A-ubiquitin (Ub) ligase complex. DDB1-CUL4ADDB2 is a cullin-RING (i.e., E3) Ub ligase (69–72). DDB2 is thought to function as a substrate adapter protein. This complex has a unique property of binding UV-damaged DNA whereby multiple substrates can be ubiquitinated, including, XPC, DDB2, and possibly histone H2A (73, 74). We speculate that the DDB1-CUL4ADDB2 E3 ligase may ubiquitinate additional proteins during infection that affect replication. If this is correct, DDB2-associated ubiquitination may play an important role in recruiting other host proteins to the RC to form complexes that contribute to DNA replication.
Replication compartments seem to be an important site for relocalization of DNA damage and repair proteins in virus replication (10, 12, 40). DDB2 is relocalized to RC during HCMV infection, as found here, and XPD and Cockayne syndrome complementation group B (CSB) relocalize to the RC in UV-irradiated HCMV-infected cells (18). In addition, PCNA and components of mismatch repair factors are recruited to EBV RCs (75). These observations suggest that functional RCs need host repair proteins for the efficient production of viral DNA.
In this work, we found that DDB2 can affect viral gene expression, so the delay in virus replication in DDB2 mutant cells may not be dependent on viral DNA replication. Although the initial level of DNA detected is much lower in XPE cells, the rate of amplification seems similar. This observation could be interpreted as indicating a role for DDB2 in the initiation of viral DNA replication. Alternately, it is possible that the defect is not in DNA replication but in some manifestation of the entry process, such as genome stability or circularization, or perhaps post-IE transcriptional defects. The answer(s) to this question awaits future study.
Here we discovered a feedback loop between DDB2 and ATM in infected cells. ATM, as an upstream factor of DDB2, decreases the transcription of DDB2, and DDB2 contributes to the accumulation of ATM. Although the DDB2-Cul4A-DDB1 complex is a downstream target of ATM (66), we do not know how ATM inhibits DDB2 transcription; maybe its role is indirect. Perhaps DDB2 functionally interacts with other phosphatidylinositol 3 (PI3)-like kinases to produce feedback loops. In support of this possibility, ATR kinase has been reported to be a master regulator of NER during the S phase of the cell cycle (76). ATM accumulation has not been observed before in any context, leading us to speculate that HCMV infection has shifted a normally tightly regulated mechanism wherein DDB2 levels are moderated in a manner that sustains “steady-state” levels of ATM.
Although XP proteins accumulate in herpesvirus RCs, contribute to viral replication, and can function in NER during infections, it is still a mystery what these proteins are doing mechanistically to facilitate the replication of herpesvirus DNA. Perhaps the DDB2-ATM loop has multiple functions that aid viral DNA replication, including facilitating the repair of abnormal DNA structures generated during replication. Because most DNA viruses require ATM activation to replicate (9, 10, 13, 14, 16, 77–79), it will be interesting to determine the mechanistic contribution(s) of DDB2 and its relationship with ATM to viral DNA replication during infection with HCMV and other viruses.
ACKNOWLEDGMENTS
We thank members of the Kowalik laboratory for commenting on the manuscript.
The research presented in this article was funded by the NIH (AI076189, to T.F.K.). A.L.B. is grateful for the generous support of the Charles H. Hood Foundation, the Burroughs Wellcome Fund, the Bill and Melinda Gates Foundation's Collaboration for AIDS Vaccine Discovery, and the University of Massachusetts Medical School's Center for AIDS Research.
