

Abstract
In infected people, the HIV-1 envelope glycoprotein (Env) constantly evolves to escape the immune response while retaining the essential elements needed to mediate viral entry into target cells. The extensive genetic variation of Env is particularly striking in the V1/V2 hypervariable domains. In this study, we investigated the trade-off, in terms of fusion efficiency, for encoding V1/V2 domains of different lengths. We found that natural variations in V1/V2 length exert a profound impact on HIV-1 entry. Variants encoding compact V1/V2 domains mediated fusion with higher efficiencies than related Envs encoding longer V1/V2 domains. By exchanging the V1/V2 domains between Envs of the same infected person or between two persons linked by a transmission event, we further demonstrated that V1/V2 domains critically influence both Env incorporation into viral particles and fusion to primary CD4 T cells and monocyte-derived dendritic cells. Shortening the V1/V2 domains consistently increased Env incorporation and fusion, whereas lengthening the V1/V2 domains decreased Env incorporation and fusion. Given that in a new host transmitted founder viruses are distinguished by compact Envs with fewer glycosylation sites, our study points to fusion and possibly Env incorporation into virions as limiting steps for transmission of HIV-1 to a new host and suggests that the length and/or the N-glycosylation profile of the V1/V2 domain influences these early steps in the HIV life cycle.
INTRODUCTION
For human immunodeficiency virus type 1 (HIV-1) to spread, it must overcome the vulnerabilities inherent in its release as a free viral particle. Like other viruses, HIV-1 has evolved multiple strategies to minimize the exposure of its free virions to the host immune system. HIV-1 manipulates synapses between infected and target cells to create a highly coordinated process of viral release and entry (1). HIV-1 has also evolved to encode an envelope protein (Env) that mediates HIV-1 entry with minimal exposure of its key protein domains.
Env triggers the fusion of viral and cellular membranes through a series of sequential conformational changes. Each Env spike is composed of three noncovalently associated heterodimers of Env subunits gp120 and gp41. These subunits are encoded by a single HIV-1 env gene that is translated into a 160-kDa polyprotein precursor and cleaved in the Golgi apparatus by a furin-like protease. Each subunit executes a specific function: gp120 binds to CD4 and a chemokine receptor, while gp41 carries the sequence for the fusion peptide. The binding of gp120 to CD4 triggers a conformational change in gp120 to expose the bridging sheet, which subsequently binds the chemokine receptor (2–5). Further conformational changes activate the fusion machinery, resulting in insertion of the fusion peptide at the N terminus of gp41 in the target cell membrane (6). The heptad repeat domains of gp41 then fold into a metastable prefusion intermediate known as the triple-stranded coiled-coil, which subsequently folds into a stable six-helix bundle that juxtaposes the viral and cellular membranes for fusion (6–10).
In the infected host, HIV-1 constantly evolves to escape the immune response while retaining the essential elements required for its replication. The HIV-1 env adaptation rate is the highest ever recorded for a single protein-coding gene (11). Following the evolutionary dynamics of 50 HIV-1 patients, Williamson noted that most fixed Env amino acid changes presented a selective advantage, with an average of one adaptive fixation event occurring every 2.5 months (11). This high adaptation rate explains how HIV-1 maintains high levels of replication, despite strong antiviral immune responses (12). This extensive genetic variation is particularly striking in the five hypervariable regions (V1 to V5) of the env gene (13, 14). The V1/V2 domain is the most variable in terms of loop length and number of glycosylation sites (15–20). V1/V2 domains range in length from 50 to 90 amino acids (aa), while the length variation of V4 and V5 loops ranges from 19 to 44 aa and 14 to 36 aa, respectively (for a review, see reference 21). In contrast, the V3 loop and C2, C3, and C4 domains show relatively little variation in length.
The trade-off, in terms of fusion efficiency, for encoding variable domains of different lengths and glycosylation patterns is difficult to characterize. Some variable domains that are functional in the context of a particular Env backbone might not be functional in another (22). Several studies have highlighted a possible connection between the V1/V2 domain and HIV-1 infection efficiency (23–26). Virions encoding chimeric BaL Env with V1/V2 domains from JR-CSF or NL4-3 replicate less efficiently in macrophages but not in lymphocyte cultures (23, 25). Elimination of glycosylation sites neighboring the V1/V2 region also compromises replication of DH12 dual-tropic primary isolates in peripheral blood mononuclear cells (PBMCs) (26). Chimeric Envs with V1 to V5 domains seem to fuse to target cells with low CCR5 levels more efficiently during the chronic phase of HIV-1 infection than during the acute phase of infection (24). In this study, the Env sequences identified during the chronic phase of infection fused to target cells contained a higher number of predicted glycosylation sites within their V1/V2 domains and were less sensitive to the fusion inhibitor enfuvirtide (T20), which served as an indicator of faster and more efficient fusion (24, 27). These studies suggested that V1/V2 domains affect HIV entry. Unfortunately, the methods used to evaluate HIV-1 fusion required the completion of other steps of the viral life cycle and utilized less physiologically relevant indicator cell lines. We previously developed a fluorescence resonance energy transfer (FRET)-based fusion assay that specifically measures the fusion step in the HIV life cycle and allows the measurement of fusion to the relevant primary cells (28–30). This versatile method, based on the transfer of BlaM-Vpr from HIV virions to target cells, has, however, not yet been used to investigate the effects of particular viral signatures, such as the length of V1/V2 domains, on HIV entry.
In this study, we investigated the effects of V1/V2 domain length on HIV-1 fusion in a FRET-based fusion assay to measure viral entry into resting and activated CD4 T cells and monocyte-derived dendritic cells (MDDCs) (28, 31). To limit the genetic difference between Env backbones, we compared Envs from the same individuals or from two individuals linked by a transmission event and engineered mutant Envs by swapping the V1/V2 domain sequences of related Envs.
MATERIALS AND METHODS
Subjects and envelope sequences.
HIV-1 envelope V1 to V4 sequences from transmission pairs 55, 109, 106, and 205 have been described previously (32–34). All subjects acquired HIV-1 through heterosexual contact and were documented to have subtype C infection. For all the Env genotypes, the entire gp160 protein was sequenced, and all sequences are accessible through GenBank. We defined the V1 and V2 regions to be the amino acids corresponding to HxB2 envelope aa 131 to 157 and 158 to 196, respectively (19, 35). For pairs 106, 109, and 205, the V1 loop was swapped, and for pair 55, the major differences were in V2 domains. The relevant institutional review boards approved all human subject protocols, and all subjects provided written informed consent before enrollment.
Cloning of WT and V1/V2 mutant Envs.
To facilitate cloning of the primary envelopes into the proviral DNA, we selected the TN6-green fluorescent protein (GFP) proviral DNA expression vector, an NL4-3-based construct modified to contain a BstEII restriction site 15 nucleotides (nt) after the signal peptide of the NL4-3 env and a NcoI site at the end of the envelope (36). The primary wild-type (WT) envelopes were amplified with the sense primer C6323+ (TTGTGGGTCACCGTCTATTATGGGG) and the antisense primer ASenvNcoI (CTGCATCCATGGTTTATTGTAAAGCTGCTTC). The PCR amplification and cloning protocols are extensively described elsewhere (37). Mutations within the V1 or V2 domain were introduced by PCR. First, a 5′ fragment (∼300 bp) and a 3′ fragment (∼2,100 bp) of env were amplified with antisense and sense primers carrying the mutations, respectively. The PCR products were then purified and combined to perform a second amplification of the entire envelope with primers C6323+ and ASenvNcoI. For the mutants with extensive insertions and deletions [mutants 106F9(M43), 106M43(F9), and 205F4.2(M6.3)], fragments of DNA were synthesized and subcloned directly into the WT envelopes (Integrated DNA Technologies).
