

Abstract
Enzyme-dead mutations in the herpes simplex virus 1 UL12 gene that abolished its endo- and exonuclease activities only slightly reduced viral replication in cell cultures. However, the UL12 null mutation significantly reduced viral replication, suggesting that a UL12 function(s) unrelated to its nuclease activities played a major role in viral replication. In contrast, the enzyme-dead mutations significantly reduced viral neurovirulence in mice, suggesting that UL12 nuclease activities were critical for viral pathogenesis in vivo.
TEXT
The herpes simplex virus 1 (HSV-1) UL12 gene encodes a nuclease with both endo- and exonuclease activities and an alkaline pH optimum in vitro (1). The UL12 amino acid sequence is well conserved in all Herpesviridae subfamilies (2, 3). Recombinant viruses that cannot express UL12 have a 100- to 1,000-fold reduction in their progeny virus titers in cell cultures, suggesting that UL12 has a critical role in viral replication in vitro (4, 5). The glycine and serine residues at UL12 positions 336 and 338, respectively, are highly conserved in UL12 homologues in other herpesviruses, and alanine substitutions for these conserved residues (G336A/S338A) in purified UL12 expressed in a baculovirus system abolished both endo- and exonuclease activities in vitro (3), indicating that these residues were essential for both UL12 nuclease activities. Transfection of cell cultures with a wild-type UL12 expression plasmid efficiently complemented growth of the UL12 null mutant virus, but transfection with an expression plasmid carrying UL12 with the G336A/S338A double mutation barely complemented growth of the UL12 null mutant virus (3), indicating that the UL12 nuclease activities were required for efficient viral replication in vitro. However, construction and characterization of a recombinant virus carrying an enzyme-dead mutation in UL12, such as the G336A/S338A double mutation, have not been reported, and therefore, the exact role(s) of the UL12 nuclease activities in HSV-1 viral replication in cell cultures has not been fully determined. Therefore, in the present study we constructed a recombinant virus carrying the G336A/S338A double mutation in UL12 and its repaired virus and characterized them together with a UL12 null mutant virus to clarify the roles of the nuclease activities of UL12 in vitro and in vivo.
Construction and characterization of recombinant viruses.
YK655 (ΔUL12), a UL12 null mutant virus in which the UL12 gene was disrupted by replacing UL12 codons 70 to 375 with a kanamycin resistance gene, was constructed by the Red-mediated mutagenesis procedure using Escherichia coli GS1783 carrying pYEbac102 (6), containing a full-length infectious HSV-1 clone, as described previously (7), except using primers 5′-GTGCCATCAAGTCCTCGTACGCGGAGGCCGTGGGGTCGCTGGGGTCCATGAGGATGACGACGATAAGTAGGG-3′ and 5′-CCCCACCCCAGACGACGTCAGCTGTGGACCCGAGCTCCCATTCGCCCGATCATGGACCCCAGCGACCCCACGGCCTCCGCGTACGAGGACTTGATGGCACCAACCAATTAACCAATTCTGATTAG-3′. YK656 (ΔUL12-repair), in which the UL12 null mutation in YK655 was repaired, was constructed by transfection of Vero (simian kidney epithelial) cells (6) with the transfer plasmid pBS-UL12 using Lipofectamine 2000 (Invitrogen) and by subsequent superinfection of the transfected cells with YK655 (ΔUL12) as described previously (8). pBS-UL12 contained the entire UL12 open reading frame bounded by the 0.5-kbp upstream and 1.1-kbp downstream UL12 flanking sequences. To construct pBS-UL12, a 1.9-kbp DNA fragment containing part of the UL12 gene and its upstream region was amplified from pBC1012 (9) by PCR using primers 5′-GCGCGGCCGCACGACCAAACCGACGTATTG-3′ and 5′-GCCTCGAGGAGGCGGGTATGGTGGACCG-3′, and a 1.6-kbp DNA fragment containing part of the UL12 