

Abstract
During infection, binding of mature poliovirus to cell surface receptors induces an irreversible expansion of the capsid, to form an infectious cell-entry intermediate particle that sediments at 135S. In these expanded virions, the major capsid proteins (VP1 to VP3) adopt an altered icosahedral arrangement to open holes in the capsid at 2-fold and quasi-3-fold axes, and internal polypeptides VP4 and the N terminus of VP1, which can bind membranes, become externalized. Cryo-electron microscopy images for 117,330 particles were collected using Leginon and reconstructed using FREALIGN. Improved rigid-body positioning of major capsid proteins established reliably which polypeptide segments become disordered or rearranged. The virus-to-135S transition includes expansion of 4%, rearrangements of the GH loops of VP3 and VP1, and disordering of C-terminal extensions of VP1 and VP2. The N terminus of VP1 rearranges to become externalized near its quasi-3-fold exit, binds to rearranged GH loops of VP3 and VP1, and attaches to the top surface of VP2. These details improve our understanding of subsequent stages of infection, including endocytosis and RNA transfer into the cytoplasm.
INTRODUCTION
Poliovirus is the causative agent of poliomyelitis and the type member of the Enterovirus genus. As such, it shares structural and functional similarities with other members of the Picornaviridae family, including the coxsackievirus B viruses, which are associated with cardiomyopathies, central nervous system (CNS) infections, and diabetes; rhinoviruses, which are the most significant cause of the common cold; echoviruses, which can cause aseptic meningitis, gastroenteritis, and respiratory diseases; enterovirus 71 (EV71) and coxsackievirus A16 (CAV16), which have recently caused epidemics in Asia of hand-foot-and-mouth disease, with a high frequency of central nervous system involvement and with high morbidity and mortality; and the foot-and-mouth disease virus, which causes devastating outbreaks of foot-and-mouth disease in livestock (1).
Mature poliovirus (which sediments at 160S) is a spherical, nonenveloped virus, with a diameter of approximately 30 nm. Its capsid consists of 60 copies each of 4 proteins (VP1 to VP4) arranged on a T=1 (pseudo-T=3) icosahedral lattice and encloses a 7,500-base positive-sense single-stranded RNA (ssRNA) viral genome (2, 3). The major proteins (VP1, VP2, and VP3) share a topology, which is an eight-stranded beta barrel. Each protein has a different set of loops connecting the strands in the barrel and unique N- and C-terminal extensions. The C-terminal extensions of the VP1 to -3 subunits are located on the exterior surface of the virus, whereas VP4 and the N-terminal extensions are intertwined on the interior of the shell, making contact with the RNA genome. VP4, which is on the inside surface of the capsid, folds into an elongated structure, due to its contacts on the inner surfaces of the capsid protein that make up the shell. The outer surface of the virus is dominated by star-shaped mesas surrounding each 5-fold axis and three-bladed propeller-like features surrounding each 3-fold axis. Each mesa is formed by 5 molecules of VP1. The hub of each propeller is centered on an icosahedral 3-fold axis and is formed primarily by the VP2 and VP3 beta barrels, while the propeller blades are formed by the EF loops of VP2, the GH loop of VP1, and the C terminus of VP1. Separating these projections are deep depressions, or canyons surrounding each 5-fold mesa, and a saddle-shaped depression across each 2-fold axis. The inner surface of the protein shell is decorated by an elaborate network formed by VP4 and the N-terminal extensions of VP1, VP2, and VP3.
During the viral life cycle, native poliovirus attaches to a cell surface receptor, Pvr/CD155 (4). At 37°C, the receptor catalyzes major conformational changes in the 160S capsid (5), leading to the formation of a poliovirus cell-entry intermediate, known as the 135S or A-particle (6, 7). In the 135S particle, both the N-terminal extension of VP1 (8, 9) and VP4 (6), which is myristoylated at its N terminus (10), become externalized. Both of these externalized polypeptides then bind to membranes (9, 11, 12) and are capable of creating pores and channels in the membrane (12, 13). Conversion of the virus to its expanded state is required for the uptake of the virus into endosomal compartments of the infected cells (14). Once within an endosome, an interaction of the externalized polypeptides of poliovirus with the endosomal membrane allows the viral RNA genome to transfer efficiently and directly from the capsid interior into the cytoplasm of the infected cell, while remaining protected from RNases (E. Gropelli, H. C. Levy, E. Sun, X. Zhuang, J. M. Hogle, C. Nicol, S. Gold, T. J. Tuthill, and D. J. Rowlands, submitted for publication). Currently, the mechanism of RNA transfer is not well understood.
There are now a number of expanded picornavirus structures at intermediate and higher resolution that sample the process at different points (e.g., crystal structures of EV71 and human rhinovirus serotype 2 [HRV2] 80S-like particles and CAV16 135S-like particles, as well as cryo-electron microscopy [cryo-EM] structures of poliovirus 80S and 135S particles) (15–19). All such expanded virus structures show that 135S conversion causes the particle to expand by ∼4% and causes large holes to form at the 2-fold axes and the quasi-3-fold axes. Recently, we have generated lower-resolution asymmetric reconstructions showing that RNA extrusion from poliovirus occurs at a quasi-3-fold axis, both in isolation (20) and in complex with membrane (21). In the latter complex, viruses that are attached to membrane show remarkable consistency in the distance and orientation of the virus, relative to the bilayer, and are separated from the membrane by two long umbilical connections (21). This consistency suggests that some of the polypeptide components that become available symmetrically (in 135S particles) will subsequently reassemble in a very precise way to form well-defined asymmetric structures for membrane anchoring and RNA transfer (21).
Previous low-resolution cryo-EM studies of the poliovirus 135S particle, at about 20-Å and 10-Å resolution, respectively, have implicated the propeller tip as the binding site for the externalized N-terminal extension of VP1 (17, 22). This result has recently been confirmed directly by solving the structure of the complex of the 135S particle with an Fab directed against residues 24 to 40 of VP1 (23). However, the limited resolution of those structures restricted our ability to model the structural transitions, particularly the movement of polypeptide segments not included in the beta barrel cores of the capsid proteins.
In the present study, we have determined a higher-resolution structure of the 135S poliovirus cell-entry intermediate by using cryo-EM and three-dimensional (3D) image reconstruction at approximately 6-Å resolution. In this new structure, the positions of the major capsid proteins and the interactions of many of the structural components are unambiguously resolved, allowing us to determine with confidence which of the polypeptide segments have become shifted, disordered, or rearranged. The reconstruction has clearly interpretable density for a significant portion of the externalized N terminus of VP1. Thus, the N terminus exits the virion at the base of the canyon near the quasi-3-fold axis and participates in a novel three-component interaction with the rearranged GH loops of VP3 and VP1. This occurs at a site corresponding to the base of the larger umbilical connection observed in membrane-associated virus. The N terminus of VP1 is then directed toward the tip of the VP2 propeller, poised at high radius for membrane interaction.
MATERIALS AND METHODS
Sample preparation.
The Mahoney strain of type 1 poliovirus was grown in HeLa cell suspension, harvested by centrifugation, and released from cell pellets by freeze-thaw lysis. The virus was then purified from the cell lysate by CsCl density gradient fractionation, as described previously (8, 17). The 135S particles were made in vitro from the native virus by heating at 50°C for 3 min in a low-salt buffer (2 mM CaCl2, 20 mM HEPES, pH 7.4).
Preparation of grids.
The concentration of the 135S poliovirus preparation was adjusted to 0.2 to 0.3 mg/ml for cryo-electron microscopy. Aliquots of 4 μl of 135S viruses were applied to glow-discharged holey carbon grids (C-flat; Protochips, Inc.), thinned by blotting, and plunge-frozen into liquid ethane (maintained at about −180°C to prepare vitrified specimens) using a manual gravity plunger. Grids were stored in liquid nitrogen before imaging.
Cryo-electron microscopy.
For single-particle analysis, data were collected on a Tecnai F20 microscope (FEI Company, Hillsboro, OR) operated at 200 kV using the automated electron microscopy data acquisition system, Leginon (24), at the National Resource for Automated Molecular Microscopy (NRAMM). A single grid of the 135S preparation was used for imaging. A total of 1,853 images (each 4,096 by 4,096 pixels) were recorded using a Tietz 4K-by-4K charge-coupled device (CCD) camera, at a nominal magnification of ×62,000 (corresponding to a pixel size of 1.37 Å), with defocus values between 0.98 and 2.0 μm and an electron dose of 15 e−/Å2.