Generation and culture of MDDCs and RA-activated CD4 T cells.
PBMCs were purified from fresh buffy coats on Ficoll gradients. CD14+ monocytes were positively selected using Miltenyi anti-CD14 magnetic beads, according to the manufacturer's instructions. Dendritic cells (DCs) were derived by culturing CD14+ monocytes (2 × 106 cells/ml) for 6 days with 25 ng/ml interleukin-4 (IL-4; R&D Systems) and 50 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Biosource) in RPMI medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (38). The medium was changed every 2 days. The phenotype of these MDDCs was verified by immunostaining with antibodies specific for DCs (CD1a and HLADR; Becton, Dickinson). To generate retinoic acid (RA)-activated CD4 T cells, the peripheral blood leukocytes (PBLs) were incubated at 2 million cells per ml in RPMI supplemented with 10% FBS and IL-2 (20 IU/ml). The cells were stimulated or not with OKT3 antibody (1 μg/ml) and/or retinoic acid (10 nM) and maintained in culture for 6 days.
Viral stocks.
HIV-1 virions containing the BlaM-Vpr chimera were produced as described previously (28, 31). Briefly, 20 × 106 293T cells were transfected by the calcium phosphate method with proviral DNA (40 μg) and the pCMV-BlaM-Vpr (20 μg) and pAdVAntage (20 μg) vectors (Promega). The medium was changed after overnight culture. Cells were then allowed to produce virions during 36 h of culture at 37°C. The virus-containing supernatant was centrifuged at low speed to remove cellular debris and ultracentrifuged at 72,000 × g for 90 min at 4°C to pellet viral particles. Viral stocks were normalized on the basis of the p24Gag content, measured by enzyme-linked immunosorbent assay (ELISA; NEN Life Science Products).
Measurement of HIV-1 fusion.
MDDCs (2 × 105) and autologous resting or activated PBLs (2 × 106) were infected with BlaM-Vpr-containing virions (500 ng of p24Gag) for 2 h at 37°C in 100 μl of RPMI medium. The cells were then washed once with CO2-independent Dulbecco modified Eagle medium (Gibco BRL) and loaded with CCF2 dye (0.5 mM; Invitrogen) for 1 h at room temperature. After two washes with CO2-independent medium, the cells were incubated for 16 h at room temperature in 200 μl of CO2-independent medium supplemented with 10% FBS and 2.5 mM probenecid, an inhibitor of anion transport. In the case of the PBL infection, the fusion assay was combined with immunostaining. Infected PBLs were washed twice in staining buffer (phosphate-buffered saline [PBS] with 2% FBS) and incubated for 30 min at room temperature with a cocktail of antibodies. For resting T cells, the cocktail contained anti-CD3 and anti-CD4 conjugated to allophycocyanin (APC)-Cy7 and phycoerythrin (PE)-Cy7, respectively. For activated T cells, the cocktail included anti-CD3, anti-CD4, anti-CD45RO, and anti-β7 conjugated to PE-Cy5.5, APC-Cy7, PE-Cy7, and APC, respectively. The cells were next washed once in PBS and fixed in a 1.2% solution of paraformaldehyde overnight. The change in emission fluorescence of CCF2 after cleavage by the BlaM-Vpr chimera was measured by flow cytometry with a four-laser BD LSRII flow cytometer (Becton, Dickinson). Data were collected using FACSDiva software (Becton, Dickinson) and analyzed with FlowJo software (TreeStar). The compensation was calculated after data collection on the basis of single-stain controls using FlowJo software. Note that, for some resting CD4 T cells from particular blood donors, very low levels of the 81A fusion (<0.05%) were observed. The results from these experiments were excluded from the analysis.
Measurement of Env incorporation.
We quantified Env incorporation by Western blotting using the ImageJ64 software developed by NIH. For Western blotting, each set of viral preparations was first normalized for p24Gag to between 1 and 10 ng/μl. Fifty microliters of these normalized viral preparations was lysed by adding 10 μl of 6× Laemmli sample buffer (375 mM Tris, pH 6.8, 60% glycerol, 0.0015% bromophenol blue, 2% β-mercaptoethanol, 10% SDS) and by boiling the samples for 10 min. Twenty microliters of the lysed viral preparations was loaded on a 15% SDS-polyacrylamide gel (Bio-Rad), which was run with the standard SDS-PAGE buffers. Transfer onto nitrocellulose membranes was performed with an iBlot system (Novex; Life Technologies). The membranes were then blocked overnight at 4°C with PBS containing 0.05% Tween 20 and 5% nonfat milk. After 3 washes, the membranes were incubated for 1 h at room temperature in a 1/10 dilution of the b13 hybridoma cell supernatant in PBS supplemented with 0.05% Tween 20. After 3 washes with PBS containing 0.05% Tween 20 at room temperature, the membrane was incubated for 1 h at room temperature with the secondary antimouse horseradish peroxidase antibody (GE Healthcare Life Science) diluted 1/5,000 in PBS containing 0.05% Tween 20 supplemented with 1% nonfat milk. The membranes were washed three times and developed using a Western Lightning ECL kit (PerkinElmer) and Hyperfilm (Amersham). Immunoblotting with the anti-p24 antibody was subsequently performed under the same conditions with a 1/20,000 dilution of p24Gag antibody from ascites. The Western blot films were scanned at 600 dots per inch and saved as GIF files. Only nonsaturating exposures were used for quantification. Band intensities were quantified with the ImageJ64 macro, which represents the band densities in histograms. The areas under the curves were calculated for each of the bands, and the ratio between gp120 and p24Gag was calculated.
Statistical analysis.
To test associations between the V1/V2 length, the number of putative N-linked glycosylation sites, and fusion or Env incorporation, statistical analyses were performed using Spearman's rank correlation in Prism software (GraphPad Software). For the length and the number of potential glycosylation sites, the number of amino acids or sequons [NX(T/S), where X is any amino acid except proline] in WT V1/V2 sequences was subtracted from the number of amino acids or sequons counted in the mutant sequences. Sequences with double sequons, such as NNSS, were counted as two sequons, as overlapping sequons with a serine in the acceptor position can be used simultaneously (39).
For the fusion phenotype, the parameters used in the statistical analysis were calculated as follows. The lab-adapted 81A, included in each set of viral preparations, was used to calculate the fusion ratio, corresponding to the percentage of cells supporting fusion for a given genotype divided by the one for 81A. Then, when replicate experiments using independent clones for the same genotype were performed [i.e., for 106F9(M43), 106M43(F9), 205F4.2(M6.3), and 205M6.3(F4.2)], the median values of these fusion ratios were calculated. When infections of the same target cells with the same viral preparations were performed in triplicate, the median values of the fusion ratios were also calculated first.
For measuring the impact of V1/V2 length on fusion mediated by WT Envs, we used the fusion relative to that for 81A (i.e., the fusion ratio). For measuring the impact of V1/V2 mutations on fusion, we divided the fusion ratio of the mutant Env by the fusion ratio of the corresponding WT Env (Mut/WT). The replicate of donor cells was then averaged to obtain the final median Mut/WT ratios used in the Spearman correlation.