gene and its downstream region was amplified from pBC1012 by PCR using primers 5′-GCCTCGAGGACCACGGCCAGGGAACACA-3′ and 5′-GCGGTACCCAACATCCGCGGCTTCATCG-3′. These DNA fragments were then sequentially cloned into pBluescript II KS(+) (Stratagene) to produce pBS-UL12. YK665 (UL12G336A/S338A), carrying the G336A/S338A double mutation in UL12, was constructed by the two-step Red-mediated mutagenesis procedure using E. coli GS1783 containing pYEbac102 as described previously (7), except using primers 5′-TACGTGTGGGGTCCTCATGGACGGTCACACGGGGATGGTCGCGGCGGCCCTGGATATTCTCGTCTGTCCAGGATGACGACGATAAGTAGGG-3′ and 5′-GGTAGCCGTGAATGTCCCGAGGACAGACGAGAATATCCAGGGCCGCCGCGACCATCCCCGTGTGACCGTCAACCAATTAACCAATTCTGATTAG-3′. YK666 (UL12GA/SA-repair), in which the UL12G336A/S338A mutation in YK665 was repaired, was constructed as described previously (7), except using primers 5′-TACGTGTGGGGTCCTCATGGACGGTCACACGGGGATGGTCGGGGCGTCCCTGGATATTCTCGTCTGTCCAGGATGACGACGATAAGTAGGG-3′ and 5′-GGTAGCCGTGAATGTCCCGAGGACAGACGAGAATATCCAGGGACGCCCCGACCATCCCCGTGTGACCGTCAACCAATTAACCAATTCTGATTAG-3′. We sequenced the ICP8 and UL12 coding regions from the YK655 (ΔUL12) and YK665 (UL12G336A/S338A) viruses but did not detect any additional and unintended changes in these coding regions except the UL12 null and UL12G336A/S338A mutations in the UL12 genes of the YK655 (ΔUL12) and YK665 (UL12G336A/S338A) viruses (data not shown). All viruses used in this study were propagated and titrated in 6-5 cells (4), which are permissive for UL12 null mutant viruses and were kindly provided by S. Weller.
To confirm that the null (ΔUL12) mutation in UL12 had no effect on expression of its neighboring genes, UL11 and UL13, and to examine the effect of the G336A/S338A mutation in UL12 on expression of the UL12 gene, Vero cells were mock infected or infected at a multiplicity of infection (MOI) of 3 for 24 h with wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK665 (UL12G336A/S338A), YK666 (UL12GA/SA-repair), or R7356, a UL13 null mutant virus, which was kindly provided by B. Roizman, and analyzed by immunoblotting with antibodies to VP22 (10), α-tubulin (DM1A; Sigma-Aldrich), UL11 (11), UL12, and UL13. Anti-UL12 antibody was generated by immunization of a rabbit with a fusion protein of maltose-binding protein (MBP) and the N-terminal domain (codons 2 to 126) of UL12 (MBP-UL12-N) that had been expressed in E. coli and purified as described previously (12). Mouse monoclonal antibody to UL13 was generated as described previously (13), except using TiterMax Gold (TiterMax USA, Inc.) and a fusion protein of MBP and the domain of UL13 containing codons 336 to 518 that had been expressed and purified as described above. As shown in Fig. 1A, Vero cells infected with wild-type HSV-1(F), YK656 (ΔUL12-repair), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) expressed UL12, but cells infected with YK655 (ΔUL12) did not. The level of UL12 expression in Vero cells infected with YK665 (UL12G336A/S338A) was similar to that in Vero cells infected with wild-type HSV-1(F) (Fig. 1A). Immunofluorescence microscopy showed that UL12 was distributed diffusely throughout the nuclei of Vero cells infected with wild-type HSV-1(F) at an MOI of 10 for 12 h, as reported previously (14), and a similar UL12 localization pattern was observed in Vero cells infected with YK665 (UL12G336A/S338A) (data not shown). These results indicated that the nuclease activities of UL12 were not required for proper expression and localization of UL12 in infected cells. Furthermore, wild-type HSV-1(F) and YK655 (ΔUL12) produced similar levels of UL11 and UL13 in infected Vero cells (Fig. 1A), indicating that the null (ΔUL12) mutation in UL12 had no effect on expression of the UL12-neighboring genes UL11 and UL13.
FIG 1.
