

Abstract
Influenza viruses are a major public health threat worldwide, and options for antiviral therapy are limited by the emergence of drug-resistant virus strains. The influenza virus glycoprotein hemagglutinin (HA) plays critical roles in the early stage of virus infection, including receptor binding and membrane fusion, making it a potential target for the development of anti-influenza drugs. Using pseudotype virus-based high-throughput screens, we have identified several new small molecules capable of inhibiting influenza virus entry. We prioritized two novel inhibitors, MBX2329 and MBX2546, with aminoalkyl phenol ether and sulfonamide scaffolds, respectively, that specifically inhibit HA-mediated viral entry. The two compounds (i) are potent (50% inhibitory concentration [IC50] of 0.3 to 5.9 μM); (ii) are selective (50% cytotoxicity concentration [CC50] of >100 μM), with selectivity index (SI) values of >20 to 200 for different influenza virus strains; (iii) inhibit a wide spectrum of influenza A viruses, which includes the 2009 pandemic influenza virus A/H1N1/2009, highly pathogenic avian influenza (HPAI) virus A/H5N1, and oseltamivir-resistant A/H1N1 strains; (iv) exhibit large volumes of synergy with oseltamivir (36 and 331 μM2 % at 95% confidence); and (v) have chemically tractable structures. Mechanism-of-action studies suggest that both MBX2329 and MBX2546 bind to HA in a nonoverlapping manner. Additional results from HA-mediated hemolysis of chicken red blood cells (cRBCs), competition assays with monoclonal antibody (MAb) C179, and mutational analysis suggest that the compounds bind in the stem region of the HA trimer and inhibit HA-mediated fusion. Therefore, MBX2329 and MBX2546 represent new starting points for chemical optimization and have the potential to provide valuable future therapeutic options and research tools to study the HA-mediated entry process.
INTRODUCTION
Influenza A viruses are members of the Orthomyxoviridae family of negative-strand RNA viruses and are the etiological agents of influenza, a contagious, acute, and febrile respiratory disease (1–3). Influenza A viruses are responsible for seasonal epidemics and have caused three pandemics in the 20th century (1918, 1957, and 1968) as well as the 2009 H1N1 pandemic. Wild aquatic birds are the natural reservoir of influenza A viruses. Pandemics occur when a “new influenza virus” emerges, due to antigenic “shift,” to which the human population is immunologically naive (1–6). Vaccination is the primary strategy for the prevention and control of seasonal influenza. Both inactivated vaccines and the live attenuated vaccine are effective in preventing influenza A virus infections (5); however, vaccine efficacy can vary depending upon several factors, including the genetic relatedness among viruses used for the vaccine and circulating strains.
Currently, there are two classes of FDA-approved drugs for treatment or chemoprophylaxis of influenza: the matrix protein 2 (M2) inhibitors amantadine and rimantadine and the neuraminidase (NA) inhibitors (NAIs) oseltamivir and zanamivir (7–9). The M2 inhibitors block the activity of the ion channel formed by M2 and thereby prevent the release of viral genome segments into the cytoplasm (7–9). However, M2 ion channel inhibitors are limited in their clinical utility for treatment of influenza A viruses since all currently circulating influenza A virus strains (including the 2009 pandemic A/H1N1 and the seasonal A/H3N2 strains) are resistant to M2 inhibitors (10). NAIs, such as oseltamivir, bind the NA protein and inhibit its enzymatic activity, thereby inhibiting the efficient release of newly synthesized viruses from infected cells (2, 11). Recently, however, significant levels of oseltamivir-resistant seasonal influenza A (H1) viruses have also been encountered; the resistance has been associated with a single-amino-acid change in the viral neuraminidase (H274Y) (12). In 2008, the CDC reported that the majority of seasonal H1N1 isolates were oseltamivir resistant (13–16). Although the majority of 2009 H1N1 pandemic isolates remain susceptible to NAIs, the possibility that the H274Y mutation could appear in the pandemic H1N1 strain and result in an oseltamivir-resistant virus is a major health concern (17–19). Therefore, new antiviral strategies, including a focus on different viral targets, cellular factors, or immune-modulating drugs, are needed. For example, T-705 (favipiravir), an inhibitor of influenza virus RNA polymerase, has been identified as a potent anti-influenza agent from in vitro and in vivo preclinical studies, with activity against a range of influenza virus strains, including H5N1 (20, 21).
Viral entry is the first essential step in the viral replication cycle; consequently, blocking of viral entry into the target cell will lead to suppression of viral infectivity and is an attractive antiviral strategy. In addition, the acute nature of influenza virus infection and the accompanying cytokine storm (22) make blocking of the viral entry process particularly attractive, since it inhibits influenza-induced cytokine pulmonary immune pathology. The influenza virus protein hemagglutinin (HA) plays a key role in viral entry. HA is responsible for binding of the virus to host cells and subsequent membrane fusion within the late endosome (11). It also plays an important role in host immune responses by harboring the major antigenic sites responsible for the generation of neutralizing antibodies. Mature HA is a homotrimer, and each monomer is composed of two disulfide-linked polypeptides, HA1 and HA2, generated by proteolytic cleavage of the primary translation product HA0 and modification by multiple glycosylations. Most of the HA1 subunit forms the head region of HA, while the HA2 subunit is the primary component of the stem region. Following binding, the virus is internalized by endocytosis. Within the low-pH (5.0 to 5.5) environment of the endosome, HA undergoes conformational rearrangements, resulting in exposure of the fusion peptide, which inserts into the endosomal target membrane of the host cell. After fusion of the viral and endosomal membranes, the viral ribonucleoproteins (RNPs) are released into the cytosol and transported into the nucleus, where replication occurs (2, 11, 23).
To identify potential influenza virus entry inhibitors, we used a high-throughput screening (HTS) assay to screen a chemical compound library composed of over 100,000 unique small molecules; two novel compounds, MBX2329 and MBX2546, were selected based on their potency, and their mechanism of action was characterized. These entry inhibitors may be developed as viable broad-spectrum therapeutic options for influenza virus infection.
MATERIALS AND METHODS
Cell lines, viruses, and plasmids.
293T, A549, Madin-Darby canine kidney (MDCK), and BHK cell lines were procured from the ATCC. The cell lines were maintained in either Dulbecco's modified Eagle's medium (DMEM) or minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (50 units/ml).
Influenza A virus subtypes A/PR/8/34 (H1N1), A/California/10/2009 (H1N1), A/Florida/21/2008 (H1N1-H275Y) (oseltamivir resistant [osc-res]), A/Hong Kong (H5N1), A/Texas/12/2007(H3N2), and B/Florida/4/2006 were used in this study. Virus stocks were prepared either in 10-day-old embryonated eggs or in a MDCK cell culture.
The work with the HPAI virus A/Hong Kong (H5N1) strain was carried out at the BSL-3+ select-agent facility at Utah State University. The BSL-3+ select-agent facility has been inspected and approved by USDA/CDC. Possession and use of select agents are monitored by the responsible university biosafety officer. Select agents are governed by standard laboratory procedures approved by the Institutional Biosafety Committee, and access to the BSL-3+ laboratory and select agents is restricted to employees that have FBI and USDA clearance and appropriate BSL-3+ training.
Plasmid vectors expressing the hemagglutinin (HA) (H5) gene from a highly pathogenic H5N1 avian influenza virus (Goose/Qinghai/59/05) (24) and the envelope proteins of Ebola virus (EBOV) Zaire (EBOV-GP) (GenBank accession number L11365) (25), vesicular stomatitis virus (VSV-G) (26), and Lassa virus (LASV) (27, 28) were described previously.
Mutagenesis of the H5 HA gene.
The alanine substitution mutations were introduced between HA1 residues 35 and 64 and between HA2 residues 1 and 20 and 75 to 127, since the variations in these regions appeared to be the basis for dividing HA into two groups, as reported previously (29). All alanine substitution mutations in the H5 HA gene were generated by site-directed mutagenesis with the Quick-Change mutagenesis kit (Agilent Technologies, Santa Clara, CA), according to the supplier's protocols. All mutations were confirmed by DNA sequencing of flanking regions. The full length of the HA gene was confirmed by DNA sequencing for those mutants that displayed defective phenotypes.
Compound library.
The chemical library screened represents a broad and well-balanced collection of ∼106,000 compounds accumulated over a number of years from a variety of distinct sources. The library achieves broad coverage across property space involving the following chemical descriptors: calculated logarithm of the n-octanol/water partition coefficient (ClogP), polar surface area (PSA), globularity (three-dimensional [3D] structure), and molecular mass (average, 394.5 Da) (29).
Pseudotyping.
Avian influenza pseudotype viruses expressing H5 HA [HIV/HA(H5)] were produced by cotransfecting 12 μg of HA(H5) with 12 μg of a replication-defective HIV vector (pNL4-3-Luc-R−E−) into 293T cells (90% confluent) in 10-cm plates with Lipofectamine 2000 (Invitrogen), as previously described (30). The supernatants containing the pseudotype viruses were collected at 48 h posttransfection, pooled, clarified from floating cells and cell debris by low-speed centrifugation, and filtered through a 0.45-μm-pore-size filter (Nalgene). The culture supernatants containing HIV/HA(H5) were either used immediately or flash frozen in aliquots and stored at −80°C until use. Pseudotype viruses bearing VSV envelope protein (HIV/VSV-G), LASV envelope protein (HIV/LASV-GP), and EBOV envelope protein (HIV/EBOV-GP) were also prepared in a similar fashion, using the same Env-deficient HIV vector, as previously described (31).