Single-particle image processing.
Of the 1,853 CCD images, a subset of 1,743 nonastigmatic, contamination-free, and artifact-free micrographs was selected based on visual inspection. Initially, the contrast transfer function (CTF) was estimated using the ACE2 (automated CTF estimation) software package (25). Seven hundred twenty-three images with an ACE CTF estimation confidence value of less than 0.8 were removed from the data set. The remaining 1,020 CCD images were then used to determine average defocus value, astigmatism, tilt angle, and tilt axis using a second program, CTFIND3 (26), in Appion (27). Semiautomated particle selection using a box width of 300 Å was done using the program FindEM (28) from Appion (27). A few representative particles were selected manually from the first micrograph and used as seeds to initiate the automated particle-picking routine. The procedure yielded 119,079 2D projection images of virus particles. Each virus particle was then extracted into a 400-by-400-pixel box to generate a stack, and entries that were clearly not virus particles were removed, using statistical filtering methods implemented in the program Appion (27). A final set of 117,330 particle images was included in the single-particle parameter refinement. A previous reconstruction at ∼10-Å resolution of the 135S particle (17) was used as a starting model to initiate the search for initial Euler orientation angles and particle origins using FREALIGN V.09 (29). Thereafter, FREALIGN was used iteratively for another 31 cycles, both for parameter refinement and to compute CTF-corrected three-dimensional reconstructions, with icosahedral symmetry imposed during each reconstruction step.
Inner spherical masks were used to remove density for the RNA genome from the interior of the virus, prior to using the masked map as the reference model for the next iteration. Fourier shell correlation (FSC) between two independent reconstructions (30) was used to estimate the resolution of the final map, which was 7.2 Å (based on an FSC criterion of 0.5) and approximately 5.5 Å (based on a 0.143 threshold, per reference 31). While portions of the map support the higher-resolution estimate, including much of the N-terminal extension of VP3, which is resolved from the nearby VP1 beta barrel, and other features within the beta barrels of the three capsid proteins, other portions, particularly the regions corresponding to the EF loops of VP2, are poorly ordered, resulting in the lower-resolution estimate at the 0.5 FSC cutoff. To improve the interpretability of the final reconstruction, average Fourier amplitudes from the final map were scaled to those derived from the X-ray coordinates of the poliovirus 73S native empty capsid assembly intermediate (PDB 1POV), using the program bampweigh (32), and then sharpened (increasing Fourier amplitudes at high resolution) with an inverse B-factor of 300 Å2, to compensate for interpolation artifacts. FREALIGN V.09 was run on the Clathrin cluster, which is a shared 40-core Linux cluster with 20 GB of RAM managed by SBGrid at Harvard Medical School (http://www.sbgrid.org).
Model building and refinement.
The pseudoatomic model consists of a central protomer (one copy each of the capsid proteins VP1, VP2, and VP3), plus a 5-fold-symmetric copy of the intertwined amino termini of VP3 (residues 1 to 12). Except as noted below, the main chain coordinates in these four objects are identical rigid-body copies of crystallographically determined molecules, derived from the poliovirus 73S native empty capsid assembly intermediate, PDB entry 1POV (33). For the purpose of defining rigid bodies, much of the N-terminal extension of VP3 (residues 13 to 42) was regarded as attached to the VP1 core (residues 73 to 207 and 234 to 290), the N-terminal hairpin of VP2 (residues 14 to 27, with reduced occupancy) and C terminus of VP1 (residues 291 to 302) were regarded as belonging to the VP3 rigid body (residues 43 to 231), and the GH loop of VP1 (residues 208 to 233) was regarded as belonging to the VP2 rigid body (residues 28 to 44 and 58 to 272). Each such rigid body was first placed into the reconstruction by eye, using COOT (34). The correct placement, assuming no deformation, was unambiguous at this resolution because density features corresponding to the alpha-helical regions of the molecules were well resolved and served as fixed pivot points for fitting the beta sheets. After this preliminary rigid-body fitting, it was confirmed that the EF loop of VP1 had maintained its conformation but had shifted relative to the main barrel, so the loop was refitted manually as a rigid body, with stereochemically reasonable hinge points, and then reattached. In addition, it was confirmed that shape-similar density features were absent at high-contour level for several polypeptide chains. In VP1, missing segments included the N terminus, residues 1 to 51; parts of the GH loop, residues 219 to 223; and the C terminus, residues 291 to 302. In VP2, missing segments included the N terminus, residues 1 to 13; the bottom loop, residues 45 to 57; the C terminus, residues 266 to 272; and unexpectedly, two segments of the large EF loop, residues 134 to 143 and 158 to 172. Later, the residue 134 to 143 segment was visualized at a lower contour level and was restored with a lowered occupancy. The core of VP3 (residues 43 to 227) was only minimally altered, relative to mature virus, with a slight reorganization of the C-terminal residues 228 to 235. The rigid-body model, constructed as described from portions of the poliovirus 73S crystal structure, accounted for most of the electron density in the capsid, except for the vicinity of the quasi-3-fold axis, where a few altered chain traces were introduced that were indicated by the density map (see below). It should be emphasized that only a small number of residues differ from their rigid-body positions, and those altered polypeptides should be regarded as highly approximate.
Automated optimization of the atomic model was accomplished using locally developed Unix C-shell scripts that carried out a sequence of Unix utilities and programs from the CCP4 package (35). In each cycle of refinement, constrained rigid-body copies of the well-ordered residues from VP1, VP2, VP3, and the VP3 beta tube were superimposed by least squares (using LSQKAB [36]) onto the central protomer that was output by the previous refinement cycle. The manually built chain traces (for a small number of residues) were not replaced during the superposition step, unless guide coordinates were required to make the subsequent refinement well behaved. Then, exact icosahedral symmetry operators were used to generate all 14 polypeptide chains immediately surrounding the four central objects, for a total of 18 chains in all. A generous mask was used to isolate the corresponding portion of the cryo-EM reconstruction, which was then enclosed in a padded minimal rectangular box and Fourier transformed. Note that none of the mask edges were in direct contact with the central protomer that was being refined. The resulting Fourier amplitudes were scaled to the Fourier transform of the model, matching a sliding window of pseudoreflections by resolution using SFTOOLS (B. Hazes, unpublished observation). The resulting scaled amplitudes and “observed” phases were used as reference standards for refining the atomic model with REFMAC5 (37). By iterating this procedure, a plausible, low-energy structure was obtained, most of whose coordinates closely resemble the crystallography-based rigid-body model. Main chain segments that refined to deviate significantly from the rigid-body model were noted. Most likely, this behavior indicates the sites where changes in position or conformation have taken place, though the present resolution is not sufficient for the exact nature of the change to be known.
Accession numbers.
The identification code for the 135S density map in the EMDataBank is EMD-5710. The Protein Data Bank identification code for coordinate modeling results is 3J48.
RESULTS
Overall appearance of the reconstructed 135S particle.
The 135S capsids appear round or angular in outline when viewed in vitreous ice (Fig. 1). A total of 117,330 particle images from 1,020 CCD images were selected for the cryo-EM single-particle reconstruction, and 99,750 particles were used in the final three-dimensional reconstruction (Fig. 2A and B). The resulting icosahedrally symmetric cryo-EM map has a resolution of approximately 5.5 Å according to the Fourier shell correlation of half-data sets at a threshold of 0.143 (and 7.2 Å for a 0.5 threshold) (Fig. 2C). The overall architecture, as expected, is very similar to previously determined low-resolution structures of the 135S particles and 80S particles of poliovirus (17, 18, 22) and of other picornaviruses (38). It also is consistent with the crystal structures of expanded virus particles from other picornaviruses (15, 16, 19).
FIG 1.