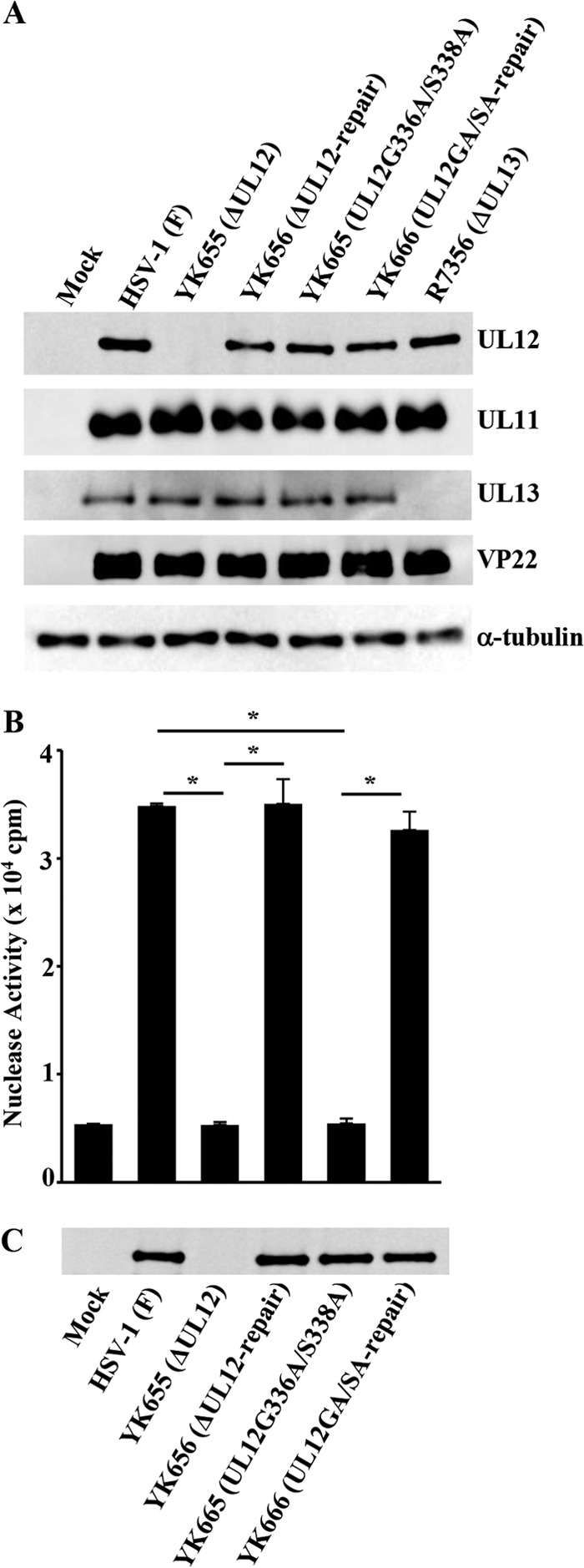
(A) Effect of the null or G336A/S338A mutation in UL12 on expression of the neighboring UL11 and UL13 genes. Vero cells were either mock infected (lane 1) or infected with wild-type HSV-1(F) (lane 2), YK655 (ΔUL12) (lane 3), YK656 (ΔUL12-repair) (lane 4), YK665 (UL12G336A/S338A) (lane 5), YK666 (UL12GA/SA-repair) (lane 6), or R7356 (ΔUL13) (lane 7) at an MOI of 3; harvested at 24 h postinfection; lysed; and analyzed by immunoblotting with antibodies to UL11, UL12, UL13, VP22, and α-tubulin. (B) Effect of the mutations in UL12 on exonuclease activity in infected Vero cells. Vero cells were mock infected or infected with each of the indicated viruses at an MOI of 3, harvested at 12 h postinfection, lysed, and assayed for exonuclease activity. Each value is the mean ± standard error of the results of triplicate experiments. Asterisks indicate statistically significant differences (P < 0.01). Data are representative of three independent experiments. (C) Immunoblots of the lysates prepared for the assays in panel B. The cell lysates were analyzed by immunoblotting with anti-UL12 antibody. Data are representative of three independent experiments.
We then assayed the exonuclease activity in Vero cells mock infected or infected at an MOI of 3 for 12 h with wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) as described previously (5, 12). In agreement with previous reports (4, 5), Vero cells that were mock infected or infected with YK655 (ΔUL12) did not contain a significant amount of exonuclease activity, but there was considerable exonuclease activity in cells infected by wild-type HSV-1(F) or YK656 (ΔUL12-repair) (Fig. 1B). The level of exonuclease activity in cells infected with YK665 (UL12G336A/S338A) was about the same as that in cells that were mock infected or infected with YK655 (ΔUL12) (Fig. 1B), confirming that the G336A/S338A mutation in UL12 abolished UL12 exonuclease activity in infected cells. The amount of UL12 protein was measured by immunoblotting, which showed no UL12 in mock- and YK655 (ΔUL12)-infected cell lysates and a similar amount of UL12 in the other infected-cell lysates (Fig. 1C).