High-throughput screening of chemical libraries.
High-throughput screening (HTS) of chemical libraries using pseudotype virus was performed essentially as described previously in 96-well plates (31). The final concentration of the test compound was 25 μM, while the final concentration of dimethyl sulfoxide (DMSO) was maintained at 1% in all wells. Low-passage A549 cell monolayers were infected with 100 μl of p24-normalized HIV/HA(H5) containing 8 μg/ml Polybrene in the presence of test compounds. After 2 h, the inoculum was removed, the cells were washed briefly, fresh medium was added, and the plates were incubated for 72 h. Prior to each screening, each batch of the viral preparation was titrated to determine the amount of virus required to infect the target cells so that a relatively high luciferase activity could be recorded while still remaining in a linear response range (105 to 106 relative luciferase units [RLU]). Infection was quantified from the luciferase activity of the infected A549 cells by using the Britelite Plus assay system (PerkinElmer) in a Wallac EnVision 2102 multilabel reader (PerkinElmer, MA). In the absence of compound, the assay showed an average luciferase signal of 1.2 × 106 ± 0.6 × 106 RLU, a signal-to-background ratio of >103, and a calculated screening window coefficient (Z′ factor) (32) of >0.5 ± 0.2. The luciferase signal standard error was ±50%, and >90% inhibition of luciferase activity at a concentration of 25 μM was used as the criterion for designating a compound a “hit.” Test compounds were in DMSO solutions with 80 compounds per plate. Controls were also included in each plate: 8 wells for 0% inhibition (DMSO only) (maximum signal = positive control) and 8 wells for 100% inhibition [e.g., bafilomycin for HIV/HA(H5)] (minimum signal = negative control). Percent inhibition was calculated as follows: 100 × {1 − [RLU in the presence of compound − RLU of the negative control]/[RLU of the positive control (without any inhibitor) − RLU of the negative control]}.
Antiviral assay.
The antiviral activity of the influenza virus inhibitors was evaluated by two methods: an enzyme-linked immunosorbent assay (ELISA) and plaque reduction. For the ELISA, MDCK cells were plated to be 80% confluent on 96-well plates. Cells were then infected with influenza virus at a virus dilution that would result in a 90% cytopathic effect (CPE) after 3 days (multiplicity of infection [MOI] of 1) from a seed stock of 1 × 106 PFU/ml in the presence of compounds. Viral replication was determined by measuring the levels of influenza virus nucleoprotein (NP) using an influenza A virus NCP ELISA kit (Photometric; Virusys Corp.). Percent protection was calculated as [1 − (mean OD450compound − mean OD450medium)/(mean OD450DMSO − mean OD450medium)] × 100%, where mean OD450compound, mean OD450medium, and mean OD450DMSO are the absorbance (optical density at 450 nm [OD450]) of compound- and virus-containing samples, the absorbance of no-virus control samples, and the absorbance of DMSO- and virus-containing samples, respectively. The 50% inhibitory concentration (IC50) is defined as the compound concentration that generates a 50% reduction in the NP concentration.
The plaque reduction assay was performed according to standard protocols (33, 34). Confluent monolayers of MDCK cells were infected with 100 PFU of virus (alone or in the presence of compounds). After 1 h of virus adsorption at 37°C, viral inoculums was replaced by a 50:50 mix of 1% Seaplaque agarose (in deionized water) and 2× MEM containing 2.5 μg/ml trypsin and compounds at desired concentrations. Plaques were counted after 3 to 5 days of incubation at 37°C by visual examination. The IC50 was calculated as the compound concentration required to reduce virus plaque numbers by 50%.
Hemagglutination assay.
The hemagglutination assay was performed as previously described (35). Four milliliters of influenza A/PR/8/34 (H1N1) virus particles was concentrated over a 30% sucrose cushion. The samples were spun at 55,000 rpm for 1 h in a Beckman SW55 rotor at 4°C. Virus pellets were resuspended in 400 μl of phosphate-buffered saline (PBS). Twofold serial dilutions were mixed with an equal volume of a 0.5% animal erythrocyte suspension (chicken red blood cells [cRBCs]; Lampire Biological Laboratories) in a U-bottomed 96-well plate in the presence or absence of the compounds (final concentration, 10 μM). In addition, we used antiserum to influenza virus H1 HA (ATCC V-301-501-552) as a control. HA titers were recorded after 2 h of incubation at 4°C. Hemagglutination assay experiments were repeated at least three times.
Hemolysis inhibition assay.
The procedure to determine the inhibitory effects of the compounds on virus-induced hemolysis at low pH was slightly adapted from a procedure described previously by Luo et al. (36). cRBCs were washed twice with PBS and resuspended to make a 2% (vol/vol) suspension in PBS, which was stored at 4°C until use. One hundred microliters of compound diluted in PBS was mixed with an equal volume of the influenza virus A/PR/8/34 (H1N1) strain (108 PFU/ml) in a 96-well plate. After incubating the virus-compound mixture at room temperature for 30 min, 200 μl of 2% chicken erythrocytes prewarmed at 37°C was added. The mixture was incubated at 37°C for another 30 min. To trigger hemolysis, 100 μl of sodium acetate (0.5 M; pH 5.2) was added and mixed well with the erythrocyte suspension. The mixture was incubated at 37°C for 30 min for HA acidification and hemolysis. To separate nonlysed erythrocytes, plates were centrifuged at the end of incubation at 1,200 rpm for 6 min. Three hundred microliters of supernatant was transferred to another flat-bottom 96-well plate. The OD540 was read on a microtiter plate reader. Percent protection was calculated as [1 − (mean OD540compound − mean OD540PBS)/(mean OD540DMSO − mean OD540PBS)] × 100%, where mean OD540compound, mean OD540PBS, and mean OD540DMSO are the absorbance of compound- and virus-containing samples, the absorbance of no-virus control samples, and the absorbance of DMSO- and virus-containing samples, respectively. IC50 is defined as the compound concentration that generates 50% maximal protection.
Cytotoxicity assay.
Cell viability was measured to determine the effect of compounds on cellular functions so that a 50% cytotoxicity concentration (CC50) could be calculated; the ratio of this value to the IC50 is referred to as the selectivity index (SI) (CC50/IC50). Cell viability was determined by measuring the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level in the cell lysate by using an AlphaScreen SureFire GAPDH assay kit (PerkinElmer).
NMR spectroscopy.
Recombinant H5 HA and NA were obtained from BEI Resources. Monoclonal antibody (MAb) C179 was obtained from TaKaRa. All nuclear magnetic resonance (NMR) experiments were performed on a Bruker 900-MHz spectrometer equipped with a cryogenic probe. WaterLOGSY (water ligand observed via gradient spectroscopy) experiments were performed as previously described (37, 38), with a relaxation delay of 2.5 s, a mixing time of 2 s, and 1,024 scans (or 5,120 scans for the competition experiments). Saturation transfer difference (STD) experiments were performed as previously described (39–41), with a relaxation delay of 2.5 s, a saturation time of 1 s, and 256 scans with “on” resonance saturation at −1.5 ppm and “off” resonance saturation at 30 ppm. Nuclear Overhauser effect (NOE) spectroscopy (NOESY) experiments were performed as previously described (42), with a mixing time of 1 s and 24 scans. Spectra were processed by NMRPipe with a relaxation delay and 5-Hz line broadening and analyzed by NMRDraw (43).
Synergy studies.
The combined efficacy of the compounds used together with either oseltamivir carboxylate or amantadine was determined by methods reported previously (44–46). Briefly, combinations of drug dilutions were added to 96-well plates containing monolayers of MDCK cells, which were subsequently infected at a low multiplicity of infection. Cell viability was determined at 72 h following infection by using Neutral Red. The efficacy of the two individual agents was used to calculate theoretical additive interactions. The calculated additive effects were then subtracted from the observed effects to reveal regions where greater-than-expected inhibition was observed. Synergy plots represent the percent inhibition above or below the expected inhibition (calculated additive inhibition) and are presented as the means of four replicates at a level of 95% confidence, which eliminates insignificant deviations from the additive surface. The volume under the surface was calculated and used as a quantitative measure of synergy. Synergy volumes of >100 μM2% are generally considered to be significant (47, 48). Potential cytotoxicity was evaluated concurrently in uninfected MDCK cells with the same exposure to compounds to ensure that the antiviral effects and synergistic interactions were specific.
RESULTS
Identification of new influenza virus entry inhibitors.