For the Env incorporation phenotype, all genotypes were analyzed twice by quantifying two independent sets of viral production by Western blotting. The pairs of mutant and WT Envs were always loaded on the same gels. First, we calculated the incorporation ratio by dividing the gp120 band intensities by the p24Gag band intensities. Then, when replicate clones of the same genotype were analyzed [i.e., for 106F9(M43), 106M43(F9), 205F4.2(M6.3) and 205M6.3(F4.2)], the average value of the incorporation ratio was calculated. Finally, the incorporation ratio of mutant Env was divided by its WT counterpart and used in statistical analysis.
Nucleotide sequence accession numbers.
The set of envelopes (from the WT and V1/V2 mutants) was sequenced, and the WT sequences are accessible through GenBank (accession numbers AY423943, AY423938, AY423939, AY423947, AY423973, AY423971, AY423980, AY423979, AY423978, AY424151, AY424152, AY424153, AY424154, AY424155, AY424163, AY424164, AY424169, AY424167, AY424165, AY424126, AY424127, AY424128, AY424129, AY424133, AY424138, AY424141, AY424140, AY424143, KF800743, KF145142, KF800748, KF145141, KF800744, KF800745, KF800746, KF800747, and GQ485416).
RESULTS
Engineering recombinant provirus encoding primary Envs with natural variations in V1/V2 length.
Cloned full-length Envs from four male-to-female transmission pairs from the Lusaka cohort of serologically discordant couples formed the basis of our study, as they display differences in the lengths of their V1/V2 variable domains (32, 34, 40). A total of 37 primary Envs, consisting of 4 or 5 Envs per person, was selected on the basis of their availability as molecular clones encoding full-length Env (Fig. 1). The V1/V2 regions of these Envs were aligned to highlight the natural variations within each transmission pair. We previously reported that Env encoded in cis from a provirus mediated fusion more efficiently than did Env from pseudotyped virions (37). We therefore amplified and subcloned these 37 Envs into the TN6-GFP molecular clone (36). All Env inserts were confirmed by sequencing. Given the position of the TN6-GFP restriction site used for cloning, the resulting viruses encode Envs of ∼840 aa, with the first 41 aa spanning the Env signal peptide and the 5 terminal aa being encoded by the NL4-3 provirus.
FIG 1.

Alignment of V1/V2 domains of WT Envs with natural variations in V1/V2. Sequences from the 37 WT Envs from the four transmission pairs of the Lusaka cohort were aligned using multiple-sequence alignments with hierarchical clustering (by use of the MultiAlin program [61]). The V1/V2 loop was identified by comparison with the sequence of HxB2. Each Env genotype is named with the number corresponding to the transmission pair, a letter corresponding to the person in the couple (F for the acutely infected female and M for the male transmitting partner), and a number corresponding to the Env clone. Note that although the Env genes from the female recipient encode identical V1 or V2 domains, they each encode a slightly different variant due to differences in other parts of the Env. The yellow areas highlight the part of the sequences with variation in the amino acid within the transmission pair.
Measuring fusion mediated by primary Envs with the virion-based fusion assay.
We measured the ability of these Envs to mediate fusion to cellular targets in our virion-based fusion assay (28, 29, 31). Cellular targets included resting or activated CD4 T cells and MDDCs. MDDCs were derived from monocytes by culturing in GM-CSF and IL-4. To generate activated CD4 T cells, peripheral blood lymphocytes (PBLs) were cultured in the presence of OKT3 antibody and retinoic acid (RA). RA-OKT3 costimulation led to the generation of CD4 T cells expressing α4β7 and high levels of CCR5 (41). This unique population of memory CD4 T cells expressing α4β7 and high levels of CCR5 has been shown to gain access to the reproductive tract tissues during a sexually transmitted infection, such as Chlamydia trachomatis infection (42). Given that sexually transmitted infections are associated with increased HIV transmission, it was important to also evaluate HIV fusion to this particular cell type.
We generated virions containing BlaM-Vpr, a protein chimera used to detect fusion, by transfecting 293T cells. Viral preparations were normalized for p24Gag levels by ELISA and used to infect the three types of target cells from healthy donors for 2 h at 37°C. Upon viral fusion, BlaM-Vpr was transferred to target cells and detected by subsequent loading of target cells with the BlaM substrate CCF2. The gating strategy and representative fusion fluorescence-activated cell sorting (FACS) plots are presented in Fig. 2A and B, respectively. As expected, levels of fusion were much higher in target MDDCs than in CD4 T cells identified by immunostaining (Fig. 2B) (29). For R5-tropic 81A, 28% of MDDCs supported fusion, while only 0.3% of resting CD4 T cells and 2.6% of activated CD4 T cells did (Fig. 2A). For WT Envs from transmission pair 109, the rates of fusion to MDDCs and resting CD4 T cells of the same blood donor ranged from 63 to 1.2% and 2.3 to 0.2%, respectively (Fig. 2B). Activated CD4 T cells supported much higher levels of HIV-1 fusion than did resting cells, ranging from 42 to 0.7%, likely due to the increased expression of CCR5 (data not shown). In each set of genotypes analyzed, we included 81A proviral DNA as a positive control, which allowed normalization between different experimental data sets.
FIG 2.

Analysis of fusion with the virion-based fusion assay. (A) Gating strategy used to measure HIV-1 fusion to MDDCs or resting or activated PBLs. For measuring fusion to CD4 T cells, PBLs were infected, allowed to fuse to CD4 T cells, and immunostained. Resting T cells were gated for CD3+ CD4+, and activated T cells were gated for CD3+ CD4+ CD45RO+ β7+. The fusion gates were always set on the uninfected samples, where usually less than 0.01% of the cells showed a shift in fluorescence. Note the increase in the number of cells in the fusion gates for 81A. FSC, forward scatter; SSC, side scatter. (B) Representative fusion FACS plot obtained with WT Envs from transmission pair 109. Viral supernatants from 293T cells transfected with WT clones of pair 109 were normalized for p24Gag levels and used to infect MDDCs and resting or activated CD4 T cells.
The virion-based fusion assay measures Env-mediated fusion with high consistency and reproducibility.
The reproducibility of our fusion measurements was assessed by performing the infection in triplicate, using target cells from multiple donors, and analyzing the fusion of independent clones encoding the same Env genotype (Fig. 3). Comparisons of replicate infections revealed very low levels of variability (Fig. 3A). Donor-to-donor variability was also acceptable when fusion was normalized to the levels obtained with the R5-tropic 81A virus. For the same viral preparation, the correlation between fusion measurements performed using target cells of two different donors was high (Fig. 3B). Finally, we obtained very consistent results between independent clones of the same Env genotype (i.e., two to three independent clones per genotype and four Env genotypes total [Fig. 3C]), highlighting the minimal influence of proviral DNA preparation, 293T transfection, and normalization to p24Gag on fusion measurements.
FIG 3.