Effect of the G336A/S338A double mutation in UL12 on viral replication and cell-cell spread in HEL, Vero, and A549 cells.
To examine the significance of the UL12 nuclease activities in regulation of HSV-1 replication in cell cultures, we analyzed viral growth in HEL (human lung fibroblast) (15), Vero, and A549 (human lung epithelial) cells infected with wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) at an MOI of 0.01 for 48 h or at an MOI of 3 for 24 h and assayed total (intracellular and extracellular) progeny virus production. The A549 cells were kindly provided by Y. Kawaoka. As shown in Fig. 2A and B, the progeny virus titer in HEL or Vero cells infected with YK655 (ΔUL12) at an MOI of 0.01 was 11,000- or 14,000-fold less than that in cells infected with wild-type HSV-1(F) and 12,000- or 5,600-fold less than that in cells infected with YK656 (ΔUL12-repair), respectively. Similarly, the progeny virus titer in A549 cells infected with YK655 (ΔUL12) at an MOI of 0.01 was 880-fold less than that in cells infected with wild-type HSV-1(F) and 220-fold less than that in cells infected with YK656 (ΔUL12-repair) (Fig. 2C). These results are in agreement with previous reports (4, 5) and further verified that UL12 was critical for viral replication in cell cultures. In contrast, the progeny virus titer in HEL or A549 cells infected with YK665 (UL12G336A/S338A) at an MOI of 0.01 was only 1.4- or 5.5-fold less than that in cells infected with wild-type HSV-1(F) and only 1.6- or 1.9-fold less than that in cells infected with YK666 (UL12GA/SA-repair), respectively (Fig. 2A and C). Similarly, the progeny virus titer in Vero cells infected with YK665 (UL12G336A/S338A) at an MOI of 0.01 was only 13-fold less than that in cells infected with wild-type HSV-1(F) and only 5.3-fold less than that in cells infected with YK666 (UL12GA/SA-repair) (Fig. 2B). Thus, the G336A/S338A double mutation in UL12 had much less effect on viral replication in HEL, Vero, and A549 cells than did the UL12 null mutation, and it barely or only slightly reduced viral replication in these cells. Similar results were also obtained with HEL, Vero, and A549 cells infected with each of these viruses at an MOI of 3 (Fig. 2D to F).
FIG 2.
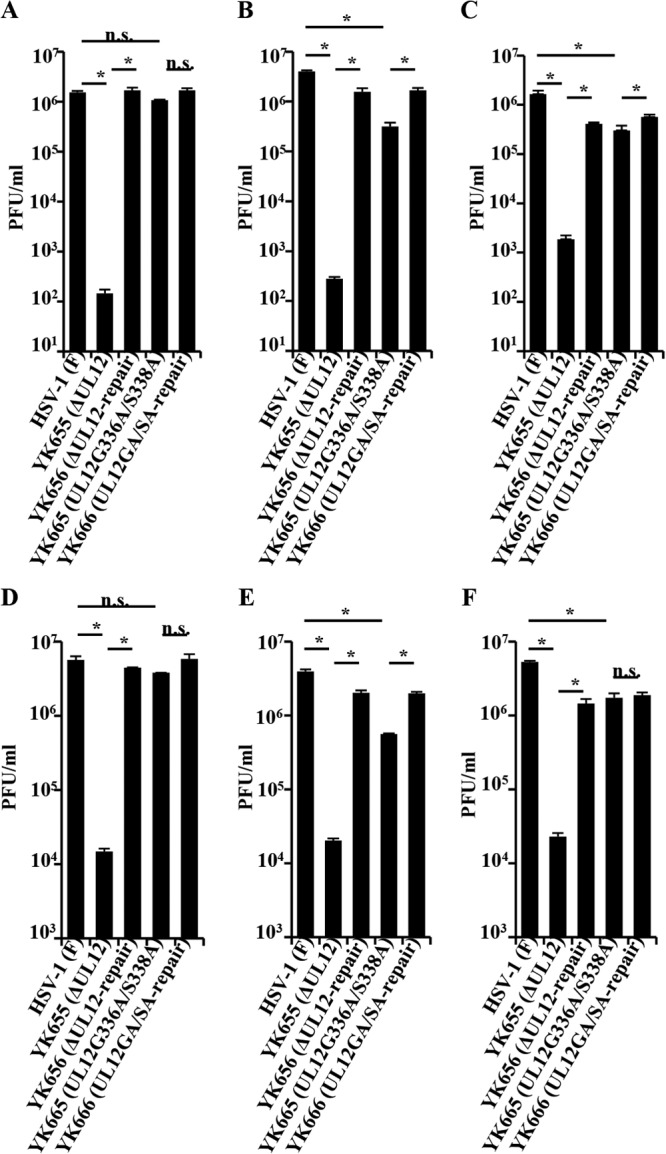
Effects of the mutations in UL12 on production of infectious viruses. HEL (A and D), Vero (B and E), and A549 (C and F) cells were infected with each of the indicated viruses at an MOI of 0.01 (A to C) or 3 (D to F). Total virus from the cell culture supernatants and infected cells was harvested at 48 (A to C) and 24 h (D to F) postinfection and was assayed on 6-5 cells. Each data point is the mean ± standard error of triplicate samples and is representative of three independent experiments. Asterisks indicate statistically significant differences (P < 0.05). n.s., not significant.