A chemically diverse small molecule library (106,440 compounds) was screened for inhibitors of the HA-mediated entry process by using influenza pseudotype virus [HIV/HA(H5)] according to previously described methods (31, 35, 49). HIV/HA(H5) contains an “HIV core” with a luciferase reporter gene (with deletions in the HIV genome making it replication deficient) and an influenza virus H5 HA “envelope” protein (24). The inhibitory effect of the small molecules on virus entry was quantified by measuring the decrease of the mean luciferase activity in the presence of the test compounds. A total of 2,038 compounds (2.03% hit rate) produced ≥90% reduction of the mean luciferase activity of the positive control [uninhibited HIV/HA(H5)] at a test concentration of 25 μM. These “hit” compounds included both influenza virus entry inhibitors and off-target hit compounds, which include (i) inhibitors of HIV replication, (ii) inhibitors of luciferase enzyme activity, and (iii) cytotoxic compounds. To specifically identify inhibitors of HA-mediated HIV/HA(H5) entry, we counterscreened the hit compounds with HIV/VSV-G. HIV/VSV-G has the same HIV backbone but expresses a different envelop protein, VSV-G. The counterscreen with HIV/VSV-G eliminated 1,897 compounds as off-target hits (Table 1). From the remaining 141 hits, 36 compounds (0.035% hit rate) were chosen based on their low cytotoxicity (CC50 of ≥25 μM) and high antiviral selectivity index (SI) (CC50/IC90 of ≥10). Two compounds, MBX2329 (molecular mass, 359.4 Da), an aminoalkyl phenol ether, and MBX2546 (molecular mass, 394.4 Da), a sulfonamide, were prioritized based on their inhibitory potency (IC90 of ≤10 μM) and chemical tractability (synthetically accessible, stable, drug-like structures) (Fig. 1A). MBX2329 and MBX2546 displayed concentration-dependent inhibitory activities, with HIV/HA(H5) displaying IC90 values of 8.6 μM and 5.7 μM, respectively (Fig. 1B and C).
TABLE 1.
Results of the screening of compound libraries
| No. of compounds screened | No. (%) of: |
|||
|---|---|---|---|---|
| Primary hits [≥90% inhibition of HIV/HA(H5) in HTS]a | Primary hits displaying ≥25% inhibition of HIV/VSV-Ga,b | Specific hits [primary hits inhibiting HIV/HA(H5) only] | Specific hits displaying SI of >10 and CC50 of >25 μMc | |
| 106,440 | 2,038 (2.03) | 1,897 (1.84) | 141 (0.14) | 36 (0.035) |
HIV/HA(H5) and HIV/VSV-G were generated by transfection of 293T cells with pNL4-3-Luc-R−E− as the HIV-l expression vector and with HA(H5) and VSV-G, respectively.
A total of 1,897 primary hits inhibited HIV/VSV-G by >90% at 25 μM, whereas the RLU values of the controls varied by ≤20%.
CC50 values were determined by measuring the endogenous GAPDH in cellular lysates by using the AlphaScreen SureFire GAPDH assay (PerkinElmer).
FIG 1.
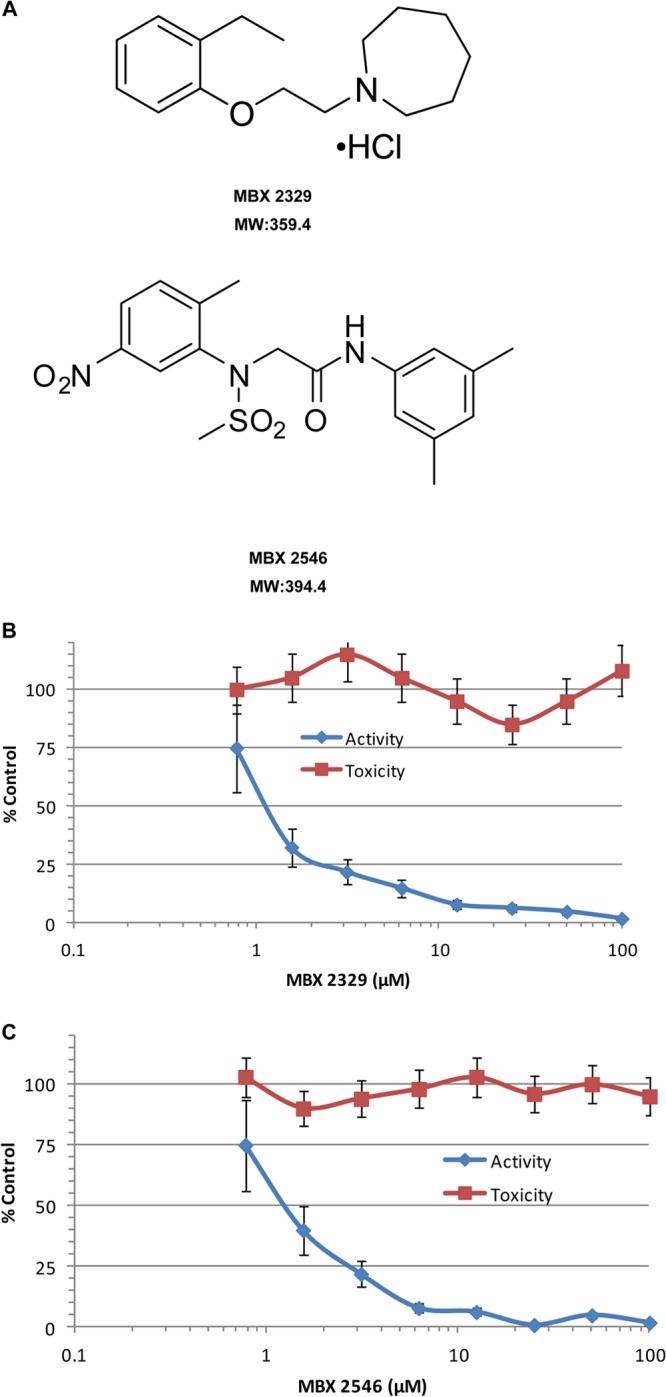
Inhibition of HIV/HA(H5) by MBX2329 and MBX2546. (A) Structures of MBX2329 and MBX2546. Chemical structures and molecular weights (MW) (in thousands) of MBX2329 and MBX2546 are shown. MBX2329 and MBX2546, with aminoalkyl phenol ether and aminoacetamide sulfonamide scaffolds, respectively, were prioritized based on potency and selectivity against HIV/HA(H5). (B and C) The inhibitory effect of compounds MBX2329 (B) and MBX2546 (C) on HIV/HA(H5) infectivity was investigated by using A549 cells as described in Materials and Methods. Three independent experiments were performed to determine the effect of the compounds.
To evaluate the spectrum of antiviral activity of MBX2329 and MBX2546, both compounds were investigated for inhibition of entry of Lassa virus (LASV) and Ebola virus (EBOV), both of which also bear type 1 envelope proteins similar to HA. The pseudotype platform was used because it provided a direct comparison of the activities of MBX2329 and MBX2546 against HIV/LASV-GP, HIV/EBOV-GP, and HIV/HA(H5). Compounds MBX2329 and MBX2546 displayed little inhibitory activity against HIV/LASV-GP (IC90 of ∼100 μM), HIV/EBOV-GP (IC90 of >100 μM), or HIV/VSV-G (IC90 of 85 to >100 μM) (Table 2), suggesting that they specifically inhibit the entry of influenza viruses.
TABLE 2.
Specificities of MBX2329 and MBX2546

Pseudotype virus was generated by cotransfection of 293T cells with pNL4-3-Luc-R−E− and the respective envelope glycoproteins. The pseudotype virus host was 293T cells; cytotoxicity (CC50) for both compounds in 293T cells was >100 μM.
MBX2329 and MBX2546 are potent subtype-specific inhibitors.
As shown in Table 3 and Fig. 2, MBX2329 inhibited influenza A H1N1 virus strains A/PR/8/34 (H1N1) (Fig. 2A), A/Florida/21/2008 (H1N1-H275Y) (oseltamivir-resistant strain) (Fig. 2C), A/Washington/10/2008 (H1N1), and A/California/10/2009 (H1N1) (2009 pandemic strain) with IC50s of between 0.29 μM and 0.53 μM. Similarly, MBX2546 inhibited influenza A H1N1 virus strains A/PR/8/34 (H1N1) and A/Florida/21/2008 (H1N1-H275Y) (oseltamivir-resistant strain) with IC50s of 0.3 μM and 5.8 μM (Fig. 2B and D and Table 3), respectively. MBX2546 also inhibited other H1N1 strains, including A/California/10/2009/H1N1 (2009 pandemic strain), with IC50s of between 0.55 μM and 1.5 μM. Both MBX2329 and MBX2546 inhibited HPAI H5N1 virus strain A/Hong Kong/H5N1 with IC50s of 5.9 μM and 3.6 μM, respectively (Table 3).
TABLE 3.
Activities of MBX2329 and MBX2546 against different influenza virus subtypesa
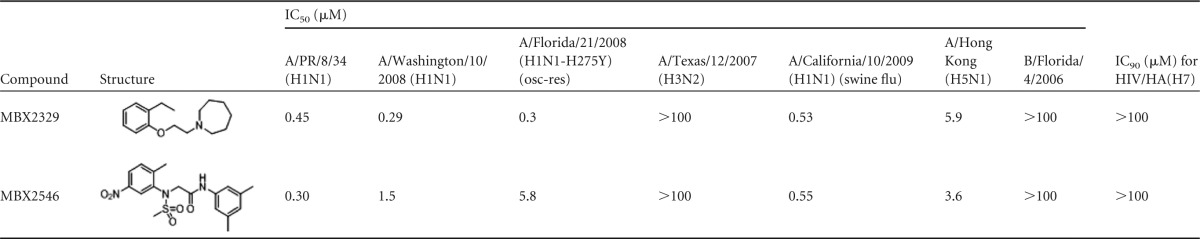
The infectious-virus host was MDCK cells; the pseudotype virus host was A549 cells. The cytotoxicity (CC50) of the two compounds versus MDCK and A549 cells was >100 μM in one or more of the four testing laboratories.