The contents of this publication are solely our responsibility and do not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 11 December 2013
REFERENCES
- 1.Peggs KS. 2004. Cytomegalovirus following stem cell transplantation: from pharmacologic to immunologic therapy. Expert Rev. Anti Infect. Ther. 2:559–573. 10.1586/14787210.2.4.559 [DOI] [PubMed] [Google Scholar]
- 2.Cannon MJ. 2009. Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. 46(Suppl 4):S6–S10. 10.1016/j.jcv.2009.09.002 [DOI] [PubMed] [Google Scholar]
- 3.Hassan J, Connell J. 2007. Translational mini-review series on infectious disease: congenital cytomegalovirus infection: 50 years on. Clin. Exp. Immunol. 149:205–210. 10.1111/j.1365-2249.2007.03454.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharjee B, Renzette N, Kowalik TF. 2012. Genetic analysis of cytomegalovirus in malignant gliomas. J. Virol. 86:6815–6824. 10.1128/JVI.00015-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobbs CS. 2011. Evolving evidence implicates cytomegalovirus as a promoter of malignant glioma pathogenesis. Herpesviridae 2:10. 10.1186/2042-4280-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, Soroceanu L, Cobbs CS. 2012. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 14:246–255. 10.1093/neuonc/nor227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. 2012. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86:854–864. 10.1128/JVI.06097-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soroceanu L, Cobbs CS. 2011. Is HCMV a tumor promoter? Virus Res. 157:193–203. 10.1016/j.virusres.2010.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariumi Y, Trono D. 2006. Ataxia-telangiectasia-mutated (ATM) protein can enhance human immunodeficiency virus type 1 replication by stimulating Rev function. J. Virol. 80:2445–2452. 10.1128/JVI.80.5.2445-2452.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.E X, Pickering MT, Debatis M, Castillo J, Lagadinos A, Wang S, Lu S, Kowalik TF. 2011. An E2F1-mediated DNA damage response contributes to the replication of human cytomegalovirus. PLoS Pathog. 7:e1001342. 10.1371/journal.ppat.1001342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.E X, Stadler BM, Debatis M, Wang S, Lu S, Kowalik TF. 2012. RNA interference-mediated targeting of human cytomegalovirus immediate early or early gene products inhibits viral replication with differential effects on cellular functions. J. Virol. 86:5660–5673. 10.1128/JVI.06338-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 280:8156–8163. 10.1074/jbc.M411405200 [DOI] [PubMed] [Google Scholar]
- 13.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 102:5844–5849. 10.1073/pnas.0501916102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5:e1000605. 10.1371/journal.ppat.1000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perfettini JL, Nardacci R, Bourouba M, Subra F, Gros L, Seror C, Manic G, Rosselli F, Amendola A, Masdehors P, Chessa L, Novelli G, Ojcius DM, Siwicki JK, Chechlinska M, Auclair C, Regueiro JR, de The H, Gougeon ML, Piacentini M, Kroemer G. 2008. Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia. PLoS One 3:e2458. 10.1371/journal.pone.0002458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarakanova VL, Leung-Pineda V, Hwang S, Yang CW, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW., IV 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. 10.1016/j.chom.2007.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castillo JP, Frame FM, Rogoff HA, Pickering MT, Yurochko AD, Kowalik TF. 2005. Human cytomegalovirus IE1-72 activates ataxia telangiectasia mutated kinase and a p53/p21-mediated growth arrest response. J. Virol. 79:11467–11475. 10.1128/JVI.79.17.11467-11475.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Dowd JM, Zavala AG, Brown CJ, Mori T, Fortunato EA. 2012. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 8:e1003038. 10.1371/journal.ppat.1003038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hang B. 2010. Formation and repair of tobacco carcinogen-derived bulky DNA adducts. J. Nucleic Acids 2010:709521. 10.4061/2010/709521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berneburg M, Lehmann AR. 2001. Xeroderma pigmentosum and related disorders: defects in DNA repair and transcription. Adv. Genet. 43:71–102. 10.1016/S0065-2660(01)43004-5 [DOI] [PubMed] [Google Scholar]