We also analyzed plaque sizes in HEL, Vero, and A549 cell monolayers infected with wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) with plaque assay conditions as described previously (16). In agreement with the growth properties of these viruses as described above, (i) YK655 (ΔUL12) produced significantly smaller plaques than did wild-type HSV-1(F) and YK656 (ΔUL12-repair) in HEL, Vero, and A549 cells; (ii) YK665 (UL12G336A/S338A) produced smaller plaques than did wild-type HSV-1(F) and YK666 (UL12GA/SA-repair) in Vero cells but larger plaques than did YK655 (ΔUL12); and (iii) YK665 (UL12G336A/S338A) produced plaques similar in size to those produced by wild-type HSV-1(F) and YK666 (UL12GA/SA-repair) in HEL and A549 cells (Fig. 3).
FIG 3.
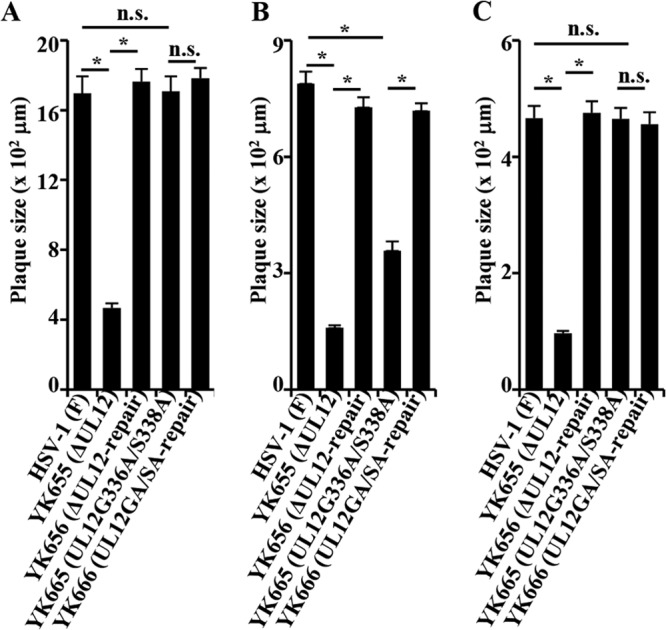
Effects of each of the mutations in UL12 on virus plaque size. HEL (A), Vero (B), and A549 (C) cells were infected with each of the indicated viruses at an MOI of 0.001, 0.0001, or 0.00001, respectively, under plaque assay conditions. The diameters of 25 single plaques for each of the indicated viruses were determined 48 h postinfection. Each data point is the mean ± standard error of the measured plaque sizes. The asterisks indicate statistical differences (P < 10−8). n.s., not significant.
Taken together, these results indicated that the UL12 nuclease activities had only a minor or no significant role in viral replication and cell-cell spread in cell cultures, suggesting that a UL12 function(s) unrelated to its nuclease activities played a major role in these viral processes.
Effect of the G336A/S338A double mutation in UL12 on viral neurovirulence in mice.