FIG 2.
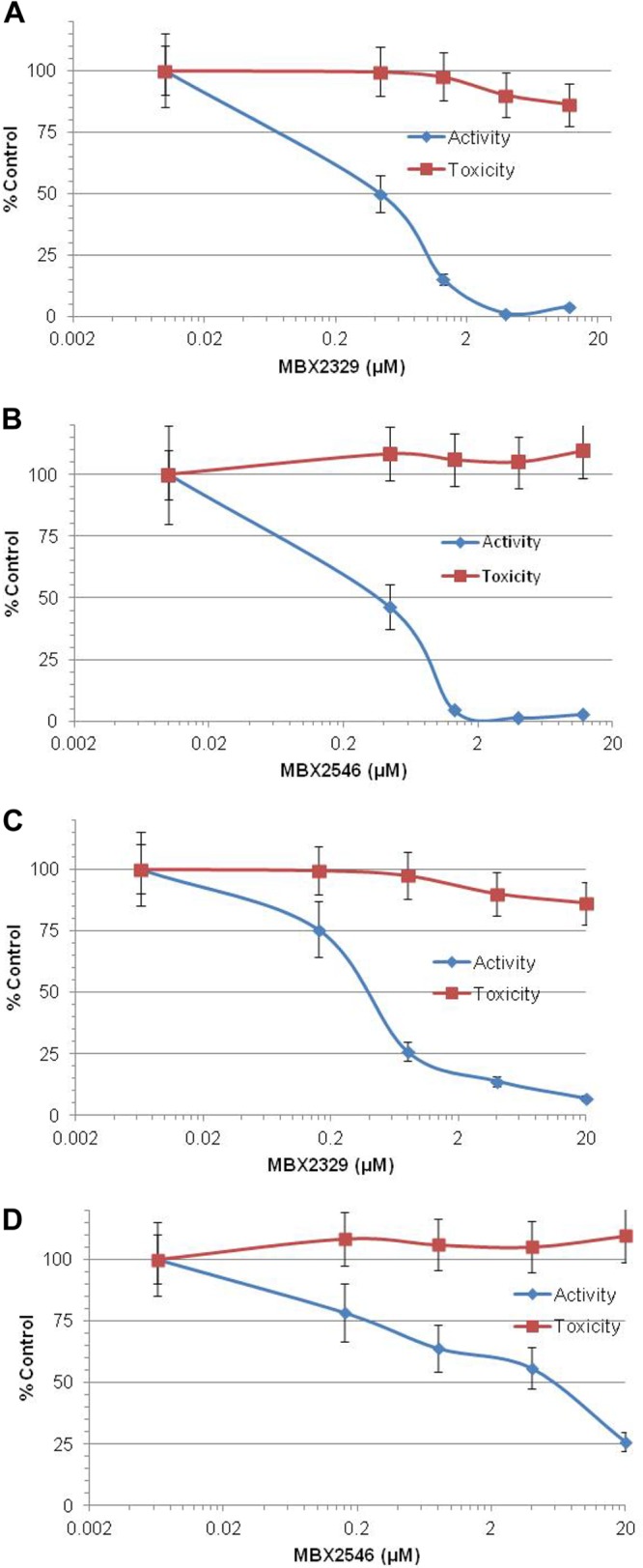
Influenza virus-inhibitory spectrum of MBX2329 and MBX2546. Shown are dose-dependent inhibitory effects of MBX2329 (A) and MBX2546 (B) on infection by influenza A virus vaccine strain A/PR/8/34 (H1N1) and of MBX2329 (C) and MBX2546 (D) on infection by oseltamivir-resistant influenza A (H1N1) virus strain A/Florida/21/2008 (H1N1-H275Y) on MDCK cells. An MOI of 1.0 was used for infection. Three independent experiments were performed to determine the effect of compounds.
MBX2329 and MBX2546 displayed significantly less activity against influenza A H3N2 virus strain A/Texas/12/2007 (H3N2) and influenza B virus strain B/Florida/4/2006 (Table 3). They did not inhibit influenza A H3N2 virus strains A/Perth/16/2009 (H3N2), A/Victoria/3/75 (H3N2), A/Panama/2007/99 (H3N2), A/Sydney/05/97 (H3N2), A/California/7/04 (H3N2), and A/Wyoming/03/2003 (H3N2) at the maximum concentration tested (data not shown). MBX2329 and MBX2546 also did not inhibit HIV/HA(H7) infection at 100 μM (Table 3). The 50% cytotoxic concentrations (CC50s) for both compounds against MDCK cells were >100 μM. In summary, MBX2329 and MBX2546 specifically inhibit influenza viruses of group 1 HA (H1 and H5 subtypes) and not group 2 HA (H3 and H7 subtypes).
MBX2329 and MBX2546 bind to the group 1 HA-specific conformational epitope in the HA stem region.
Specific inhibition of influenza viruses with group 1 HA by MBX2329 and MBX2546 suggests that they interact with group 1 HA. To verify that HA is the target of the compounds, we investigated the binding of MBX2329 and MBX2546 to recombinant H5 HA (a group 1 HA) using WaterLOGSY NMR spectroscopy, which is designed to detect binding of small molecules to high-molecular-mass targets (37). Recombinant NA was used as the specificity control. In Fig. 3A, the top spectrum corresponds to the 1D NMR spectrum of the downfield region of MBX2329, with the aromatic resonances of the compound being denoted by red arrows. The second spectrum corresponds to the WaterLOGSY spectrum observed for MBX2329 in the absence of HA (i.e., a control experiment), and the third spectrum corresponds to the WaterLOGSY spectrum observed for MBX2329 in the presence of H5 HA. The relatively strong positively phased resonances of MBX2329 in the presence of H5 HA indicate that it is binding to HA. Conversely, the absence of the signals in the fourth spectrum, which corresponds to the WaterLOGSY experiment in the presence of NA, suggests that MBX2329 is not binding to NA.
FIG 3.

MBX2329 and MBX2546 bind to the conserved conformational epitope in the HA stem region. (A and B) Detection of binding of MBX2329 (A) and MBX2546 (B) to recombinant H5 HA by WaterLOGSY NMR. Red arrows represent the aromatic peaks of MBX2329, while green arrows represent the aromatic peaks of MBX2546. For this set of experiments, the conditions were 50 μM MBX2329 or MBX2546 with or without 0.2 μM HA in 20 mM PBS (pH 7.2) at 25°C, using a 900-MHz spectrometer with a mixing time of 2 s. (C and D) Competition WaterLOGSY of MBX2329 (C) and MBX2546 (D) binding to H5 HA in the absence (top spectrum) and in the presence (bottom spectrum) of MAb C179. For this set of experiments, the conditions were 20 μM MBX2329 or 10 μM MBX2546 with or without 0.2 μM HA and with or without 0.4 μM C179 in 50 mM PBS (pH 7.3) at 25°C, using a 900-MHz spectrometer with a mixing time of 2 s.
We characterized the binding properties of MBX2546 in a similar manner (Fig. 3B). The top spectrum corresponds to the 1D NMR spectrum of the downfield region of MBX2546, with the aromatic resonances of the compound being denoted by green arrows. The second spectrum corresponds to the WaterLOGSY spectrum observed for MBX2546 in the absence of HA (i.e., a control experiment), and the third spectrum corresponds to the WaterLOGSY spectrum observed for MBX2546 in the presence of H5 HA. In the presence of H5 HA, the increased intensity of the positively phased resonances of MBX2546 clearly indicates that it is binding to HA. In contrast, in the presence of NA (Fig. 3B, bottom spectrum), the relatively weak signals, which resemble the no-protein control, suggest that MBX2546 is not binding to NA.
To further determine the region of HA binding of MBX2329 and MBX2546, a WaterLOGSY-based competition assay was performed with a monoclonal antibody (C179). C179 was previously shown to bind to a group 1 HA conformational epitope in the stem region formed by amino acid positions 318 to 322 in the HA1 subunit and by amino acid positions 47 to 58 in HA2 (50). Overlapping of binding sites would lead to a decrease in the signal of the compounds in the binding assay. As shown in Fig. 3C, the addition of an equivalent amount of MAb C179, with respect to HA, significantly decreased the binding of MBX2329. The average reduction of MBX2329 resonance intensities was 52% ± 11% (n = 8; P = 0.001), suggesting that the antibody is displacing the compound. Likewise, an equivalent amount of MAb C179 similarly decreased the binding of MBX2546 to H5 HA observed by WaterLOGSY (Fig. 3D), with an average reduction of the MBX2546 resonance intensities of 81% ± 5% (n = 8; P = 0.0001). The results once again suggest that the antibody is also displacing MBX2546. These results are consistent with the notion that both inhibitors inhibit influenza A viruses with group 1 HA by binding to a group 1 HA-specific conformational epitope in the stem region of HA.