- 21.de Laat WL, Jaspers NG, Hoeijmakers JH. 1999. Molecular mechanism of nucleotide excision repair. Genes Dev. 13:768–785. 10.1101/gad.13.7.768 [DOI] [PubMed] [Google Scholar]
- 22.Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. 2007. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 145:1388–1396. 10.1016/j.neuroscience.2006.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraemer KH, Lee MM, Andrews AD, Lambert WC. 1994. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch. Dermatol. 130:1018–1021. 10.1001/archderm.1994.01690080084012 [DOI] [PubMed] [Google Scholar]
- 24.Kraemer KH, Levy DD, Parris CN, Gozukara EM, Moriwaki S, Adelberg S, Seidman MM. 1994. Xeroderma pigmentosum and related disorders: examining the linkage between defective DNA repair and cancer. J. Investig. Dermatol. 103:96S–101S. 10.1111/1523-1747.ep12399329 [DOI] [PubMed] [Google Scholar]
- 25.Friedberg EC. 2001. How nucleotide excision repair protects against cancer. Nat. Rev. Cancer 1:22–33. 10.1038/35094000 [DOI] [PubMed] [Google Scholar]
- 26.Hwang BJ, Toering S, Francke U, Chu G. 1998. p48 activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol. Cell. Biol. 18:4391–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. 2000. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell 5:737–744. 10.1016/S1097-2765(00)80252-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hwang BJ, Ford JM, Hanawalt PC, Chu G. 1999. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. U. S. A. 96:424–428. 10.1073/pnas.96.2.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiyanov P, Hayes SA, Donepudi M, Nichols AF, Linn S, Slagle BL, Raychaudhuri P. 1999. The naturally occurring mutants of DDB are impaired in stimulating nuclear import of the p125 subunit and E2F1-activated transcription. Mol. Cell. Biol. 19:4935–4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu G, Chang E. 1988. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242:564–567. 10.1126/science.3175673 [DOI] [PubMed] [Google Scholar]
- 31.Fujiwara Y, Masutani C, Mizukoshi T, Kondo J, Hanaoka F, Iwai S. 1999. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem. 274:20027–20033. 10.1074/jbc.274.28.20027 [DOI] [PubMed] [Google Scholar]
- 32.Keeney S, Chang GJ, Linn S. 1993. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J. Biol. Chem. 268:21293–21300 [PubMed] [Google Scholar]
- 33.Reardon JT, Nichols AF, Keeney S, Smith CA, Taylor JS, Linn S, Sancar A. 1993. Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. J. Biol. Chem. 268:21301–21308 [PubMed] [Google Scholar]
- 34.Treiber DK, Chen Z, Essigmann JM. 1992. An ultraviolet light-damaged DNA recognition protein absent in xeroderma pigmentosum group E cells binds selectively to pyrimidine (6-4) pyrimidone photoproducts. Nucleic Acids Res. 20:5805–5810. 10.1093/nar/20.21.5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80:859–868. 10.1016/0092-8674(95)90289-9 [DOI] [PubMed] [Google Scholar]
- 36.Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. 2000. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 14:349–359. 10.1101/gad.14.3.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bessho T, Sancar A, Thompson LH, Thelen MP. 1997. Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J. Biol. Chem. 272:3833–3837. 10.1074/jbc.272.6.3833 [DOI] [PubMed] [Google Scholar]
- 38.Kulaksiz G, Reardon JT, Sancar A. 2005. Xeroderma pigmentosum complementation group E protein (XPE/DDB2): purification of various complexes of XPE and analyses of their damaged DNA binding and putative DNA repair properties. Mol. Cell. Biol. 25:9784–9792. 10.1128/MCB.25.22.9784-9792.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. 1995. Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270:2415–2418. 10.1074/jbc.270.6.2415 [DOI] [PubMed] [Google Scholar]