To examine the significance of UL12 and its nuclease activities in HSV-1 pathogenesis in vivo, 3-week-old female mice were infected intracerebrally with 10-fold serial dilutions of YK655 (ΔUL12), YK656 (ΔUL12-repair), YK665 (UL12G336A/S336A), or YK666 (UL12GA/SA-repair) in groups of 6 per dilution. Mortality was monitored for 14 days postinfection, and 50% lethal dose (LD50) values were determined by the Behrens-Karber method. These animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval number 19-26). The LD50 values of YK655 (ΔUL12) and YK656 (ΔUL12-repair) were 104.8 and 101.7, respectively, showing that the LD50 of YK655 (ΔUL12) was 1,300-fold greater than that of YK656 (ΔUL12-repair). The LD50 values of YK665 (UL12G336A/S336A) and YK666 (UL12GA/SA-repair) were 103.5 and 101.5 (data not shown), respectively, showing that the LD50 of YK665 (UL12G336A/S336A) was 100-fold greater than that of YK666 (UL12GA/SA-repair). These results indicated that the nuclease activities of UL12 played a major role in viral virulence in mice following intracerebral inoculation.
The nuclease activities of UL12 have been thought to be important for UL12 in promoting viral replication in cell cultures, based on reports showing that (i) UL12 null mutant virus exhibited a 100- to 1,000-fold reduction in progeny virus titer in cell cultures (4, 5) and (ii) wild-type UL12 efficiently complemented growth of the UL12 null mutant virus but not of the UL12 enzyme-dead mutant (G336A/S338A) (3). A model has been proposed suggesting that UL12 in conjugation with ICP8, an HSV-1 single-strand DNA (ssDNA) binding protein, functions as a recombinase similar to the well-studied bacteriophage λ Red recombinase (17–19). The λ Red recombinase mediates DNA strand exchange and recombination and is composed of two components: exonuclease Redα, which shares homology to UL12, and the ssDNA binding protein Redβ (20, 21). The λ Red recombination system has been reported to play an important role in formation of the viral DNA concatemers necessary for encapsidation and production of infectious progeny virus (22, 23). In support of this model, it has been shown that UL12 interacts with ICP8 in infected cells (24) and that UL12 together with ICP8 can mediate both a strand exchange reaction in vitro and single-strand annealing, which is one of the pathways for repairing chromosomal double-strand breaks in some cell types when these viral proteins are transiently overexpressed (19). The formation of concatemeric viral DNA is also an essential step in HSV-1 replication (1), and therefore, UL12 and ICP8 may function as a two-component recombinase analogous to the λ Red recombinase and play a role in formation of viral DNA concatemers.
As described above, most of the molecular mechanisms for UL12 functions proposed so far are based on the nuclease activities of UL12. However, we have presented data here showing that UL12 nuclease activities had only a minor or no significant role in viral replication in cell cultures, indicating that an undefined activity(ies) of UL12, unrelated to its nuclease activities, played a major role. The discrepancy between the previous report showing that the UL12 enzyme-dead mutant (G336A/S338A) was not able to complement growth of the UL12 null mutant virus in Vero cells and our observation in this study that YK665 (UL12G336A/S336A) replicated 1,100- or 28-fold more efficiently than did YK655 (ΔUL12) in Vero cells at an MOI of 0.01 or 3, respectively, might be due to poor efficiency of the complementation of growth of the UL12 null mutant virus in the previous report. It has been reported to be difficult to complement growth of the UL12 null mutant virus to wild-type levels of virus production, even in cell lines that express wild-type levels of UL12 (4, 5), suggesting that strict regulation of UL12 expression in infected cells is required for efficient viral replication. In the previous report (3), the complementation of growth of the UL12 null mutant virus was performed essentially by transient transfection that was expected to support much less efficient viral growth than complementing cell lines, since not all cells are transfected. As demonstrated in this study, the UL12 enzyme-dead mutation (G336A/S338A) impaired viral replication in Vero cells, and therefore, the poor complementation efficiency might result in no production of infectious virus progeny.