Cryo-electron micrograph showing a field of 135S particles in vitreous ice. The 135S particles are filled by RNA and, depending on orientation, can appear round (B) or angular (C and arrows in panel A). A small number of 80S empty capsids are present (D and asterisks in panel A). Scale bars are included. This representative CCD image showing 135S particles was recorded at −2.18-μm defocus and an ×62,000 nominal magnification, using a Tecnai F20 microscope (FEI).
FIG 2.

Reconstruction of poliovirus 135S particles. (A and B) Surface rendering colored by radial position, showing the outer (A) and inner (B) surfaces. The map has been sharpened and displayed at high contour to emphasize higher-resolution details. In many places, individual polypeptide chains are resolved, which facilitates the rigid-body fitting of pseudoatomic models. Note the hole at the icosahedral 2-fold axis (center), where red-colored density is visible from the exterior. Labels indicate a mesa and a propeller tip, which are major projections from the outer surface. Separating the projections are canyons surrounding each 5-fold mesa and a saddle-shaped depression crossing the 2-fold axis. Symmetry axes are indicated by numbers. (C) A Fourier shell correlation (FSC) was calculated between randomly selected half-data sets. This curve suggests a resolution of ∼7 Å (at 0.5 correlation) and that there is meaningful information at ∼5.5 Å (the resolution at which FSC falls below 0.143).
Overall structural changes that accompany expansion.
At the present resolution, the electron density map (Fig. 3A and 4) shows clearly where the beta sheets are located. It shows several alpha-helices as resolved from the remainder of the beta barrel cores, and it traces the path of a number of well-resolved polypeptide chains outside the beta barrels (Fig. 3A). With only a few exceptions (which are depicted in Fig. 3B), rigid-body transformations of crystallographic models (specifically, three decorated beta barrels and one VP3 beta tube; see Materials and Methods) fit well to features of the electron density map. This indicates that the core structures of the capsid proteins are generally well preserved, and not deformed, upon the 160S-to-135S transition.
FIG 3.

The quality of the reconstructed density map makes rigid-body fitting of the major capsid proteins unambiguous. (A) Density features on the inner surface of the capsid are shown, with symmetry axes indicated by numbers and clipping planes applied for clarity. After rigid-body fitting of the major capsid proteins, segments of the backbone traces of VP1 (blue), VP2 (yellow), and VP3 (red) are clearly shape-similar to tubular features in the sharpened density map. Alpha-helices (arrows) are often well resolved and serve as fixed pivot points for positioning the beta sheets unambiguously. (B) Docked models from the crystal structure of mature (160S) poliovirus (PV) (PDB 1HXS) were compared with the reconstruction of 135S particles, in order to determine which polypeptide segments had become shifted (magenta), rearranged significantly (cyan), or disordered (orange) during virus expansion.
FIG 4.

Upon poliovirus expansion, the topography of the outer surface changes dramatically. (A) A portion of the outer surface of the 135S reconstruction (right), viewed along a 2-fold axis, is compared with the corresponding density (left), calculated from the crystallographic model of mature poliovirus to 6 Å. Models of VP1, VP2, and VP3 are blue, yellow, and red, respectively, and symmetry axes are numbered. Shifts of the beta barrels cause a large hole to open at the 2-fold axis in 135S particles, as seen in a closeup comparison view. (B) The opening of a smaller hole at the quasi-3-fold (yellow asterisk) is associated with the shift or rearrangement nearby loops (as labeled). In the 135S reconstruction, in the upper right of the panel, the disordering of density for the VP2 propeller tip is dramatic.
It should be emphasized that a map at the present resolution cannot be a perfect match to the model, due both to phase error and to Fourier series termination, unless the model is overparameterized inappropriately. Frequently, short stretches of density for central strands of beta sheets are interrupted in a way that would be precluded by the beta hydrogen bonding pattern, indicating that it is the map, and not the model, that is the less reliable. Therefore, we remodel only longer stretches, where the lack-of-fit of the native conformation is unambiguous, and unassigned density is available that can be assigned to the residue range with some confidence. We name each such residue range and caution the reader that the resulting chain trace is highly approximate. In our figures, we choose to show a higher contour level, where the overall correlation of the rigid-body model with density is apparent, and where some of the chains are resolved, in order to demonstrate that the rigid-body assumption is generally good, though not perfect. At a lower contour level than the one chosen, the model is enclosed, but none of the individual chains can be traced.
As was noted in the previous low-resolution structures of poliovirus 135S particles (17, 22), the particles have expanded by ∼4%. Note, however, that the expansion is not uniform. In the pseudoatomic models of 135S particles, the center of mass of VP1 has shifted radially outward by 5%, relative to the VP1 position in the 160S capsid, whereas the VP2 and VP3 subunits have shifted outwards by only 3% relative to their native positions in the 160S capsid. Additionally, the VP1 beta barrel exhibits a “hinge” motion so that its top surface flattens. To accomplish this, many residues at the “open” end of the VP1 beta barrel (including its CD helix, GH loop, and C-terminal extension) remained tethered to the VP2 and VP3 barrels, while residues at the narrow end of the barrel remained clustered together at the 5-fold axis. A similar hinge movement has been seen in high-resolution structures of expanded forms of HRV2, EV71, and CAV16 (15, 16, 19). Nonuniform expansion has also been seen in the structure of the complex of 160S virions with an Fab directed against the N terminus of VP1 at 37° (39). In that instance, expansion at the 5-fold axes was 7%, and expansion at the 3-fold axes was only 1%, which would suggest that the outward shift of VP1 accounted for most of the expansion, while the radial shift of VP3 was much less.
Discrepancies between the map and model indicate the sites of changes in detail.
After rigid-body fitting of VP1, VP2, and VP3, all polypeptide segments that did not occupy consistently high density were removed from the “omit” model and subsequently classified as disordered, shifted, or rearranged (Fig. 3B). Here, we apply the term “rearranged” to a chain that is visible in the structure of mature virus but appears in a different conformation in the expanded virus particle. We use “shifted” to suggest, instead, that the conformations are similar but that the segments differ noticeably in location when the coordinates of the mature virus were superimposed on expanded virus by least squares. “Disordered” residues lie outside a chosen contour, and no shape-similar density feature appears when the contour level is lowered.
The capsids of expanded viruses have holes at the 2-fold and quasi-3-fold symmetry axes.
Within the boundaries of the kite-shaped biological protomer (with neighboring 5-fold, 3-fold, and two 2-fold axes at its corners), the side-by-side contacts of the beta barrels tend to remain intact. As a result, the expanded virus shell does not form significant holes within the boundaries of individual protomers. On the other hand, virus expansion causes significant disruption of the interfaces between neighboring protomers and the formation of large holes at the 2-fold and quasi-3-fold axes. These holes are particularly evident when the reconstruction is viewed at high contour (Fig. 4). The positions of these holes are highly relevant to the question of how internal polypeptides become externalized and how and where genomic RNA becomes unwound and passes through the capsid.
One large hole is located at the 2-fold axis and disrupts a tight association of the helices connecting the C and D strands of two symmetry-related copies of VP2 in mature poliovirions (Fig. 4A). The disordering of the nearby bottom loop of VP2 (residues 45 to 57, see below) contributes to enlarging this hole.
A second, smaller opening occurs nearby, at the base of the canyon, at the quasi-3-fold axis, which is located at the center of the 5-3-3 icosahedral triangle (Fig. 4B). Inspection of the atomic model near the quasi-3-fold (Fig. 5A) shows that the opening of this hole is caused by shifts of the capsid proteins away from one another, together with the disordering or rearrangement of a number of polypeptides in the interface between biological protomers. In particular, the GH loop of VP3 rearranges to form a radially oriented beta structure, which is also seen in high-resolution crystal structures of expanded viruses (15, 16, 19). In comparison, in the mature 160S virion, a flatter conformation of this loop lies close to the virus surface and occupies most of the quasi-3-fold interface, keeping it closed (2). As observed for poliovirus 80S particles (18), the size of the quasi-3-fold hole appears to be large enough to allow the externalization of VP4 and the N-terminal extension of VP1 to occur. However, without further expansion, the hole is clearly too small for the passage of the viral genome, such as that observed in structures caught in the act of RNA release (20, 21).
FIG 5.