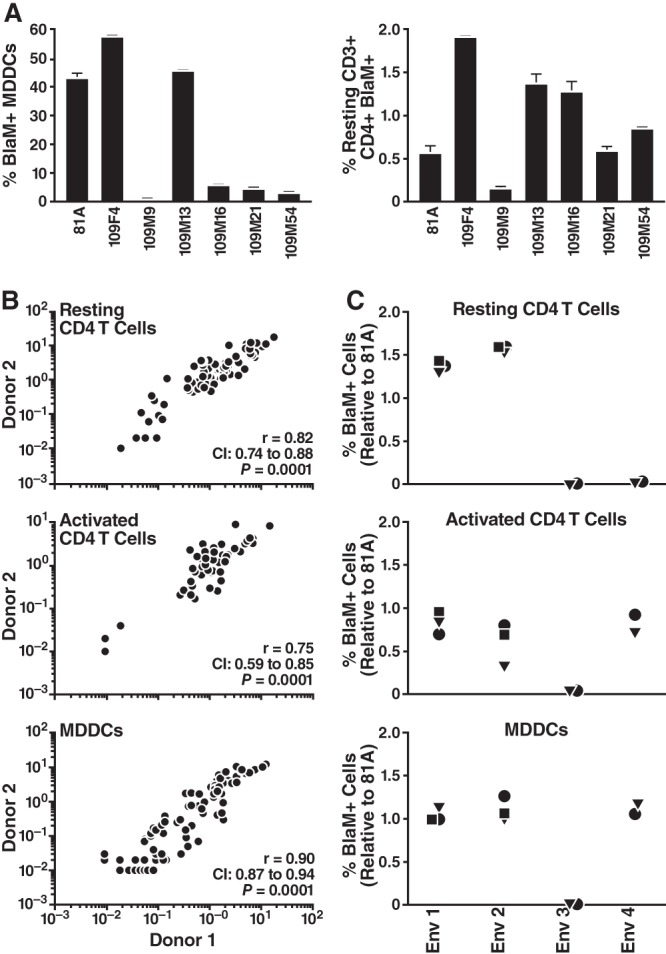
Reproducibility of fusion measurements. (A) Triplicate infections were performed with the same viral preparation with MDDCs or CD4 T cells. Error bars represent standard deviations. (B) Donor-to-donor target cell variability. Results of experiments in which the same viral preparations were used to infect the cells of two donors are presented. To account for changes in HIV-1 susceptibility, the fusion ratio corresponding to the percentage of cells supporting infection was divided by the fusion measurement for 81A. (C) Variability due to different plasmid preparation, virion production, quantification, and infection. For four genotypes (Env 1 to Env 4), two or three clones (represented by different symbols) were tested simultaneously using the same donor target cells. Note the similarity of the fusion phenotypes for identical genotypes.
Natural variations in V1/V2 length significantly affect HIV-1 fusion to resting or activated CD4 T cells and MDDCs.
First, we assessed the ability of the 37 WT Envs to mediate fusion. With each set of viral preparations, 81A was included as an internal control to normalize fusion measurements and to permit comparisons between experiments. Each Env was tested at least three times using targets cells from three different healthy donors. The median value for the replicates of fusion relative to the value for 81A was calculated and used in the statistical analysis. The lengths of V1/V2 from 37 Envs from subtype C HIV-1 ranged from 61 to 100 aa (Fig. 1). The relationship between the V1/V2 length and fusion was tested using Spearman's rank correlation coefficient. We found that the V1/V2 length inversely correlated to fusion to resting or activated CD4 T cells and MDDCs (Fig. 4). The results of statistical analysis after removal of the results for 106M43, which encoded a particularly long V1 of 100 aa, remained significant for activated CD4 T cells and MDDCs, while in resting CD4 T cells, the P values dropped to 0.052. For activated CD4 T cells, the statistical significance was maintained even when analysis was performed with data for any combination of three transmission pairs, indicating that the correlation was not skewed by a particular set of Envs. Our results suggest that natural variants of Envs encoding compact V1/V2 domains mediate HIV entry more efficiently than Envs with longer V1/V2 domains.
FIG 4.
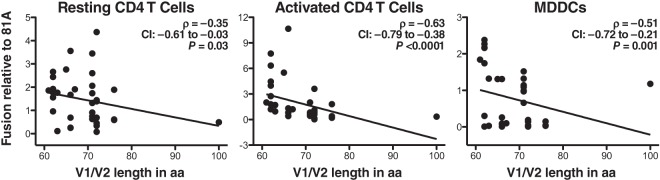
Correlation between fusion and V1/V2 length of WT Envs. Fusion mediated by the 37 WT Envs corresponding to four transmission pairs was tested by the fusion assay with resting or activated CD4 T cells and MDDCs. The ratio between WT Env fusion and 81A fusion was first calculated. Then, results for replicate infections of the same target cells with the same viral preparation and replicate donors were successively averaged, as described in Materials and Methods. The median values of fusion relative to the value for 81A were used for statistical analysis. The correlation between change in length and fusion was analyzed using Spearman's rank correlation. CI, confidence interval. Best-fit lines were added to highlight the negative correlations.
Engineering recombinant provirus encoding Envs with different V1 and V2 domains.
To specifically demonstrate the link between V1/V2 length and fusion, we engineered mutants by swapping the V1 or V2 domain sequences within Envs of the same individual or between two individuals linked by a transmission event. We selected all the WT Envs encoding V1 or V2 domains longer than the Envs from the newly infected partner. These eight Envs (55M2.9, 55M32A, 106M43, 109M9, 109M16, 109M21, 109M54, and 205M6.3) were mutated to encode the compact Env found in the acutely infected partner (Fig. 5). We created an additional ninth long-to-compact mutant [205M6.3(M4.1)] because 205M4.1, a WT Env from the chronically infected partner, encoded a domain much shorter than the one from the acutely infected partner (205F4.2). Then, we designed primers to insert into the short Env from the acutely infected partner the long V1 or V2 sequences from the identified long Env from the chronically infected partner. Additionally, we designed a set of mutants where the length of V1/V2 was identical but 2 aa were exchanged [55F4a(M4.5) and 55M4.5(F4a)]. A total of 19 mutants were successfully cloned and confirmed by sequencing.
FIG 5.
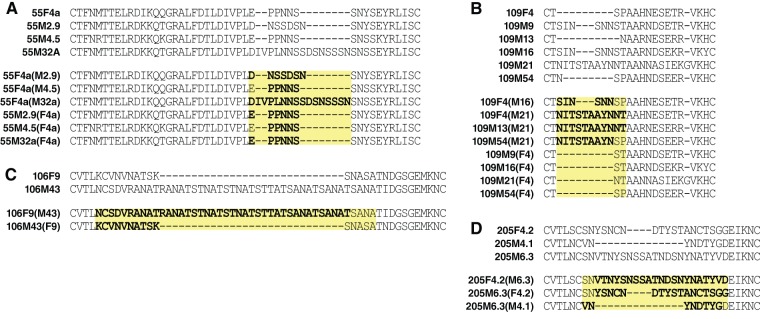
Alignment of WT and mutant Envs. Mutations affecting the V1 or V2 domain were designed by swapping amino acids between Envs of a transmission pair and/or the same infected individual. (A to D) WT and mutant Envs of each of the four transmission pairs of the Lusaka cohort, respectively. The V1/V2 mutants were named with the Env WT backbone, followed in parentheses by the clone name from which V1 or V2 was copied. Mutations are highlighted in bold.
Changes in the V1/V2 length inversely correlate with Env fusion.
We next assessed the virion fusion levels mediated by our isogenic Envs. For each mutant, we determined the changes in V1/V2 length by subtracting the number of amino acids of the WT genotype from the number of amino acids of the mutant genotype (Mut − WT) and analyzing the fold changes in fusion by dividing the fusion measurements of the mutant by those of the WT (Mut/WT). Statistical analysis, using Spearman's rank correlation coefficient, revealed a significant inverse correlation between the changes in length and changes in fusion (Fig. 6). Specifically, shortening the V1/V2 region consistently increased viral fusion, whereas lengthening the V1/V2 region decreased viral fusion.
FIG 6.