We next investigated whether MBX2329 and MBX2546 bind to overlapping sites on HA using a WaterLOGSY-based competition assay. In this experiment, equal amounts of compound were added in the WaterLOGSY experiment, with the notion that overlapping binding sites would lead to a decrease in the signal of one or both compounds. Based on the absence of a change in signal intensity (Fig. 4A), the compounds appear to be able to simultaneously bind to HA, suggesting that they bind to different regions of HA. Furthermore, we used a NOESY experiment to examine the relative proximity of the compound binding sites. In this experiment, the absence of intermolecular NOEs in the presence of H5 HA (Fig. 4B) further suggests that MBX2329 and MBX2546 bind to sites that are >6 Å apart. We further examined the binding epitopes of MBX2329 and MBX2546 for H5 HA by an STD NMR experiment, which identifies 1H in closest proximity to the protein surface (39, 41). The relative STD for the interaction between MBX2329 and H5 HA is shown in Fig. 4C. In this representation, the red spheres and numbers represent the relative STD signal. The contact is relatively uniform, with the aromatic ring of MBX2329 in closest contact. Similarly, Fig. 4D shows the relative STD for the interaction between MBX2546 and H5 HA. In this representation, the green spheres and numbers represent the relative STD signal (the gray spheres represent STD signals that are too weak to be quantified). In this case, the contact is relatively nonuniform, with the most important contacts being in the center of the molecule.
FIG 4.

MBX2329 and MBX2546 bind to nonoverlapping sites on HA. (A) Competition assay performed between MBX2329 and MBX2546 for binding to influenza virus H5 HA by WaterLOGSY NMR. The top spectrum represents the sum of the WaterLOGSY of MBX2329 and H5 HA with the WaterLOGSY of MBX2546 and H5 HA. The bottom spectrum represents the WaterLOGSY signal in the presence of MBX2329, MBX2546, and H5 HA. The experimental conditions were 50 μM MBX2329 and/or 20 μM MBX2546 plus 0.2 μM HA in 20 mM PBS (pH 7.2) at 25°C, using a 900-MHz spectrometer with a mixing time of 1 s. (B) Two-dimensional NOESY of MBX2329 and MBX2546 in the presence of H5 HA. The lines represent the intramolecular NOE connectivities. The experimental conditions were 50 μM MBX2329, 50 μM MBX2546, and 0.2 μM HA in 20 mM PBS (pH 7.2) at 25°C, using a 900-MHz spectrometer with a mixing time of 1 s. (C and D) STD NMR studies to characterize the binding epitopes of MBX2329 (C) and MBX2546 (D) for H5 HA. The numbers and sizes of the spheres represent the intensity of the STD signal, which is related to the distance to the protein surface. For this set of experiments, the conditions were 50 μM MBX2329/MBX2546 with or without 0.2 μM HA in 20 mM PBS (pH 7.2) at 25°C, using a 900-MHz spectrometer with a saturation time of 1 s. (E) Effect of MBX2329 and MBX2546 on the infectivity of HIV/HA(H5) mutants. A549 cells were infected with mutant or wild-type HIV/HA(H5) with either MBX2329 or MBX2546 at 6.25 μM. Inhibition of infection by HIV/HA(H5) (or its mutants) was detected as a reduced luciferase signal. Each mutant was tested in triplicate; error bars indicate standard deviations. WT, wild type.
The conformational antigenic epitope (amino acid positions 318 to 322 in the HA1 subunit and positions 47 to 58 in HA2) recognized by MAb C179 is in the stem region and is specific for influenza virus with group 1 HA. To further explore the potential roles of amino acids in the group 1 HA-specific region in binding to MBX2329 and MBX2546, we generated HIV/HA(H5) carrying single-amino-acid substitutions by alanine scanning mutagenesis and examined the sensitivity of these mutants to MBX2329 and MBX2546. As shown in Fig. 4E, at 6.25 μM, the HIV/HA(H5) mutants bearing either a K51A mutation in HA1 or a G16A mutation in HA2 were less susceptible to inhibition by MBX2329, suggesting that MBX2329 interacts with amino acid residues K51 in HA1 and G16 in HA2. Interestingly, none of the mutants was resistant to MBX2546 at the same concentration, further suggesting that they bind at different sites near the conformational epitope recognized by C179. Therefore, taken together, we conclude that (i) both MBX2329 and MBX2546 bind to HA near the group 1 HA-specific conformational epitope in the HA stem region and (ii) the binding sites are not overlapping in the stem region of trimeric HA. The results are consistent with the notion that these inhibitors block HA-mediated membrane fusion (see below).
MBX2329 and MBX2546 inhibit HA-mediated fusion.
Based on the results described above, both MBX2329 and MBX2546 bind to the HA stem region, which is the target for group 1 HA-specific antibodies that disrupt the HA-mediated membrane fusion process (50–55). To investigate the role of these inhibitors in HA-mediated fusion, we performed hemagglutination and hemolysis assays.
The hemagglutination assay was performed to determine whether MBX2329 and MBX2546 prevented the binding of virus with cell surface receptors containing sialic acid (SA). Briefly, 10-fold serial dilutions of concentrated influenza A/PR/8/34 (H1N1) virus particles were mixed with chicken red blood cells (cRBCs) using virus-only wells without an inhibitor as the positive control and wells lacking both virus and the inhibitor as the negative control. In addition, we used the antiserum to influenza virus H1 HA (ATCC V-301-501-552) at two different dilutions (1:10 and 1:25) as controls. The results of the hemagglutination experiment in the presence of either MBX2329 or MBX2546 were similar to those of the positive control (without any compound), as shown in Fig. 5A. Therefore, the results suggest that neither MBX2329 nor MBX2546 inhibits the binding of influenza virus to cRBCs.
FIG 5.

MBX2329 and MBX2546 inhibit fusion of HA to the endosomal membrane. (A) Hemagglutination assay using cRBCs. Following incubation of MBX2546, MBX2329, or antiserum to influenza virus H1 HA (ATCC V-301-501-552) with antiserum to influenza A virus strain A/PR8/34 (H1N1), a suspension of freshly prepared cRBCs was added, and inhibition of hemagglutination was investigated as described in Materials and Methods. Three independent experiments were performed to determine the effect of compounds. (B) Inhibition of HA-mediated hemolysis of cRBCs by MBX2546 and MBX2329. Following incubation of influenza A virus A/PR/8/34 (H1N1) with MBX2546 or MBX2329, a suspension of freshly prepared chicken erythrocytes was added, and the degree of hemolysis (y axis) was detected at pH 5.2 by measuring the OD540 as described in Materials and Methods. Wells lacking the virus were used as controls to determine the effect of compounds on cRBCs.
The hemolysis assay was performed by using influenza A virus A/PR/8/34 (H1N1) to determine the effect of MBX2329 and MBX2546 on fusion. To trigger hemolysis, the virus-cell suspension was acidified (pH 5.2) briefly to initiate HA conformational changes that lyse cRBCs to release hemoglobin. Wells lacking the virus were used as controls to determine the effect of compounds on cRBCs. Both MBX2329 and MBX2546 inhibited acid-induced hemolysis in a dose-dependent manner (Fig. 5B), with IC50s of 2.1 μM and 1.56 μM for MBX2329 and MBX2546, respectively. Therefore, taken together, the results indicate that MBX2329 and MBX2546 inhibit fusion of the virus with the endosomal membrane. Here we also used antiserum to influenza virus H1 HA as a control (data not shown).
MBX2329 and MBX2546 exhibit strong synergy with oseltamivir.
Finally, the synergistic efficacy of MBX2329 and MBX2546 in combination with oseltamivir or amantadine was evaluated by using influenza A(H1N1) virus strain A/California/10/2009 according to previously described methods (45, 46). Both MBX2329 and MBX2546 in combination with oseltamivir displayed marked synergistic inhibition of influenza virus infection (331 ± 112 μM2% for MBX2329 and 36 ± 2.8 μM2% for MBX2546), as shown by plotting the concentration versus synergy (Fig. 6A and B). The large volumes of synergy produced by the combination were statistically significant, as indicated by the values at the 95% confidence level (Table 4). Cytotoxicity was also evaluated with the same experimental design used for the combined efficacy study to evaluate synergistic cytotoxicity. These studies used the same MDCK cell monolayers and the same drug exposures as those used for the antiviral studies. At the concentrations used in these studies, no significant cytotoxicity was observed with either oseltamivir, MBX2329, or MBX2546 (data not shown). Strikingly, the observed synergy was restricted to the combination of the HA inhibitors and oseltamivir; no significant synergy was observed with the combination of HA inhibitors and amantadine (Table 4).
FIG 6.