Pseudoatomic model in the vicinity of the quasi-3-fold hole. (A) Overview, to provide context. (B) Zoomed-in view. VP2 is yellow, and VP3 is red. Two symmetry copies of VP1 are purple and blue, with the rebuilt N-terminal extension of the blue copy shown in cyan. Following the example of the crystal structure of the 135S-like particles of coxsackie virus A16, a short stretch of the N terminus of VP1 (labeled in cyan) forms a third beta strand alongside the two-stranded beta sheet (labeled in red) that is created by the rearrangement of the GH loop of VP3. The distal end of the GH loop of VP1 (labeled in purple) has become reoriented radially and leans rightward to contact the 3-stranded beta structure. Hypothetically, these three polypeptide segments may serve as a nucleus for building additional ordered structure atop the VP2 beta barrel, in order for the uncoating process to proceed further, upon membrane interaction. (C) In the vicinity of the quasi-3-fold axis, a section of the reconstruction shows that strong linear electron density features are present that correspond to proximal portions of the GH loops of VP3 (red) and VP1 (blue), which have both adopted an extended conformation that is rearranged relative to mature virus. The externalized N-terminal extension of VP1 extends toward the viewer through the indicated density feature (yellow arrow).
Polypeptide rearrangement on the inner surface.
The structure of the 160S mature poliovirion is held together and stabilized through a network of interactions on the inner surface of the capsid that includes intraprotomer, intrapentamer, and interpentamer contacts (2, 40). The full-length VP4 (69 residues) and the N termini of VP1, VP2, and VP3 interact extensively with one another, providing the virus particle with overall stabilization. The inner surface of the 135S particle (Fig. 2 and 3A) is very different from that of the mature virion. The differences are primarily a consequence of the fact that several peptide segments that are well ordered on the inner surface of the mature virion become rearranged, disordered, or externalized in the thinner 135S capsids (Fig. 3B) (17).
Modeling VP2.
When VP2 from 135S particles is superimposed on VP2 from 160S particles, the beta barrel of VP2 clearly remains in place, but the conformation of the N-terminal beta-hairpin (residues 14 to 27), which participates in interpentameric contacts below the VP3 beta barrel in the virion, is significantly displaced. Additionally, density for the bottom loop (residues 45 to 57) is missing, just as in the crystal structure of the 73S native empty capsid assembly intermediate (33), in which the N terminus of VP1 is also disordered. The electron density map suggests several other localized rearrangements in VP2. These include the HI loop (approximately in residues 240 to 244) in the vicinity of the 3-fold symmetry axis, as well as the large double loop between beta strands E and F of VP2, wherein the density for residues 134 to 143 has weakened (suggesting that it is flexible) and residues 158 to 172 have become disordered. The disordering of the EF loop of VP2 results in the disappearance of the density for the tips of the blades of the propeller-like features (Fig. 4B, upper right). The disordering of this loop appears to occur in concert with the disordering or rearrangement of several polypeptides that support the EF loop in virions, including the C terminus of VP1 (along the western edge of the EF loop), the GH loop of VP1 (along the eastern edge), and the C terminus of VP2 (residues 266 to 272, along the southern edge). In 135S particles, the C terminus of VP1 becomes disordered (see below), the GH loop of VP1 rearranges (see below), and there is no visible density corresponding to the native conformation of the C terminus of VP2 all the way back to its junction with the VP2 beta barrel at residue 265. Instead, just south of this junction, a pair of relatively strong, but disconnected, density features are visible in the 135S map, within the 2-fold hole. Due to the proximity of these features to the junction with the C-terminal end of the VP2 beta barrel, we surmise that the hole might be a preferred binding site for the disordered VP2 C-terminal extension. It is not yet clear whether keeping the 2-fold hole partially plugged in 135S particles is functionally relevant.
The two-loop “gate.”
The observation that the base of the VP2 C terminus (i.e., at residue 265) remains ordered is relevant to the structural integrity of the icosahedral capsid. This junction appears to remain firmly clamped between two folds of the EF loop of VP3 from a neighboring protomer (Fig. 5A) in both mature and expanded virions. This persistent interprotomer contact between the VP3 EF loop and the VP2 C terminus is also notable for creating a “gate” between the 2-fold and quasi-3-fold holes that persists during the 135S expansion. The gate is much more substantial in mature poliovirus, where it also includes the GH loop of VP3 (residues 169 to 189) and the bottom loop of VP2 (residues 45 to 57), which both rearrange upon virus expansion. In our current model for virus expansion (41), the VP1 N-terminal extension most likely emerges through the 2-fold hole (wherein polypeptide externalization is reversible) and transfers through the gate into the quasi-3-fold hole, where the exposed polypeptide becomes securely anchored by neighboring structures (see below).
Modeling VP3.
The four-body rigid-body model, including only well-ordered residues from the poliovirus 73S crystal structure, accounted for most of the electron density in the capsid but notably failed to account for a number of density features in the vicinity of the quasi-3-fold axis. Density for the GH loop of VP3 was clearly not a good fit for the loop conformation that was seen in the mature poliovirus, where a flattish loop path close to the surface helps to keep the quasi-3-fold hole plugged (Fig. 4B, on the left). A better fit to the density was obtained from the conformation seen in 80S particles of EV71 (16) and in poliovirus (M. Wolf, D. J. Filman, and J. M. Hogle, unpublished data), where the GH loop of VP3 becomes elongated into a radially oriented two-stranded beta structure, but it was clear that two beta strands were insufficient to account for the entire volume of density that was present. Additionally, the two strands of the 80S hairpin would have to be twisted by about 90 degrees for both strands of the hairpin to fit within our current 135S density envelope. The density was a much better fit for models derived from the recently published structure of a 135S-like particle from coxsackievirus A16 (19). In our current model, residues 169 to 175 and 181 to 188 of VP3 and residues 51 to 72 of VP1 are adapted from a superposed copy of the CAV16 atomic model. For the most part, these three polypeptide segments appear to move together as part of the VP3 rigid body. To complete the GH loop of VP3, residues 176 to 180 at the distal end were fitted manually to lie within a low-density contour, as a highly approximate chain tracing. Fortunately, due to the shortness of this missing loop, there was only one available path to choose. Notably, formation of the three-stranded structure has now been observed in both of the known structures of picornavirus 135S particles, but in none of the 80S (15, 16) or 73S (33) structures, all of which lack density for the N-terminal extension of VP1.
In VP3, some additional differences between the 135S and 160S models (Fig. 3B) are due to the continued noncovalent attachment of certain polypeptide chains to neighboring beta barrels, as the barrels separate during virus expansion. Thus, in 135S particles, both the N-terminal extension of VP3 (residues 13 to 42) and the C-terminal polypeptide of VP3 (residues 227 to 235) travel together with the VP1 rigid body. As a result, both chains become displaced, relative to the VP3 beta barrel, without requiring significant changes in the intermolecular interactions that the polypeptides make. Additionally, the VP3 beta tube (five intertwined copies of residues 1 to 12) is seen to rise along the 5-fold axis, to remain in contact with the underside of VP1.
Modeling VP1.
Compared to its position in the 160S capsid, the center of mass of VP1 has moved outward radially by ∼6.5 Å. The tips of the DE and HI loops nearest to the 5-fold axis have moved out by a distance of 5.2 Å and 6.2 Å, respectively. The BC loop, which is located on top of the mesa, but farthest from the 5-fold axis, has moved by 7.4 Å but also appears likely to be deformed, as much of the native conformation of the loop would protrude through the density envelope. Overall, the VP1 wedge moves outward by about 5 Å along the axis and by 6.6 Å in the middle. These movements suggest an “umbrella-like” motion, wherein the part of the barrel nearest the canyon rises more than the part near the axis, causing the 135S particle to be more angular in profile than the 160S particle and resulting in an elevation and a broadening of the mesa. A similar hinge motion for VP1, and a radial expansion of the capsid protein subunits, was seen in the crystal structures of 80S particles of EV71 (16) and HRV2 (15) and in the 135S particle of CAV16 (19).