- 40.Luo MH, Rosenke K, Czornak K, Fortunato EA. 2007. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 81:1934–1950. 10.1128/JVI.01670-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 42.Gault E, Michel Y, Dehee A, Belabani C, Nicolas JC, Garbarg-Chenon A. 2001. Quantification of human cytomegalovirus DNA by real-time PCR. J. Clin. Microbiol. 39:772–775. 10.1128/JCM.39.2.772-775.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanfler J, Kreuzer KA, Laurisch K, Rayes N, Neuhaus P, Schmidt CA, Oettle H. 2003. Quantitation of cytomegalovirus (hCMV) DNA and beta-actin DNA by duplex real-time fluorescence PCR in solid organ (liver) transplant recipients. Med. Microbiol. Immunol. 192:197–204. 10.1007/s00430-002-0166-6 [DOI] [PubMed] [Google Scholar]
- 44.Li R, Zhu J, Xie Z, Liao G, Liu J, Chen MR, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400. 10.1016/j.chom.2011.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van Driel R. 2007. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J. Cell Sci. 120:2706–2716. 10.1242/jcs.008367 [DOI] [PubMed] [Google Scholar]
- 46.Molinier J, Lechner E, Dumbliauskas E, Genschik P. 2008. Regulation and role of Arabidopsis CUL4-DDB1A-DDB2 in maintaining genome integrity upon UV stress. PLoS Genet. 4:e1000093. 10.1371/journal.pgen.1000093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penfold ME, Mocarski ES. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46–61. 10.1006/viro.1997.8848 [DOI] [PubMed] [Google Scholar]
- 48.Colletti KS, Xu Y, Cei SA, Tarrant M, Pari GS. 2004. Human cytomegalovirus UL84 oligomerization and heterodimerization domains act as transdominant inhibitors of oriLyt-dependent DNA replication: evidence that IE2-UL84 and UL84-UL84 interactions are required for lytic DNA replication. J. Virol. 78:9203–9214. 10.1128/JVI.78.17.9203-9214.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Y, Cei SA, Huete AR, Pari GS. 2004. Human cytomegalovirus UL84 insertion mutant defective for viral DNA synthesis and growth. J. Virol. 78:10360–10369. 10.1128/JVI.78.19.10360-10369.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, Cei SA, Rodriguez Huete A, Colletti KS, Pari GS. 2004. Human cytomegalovirus DNA replication requires transcriptional activation via an IE2- and UL84-responsive bidirectional promoter element within oriLyt. J. Virol. 78:11664–11677. 10.1128/JVI.78.21.11664-11677.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derveaux S, Vandesompele J, Hellemans J. 2010. How to do successful gene expression analysis using real-time PCR. Methods 50:227–230. 10.1016/j.ymeth.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 52.Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 85:4841–4852. 10.1128/JVI.02344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qin Q, Penkert RR, Kalejta RF. 2013. Heterologous viral promoters incorporated into the human cytomegalovirus genome are silenced during experimental latency. J. Virol. 87:9886–9894. 10.1128/JVI.01726-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pandita TK, Lieberman HB, Lim DS, Dhar S, Zheng W, Taya Y, Kastan MB. 2000. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 19:1386–1391. 10.1038/sj.onc.1203444 [DOI] [PubMed] [Google Scholar]
- 55.Yajima H, Lee KJ, Zhang S, Kobayashi J, Chen BP. 2009. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J. Mol. Biol. 385:800–810. 10.1016/j.jmb.2008.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Millhouse S, Su YH, Zhang X, Wang X, Song BP, Zhu L, Oppenheim E, Fraser NW, Block TM. 2010. Evidence that herpes simplex virus DNA derived from quiescently infected cells in vitro, and latently infected cells in vivo, is physically damaged. J. Neurovirol. 16:384–398. 10.3109/13550284.2010.515651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. 2011. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 7:e1001344. 10.1371/journal.ppat.1001344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renzette N, Gibson L, Bhattacharjee B, Fisher D, Schleiss MR, Jensen JD, Kowalik TF. 2013. Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. PLoS Genet. 9:e1003735. 10.1371/journal.pgen.1003735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hyman RW, Oakes JE, Kudler L. 1977. In vitro repair of the preexisting nicks and gaps in herpes simplex virus DNA. Virology 76:286–294. 10.1016/0042-6822(77)90303-8 [DOI] [PubMed] [Google Scholar]