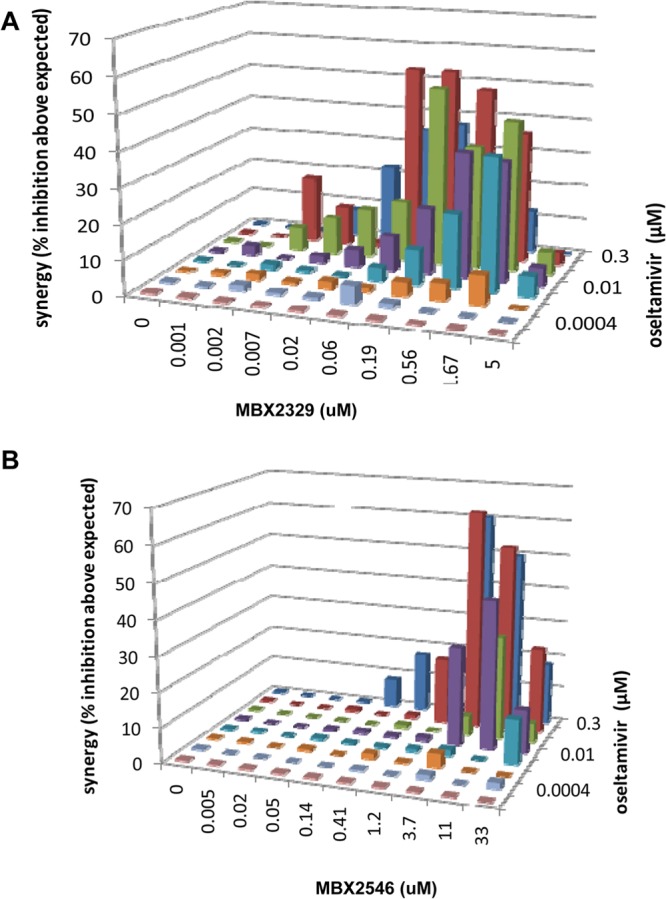
Synergy of double combinations of oseltamivir and MBX2329 or MBX2546 carboxylate against 2009 pandemic influenza A virus strain A/California/10/2009 (H1N1). Shown are plots of synergy volume for double combinations of oseltamivir (at 0.1 μg/ml) and MBX2329 (A) or MBX2546 (B) against 2009 pandemic influenza A virus strain A/California/10/2009 (H1N1), as determined by a neutral red assay. Synergy volume data are presented as the means of 18 replicates from 6 experiments with 95% confidence intervals.
TABLE 4.
Combined efficacy of HA inhibitors with oseltamivir or amantadine against influenza virus A/California/10/2009
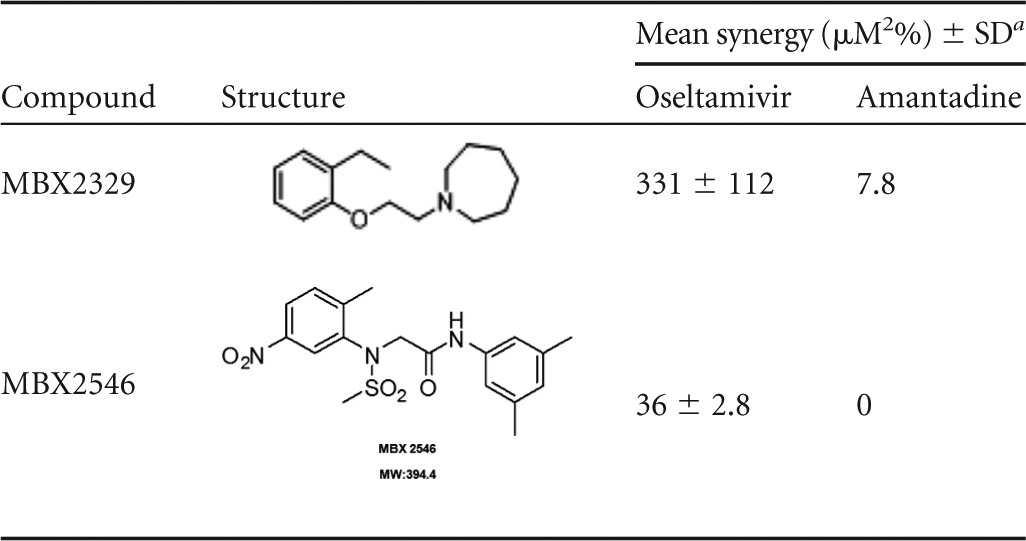
The efficacy of the compounds was determined in combination with oseltamivir or amantadine. Volumes shown represent a minimal estimate of synergy at the 95% confidence level.
DISCUSSION
HA plays an important role in the early stages of viral infection by facilitating influenza virus entry into host cells by controlling two critical aspects of entry: receptor binding and membrane fusion. In this study, we describe two small molecules, MBX2329 and MBX2546, with aminoalkyl phenol ether and aminoacetamide sulfonamide scaffolds, respectively, that inhibit multiple influenza A viruses, including the 2009 pandemic influenza virus A/H1N1, high pathogenic avian influenza (HPAI) virus A/H5N1, and oseltamivir-resistant A/H1N1 strains, in a potent (IC50 of 0.47 to 5.8 μM) and selective (CC50 of >100 μM) manner in vitro. Mechanistic studies indicate that these compounds bind to a conserved epitope in the HA stem region, which has been implicated in the HA-mediated membrane fusion process (Fig. 7). Furthermore, we have demonstrated that these inhibitors are highly synergistic with oseltamivir. Therefore, further optimization of these small molecule inhibitors as potential therapeutic agents, either individually or in combination with existing anti-influenza virus treatments, appears warranted.
FIG 7.
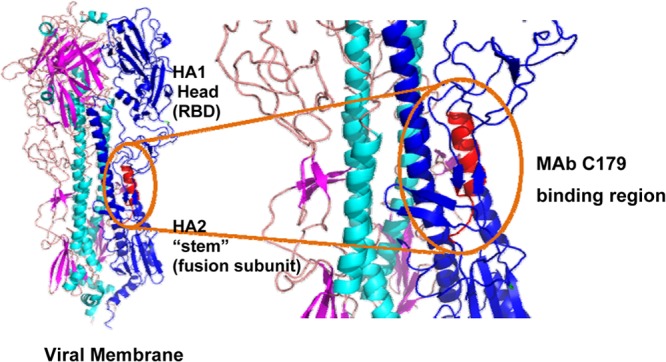
Putative MBX2329 and MBX2546 binding region in HA. HA is a trimer consisting of three identical copies of the HA protein (the three monomers are shown in blue). Each monomer contains two subunits, HA1 and HA2. HA1 is the receptor binding domain (RBD) and contains the sialic acid binding pocket. HA2 contains the membrane fusion machinery. MAb C179 blocks membrane fusion. MAb C179 binds the HA stem and interacts with residues from the N- and C-terminal regions of HA1 (residues 38, 40, 42, 291 to 293, and 318) and the N-terminal portion of HA2 (residues 18 to 21, 38, 41 to 43, 45, 46, 52, and 56), including helix A. The putative binding sites of MBX2329 and MBX2546 overlap this region.
MBX2329 and MBX2546 were selected as HA-specific entry inhibitors from a library of >100,000 small molecules because they are (i) potent (IC90 of 8.6 μM and 5.7 μM, respectively); (ii) selective (CC50 of >100 μM), yielding selectivity index (SI) values of >20 to 200; and (iii) chemically tractable (synthetically accessible, stable structures with drug-like properties) (Fig. 1A to C). Considering the overall similarity between class I envelope proteins, we tested the activity of the two hit compounds against a series of prototypic RNA viruses bearing class I fusion proteins and influenza virus strains from different subtypes. In contrast to the potent inhibition of HIV/HA(H5), MBX2329 and MBX2546 were inactive against HIV/LASV-GP, and HIV/EBOV-GP (Table 2), demonstrating the specificity of the two inhibitors for influenza virus. MBX2329 and MBX2546 strongly inhibited influenza viruses with H1 and H5 subtypes (Table 3). Both of these subtypes are members of group 1 HAs, one of the two groups of influenza A virus HA. Both groups have very similar overall structures, with HA1 forming a membrane-distal domain that contains the receptor binding subdomains and the HA2 polypeptide forming the fusion subdomain and the stem of the trimers (10). However, the rotation of the membrane-distal subdomains relative to the central stem varied between the different HAs, and the variation is the basis for dividing HA into two groups. The broad activity of MBX2329 and MBX2546 against influenza viruses with group 1 HA concomitant with the lack of inhibition of influenza A viruses with group 2 HA (H3 and H7 subtypes) provided the first clue that the compounds interact with the conserved conformational epitope in the stem region of group 1 HA that is involved in fusion.
Several lines of evidence confirmed that MBX2329 and MBX2546 bind to the group 1 HA-specific conserved epitope in the HA stem that is involved in fusion. First, the binding of MBX2329 and MBX2546 to H5 HA and the lack of apparent binding to N1 NA by WaterLOGSY NMR spectroscopy binding studies suggest that the compounds bind specifically to HA (Fig. 3A and B). Second, both MBX2329 and MBX2546 compete with MAb C179 (Fig. 3C). MAb C179 binds to the HA stem recognizing a unique group 1 HA epitope. The antibody interacts with residues from the N- and C-terminal regions of HA1 (residues 38, 40, 42, 291 to 293, and 318) and the N-terminal portion of HA2 (residues 18 to 21, 38, 41 to 43, 45, 46, 52, and 56), including helix A (56), as shown schematically in Fig. 7. Therefore, the competition suggests that both compounds bind to this conserved epitope. An alternative explanation may be that binding of MAb C179 could result in a small conformational change that displaces the compound regardless of whether it binds in the region of the epitope. However, the structure of C179 in complex with an H2 HA (belonging to group 1 HAs, like H5 HA) suggests that binding of C179 does not cause any conformational changes within HA (56). Therefore, the overlap between the compound and MAb C179 binding sites remains the most plausible explanation. Furthermore, the G16A mutation in HA2 rendered the HIV/HA(H5) viruses resistant to MBX2329. Amino acid residue G16 of HA2 is located very close to the conformational antigenic epitope for MAb C179. Third, the compounds have no effect on HA-mediated hemagglutination of cRBCs, suggesting that they do not inhibit binding of HA to SA residues (Fig. 5A). Strikingly, as shown in Fig. 5B, both MBX2329 and MBX2546 blocked hemolysis in a low-pH environment in a dose-dependent manner. Since virus absorption and HA conformational changes are two key events required for hemolysis, the results suggest that MBX2329 and MBX2546 interfere with the fusion step during viral entry. Interestingly, MAb C179 was also shown to have no effect on virus attachment (50) but acts by blocking membrane fusion. Therefore, these results support the hypothesis that both MBX2329 and MBX2546 bind in the highly conserved epitope of group 1 HA that is involved in fusion.