There also is evidence that the EF loop of VP1 (residues 162 to 179) undergoes a slight tilting and a lateral shift relative to the rest of the VP1 barrel (Fig. 3B). This loop movement might be required so that the EF loop can continue to maintain its contacts with both of the VP1 barrels that are located on either side of it, even as the barrels are situated further apart in the expanded 135S particles than in 160S virions. Indeed, the forced separation of the EF loop from its own beta barrel is hypothesized to be an important aspect of the mechanism of the receptor-induced 160S-to-135S transition (M. Strauss, D. J. Filman, N. Cheng, D. M. Belnap, and J. M. Hogle, unpublished data).
Polypeptides involved in externalizing the N terminus of VP1.
In 135S particles, the N-terminal extension of VP1 is externalized and is capable of binding to specific Fabs (23) or to membrane (9, 11). Our previous 10-Å reconstruction of the 135S particle clearly showed that the N terminus did not exit at the 5-fold axis, as previously suggested, and led to the conjecture based on genetic data that the N terminus was instead released from the base of the canyon near the quasi-3-fold axis (17). However, the limited resolution of this reconstruction did not allow us to directly visualize the site of externalization. Recently, the crystal structure of 135S particles from CAV16 (19) showed how the proximal end of the VP1 N-terminal extension rearranges to exit through the capsid at the quasi-3-fold hole. The present cryo-EM structure of 135S particles confirms that a similar rearrangement of the N-terminal extension occurs in poliovirus and that additional polypeptides rearrange to contact the extension and stabilize its exit point (Fig. 5A and B). In particular, residues corresponding to 59 to 68 from the N-terminal extension of VP1 are reordered in 135S particles from both CAV16 (19) and poliovirus. As this polypeptide extends away from the VP1 beta barrel, several residues (63 to 72) extend across the inner surface of the capsid until they reach the southern end of the quasi-3-fold hole. Each N terminus then turns radially outward, with residues 59 to 62 interacting with the southern edge of the reorganized GH loop of VP3 in its extended conformation (Fig. 5A).
Quasi-3-fold anchor.
The exiting N-terminal extension, in its new arrangement, is further anchored by extensive contacts with the GH loop of VP2 (residues 216 to 223), together with parts of the D strand of the VP2 barrel (residues 100 to 101), the N terminus of VP3 (residues 29 to 32), and the end of the G strand from the neighboring copy of VP1 (residues 201 to 202). The extended conformation of the VP3 GH loop is supported by residues from the EF loop of the neighboring VP1 (residues 162 to 163) and by the “doorstop” linker region of the neighboring VP1 GH loop (residues 233 to 236, just prior to the H strand), which normally controls access to the stability-regulating “pocket factor” lipid that occupies the center of the VP1 beta barrel in mature virions (40). Note that as the VP1 and VP2 barrels move apart during the 160S-to-135S transition, this linker segment provides the slack that allows parts of the VP1 GH loop to remain bound to the top of the VP2 barrel. During the transition, the linker shifts to slightly lower radius in the quasi-3-fold hole (which is apparent in the density map) while making contact with the newly rearranged GH loop of VP3 and N-terminal extension of VP1 and helping to hold the new structural features in place.
The polypeptide segments that clamp the GH loop of VP3 and the N terminus of VP1, for the most part, are well ordered, and most are in their native conformations, so that rigid-body movements of the native beta barrels serve to bring them into the proper arrangement. The final contact is more tenuous, involving the distal end of the GH loop of the VP1 (residues 213 to 227) that belongs to the neighboring protomer to the west. Evidence from the poliovirus 135S map, as detailed below, indicates that the loop has reoriented to project radially outward while tilting sideward to contact the quasi-3-fold site (Fig. 5). Presumably, this additional loop rearrangement cannot occur until after the GH loop of VP3 and the N terminus of VP1 have rearranged to receive it.
Extreme N terminus of VP1.
Once the N terminus of VP1 leaves its binding site in the quasi-3-fold hole, there are a number of directions that it could travel. A pathway through weak density does exist for residues 51 to 58 that extends across the top of the VP2 beta barrel and reaches the large double loop in VP2 that connects beta strands E and F. This path is included in the model as a highly approximate chain trace. This possible path would be in agreement with the observation that an Fab from an antibody recognizing residues 24 to 40 of VP1 binds at this propeller tip (23). Additional confirmation of the location of the VP1 N terminus was obtained in our previous lower-resolution cryo-EM studies of the 135S poliovirus cell-entry intermediate (17), where removal of the N-terminal helix using V8 protease created a significant-difference electron density feature at the propeller tip.
Our current proposal for the path of the VP1 N-terminal extension differs from our previous suggestion (17), which held that residues 42 to 52 (a predicted amphipathic helix) might be responsible for creating the tubular density feature (Fig. 6, open arrowhead) that appears to bridge across the canyon between the mesa and the propeller tip in our lower-resolution maps. In the present higher-resolution map of 135S virions, in the corresponding location, there are no interpretable density features above the noise level that would help to explain the lower-resolution observation.
FIG 6.
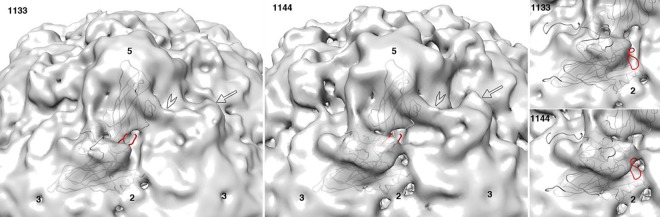
Lower-resolution maps validate the atomic model. To verify that the newly built conformation of the GH loop of VP1 is real and not an artifact of high-frequency noise, we displayed the pseudoatomic model, superimposed on two independent lower-resolution reconstructions of poliovirus 135S particles (EMDB entries 1133 and 1144) that were previously solved by Bubeck et al. at 10-Å resolution (17). Comfortably above the noise level, both 135S maps show extra density features (which are not present in 160S particles), located atop the VP2 beta barrel (arrow) and bridging across the canyon (open arrowhead). Importantly, these density features were more pronounced in the 1144 population of particles, though the two populations were produced using the same protocol and processed identically using PFT2 (42) and EM3DR2 (http://people.chem.byu.edu/belnap/). Observe that the 1144 map (middle panel) has a density projection that covers most of the newly built GH loop of VP1 (red). In contrast, coverage of the GH loop is poor in the 1133 map (left panel). For comparison (right), the 160S and 73S conformations of the VP1 GH loop (red), the VP1 C terminus (gray), and the tip of the EF loop of VP2 (gray) all clearly lie outside the isocontour surface in both maps.
Rearrangement of the GH loop of VP1.
In the present higher-resolution maps, polypeptides from VP1 have been observed to form two structures that were not present in mature virus. As noted above, one is an ordered redirection of a portion of the VP1 N-terminal extension (residues 58 to 67) that leads to its externalization through the quasi-3-fold hole. The other involves a significant reorientation of the distal end of the GH loop of VP1. Thus, the rearranged GH loop of VP1 no longer lies flat across the top of the VP2 beta barrel, supporting the eastern edge of the EF loop of VP2, as it does in 160S particles. Instead, the GH loop now projects outward and leans sideways, making contacts with the externalized N-terminal extension of VP1 and the rearranged GH loop of VP3, both of which belong to a neighboring protomer (Fig. 5).
It is important to explain how the rearrangement of the GH loop of VP1 was found, because modeling a new detailed structure into a map at this resolution is normally not feasible. Early on, while checking the agreement of the complete rigid-body-docked atomic model with the map, we noticed that the map included two strong, parallel, radially oriented tubes of unoccupied density protruding from the top surface of the VP2 envelope. At the time, we had no indication that we would be able to identify the polypeptide segments to build into them. Some time later, we removed all polypeptide segments whose native conformation caused them to lie outside the chosen isocontour envelope. This yielded an “omit” model composed exclusively of reliable components that appeared not to change conformation (gray tubes in Fig. 3B). Remarkably, the two parallel density tubes joined to the density envelope very close to the sites where the distal end of the GH loop of VP1 in the atomic model had been truncated: one tube at the site of residue 214 and the other tube near residue 229. Initially, we had assumed that residues 213 to 228 from the distal end of the GH loop of VP1 were disordered because their native conformation placed them outside the contour envelope. However, the observed density tubes demonstrated that the distal end of the GH loop must instead be rearranged in a well-defined way (Fig. 5).