Our present study suggested that elucidation of the mechanisms(s) by which UL12 acts in viral replication in cell cultures required characterization of a still-unidentified function(s) of UL12 that is independent of its nuclease activities. At present, we can only speculate about the unidentified function(s) of UL12, unrelated to its nuclease activities, since no functions of UL12 that explain the critical contribution of UL12 to the production of infectious progeny virus have been identified. Thus, it has been reported that the UL12 null mutation had no or only modest effects on viral DNA and protein synthesis and cleavage and packaging of viral DNA (3, 5, 25). Whereas Shao et al. reported that UL12 was critical for efficient nuclear egress of nucleocapsids (4), Porter and Stow demonstrated that the UL12 null mutation did not greatly impair nuclear egress of nucleocapsids (25). Interestingly, both of the groups observed that the levels of DNase-resistant viral DNAs from nuclear fractions of Vero cells infected with UL12 null mutant viruses were apparently impaired, compared to those of DNase-resistant viral DNAs from nuclear fractions of wild-type HSV-1-infected cells (4, 25). It has been shown that viral DNA in HSV-1-infected cells that is in viral replication forms is sensitive to DNase digestion, whereas unit-length and encapsidated viral DNA is resistant to DNase digestion (4). Taken together with the report showing that the UL12 null mutation had no obvious effect on packaging of viral DNA into capsids (3), these observations suggested that capsid formation was impaired in the absence of UL12. Thus, there might be a possibility that UL12 is involved in efficient formation of capsids in infected cells. In agreement with this hypothesis, our preliminary electron microscopic analyses showed that the number of nuclear capsids in Vero cells infected with YK655 (ΔUL12) was apparently lower than that of nuclear capsids in Vero cells infected with YK656 (ΔUL12-repair) (data not shown). Further studies to unveil the unidentified function(s) of UL12 that is independent of its nuclease activities will be needed, and such studies are under way in this laboratory.
Although the nuclease activities of UL12 appeared to play only a minor role in viral replication in cell cultures, we demonstrated here that the nuclease activities of UL12 played a major role in neurovirulence in mice following intracerebral inoculation. Therefore, we note that the previously proposed functions of UL12 based on its nuclease activities may be important for HSV-1 pathogenesis in vivo. The contradiction between the minor role of the UL12 nuclease activities in viral replication in cell cultures and their important role in neurovirulence in mice was of interest, but similar observations have been observed with other HSV-1 functions. Thus, it has been reported that the kinase-dead mutation in HSV-1 protein kinase Us3 had no effect on or slightly (approximately 10-fold) impaired viral replication in cell cultures, depending on cell types (7, 26). In contrast, the mutation in Us3 was shown to considerably (1,000-fold) reduce neurovirulence in mice following intracerebral inoculation (27), indicating that, like the UL12 nuclease activities, the Us3 kinase activity played no or a minor role in viral replication in cell cultures, whereas the Us3 kinase activity was critical for viral neurovirulence in mice. One may argue that a host species-specific factor(s) is involved in these contradictions. However, this is unlikely, based on the observation that growth properties of YK655 (ΔUL12) and YK665 (UL12G336A/S338A) in mouse NIH 3T3 cells were similar to those in Vero cells (data not shown). Conceivably, a cellular activity(ies) that functions like the UL12 nuclease activities may exist and HSV-1 may encode UL12 to compensate for the low level of the cellular activity(ies) if that is the situation in its host cells, e.g., neurons in the brains of mice.
The HSV-1 UL12 gene encodes not only full-length UL12 but also UL12.5, an amino-terminally truncated protein that initiates at UL12 codon 127 (2, 28, 29). Although UL12.5 retains the UL12 nuclease and strand exchange activities in vitro when expressed in a baculovirus expression system (2, 30), a recombinant virus expressing UL12.5 at the wild-type level but incapable of expressing UL12 showed viral replication and cell-cell spread in cell cultures at levels almost identical to those of the UL12 null mutant virus (2). These results suggested that the role of UL12.5 in viral replication in cell cultures is almost negligible. However, a role of UL12.5 in viral pathogenesis in vivo has not been determined thus far. Therefore, it is possible that the reduced viral neurovirulence in mice due to the G336A/S338A mutation in UL12 was a result of the inactivation of both UL12 and UL12.5 nuclease activities.
ACKNOWLEDGMENTS
We thank Sandra Weller, Yoshihiro Kawaoka, and Bernard Roizman for providing 6-5 cells, A649 cells, and the UL13 null mutant virus R7356, respectively. We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers, Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases, and a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan and by grants from the Takeda Science Foundation, the Naito Foundation, the Sumitomo Foundation, and the Tokyo Biochemical Research Foundation. H.F. was supported by research fellowships from JSPS for Young Scientists.