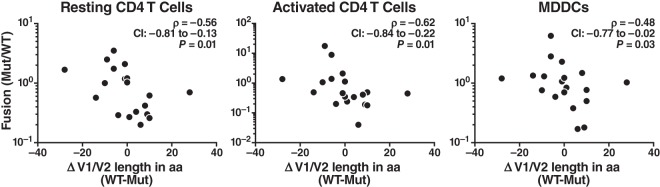
Correlation between changes in fusion and in V1/V2 length. The ratio between fusion of the mutant and that of the corresponding WT clone was calculated for the 19 V1/V2 mutants. Results for replicates of clones, replicate infections of the same target cells with the same viral preparation, and replicate donors were successively averaged, as described in Materials and Methods. The median values of Mut/WT were used for statistical analysis. The correlation between change in length and fusion was analyzed using Spearman's rank correlation.
Changes in V1/V2 length inversely correlate with Env incorporation.
Next, we tested whether V1/V2 length changes also affected Env incorporation into virions. For each isogenic pair of virions, the amount of Env incorporation into virions was measured by Western blotting with the b13 antibody (43), which recognizes an epitope within the C2 domain of Env glycoproteins. This linear epitope was present in all clones. Input was normalized using anti-p24Gag (44). The reproducibility of the Env incorporation measurements was assessed by analyzing multiple proviral clones of the same genotype in parallel (data not shown). All of the genotypes from a given transmission pair were measured on the same Western blot, with transfections, viral production, and concentration performed simultaneously. Two independent sets of viral preparations were quantified and the results were averaged.
The fold change in Env incorporation (Mut/WT) was then compared to the changes in V1/V2 length (Mut − WT). The calculation is illustrated in Fig. 7 for transmission pair 109. After calculating Env incorporation by the ratio of the intensities between the Env and p24 bands (Fig. 7A), the ratio of Env incorporation between each mutant and its WT counterpart was calculated, and the results are indicated in Fig. 7A. This Mut/WT ratio was plotted against the changes in length (Fig. 7B). For the set of 19 mutants, we found that the amount of Env incorporated into virions significantly inversely correlated with V1/V2 length differences (Fig. 8). Exchanging V1/V2 domains with more compact domains consistently increased Env incorporation, whereas switching to longer V1/V2 domains decreased Env incorporation. This correlation was maintained when the data for two mutants [109F4(M16) and 109M54(M21)] with particularly small amounts of Env were removed from the analysis (Spearman r = −0.76, confidence interval = −0.90 to −0.42, P < 0.001).
FIG 7.
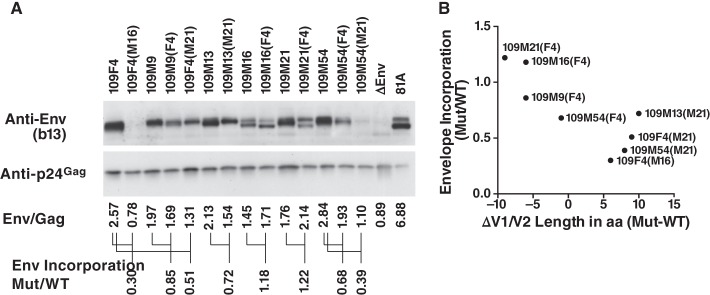
Measurement of Env incorporation. Viral preparations of WT and V1/V2 mutants were lysed, and 100 ng of p24Gag from the viral lysate was analyzed by Western blotting. The b13 antibody was used to measure the incorporation of Env into virions, while the p24Gag antibody was used to control for the virion input. (A) Viral incorporation for the set of WT and V1 mutant Envs corresponding to pair 109. The ratio between the intensities of the Env and p24Gag bands was first calculated for all the genotypes. Then, the fold change in Env incorporation between the mutant and its corresponding WT was calculated by dividing the ratio of Env/p24 obtained for the mutant by the one obtained for the corresponding WT. These fold changes are given for each mutant of transmission pair 109. (B) Relationship between changes in Env incorporation and V1/V2 length. The Mut/WT fold change in Env incorporation was then graphed against the changes in Env length measured by subtracting the number of amino acids of the WT sequence from the number of amino acids of the mutant (WT − Mut).
FIG 8.

Relationship between changes in V1/V2 loop length, Env incorporation, and fusion to MDDCs and CD4 T cells. (A) Correlation between V1/V2 length and envelope incorporation. The ratio between the intensity of the Env and p24Gag signal was measured for the entire set of WT and mutant genotypes. This quantification was performed twice using two sets of viral production. The change in envelope incorporation (Mut/WT) was calculated for each set, averaged, and plotted versus the changes in length (WT − Mut). (B) Link between envelope incorporation (Mut/WT) and fusion to resting or activated CD4 T cells and MDDCs (Mut/WT). (C) Effect of Env titration on fusion to MDDCs and CD4 T cells. Proviral DNA of the 109F4 WT envelope was cotransfected into 293T cells with NL4-3 ΔEnv at the indicated ratio and with BlaM-Vpr. After normalization by the use of the p24Gag signal, MDDCs and CD4 T cells were infected with the equivalent of 500 ng of p24Gag for 2 h. The level of fusion was reported relative to the level of fusion obtained with 100% WT 109F4. Envelope incorporation was measured by Western blotting with the b13 antibody, and p24Gag was used to control for the amount of input virions.
Changes in Env incorporation correlate with fusion to CD4 T cells.
Decreasing the V1/V2 length on Env could enhance HIV-1 entry by increasing the amount of Env packaged into the virus and/or by heightening Env fusogenicity. In CD4 T cells, the changes in Env incorporation incurred by V1/V2 mutations correlated with changes in fusion (Fig. 8B), suggesting that Env incorporation contributes to greater fusion and, ultimately, more efficient entry into this target cell type. In MDDCs, changes in Env incorporation tended to correlate with changes in fusion, although this tendency was not statistically significant (Fig. 8B). To further investigate this, we artificially titrated the amount of a given Env (109F4) on virions (Fig. 8C). 293T cells were transfected with decreasing amounts of TN-GFP-109F4 and increasing amounts of ΔEnv proviral DNA. Western blot analysis confirmed the Env titration in these viral preparations. The efficiency of viral entry was then measured by the fusion assay with the same viral input measured by p24Gag. We found that increasing the amount of Env on the virus also increased fusion to MDDCs in a dose-dependent manner, suggesting that Env incorporation also influences fusion to MDDCs.
Changes in the number of N-glycosylation sites inversely correlate with changes in Env incorporation and fusion.
Changes in V1/V2 length are accompanied by changes in the number of potential glycosylation sites identified by the NX(T/S) amino acid sequence, where X is any amino acid except proline (Fig. 9A). Interestingly, the glycosylation changes correlated with both the changes of Env incorporated into virions (Fig. 9B) and the changes in fusion to the three cell types (Fig. 9C). Figure 9D summarizes the relationship between V1/V2 length, number of glycosylation sites, Env incorporation, and fusion. These four parameters were significantly correlated, except for the relationship between Env incorporation and MDDC fusion, which correlated only in 109F4 titration experiments.
FIG 9.

Link between changes in the number of potential glycosylation sites in the V1/V2 domain, V1/V2 length, Env incorporation, and fusion to MDDCs and CD4 T cells. Correlations between changes in the number of potential glycosylation sites identified by NX(T/S) and changes in V1/V2 length (A), Env incorporation (B), and fusion to MDDCs or resting or activated CD4 T cells (C) were determined. (D) Summary of the correlation between the changes incurred by the mutations for the four variables. Note that the four parameters were significantly correlated (P < 0.05, Spearman's test) for all comparisons except the relationship between Env incorporation and MDDC fusion (the changes in Env incorporation levels did not correlate with fusion).