In the current model, the stems of the reoriented GH loop (residues 213 to 218 and 224 to 226) have been model built and refined as a partially occupied chain trace. This is intended to indicate the path of the polypeptide, rather than providing reliable atomic coordinates. The extreme distal end of the loop has been omitted, for lack of a clear density path. Note that two intermediate segments of the GH loop (residues 208 to 212 and 227 to 229) continue to bind to the top of the VP2 beta barrel, in a way similar to what is seen in the 73S and 160S crystal structures (2, 33).
DISCUSSION
Evidence for the rearranged GH loop of VP1 in lower-resolution studies.
Our earlier publication of the 10-Å structure of the 135S particle included two independent reconstructions (EMDB entries 1133 and 1144), both generated using PFT2 (42) and EM3DR2 (http://people.chem.byu.edu/belnap/). When viewed at a lower contour level, the 1144 reconstruction shows extra density features on the top of the VP2 beta barrel that do not appear in the 1133 reconstruction (Fig. 6), even though there were no deliberate differences in sample preparation or data processing. Note that in both reconstructions, only a small percentage of the boxed particles were rejected, which ensures that each of the two reconstructions was actually representative of its population. The density features that appear in the 1144 map (Fig. 6) include a broad triangular area with a pronounced longitudinal ridge (indicated by an arrow), extending across the top of the VP2 beta barrel, as well as a large tubular density feature (indicated by an arrowhead) that appears to cross the canyon bridging the points of the 5-fold mesa and the tip of the propeller (17). In fitting models to this map, we noted that although we could readily fit the VP1 and VP3 rigid bodies into the density, there was no single structure for VP2 that would explain the density surrounding the propeller. The model that we settled on placed the top surface of the VP2 barrel in a position that turned out to be essentially correct but left the EF loop of VP2 protruding through the contour surface, as well as leaving an outward-projecting blade of unfilled density, corresponding to the longitudinal ridge (Fig. 6, rightmost panels). This density feature was clearly real, icosahedrally reinforced, and strong. In the original low-resolution study, it was impossible to decide whether VP2 was present in multiple orientations or did not actually behave as a rigid body. Now, with higher-resolution information available, it is clear that most of the VP2 beta barrel is rigid but that numerous polypeptides around the propeller tip become disordered, while other polypeptides (including the GH loop of VP1) rearrange to account for the previously unfilled low-resolution density features.
Although our newest model for the 135S structure accounts for some of the extra density in the 1144 reconstruction, much of the density is still unexplained. In particular, we have no good way to model the tubular density feature that appears to span the canyon in lower-resolution maps (Fig. 6, arrowhead) and which we had previously ascribed incorrectly to a helix corresponding to residues 45 to 54 from the N terminus of VP1. In both 1144 and the present reconstruction, and with the beta barrels placed correctly, the top of VP2 provides the only strong density connection between the quasi-3-fold hole and the “tubular” region. In contrast, the postulated helix assignment would have approached the tube area from the opposite direction.
Uncoating of picornavirions may involve a succession of stepwise changes.
The fact that the extra density was observed in one 135S reconstruction (EMDB 1144) and not in another (EMDB 1133) lends support to the idea that those two previous 135S reconstructions may have sampled different stages along the uncoating pathway. We hypothesize that the EMDB 1144 reconstruction, with extra density atop VP2 (Fig. 6, arrow), is more similar to the population of 135S particles in our current higher-resolution reconstruction, where new density was seen for the reoriented GH loop of VP1. In contrast, the EMDB 1133 reconstruction, which lacks that extra density, may represent an earlier stage of the uncoating pathway that is closer to that of the 135S-like particles of CAV16 (19). Thus, in CAV16, virus expansion caused structural changes in the quasi-3-fold hole but not on the top of the VP2 beta barrel. Correspondingly, more of the polypeptide remained ordered and in a virion-like conformation, including most of the GH loop of VP1 and all of the EF loop of VP2 (Fig. 7). These observations support the idea that uncoating may be a multistep process, with each 135S structure sampling a different stage along a continuum of states.
FIG 7.

The conformational changes seen in the expanded structures of other picornaviruses (such as the CAV16 135S particle [19]) (B) are less extensive than the ones seen in poliovirus 135S particles (A). Main chain traces from crystal structures of mature virions are shown for capsid proteins VP1, VP2, and VP3. The locations of polypeptide segments that become shifted, rearranged significantly, or disordered are colored magenta, cyan, and orange, respectively. The greater extent of changes in poliovirus 135S particles (A) might be the result of inherent lesser stability or simply greater heterogeneity in cryo-EM preparations, versus the preparations of expanded forms of other viruses that have been crystallized (15, 16, 19). In either case, our hypothesis is that much of the poliovirus population must be relatively further along the uncoating pathway. Panel A is Fig. 3B, repeated here for clarity.
A situation similar to that was previously demonstrated for cryo-EM structures of 80S particles (18), where a “later” population of particles (which had been incubated longer) had tended to develop large, visible density features on the inner surface of the capsid, near the 2-fold axis. These features were considerably smaller in the “earlier” population, where the features had less time to develop. We suspect that, in a similar way, the extra density features seen at low contour level, both in the current higher-resolution 135S map and in the previous EMDB 1144 reconstruction, may indicate that those images were derived from a later, and perhaps more conformationally heterogeneous, population of virus particles.
Taken together, the maps and the comparisons with the CAV16 135S particle suggest that the rearrangement of 135S particles is likely to be a multistep process, sampled at different points along the uncoating trajectory by different virus preparations and by different picornavirus structures. In that context, the partial ordering of the GH loop of VP1 and the development of additional unexplained density on the top of the VP2 beta barrel place the present poliovirus reconstruction further along the trajectory than the states that the other picornavirus structures represent. It is not yet clear whether this is due to inherent differences in the type of virus or to the use of cryo-EM rather than crystallography.
Polypeptide segments that rearrange are displayed at high radius, in a large circle around the canyon.
Among the polypeptides that become disordered or rearranged in the 135S transition of poliovirus, there are a number of loops that are normally exposed on the outer surface. This is consistent with the observations (i) that the capsids of 135S particles are immunologically distinct from those of the mature 160S virions and from the 73S native-antigenic empty capsids (33) and (ii) that the regions of the outer surface that exhibit the greatest structural variability, when picornavirus structures are compared, are exposed loops that include some of the most important immunogenic sites (43, 44).
Additionally, many of the polypeptide segments that moved or became disordered (Fig. 3B) were those responsible for stabilizing the mature 160S particle, frequently via the binding of loops and N- and C-terminal extensions to the inner and outer surfaces of neighboring proteins. Importantly, when the sites of rearrangement are displayed on the outer surface of the icosahedrally symmetric capsid, a striking pattern is observed (Fig. 8). The components that rearrange are displayed, for the most part, at high radius and form a large circle that extends longitudinally over the top of each VP2 beta barrel, across the top of VP3, and into the next quasi-3-fold axis. Notably, the changed areas tend to exclude the canyon and involve only minimal changes to the 5-fold mesa.
FIG 8.
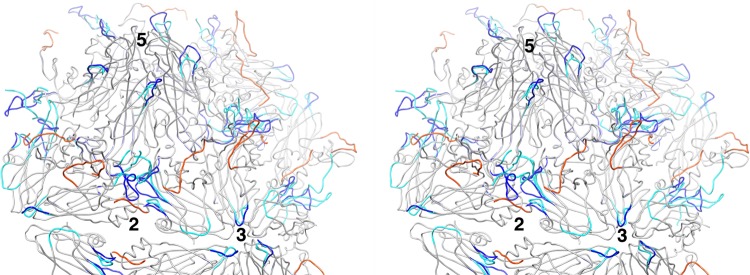
Stereodiagram shows the location on the poliovirus surface of polypeptides that become disordered or rearranged during the 160S-to-135S expansion. A portion of the atomic model is shown. The parts of the structure that remain unchanged and are common in 160S and 135S particles are colored gray. Polypeptide segments in the 160S conformation that become disordered in the 135S particles are colored in orange, and those rearranged are shown in dark blue. Rearranged segments, as seen in 135S particles, are cyan. Observe that extensive changes occur in a continuous ring of altered polypeptides that surrounds the canyon, while the mesa shows few such changes. Rearranged polypeptides, displayed at high radius atop the VP2 beta barrel, may be poised for interaction with membranes and/or formation of the quasi-3-fold umbilicus (21) that is implicated in RNA transfer. Numbers indicate the positions of icosahedral symmetry axes.