The known structural features of HA also support the conclusion that MBX2329 and MBX2546 inhibit HA-mediated fusion by binding to this conserved epitope of HA. Structural differences among HAs are located mainly in three domains. The first domain is the antigenic region on the surface of the HA1 globular heads. This domain is the most variable region and is primarily responsible for the antigenic differences among the HA subtypes, and binding to this domain does not affect virus entry. The second domain is the receptor binding domain that is involved in the interaction with cell surface SAs. Since MBX2329 and MBX2546 do not block receptor binding measured by the hemagglutination assay (Fig. 5A), they are unlikely to bind in the receptor binding domain. The third region is in the stem region near the hydrophobic pocket that contains the fusion peptide at the N terminus of HA2. In addition to C179, several novel group 1 HA-specific human antibodies, CR6261 and F10 (53–55, 57), have been discovered. These antibodies have very similar patterns of reactivity and neutralization when tested, and they compete with C179. Like C179, CR6261 and F10 exhibit broad activity against group 1 HAs, including the H1, H2, H5, and H9 subtypes. Crystal structures of C179 bound to H2 HA, CR6261 bound to H1 and H5 HA, and F10 bound to H5 HA revealed that these antibodies recognize conserved epitopes in the stem region of HA (51, 54). The epitope lies close to the virus membrane and consists of an α-helix from HA2 and adjacent loops derived from HA1 (Fig. 7). The binding of C179, CR6261, and F10 inhibits key conformational changes in HA that drive the fusion of the viral and endosomal membranes. Since both MBX2329 and MBX2546 compete with C179, we hypothesize that a similar interaction between the compounds and amino acid residues in this conserved epitope results in inhibition of key conformational changes in HA that drive fusion. Clearly, further structural studies are needed to investigate these possibilities.
Several small molecule inhibitors that block the HA-mediated fusion process have been identified (33, 34, 36, 58–60), and all of them display subtype-dependent activities. Importantly, both MBX2329 and MBX2546 exhibit increased potencies compared to previously described antivirals. Like MBX2329 and MBX2546, these inhibitors appear to bind directly to HA and block the HA conformational change. These inhibitors include BMY 27709 (36), 180299 (a podocarpic acid derivative) (61), tert-butyl hydroquinone (TBHQ) (60), a series of N-substituted piperidine derivatives (33), N-(1-thia-4-azaspiro[4.5]decan-4-yl)carboxamides (34), and stachyflin (62, 63). The N-(1-thia-4-azaspiro[4.5]decan-4-yl)carboxamide compounds and TBHQ target influenza A/H3 viruses, while 180299 displayed only strain-specific inhibition of influenza A/H1 viruses. Interestingly, the piperidines exhibited higher potency against influenza A/H1 viruses with mutated M2 than against wild-type virus, suggesting the possibility of a different mechanism of action, which would preclude them from being used clinically. Moreover, except for TBHQ, the binding sites of these inhibitors on HA are unknown, although drug resistance profiling and other studies suggest that these molecules bind to HA2 protein. These studies, together with the results described in this report, demonstrate the feasibility and potential of using small molecules as entry inhibitors to block infection by influenza viruses.
Interestingly, competition studies showed that MBX2329 and MBX2546 do not compete with each other (Fig. 4A) and appear to simultaneously bind to different regions of HA. These results were further supported by NOESY experiments, where the absence of signal in the NOESY experiment suggested that the binding sites for MBX2329 and MBX2546 on HA are >6 Å apart (Fig. 4B). It should be noted that while the absence of signals in the NOESY experiment does not prove that the compounds bind to distant sites, it is nevertheless consistent with this notion. This hypothesis is also supported by studies on the resistant mutants generated in the current study. HIV/HA(H5) mutants with the K51A mutation in HA1 or the G16A mutation in HA2 were resistant to MBX2329 but not to MBX2546. This result is again consistent with the postulate that they bind at different sites. The STD NMR data provide further input on their binding interaction. The most important interactions of MBX2329 with H5 HA in the bound state occur through the aromatic ring (Fig. 4C). The smaller interactions of the aliphatic ring imply that this moiety could be modified somewhat (Fig. 4C). Interestingly, the most important contacts of MBX2546 with H5 HA were with the anilide aromatic ring in the center of the molecule (Fig. 4D). The STD NMR data also suggest that the two methyl groups at the 3- and 5-positions of the second aromatic ring of MBX2546 are relatively distant from the HA(H5) protein surface and hence could be removed or modified to increase solubility or make new types of contacts.
The exact mechanism by which MBX2329/MBX2546 inhibits the low-pH-induced fusion of to the group 1 HAs with the endosomal membrane needs further investigation. Based on studies with MAb CR6261, most of the amino acid residues in this conserved epitope are solvent exposed in the prefusion state and accessible to antibodies. Interestingly many of the epitope amino acid residues are also accessible to interactions in the postfusion state due to functional constraints on the protein sequence. We propose that both MBX2329 and MBX2546 interact with this highly conserved epitope in both the prefusion and postfusion states and inhibit key conformational changes in HA that drive the fusion of the viral and endosomal membranes. This results in blocking of the viral RNA from the endosome, and presumably, the virus particle is degraded in the lysosome.
Another interesting aspect of the antiviral activity of MBX2329 and MBX2546 is the synergistic inhibition of viral replication in combination with oseltamivir (Fig. 5A and B). No significant synergy was observed with the combination of HA inhibitors and amantadine (Table 4). To be viable, influenza virus strains must exhibit a balance between the HA-mediated entry process (early step) and NA-mediated egress activity (late step). Therefore, observed synergy was likely due to the simultaneous disruption of NA-mediated egress activities by oseltamivir (late step) and HA-mediated entry activity by MBX2329/MBX2546 (early step). In contrast, both MBX2329/MBX2546 and amantadine act during the virus entry process and do not produce a synergistic increase in antiviral activity. We are aware that the 2009 pandemic influenza A virus strain A/California/10/2009 (H1N1) is resistant to the amantadines. The synergistic effect is the increase in activity above the theoretical additive interactions calculated from the concentration-response curves of the single agent. Therefore, it is possible to calculate the synergy between MBX2329/MBX2546 and amantadine, even though the strains are resistant to it. Moreover, previous reports (45) have shown that amantadine-resistant influenza virus displayed synergy when amantadine was used in combination with oseltamivir and ribavirin. Further characterization of their combined efficacy with oseltamivir will be evaluated with oseltamivir-resistant strains of H1N1 influenza virus, since these drug-resistant strains sometimes represent the majority of all H1N1 isolates circulating in the population. For full coverage of influenza virus strains in the clinic, it may be necessary to combine two or more different types of small molecule inhibitors or antibodies against HA for a full coverage of the anti-influenza spectrum for potential clinical use.
In summary, we have identified two novel influenza virus inhibitors, MBX2329 and MBX2546, that could serve as starting points for the development of a therapeutic agent and can also be used as chemical tools for exploring the molecular mechanism of the low-pH-induced HA conformational change.
ACKNOWLEDGMENTS
We thank Michael Farzan from Harvard Medical School for providing plasmid vectors expressing the hemagglutinin (H7) gene and the envelope proteins of Lassa virus.