DISCUSSION
In the present study, we found that V1/V2 domains critically influence both Env incorporation into viral particles and fusion to primary CD4 T cells and MDDCs. Shortening the V1/V2 domains consistently increased fusion and Env incorporation, whereas lengthening the V1/V2 domains decreased fusion and Env incorporation. Natural variation in V1/V2 length also impacted viral entry. WT Envs encoding compact V1/V2 domains mediated fusion with efficiencies higher than those for related Envs encoding longer V1 or V2.
Our observations contrast with those from previous mutagenesis studies that reported that deletions of V1/V2 were accompanied by modest (45, 46) to stronger (47, 48) defects in viral replication, which could be compensated for by a few adaptive mutations in gp120 or gp40. In our approach, the deletions were smaller and guided by existing sequences corresponding to those of other viruses found in the same individual or partner. This strategy likely alleviated the need for compensatory mutations, although the strength of the effects seemed to be context dependent. For example, although mutations to create 109M9(F4) and 109M16(F4) resulted in the same decrease in V1 length, the effects were much stronger in the context of 109M9 than in the context of 109M16. The nature of the sequence inserted into Env also seemed to influence Env incorporation. Insertions of sequences into the V1 domain of 109F4 led to reduced Env incorporation that was more pronounced for 109F4(M16) than for 109F4(M21), even though the M16 insert was smaller. Nevertheless, despite a noticeable influence of the Env backbone, our analysis of 19 isogenic clones derived from four transmission pairs revealed a statistically significant inverse correlation between changes in V1/V2 length and changes in Env incorporation and fusion. These correlations were not biased by one or two pairs, given that in each transmission pair, at least one long-to-compact mutation and one compact-to-long mutation led to a gain or a loss of function, respectively.
Our study highlights the importance of V1/V2 domains in Env biogenesis. Using the b13 antibody, which detects all forms of HIV Env at the surface (i.e., gp120 in the trimeric spike, monomeric gp120/gp140, and gp160), we found that the length of the V1/V2 domain Env inversely correlated to Env packaging onto the surface of virions. In two cases [109M21(F4) and 109M16(F4)], the mutation seemed to affect the cleavage of gp160 into functional gp120. In general, Env incorporation is relatively low, with only 7 to 14 spikes, on average, present on the HIV virion surface (49). Many factors govern Env incorporation into HIV-1 virions, including the expression of properly folded Env and the gp41/p55Gag interaction. Since our study compared isogenic clones with identical gp41 and p55Gag sequences, V1/V2 exchanges and the concomitant modifications in V1/V2 glycosylation likely affect Env folding in the producer cell. Interestingly, decreasing the number of potential N-glycosylation sites in the V1/V2 region resulted in increased Env incorporation. Given the exceptional density of glycosylation in HIV-1 Env, our results suggest that there is a threshold of glycans that HIV Env can tolerate to ensure proper folding and exposure of functional domains during the entry process. Consistent with this theory, HIV-1 seems to have adopted an evolving glycan shield strategy that involves the rearrangement, rather than the addition, of glycans to evade neutralizing antibody responses (50). Moreover, glycosylation is an energy-dependent process, and therefore, higher glycosylation could result in slower folding kinetics and slower incorporation into virions. Compared to the folding of other glycoproteins, Env folding is slow, and cleavage of the signal peptide occurs only when the protein is fully synthesized. These unusual features point to Env glycosylation and folding as limiting and delicate steps in the HIV-1 life cycle, steps that could also be influenced by the nature of V1/V2.
Although our study included only a limited number of WT Envs from subtype C infections, we found that the natural variation in V1/V2 length significantly influences HIV fusion. Variants encoding Envs with shorter V1/V2 domains were more fusogenic than variants with longer V1/V2 domains. Our results could explain the genetic bottleneck observed during transmission to a new host (for a review, see reference 51). HIV-1 variants encoding Envs with shorter and less glycosylated variable domains were selected for during transmission, suggesting that compact Envs confer a greater transmissibility or fitness advantage in establishing new infection. Until recently, no clear functional phenotype could be attributed to the founder Env (24, 34, 40, 52–54). In fact, the only phenotype reported for compact Envs was increased sensitivity to neutralizing antibodies in linked donor plasma, which does not explain the selective advantage in a new host where no HIV-1 immune response has developed. Recently, Parrish et al. (55) reported that transmitted founder virions had increased fitness and packaged more Env than variants isolated during the chronic phase of infection. While our analysis of the isogenic clones revealed an inverse correlation between V1/V2 length changes and fusion, the analysis of 37 WT Envs failed to reveal any significant correlation between V1/V2 length and Env incorporation (data not shown). The number of transmission pairs studied here was also too small to compare the fusogenicity of transmitted and nontransmitted Envs. Further studies with larger sets of Envs from different HIV subtypes will be needed to validate and extend our observation to other subtypes.
Our findings have important implications for vaccine development, as V1/V2 domains are often targeted by neutralizing antibodies (56). In the RV144 Thai vaccine trial, the levels of antibodies directed against V1/V2 inversely correlated with infection risk (57, 58). Consistent with these findings, macaques immunized with a similar regimen and challenged with repeated low doses of simian immunodeficiency virus mac251 mounted a statistically significant antibody response to V1/V2 that was inversely correlated with infection (59). In superinfection studies, anti-V1/V2 antibodies to the primary virus were associated with a decreased risk of superinfection by a second virus of the same subtype (60). These V1/V2 antibodies could restrict the HIV-1 species with V1/V2 domains conducive to high-level Env incorporation and high-level fusion (i.e., the compact V1/V2 Env), characteristics which seem to be the best fit to establish the infection in a new host. Once the infection is established, cell-to-cell spread of HIV-1 could alleviate the need for highly fusogenic virions and lead to the growth of virions encoding longer V1/V2 domains.
Understanding the phenotypic properties of HIV-1 variants capable of establishing an infection in a new host is key for the development of an effective prophylactic vaccine and female-controlled microbicides. Increased Env incorporation into virions and replication fitness are emerging as central phenotypic features of the transmitted founder viruses. Our study uniquely identifies the V1/V2 domain of Env as exerting a major influence on HIV fusion and, possibly, on Env incorporation. Further studies to evaluate the impact of V1/V2 length and the N-glycosylation profile on Env biogenesis and viral spread in mucosal tissue will bring key insights that will facilitate the development of candidate vaccines with greater breadth and efficacy.
ACKNOWLEDGMENTS
This study was supported by funding from the National Institutes of Health (P01 AI083050). UCSF-GIVI CFAR provided infrastructure support (P30 AI27763). These studies were also made possible by grant RR 18928-01 from the NIH National Center for Research Resources.
We thank G. Lewis for the gift of the b13 antibody, S. Allen and J. Mulenda for establishing the Lusaka cohort, K. Eilertson of the Gladstone Bioinformatic Core, and M. Gesner of the Gladstone Flow Core for their contributions; G. Howard and A. Lucido for editorial assistance; T. Roberts for graphic arts; and S. Cammack and R. Givens for administrative assistance. Thanks go to W. Yonemoto, B. Webster, and the members of the Women's HIV-1 Program for discussions. We also thank the staff, interns, and subjects enrolled in the Lusaka cohort and the healthy blood donors.