A model for membrane attachment, internalization, and genome translocation.
To develop an updated model for the sequence of polypeptide rearrangements that occur during poliovirus uncoating, we have assumed that the known native and expanded structures of poliovirus, poliovirus complexes (with receptor, Fabs, or membrane), and other picornaviruses are each representative of specific states along a common trajectory (Fig. 9). Along the trajectory, virus expands, RNA content is diminished, and polypeptide order tends to decrease, except when nonicosahedral structures for membrane interaction and RNase-protected RNA transfer are being built.
FIG 9.
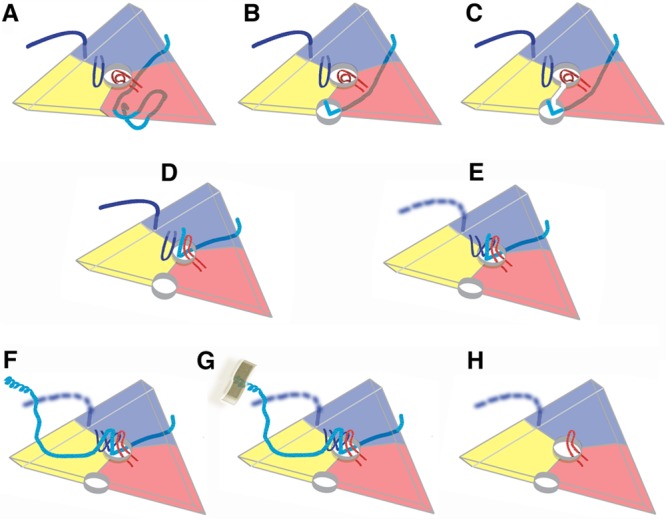
A proposed sequence of polypeptide rearrangements during poliovirus uncoating is obtained by placing known picornavirus structures along a timeline. This simplified diagram represents a 5-3-3 icosahedral triangle, viewed from the outside of the virion, with the beta barrels of VP1, VP2, and VP3 shown in blue, yellow, and red, respectively. (A) In mature 160S virions, no hole is evident at the 2-fold axis, as capsid protein subunits fit together closely. The C terminus and GH loop of VP1 (both dark blue) bind atop VP2, on either side of the propeller. The quasi-3-fold hole (center) is blocked by the coiled GH loop of VP3 (red). The N terminus of VP1 (cyan) belongs to the symmetry-related VP1 whose beta barrel (not shown) is located to the right of the 5-3-3 triangle and binds to the inner surface of the capsid (2, 40). (B) In “breathing” expanded virions, subunits move apart reversibly. The N-terminal extensions of VP1 are temporarily exposed through the resulting 2-fold holes, where Fabs can trap them (39). (C) We surmise that the N-terminal extension of VP1 can shift from the 2-fold hole to the quasi-3-fold hole when the “gate” between the holes is open transiently. (D) The GH loop of VP3 (red) is rearranged to form a radially oriented hairpin. This opens the quasi-3-fold hole and, as seen in the structure reported here and in the coxsackievirus A16 135S structure (19), helps to form a binding site for the exposed N-terminal extension of VP1 to lock into place. At this 135S stage, RNA is still present, but VP4 has been released. (E) In poliovirus 135S particles, both the C terminus and the tips of the GH loop of VP1 (dark blue) become disordered. However, there is evidence in the density that at least some of the copies of the VP1 GH loop rearrange and bind to other rearranged polypeptides in the quasi-3-fold area, including the N terminus of VP1 and the exposed GH loop of VP3. This completely blocks the hole and may provide a nucleus for building asymmetric structures later on. (F) The long N terminus of VP1 migrates across the top of VP2. Its amphipathic helices, displayed at high radius at the propeller tip, are not well ordered and visible but are detectable using specific Fabs and V8-protease difference maps (23). This diagram represents the structure in most of the 60 icosahedrally related copies. (G) On the cell surface, one or two of the membrane-facing copies of the VP1 N terminus will bind to membrane. In our proposal, this tethering serves as a trigger for the asymmetric self-assembly of a large umbilical connection between virus and membrane, as previously visualized by Strauss et al. (21). The connector is believed to be involved in RNase-protected RNA transfer and most likely includes contributions from VP4 and various disordered N- and C-terminal extensions. (H) After some or all RNA exits, the expanded 80S empty capsid remains, showing 2-fold and quasi-3-fold holes. The GH loop of VP3 remains rearranged. In the absence of RNA and membranes, the N terminus of VP1 becomes disordered and retracted into the capsid interior (15, 16, 23). In the EV71 and HRV2 80S particles, the C terminus and GH loop of VP1 show a greater degree of order than they do in poliovirus (15, 16).
Cryo-EM reconstructions have been determined for various states of the poliovirus capsid bound to the Fabs of antibodies that are directed against residues 20 to 40 of VP1 (45) and residues 39 to 55 of VP1 (46). These structures have demonstrated that the N-terminal extension of VP1 is externalized at the 2-fold axes in “breathing” particles (39) and shifts to the tips of the propellers in the 135S particles (23). These observations can be considered together with the observations from expanded virus structures concerning the site of egress of the externalized N-terminal extension of VP1.
Based on these data, we propose the following model for the events leading to internalization of the virus and translocation of its genome across the endosomal membrane and into the cytoplasm. We propose that soon after the virus attaches to receptor, several of the N-terminal extensions of VP1 (each up to 70 residues in length) will exit reversibly at the 2-fold axes (similar to the action in breathing virus). Due to receptor binding, one or more of the extensions will become exposed on the side of the virus that faces the cell membrane, close enough for their amphipathic helices to insert into the membrane. These copies will serve as anchors that keep the virus attached to the membrane after the virus has detached from the receptor during the 160S-to-135S transition. During this transition, VP4 and the other N-terminal extensions of VP1 will become externalized, and the N-terminal extensions of VP1 will pass through the two-loop gates that separate the holes at the 2-fold and quasi-3-fold axes, in a gear-shift-like movement. Once the N-terminal extension has transferred into the quasi-3-fold hole, it will be clamped securely into place by its contacts with the polypeptide chains that surround it (including the loops of the gate and the rearranged GH loops of VP3 and VP1), thereby making the formation of the 135S particle irreversible.
Interactions of the VP1 N termini will also direct their terminal amphipathic helices to bind at high radius at the tips of the VP2 propellers, where they contribute to the circle of rearranged polypeptides. We propose that the display at high radius facilitates the insertion of multiple copies of the helices into the membrane early in infection. This may induce a curvature in the membrane that ultimately triggers endocytosis, perhaps explaining the observation that the endocytosis of poliovirus (14) and of CB3 (47) can occur only after the 135S particle has been formed.
Our model suggests that several of the quasi-3-fold holes will include a three-component structure, formed on the outer surface of the virus by the exiting N terminus of VP1 and by the GH loops of VP1 and VP3. This three-component structure lines what will become the site of RNA egress from the capsid shell. Based on its location, one copy of this complex should participate in forming the base of the quasi-3-fold umbilicus that is observed in the RNA translocation complex (21) and should help to recruit additional components to build the remainder of the umbilical connector in a well-defined way. These components could include VP4 and disordered N- and C-terminal polypeptides from the high-radius ring. The resulting umbilical structures may help to catalyze the unwinding of RNA secondary structures at physiological temperature, which is essential for permitting one end of the ssRNA genome to exit through the portal in the capsid. We propose that the same umbilical structure is also responsible for protecting the genome from RNase during RNA translocation (Gropelli et al., submitted). This permits the RNA to safely and efficiently traverse the large distance observed between the virus and the endosomal membrane (21) and to pass through the membrane and into the cytoplasm, which is sufficient for infection.
ACKNOWLEDGMENTS
This work was supported by the grant NIH AI020566 (to J.M.H.). Electron microscopy data collection and processing were performed at the National Resource for Automated Molecular Microscopy, which is supported by NIH grant P41GM103310.
We thank David Stuart for sharing data prior to publication.