- 60.Wilkinson DE, Weller SK. 2003. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life 55:451–458. 10.1080/15216540310001612237 [DOI] [PubMed] [Google Scholar]
- 61.Lu CC, Chen YC, Wang JT, Yang PW, Chen MR. 2007. Xeroderma pigmentosum C is involved in Epstein Barr virus DNA replication. J. Gen. Virol. 88:3234–3243. 10.1099/vir.0.83212-0 [DOI] [PubMed] [Google Scholar]
- 62.Fitch ME, Nakajima S, Yasui A, Ford JM. 2003. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem. 278:46906–46910. 10.1074/jbc.M307254200 [DOI] [PubMed] [Google Scholar]
- 63.Wakasugi M, Kawashima A, Morioka H, Linn S, Sancar A, Mori T, Nikaido O, Matsunaga T. 2002. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem. 277:1637–1640. 10.1074/jbc.C100610200 [DOI] [PubMed] [Google Scholar]
- 64.Wakasugi M, Shimizu M, Morioka H, Linn S, Nikaido O, Matsunaga T. 2001. Damaged DNA-binding protein DDB stimulates the excision of cyclobutane pyrimidine dimers in vitro in concert with XPA and replication protein A. J. Biol. Chem. 276:15434–15440. 10.1074/jbc.M011177200 [DOI] [PubMed] [Google Scholar]
- 65.Colton SL, Xu XS, Wang YA, Wang G. 2006. The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J. Biol. Chem. 281:27117–27125. 10.1074/jbc.M602826200 [DOI] [PubMed] [Google Scholar]
- 66.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316:1160–1166. 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- 67.Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F. 2001. ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J. 20:2004–2014. 10.1093/emboj/20.8.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang ZG, Wu XH, Friedberg EC. 1991. Nucleotide excision repair of DNA by human cell extracts is suppressed in reconstituted nucleosomes. J. Biol. Chem. 266:22472–22478 [PubMed] [Google Scholar]
- 69.Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. 2006. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443:590–593. 10.1038/nature05175 [DOI] [PubMed] [Google Scholar]
- 70.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. 2003. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113:357–367. 10.1016/S0092-8674(03)00316-7 [DOI] [PubMed] [Google Scholar]
- 71.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425–479. 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 72.Shiyanov P, Nag A, Raychaudhuri P. 1999. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 274:35309–35312. 10.1074/jbc.274.50.35309 [DOI] [PubMed] [Google Scholar]
- 73.Guerrero-Santoro J, Kapetanaki MG, Hsieh CL, Gorbachinsky I, Levine AS, Rapic-Otrin V. 2008. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 68:5014–5022. 10.1158/0008-5472.CAN-07-6162 [DOI] [PubMed] [Google Scholar]
- 74.Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. 2006. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc. Natl. Acad. Sci. U. S. A. 103:2588–2593. 10.1073/pnas.0511160103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daikoku T, Kudoh A, Sugaya Y, Iwahori S, Shirata N, Isomura H, Tsurumi T. 2006. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J. Biol. Chem. 281:11422–11430. 10.1074/jbc.M510314200 [DOI] [PubMed] [Google Scholar]
- 76.Auclair Y, Rouget R, Drobetsky EA. 2009. ATR kinase as master regulator of nucleotide excision repair during S phase of the cell cycle. Cell Cycle 8:1865–1871. 10.4161/cc.8.12.8800 [DOI] [PubMed] [Google Scholar]
- 77.Ariumi Y, Kuroki M, Dansako H, Abe K, Ikeda M, Wakita T, Kato N. 2008. The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J. Virol. 82:9639–9646. 10.1128/JVI.00351-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baer A, Austin D, Narayanan A, Popova T, Kainulainen M, Bailey C, Kashanchi F, Weber F, Kehn-Hall K. 2012. Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication. J. Biol. Chem. 287:7399–7410. 10.1074/jbc.M111.296608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. 2005. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 280:40195–40200. 10.1074/jbc.C500400200 [DOI] [PubMed] [Google Scholar]