Footnotes
Published ahead of print 11 December 2013
REFERENCES
- 1.Mothes W, Sherer N, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368. 10.1128/JVI.00443-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizzuto C, Sodroski J. 2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retroviruses 16:741–749. 10.1089/088922200308747 [DOI] [PubMed] [Google Scholar]
- 4.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711. 10.1038/31514 [DOI] [PubMed] [Google Scholar]
- 5.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. 10.1126/science.280.5371.1884 [DOI] [PubMed] [Google Scholar]
- 6.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. 10.1038/nsmb.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallo SA, Puri A, Blumenthal R. 2001. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry 40:12231–12236. 10.1021/bi0155596 [DOI] [PubMed] [Google Scholar]
- 8.Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–684. 10.1016/S0092-8674(00)81430-0 [DOI] [PubMed] [Google Scholar]
- 9.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413–423. 10.1083/jcb.151.2.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. 10.1016/S0092-8674(00)80205-6 [DOI] [PubMed] [Google Scholar]
- 11.Williamson S. 2003. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20:1318–1325. 10.1093/molbev/msg144 [DOI] [PubMed] [Google Scholar]
- 12.Ho DD. 1995. Time to hit HIV, early and hard. N. Engl. J. Med. 333:450–451. 10.1056/NEJM199508173330710 [DOI] [PubMed] [Google Scholar]
- 13.Holmes EC, Zhang LQ, Simmonds P, Ludlam CA, Brown AJ. 1992. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc. Natl. Acad. Sci. U. S. A. 89:4835–4839. 10.1073/pnas.89.11.4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Starcich BR, Hahn BH, Shaw GM, McNeely PD, Modrow S, Wolf H, Parks ES, Parks WP, Josephs SF, Gallo RC, Wong-Staal F. 1986. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45:637–648. 10.1016/0092-8674(86)90778-6 [DOI] [PubMed] [Google Scholar]
- 15.Masciotra S, Owen SM, Rudolph D, Yang C, Wang B, Saksena N, Spira T, Dhawan S, Lal RB. 2002. Temporal relationship between V1V2 variation, macrophage replication, and coreceptor adaptation during HIV-1 disease progression. AIDS 16:1887–1898. 10.1097/00002030-200209270-00005 [DOI] [PubMed] [Google Scholar]
- 16.Palmer C, Balfe P, Fox D, May JC, Frederiksson R, Fenyo EM, McKeating JA. 1996. Functional characterization of the V1V2 region of human immunodeficiency virus type 1. Virology 220:436–449. 10.1006/viro.1996.0331 [DOI] [PubMed] [Google Scholar]
- 17.Shioda T, Oka S, Xin X, Liu H, Harukuni R, Kurotani A, Fukushima M, Hasan MK, Shiino T, Takebe Y, Iwamoto A, Nagai Y. 1997. In vivo sequence variability of human immunodeficiency virus type 1 envelope gp120: association of V2 extension with slow disease progression. J. Virol. 71:4871–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chohan B, Lang D, Sagar M, Korber B, Lavreys L, Richardson B, Overbaugh J. 2005. Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J. Virol. 79:6528–6531. 10.1128/JVI.79.10.6528-6531.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagar M, Wu X, Lee S, Overbaugh J. 2006. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J. Virol. 80:9586–9598. 10.1128/JVI.00141-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitrinos KM, Hoffman NG, Nelson JA, Swanstrom R. 2003. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J. Virol. 77:6811–6822. 10.1128/JVI.77.12.6811-6822.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 410:582–608. 10.1016/j.jmb.2011.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laird ME, Igarashi T, Martin MA, Desrosiers RC. 2008. Importance of the V1/V2 loop region of simian-human immunodeficiency virus envelope glycoprotein gp120 in determining the strain specificity of the neutralizing antibody response. J. Virol. 82:11054–11065. 10.1128/JVI.01341-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toohey K, Wehrly K, Nishio J, Perryman S, Chesebro B. 1995. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology 213:70–79. 10.1006/viro.1995.1547 [DOI] [PubMed] [Google Scholar]
- 24.Etemad B, Fellows A, Kwambana B, Kamat A, Feng Y, Lee S, Sagar M. 2009. Human immunodeficiency virus type 1 V1-to-V5 envelope variants from the chronic phase of infection use CCR5 and fuse more efficiently than those from early after infection. J. Virol. 83:9694–9708. 10.1128/JVI.00925-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter BL, Wehrly K, Swanstrom R, Platt E, Kabat D, Chesebro B. 2005. Role of low CD4 levels in the influence of human immunodeficiency virus type 1 envelope V1 and V2 regions on entry and spread in macrophages. J. Virol. 79:4828–4837. 10.1128/JVI.79.8.4828-4837.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogert RA, Lee MK, Ross W, Buckler-White A, Martin MA, Cho MW. 2001. N-linked glycosylation sites adjacent to and within the V1/V2 and the V3 loops of dualtropic human immunodeficiency virus type 1 isolate DH12 gp120 affect coreceptor usage and cellular tropism. J. Virol. 75:5998–6006. 10.1128/JVI.75.13.5998-6006.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves JD, Gallo SA, Ahmad N, Miamidian JL, Harvey PE, Sharron M, Pohlmann S, Sfakianos JN, Derdeyn CA, Blumenthal R, Hunter E, Doms RW. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 99:16249–16254. 10.1073/pnas.252469399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20:1151–1154. 10.1038/nbt745 [DOI] [PubMed] [Google Scholar]
- 29.Cavrois M, Neidleman J, Kreisberg JF, Fenard D, Callebaut C, Greene WC. 2006. Human immunodeficiency virus fusion to dendritic cells declines as cells mature. J. Virol. 80:1992–1999. 10.1128/JVI.80.4.1992-1999.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavrois M, Neidleman J, Kreisberg JF, Greene WC. 2007. In vitro derived dendritic cells trans-infect CD4 T cells primarily with surface-bound HIV-1 virions. PLoS Pathog. 3:e4. 10.1371/journal.ppat.0030004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavrois M, Neidleman J, Bigos M, Greene WC. 2004. Fluorescence resonance energy transfer-based HIV-1 virion fusion assay. Methods Mol. Biol. 263:333–344. 10.1385/1-59259-773-4:333 [DOI] [PubMed] [Google Scholar]
- 32.Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S, Hunter E. 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303:2019–2022. 10.1126/science.1093137 [DOI] [PubMed] [Google Scholar]