Footnotes
Published ahead of print 20 November 2013
REFERENCES
- 1.Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr. Top. Microbiol. Immunol. 343:43–89. 10.1007/82_2010_37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogle JM, Chow M, Filman DJ. 1985. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229:1358–1365. 10.1126/science.2994218 [DOI] [PubMed] [Google Scholar]
- 3.Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 56:677–702. 10.1146/annurev.micro.56.012302.160757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendelsohn CL, Wimmer E, Racaniello VR. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855–865. 10.1016/0092-8674(89)90690-9 [DOI] [PubMed] [Google Scholar]
- 5.Tsang SK, McDermott BM, Racaniello VR, Hogle JM. 2001. Kinetic analysis of the effect of poliovirus receptor on viral uncoating: the receptor as a catalyst. J. Virol. 75:4984–4989. 10.1128/JVI.75.11.4984-4989.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Sena J, Mandel B. 1977. Studies on the in vitro uncoating of poliovirus. II. Characteristics of the membrane-modified particle. Virology 78:554–566 [DOI] [PubMed] [Google Scholar]
- 7.Fenwick ML, Cooper PD. 1962. Early interactions between poliovirus and ERK cells. Some observations on the nature and significance of the rejected particles. Virology 18:212–223 [DOI] [PubMed] [Google Scholar]
- 8.Curry S, Chow M, Hogle JM. 1996. The poliovirus 135S particle is infectious. J. Virol. 70:7125–7131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fricks CE, Hogle JM. 1990. Cell-induced conformational change of poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow M, Newman JFE, Filman D, Hogle JM, Rowlands DJ, Brown F. 1987. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature 327:482–486. 10.1038/327482a0 [DOI] [PubMed] [Google Scholar]
- 11.Tuthill TJ, Bubeck D, Rowlands DJ, Hogle JM. 2006. Characterization of early steps in the poliovirus infection process: receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J. Virol. 80:172–180. 10.1128/JVI.80.1.172-180.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danthi P, Tosteson M, Li QH, Chow M. 2003. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J. Virol. 77:5266–5274. 10.1128/JVI.77.9.5266-5274.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tosteson MT, Wang H, Naumov A, Chow M. 2004. Poliovirus binding to its receptor in lipid bilayers results in particle-specific, temperature-sensitive channels. J. Gen. Virol. 85:1581–1589. 10.1099/vir.0.19745-0 [DOI] [PubMed] [Google Scholar]
- 14.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol. 5:e183. 10.1371/journal.pbio.0050183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garriga D, Pickl-Herk A, Luque D, Wruss J, Caston JR, Blaas D, Verdaguer N. 2012. Insights into minor group rhinovirus uncoating: the X-ray structure of the HRV2 empty capsid. PLoS Pathog. 8:e1002473. 10.1371/journal.ppat.1002473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 19:424–429. 10.1038/nsmb.2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bubeck D, Filman DJ, Cheng N, Steven AC, Hogle JM, Belnap DM. 2005. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 79:7745–7755. 10.1128/JVI.79.12.7745-7755.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy HC, Bostina M, Filman DJ, Hogle JM. 2010. Catching a virus in the act of RNA release: a novel poliovirus uncoating intermediate characterized by cryo-electron microscopy. J. Virol. 84:4426–4441. 10.1128/JVI.02393-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren J, Wang X, Hu Z, Gao Q, Sun Y, Li X, Porta C, Walter TS, Gilbert RJ, Zhao Y, Axford D, Williams M, McAuley K, Rowlands DJ, Yin W, Wang J, Stuart DI, Rao Z, Fry EE. 2013. Picornavirus uncoating intermediate captured in atomic detail. Nat. Commun. 4:1929. 10.1038/ncomms2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bostina M, Levy H, Filman DJ, Hogle JM. 2011. Poliovirus RNA is released from the capsid near a twofold symmetry axis. J. Virol. 85:776–783. 10.1128/JVI.00531-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss M, Levy H, Bostina M, Filman DJ, Hogle JM. 2013. RNA transfer from poliovirus 135S particles across membranes is mediated by long umbilical connectors. J. Virol. 87:3903–3914. 10.1128/JVI.03209-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 74:1342–1354. 10.1128/JVI.74.3.1342-1354.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J, Cheng N, Chow M, Filman DJ, Steven AC, Hogle JM, Belnap DM. 2011. An externalized polypeptide partitions between two distinct sites on genome-released poliovirus particles. J. Virol. 85:9974–9983. 10.1128/JVI.05013-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, Jensen GJ. 2009. Fully automated, sequential tilt-series acquisition with Leginon. J. Struct. Biol. 167:11–18. 10.1016/j.jsb.2009.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mallick SP, Carragher B, Potter CS, Kriegman DJ. 2005. ACE: automated CTF estimation. Ultramicroscopy 104:8–29. 10.1016/j.ultramic.2005.02.004 [DOI] [PubMed] [Google Scholar]
- 26.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142:334–347. 10.1016/S1047-8477(03)00069-8 [DOI] [PubMed] [Google Scholar]
- 27.Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. 2009. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166:95–102. 10.1016/j.jsb.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roseman AM. 2004. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145:91–99. 10.1016/j.jsb.2003.11.007 [DOI] [PubMed] [Google Scholar]
- 29.Grigorieff N. 2007. FREALIGN: high-resolution refinement of single particle structures. J. Struct. Biol. 157:117–125. 10.1016/j.jsb.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 30.van Heel M, Schatz M. 2005. Fourier shell correlation threshold criteria. J. Struct. Biol. 151:250–262. 10.1016/j.jsb.2005.05.009 [DOI] [PubMed] [Google Scholar]
- 31.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333:721–745. 10.1016/j.jmb.2003.07.013 [DOI] [PubMed] [Google Scholar]
- 32.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157:3–18. 10.1016/j.jsb.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 33.Basavappa R, Syed R, Flore O, Icenogle JP, Filman DJ, Hogle JM. 1994. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9Å resolution. Protein Sci. 3:1651–1669. 10.1002/pro.5560031005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 35.Collaborative Computational Project, Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760–763. 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W. 1976. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 72:922–923 [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 38.Hewat EA, Neumann E, Blaas D. 2002. The concerted conformational changes during human rhinovirus 2 uncoating. Mol. Cell 10:317–326. 10.1016/S1097-2765(02)00603-2 [DOI] [PubMed] [Google Scholar]
- 39.Lin J, Lee LY, Roivainen M, Filman DJ, Hogle JM, Belnap DM. 2012. Structure of the Fab-labeled “breathing” state of native poliovirus. J. Virol. 86:5959–5962. 10.1128/JVI.05990-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filman DJ, Syed R, Chow M, Macadam AJ, Minor PD, Hogle JM. 1989. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 8:1567–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hogle JM. 2012. A 3D framework for understanding enterovirus 71. Nat. Struct. Mol. Biol. 19:367–368. 10.1038/nsmb.2276 [DOI] [PubMed] [Google Scholar]
- 42.Baker TS, Cheng RH. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J. Struct. Biol. 116:120–130. 10.1006/jsbi.1996.0020 [DOI] [PubMed] [Google Scholar]
- 43.Minor PD. 1986. Antigenic structure of poliovirus. Microbiol. Sci. 3:141–144 [PubMed] [Google Scholar]
- 44.Page GS, Mosser AG, Hogle JM, Filman DJ, Rueckert RR, Chow M. 1988. Three-dimensional structure of the poliovirus serotype 1 neutralizing determinants. J. Virol. 62:1781–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chow M, Yabrov R, Bittle J, Hogle JM, Baltimore D. 1985. Synthetic peptides from four separate regions of the poliovirus capsid protein VP1 induce neutralizing antibodies. Proc. Natl. Acad. Sci. U. S. A. 82:910–914. 10.1073/pnas.82.3.910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roivainen M, Piraninen L, Rysa T, Narvanen A, Hovi T. 1993. An immunodominant N-terminal region of VP1 protein of poliovirion that is buried in crystal structure can be exposed in solution. Virology 195:762–765 [DOI] [PubMed] [Google Scholar]
- 47.Coyne CB, Shen L, Turner JR, Bergelson JM. 2007. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2:181–192. 10.1016/j.chom.2007.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]