Footnotes
Published ahead of print 11 December 2013
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 2.Martinez R, Shao L, Bronstein JC, Weber PC, Weller SK. 1996. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 215:152–164. 10.1006/viro.1996.0018 [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JN, Weller SK. 1998. The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology 244:442–457. 10.1006/viro.1998.9129 [DOI] [PubMed] [Google Scholar]
- 4.Shao L, Rapp LM, Weller SK. 1993. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology 196:146–162. 10.1006/viro.1993.1463 [DOI] [PubMed] [Google Scholar]
- 5.Weller SK, Seghatoleslami MR, Shao L, Rowse D, Carmichael EP. 1990. The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: isolation and characterization of a lacZ insertion mutant. J. Gen. Virol. 71:2941–2952. 10.1099/0022-1317-71-12-2941 [DOI] [PubMed] [Google Scholar]
- 6.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arii J, Wang J, Morimoto T, Suenaga T, Akashi H, Arase H, Kawaguchi Y. 2010. A single-amino-acid substitution in herpes simplex virus 1 envelope glycoprotein B at a site required for binding to the paired immunoglobulin-like type 2 receptor alpha (PILRalpha) abrogates PILRalpha-dependent viral entry and reduces pathogenesis. J. Virol. 84:10773–10783. 10.1128/JVI.01166-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956. 10.1128/JVI.79.11.6947-6956.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koshizuka T, Kawaguchi Y, Goshima F, Mori I, Nishiyama Y. 2006. Association of two membrane proteins encoded by herpes simplex virus type 2, UL11 and UL56. Virus Genes 32:153–163. 10.1007/s11262-005-6871-7 [DOI] [PubMed] [Google Scholar]
- 12.Sagou K, Uema M, Kawaguchi Y. 2010. Nucleolin is required for efficient nuclear egress of herpes simplex virus type 1 nucleocapsids. J. Virol. 84:2110–2121. 10.1128/JVI.02007-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783. 10.1128/JVI.00103-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banks LM, Halliburton IW, Purifoy DJ, Killington RA, Powell KL. 1985. Studies on the herpes simplex virus alkaline nuclease: detection of type-common and type-specific epitopes on the enzyme. J. Gen. Virol. 66:1–14. 10.1099/0022-1317-66-1-1 [DOI] [PubMed] [Google Scholar]
- 15.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J. Virol. 87:9323–9332. 10.1128/JVI.01581-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka M, Nishiyama Y, Sata T, Kawaguchi Y. 2005. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: the activity is not essential for optimal expression of UL41 and ICP0. Virology 341:301–312. 10.1016/j.virol.2005.07.010 [DOI] [PubMed] [Google Scholar]
- 17.Reuven NB, Staire AE, Myers RS, Weller SK. 2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 77:7425–7433. 10.1128/JVI.77.13.7425-7433.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reuven NB, Willcox S, Griffith JD, Weller SK. 2004. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 342:57–71. 10.1016/j.jmb.2004.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 8:e1002862. 10.1371/journal.ppat.1002862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bujnicki JM, Rychlewski L. 2001. The herpesvirus alkaline exonuclease belongs to the restriction endonuclease PD-(D/E)XK superfamily: insight from molecular modeling and phylogenetic analysis. Virus Genes 22:219–230. 10.1023/A:1008131810233 [DOI] [PubMed] [Google Scholar]
- 21.Stahl MM, Thomason L, Poteete AR, Tarkowski T, Kuzminov A, Stahl FW. 1997. Annealing vs. invasion in phage lambda recombination. Genetics 147:961–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63:751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo Piano A, Martinez-Jimenez MI, Zecchi L, Ayora S. 2011. Recombination-dependent concatemeric viral DNA replication. Virus Res. 160:1–14. 10.1016/j.virusres.2011.06.009 [DOI] [PubMed] [Google Scholar]
- 24.Vaughan PJ, Banks LM, Purifoy DJ, Powell KL. 1984. Interactions between herpes simplex virus DNA-binding proteins. J. Gen. Virol. 65:2033–2041. 10.1099/0022-1317-65-11-2033 [DOI] [PubMed] [Google Scholar]
- 25.Porter IM, Stow ND. 2004. Virus particles produced by the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12 contain abnormal genomes. J. Gen. Virol. 85:583–591. 10.1099/vir.0.19657-0 [DOI] [PubMed] [Google Scholar]
- 26.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412. 10.1128/JVI.78.1.399-412.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162. 10.1128/JVI.01447-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa RH, Draper KG, Banks L, Powell KL, Cohen G, Eisenberg R, Wagner EK. 1983. High-resolution characterization of herpes simplex virus type 1 transcripts encoding alkaline exonuclease and a 50,000-dalton protein tentatively identified as a capsid protein. J. Virol. 48:591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. 2007. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 8:188–193. 10.1038/sj.embor.7400878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reuven NB, Antoku S, Weller SK. 2004. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J. Virol. 78:4599–4608. 10.1128/JVI.78.9.4599-4608.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]