- 33.Haaland RE, Hawkins PA, Salazar-Gonzalez J, Johnson A, Tichacek A, Karita E, Manigart O, Mulenga J, Keele BF, Shaw GM, Hahn BH, Allen SA, Derdeyn CA, Hunter E. 2009. Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog. 5:e1000274. 10.1371/journal.ppat.1000274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander M, Lynch R, Mulenga J, Allen S, Derdeyn CA, Hunter E. 2010. Donor and recipient Envs from heterosexual human immunodeficiency virus subtype C transmission pairs require high receptor levels for entry. J. Virol. 84:4100–4104. 10.1128/JVI.02068-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modrow S, Hahn BH, Shaw GM, Gallo RC, Wong-Staal F, Wolf H. 1987. Computer-assisted analysis of envelope protein sequences of seven human immunodeficiency virus isolates: prediction of antigenic epitopes in conserved and variable regions. J. Virol. 61:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann T, Hagmann I, Lohrengel S, Heil ML, Derdeyn CA, Krausslich HG, Dittmar MT. 2005. T20-insensitive HIV-1 from naive patients exhibits high viral fitness in a novel dual-color competition assay on primary cells. Virology 333:251–262. 10.1016/j.virol.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 37.Cavrois M, Neidleman J, Galloway N, Derdeyn CA, Hunter E, Greene WC. 2011. Measuring HIV fusion mediated by envelopes from primary viral isolates. Methods 53:34–38. 10.1016/j.ymeth.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sallusto F, Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118. 10.1084/jem.179.4.1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddy A, Gibbs BS, Liu YL, Coward JK, Changchien LM, Maley F. 1999. Glycosylation of the overlapping sequons in yeast external invertase: effect of amino acid variation on site selectivity in vivo and in vitro. Glycobiology 9:547–555. 10.1093/glycob/9.6.547 [DOI] [PubMed] [Google Scholar]
- 40.Isaacman-Beck J, Hermann EA, Yi Y, Ratcliffe SJ, Mulenga J, Allen S, Hunter E, Derdeyn CA, Collman RG. 2009. Heterosexual transmission of human immunodeficiency virus type 1 subtype C: macrophage tropism, alternative coreceptor use, and the molecular anatomy of CCR5 utilization. J. Virol. 83:8208–8220. 10.1128/JVI.00296-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinelli E, Tharinger H, Frank I, Arthos J, Piatak M, Jr, Lifson JD, Blanchard J, Gettie A, Robbiani M. 2011. HSV-2 infection of dendritic cells amplifies a highly susceptible HIV-1 cell target. PLoS Pathog. 7:e1002109. 10.1371/journal.ppat.1002109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly KA. 2003. Cellular immunity and Chlamydia genital infection: induction, recruitment, and effector mechanisms. Int. Rev. Immunol. 22:3–41. 10.1080/08830180305229 [DOI] [PubMed] [Google Scholar]
- 43.Abacioglu YH, Fouts TR, Laman JD, Claassen E, Pincus SH, Moore JP, Roby CA, Kamin-Lewis R, Lewis GK. 1994. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res. Hum. Retroviruses 10:371–381. 10.1089/aid.1994.10.371 [DOI] [PubMed] [Google Scholar]
- 44.Cavrois M, Neidleman J, Yonemoto W, Fenard D, Greene WC. 2004. HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology 328:36–44. 10.1016/j.virol.2004.07.015 [DOI] [PubMed] [Google Scholar]
- 45.Cao J, Sullivan N, Desjardin E, Parolin C, Robinson J, Wyatt R, Sodroski J. 1997. Replication and neutralization of human immunodeficiency virus type 1 lacking the V1 and V2 variable loops of the gp120 envelope glycoprotein. J. Virol. 71:9808–9812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatatos L, Wiskerchen M, Cheng-Mayer C. 1998. Effect of major deletions in the V1 and V2 loops of a macrophage-tropic HIV type 1 isolate on viral envelope structure, cell entry, and replication. AIDS Res. Hum. Retroviruses 14:1129–1139. 10.1089/aid.1998.14.1129 [DOI] [PubMed] [Google Scholar]
- 47.Johnson WE, Morgan J, Reitter J, Puffer BA, Czajak S, Doms RW, Desrosiers RC. 2002. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J. Virol. 76:2075–2086. 10.1128/jvi.76.5.2075-2086.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fox DG, Balfe P, Palmer CP, May JC, Arnold C, McKeating JA. 1997. Length polymorphism within the second variable region of the human immunodeficiency virus type 1 envelope glycoprotein affects accessibility of the receptor binding site. J. Virol. 71:759–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu P, Chertova E, Bess J, Jr, Lifson JD, Arthur LO, Liu J, Taylor KA, Roux KH. 2003. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. U. S. A. 100:15812–15817. 10.1073/pnas.2634931100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. 10.1038/nature01470 [DOI] [PubMed] [Google Scholar]
- 51.Shaw GM, Hunter E. 2012. HIV transmission. Cold Spring Harb. Perspect. Med. 2:a006965. 10.1101/cshperspect.a006965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzalez JF, Salazar MG, Decker JM, Parrish EH, Berg A, Hopper J, Hora B, Kumar A, Mahlokozera T, Yuan S, Coleman C, Vermeulen M, Ding H, Ochsenbauer C, Tilton JC, Permar SR, Kappes JC, Betts MR, Busch MP, Gao F, Montefiori D, Haynes BF, Shaw GM, Hahn BH, Doms RW. 2012. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin alpha4beta7. PLoS Pathog. 8:e1002686. 10.1371/journal.ppat.1002686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li M, Salazar-Gonzalez JF, Derdeyn CA, Morris L, Williamson C, Robinson JE, Decker JM, Li Y, Salazar MG, Polonis VR, Mlisana K, Karim SA, Hong K, Greene KM, Bilska M, Zhou J, Allen S, Chomba E, Mulenga J, Vwalika C, Gao F, Zhang M, Korber BT, Hunter E, Hahn BH, Montefiori DC. 2006. Genetic and neutralization properties of subtype C human immunodeficiency virus type 1 molecular env clones from acute and early heterosexually acquired infections in southern Africa. J. Virol. 80:11776–11790. 10.1128/JVI.01730-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilen CB, Parrish NF, Pfaff JM, Decker JM, Henning EA, Haim H, Petersen JE, Wojcechowskyj JA, Sodroski J, Haynes BF, Montefiori DC, Tilton JC, Shaw GM, Hahn BH, Doms RW. 2011. Phenotypic and immunologic comparison of clade B transmitted/founder and chronic HIV-1 envelope glycoproteins. J. Virol. 85:8514–8527. 10.1128/JVI.00736-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Jr, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U. S. A. 110:6626–6633. 10.1073/pnas.1304288110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lavine CL, Lao S, Montefiori DC, Haynes BF, Sodroski JG, Yang X. 2012. High-mannose glycan-dependent epitopes are frequently targeted in broad neutralizing antibody responses during human immunodeficiency virus type 1 infection. J. Virol. 86:2153–2164. 10.1128/JVI.06201-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karasavvas N, Billings E, Rao M, Williams C, Zolla-Pazner S, Bailer RT, Koup RA, Madnote S, Arworn D, Shen X, Tomaras GD, Currier JR, Jiang M, Magaret C, Andrews C, Gottardo R, Gilbert P, Cardozo TJ, Rerks-Ngarm S, Nitayaphan S, Pitisuttithum P, Kaewkungwal J, Paris R, Greene K, Gao H, Gurunathan S, Tartaglia J, Sinangil F, Korber BT, Montefiori DC, Mascola JR, Robb ML, Haynes BF, Ngauy V, Michael NL, Kim JH, de Souza MS. 2012. The Thai phase III HIV type 1 vaccine trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res. Hum. Retroviruses 28:1444–1457. 10.1089/aid.2012.0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366:1275–1286. 10.1056/NEJMoa1113425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. 2013. Antibodies with high avidity to the gp120 envelope protein in protection from SIVmac251 acquisition in an immunization regimen that mimics the RV-144 Thai trial. J. Virol. 87:1708–1719. 10.1128/JVI.02544-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Basu D, Kraft CS, Murphy MK, Campbell PJ, Yu T, Hraber PT, Irene C, Pinter A, Chomba E, Mulenga J, Kilembe W, Allen SA, Derdeyn CA, Hunter E. 2012. HIV-1 subtype C superinfected individuals mount low autologous neutralizing antibody responses prior to intrasubtype superinfection. Retrovirology 9:76. 10.1186/1742-4690-9-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881–10890. 10.1093/nar/16.22.10881 [DOI] [PMC free article] [PubMed] [Google Scholar]