This research was supported by DHHS/NIH grants 1R43AI072861-01A2, 3R43AI072861-01A2S1, 2R44AI072861-03A1, and 1R21AI101676-01.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Russell CJ, Webster RG. 2005. The genesis of a pandemic influenza virus. Cell 123:368–371. 10.1016/j.cell.2005.10.019 [DOI] [PubMed] [Google Scholar]
- 2.Tscherne DM, Garcia-Sastre A. 2011. Virulence determinants of pandemic influenza viruses. J. Clin. Invest. 121:6–13. 10.1172/JCI44947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yen HL, Webster RG. 2009. Pandemic influenza as a current threat. Curr. Top. Microbiol. Immunol. 333:3–24. 10.1007/978-3-540-92165-3_1 [DOI] [PubMed] [Google Scholar]
- 4.Brockwell-Staats C, Webster RG, Webby RJ. 2009. Diversity of influenza viruses in swine and the emergence of a novel human pandemic influenza A (H1N1). Influenza Other Respi. Viruses 3:207–213. 10.1111/j.1750-2659.2009.00096.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horimoto T, Kawaoka Y. 2009. Designing vaccines for pandemic influenza. Curr. Top. Microbiol. Immunol. 333:165–176. 10.1007/978-3-540-92165-3_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith GJ, Bahl J, Vijaykrishna D, Zhang J, Poon LL, Chen H, Webster RG, Peiris JS, Guan Y. 2009. Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 106:11709–11712. 10.1073/pnas.0904991106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das K, Aramini JM, Ma LC, Krug RM, Arnold E. 2010. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 17:530–538. 10.1038/nsmb.1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 5:1015–1025. 10.1038/nrd2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Clercq E, Neyts J. 2007. Avian influenza A (H5N1) infection: targets and strategies for chemotherapeutic intervention. Trends Pharmacol. Sci. 28:280–285. 10.1016/j.tips.2007.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Vries E, Stelma FF, Boucher CA. 2010. Emergence of a multidrug-resistant pandemic influenza A (H1N1) virus. N. Engl. J. Med. 363:1381–1382. 10.1056/NEJMc1003749 [DOI] [PubMed] [Google Scholar]
- 11.Skehel J. 2009. An overview of influenza haemagglutinin and neuraminidase. Biologicals 37:177–178. 10.1016/j.biologicals.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 12.Baz M, Abed Y, Simon P, Hamelin ME, Boivin G. 2010. Effect of the neuraminidase mutation H274Y conferring resistance to oseltamivir on the replicative capacity and virulence of old and recent human influenza A(H1N1) viruses. J. Infect. Dis. 201:740–745. 10.1086/650464 [DOI] [PubMed] [Google Scholar]
- 13.Lackenby A, Democratis J, Siqueira MM, Zambon MC. 2008. Rapid quantitation of neuraminidase inhibitor drug resistance in influenza virus quasispecies. Antivir. Ther. 13:809–820 http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18445375 [PubMed] [Google Scholar]
- 14.Lackenby A, Hungnes O, Dudman SG, Meijer A, Paget WJ, Hay AJ, Zambon MC. 2008. Emergence of resistance to oseltamivir among influenza A(H1N1) viruses in Europe. Euro Surveill. 13(5):pii=8026 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8026 [DOI] [PubMed] [Google Scholar]
- 15.Lackenby A, Thompson CI, Democratis J. 2008. The potential impact of neuraminidase inhibitor resistant influenza. Curr. Opin. Infect. Dis. 21:626–638. 10.1097/QCO.0b013e3283199797 [DOI] [PubMed] [Google Scholar]
- 16.Le QM, Kiso M, Someya K, Sakai YT, Nguyen TH, Nguyen KH, Pham ND, Ngyen HH, Yamada S, Muramoto Y, Horimoto T, Takada A, Goto H, Suzuki T, Suzuki Y, Kawaoka Y. 2005. Avian flu: isolation of drug-resistant H5N1 virus. Nature 437:1108. 10.1038/4371108a [DOI] [PubMed] [Google Scholar]
- 17.Chen LF, Dailey NJ, Rao AK, Fleischauer AT, Greenwald I, Deyde VM, Moore ZS, Anderson DJ, Duffy J, Gubareva LV, Sexton DJ, Fry AM, Srinivasan A, Wolfe CR. 2011. Cluster of oseltamivir-resistant 2009 pandemic influenza A (H1N1) virus infections on a hospital ward among immunocompromised patients—North Carolina, 2009. J. Infect. Dis. 203:838–846. 10.1093/infdis/jiq124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta T, McGrath E, Bheemreddy S, Salimnia H, Abdel-Haq N, Ang JY, Lum L, Chandrasekar P, Alangaden GJ. 2010. Detection of oseltamivir resistance during treatment of 2009 H1N1 influenza virus infection in immunocompromised patients: utility of cycle threshold values of qualitative real-time reverse transcriptase PCR. J. Clin. Microbiol. 48:4326–4328. 10.1128/JCM.01190-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheu TG, Deyde VM, Garten RJ, Klimov AI, Gubareva LV. 2010. Detection of antiviral resistance and genetic lineage markers in influenza B virus neuraminidase using pyrosequencing. Antiviral Res. 85:354–360. 10.1016/j.antiviral.2009.10.022 [DOI] [PubMed] [Google Scholar]
- 20.Moss RB, Davey RT, Steigbigel RT, Fang F. 2010. Targeting pandemic influenza: a primer on influenza antivirals and drug resistance. J. Antimicrob. Chemother. 65:1086–1093. 10.1093/jac/dkq100 [DOI] [PubMed] [Google Scholar]
- 21.Sambhara S, Poland GA. 2010. H5N1 avian influenza: preventive and therapeutic strategies against a pandemic. Annu. Rev. Med. 61:187–198. 10.1146/annurev.med.050908.132031 [DOI] [PubMed] [Google Scholar]
- 22.Oldstone MB, Teijaro JR, Walsh KB, Rosen H. 2013. Dissecting influenza virus pathogenesis uncovers a novel chemical approach to combat the infection. Virology 435:92–101. 10.1016/j.virol.2012.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. 10.1146/annurev.biochem.69.1.531 [DOI] [PubMed] [Google Scholar]
- 24.Guo Y, Rumschlag-Booms E, Wang J, Xiao H, Yu J, Wang J, Guo L, Gao GF, Cao Y, Caffrey M, Rong L. 2009. Analysis of hemagglutinin-mediated entry tropism of H5N1 avian influenza. Virol. J. 6:39. 10.1186/1743-422X-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manicassamy B, Wang J, Jiang H, Rong L. 2005. Comprehensive analysis of Ebola virus GP1 in viral entry. J. Virol. 79:4793–4805. 10.1128/JVI.79.8.4793-4805.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basu A, Kanda T, Beyene A, Saito K, Meyer K, Ray R. 2007. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 81:3933–3941. 10.1128/JVI.02622-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, Li W, Nagel J, Schmidt PJ, Nunberg JH, Andrews NC, Farzan M, Choe H. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96. 10.1038/nature05539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radoshitzky SR, Kuhn JH, Spiropoulou CF, Albariño CG, Nguyen DP, Salazar-Bravo J, Dorfman T, Lee AS, Wang E, Ross SR, Choe H, Farzan M. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc. Natl. Acad. Sci. U. S. A. 105:2664–2669. 10.1073/pnas.0709254105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC, de Jong JC, Kelso A, Klimov AI, Kageyama T, Komadina N, Lapedes AS, Lin YP, Mosterin A, Obuchi M, Odagiri T, Osterhaus AD, Rimmelzwaan GF, Shaw MW, Skepner E, Stohr K, Tashiro M, Fouchier RA, Smith DJ. 2008. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine 26(Suppl 4):D31–D34 . [DOI] [PubMed] [Google Scholar]
- 30.Rumschlag-Booms E, Guo Y, Wang J, Caffrey M, Rong L. 2009. Comparative analysis between a low pathogenic and a high pathogenic influenza H5 hemagglutinin in cell entry. Virol. J. 6:76. 10.1186/1743-422X-6-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, Butler MM, Peet NP, Majgier-Baranowska H, Williams JD, Patel I, Moir DT, Bavari S, Ray R, Farzan MR, Rong L, Bowlin TL. 2011. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 85:3106–3119. 10.1128/JVI.01456-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 33.Plotch SJ, O'Hara B, Morin J, Palant O, LaRocque J, Bloom JD, Lang SA, Jr, DiGrandi MJ, Bradley M, Nilakantan R, Gluzman Y. 1999. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J. Virol. 73:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanderlinden E, Goktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 84:4277–4288. 10.1128/JVI.02325-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Tscherne DM, McCullough C, Caffrey M, Garcia-Sastre A, Rong L. 2012. Residue Y161 of influenza virus hemagglutinin is involved in viral recognition of sialylated complexes from different hosts. J. Virol. 86:4455–4462. 10.1128/JVI.07187-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo G, Torri A, Harte WE, Danetz S, Cianci C, Tiley L, Day S, Mullaney D, Yu KL, Ouellet C, Dextraze P, Meanwell N, Colonno R, Krystal M. 1997. Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J. Virol. 71:4062–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. 2001. WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J. Biomol. NMR 21:349–359. 10.1023/A:1013302231549 [DOI] [PubMed] [Google Scholar]
- 38.Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A. 2011. The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J. Biol. Chem. 286:43352–43360. 10.1074/jbc.M111.304089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Celigoy J, Ramirez B, Tao L, Rong L, Yan L, Feng YR, Quinnan GV, Broder CC, Caffrey M. 2011. Probing the HIV gp120 envelope glycoprotein conformation by NMR. J. Biol. Chem. 286:23975–23981. 10.1074/jbc.M111.251025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCullough C, Wang M, Rong L, Caffrey M. 2012. Characterization of influenza hemagglutinin interactions with receptor by NMR. PLoS One 7:e33958. 10.1371/journal.pone.0033958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer B, Peters T. 2003. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl. 42:864–890. 10.1002/anie.200390233 [DOI] [PubMed] [Google Scholar]
- 42.Hakansson-McReynolds S, Jiang S, Rong L, Caffrey M. 2006. Solution structure of the severe acute respiratory syndrome-coronavirus heptad repeat 2 domain in the prefusion state. J. Biol. Chem. 281:11965–11971. 10.1074/jbc.M601174200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6:277–293 [DOI] [PubMed] [Google Scholar]
- 44.Nguyen HT, Sheu TG, Mishin VP, Klimov AI, Gubareva LV. 2010. Assessment of pandemic and seasonal influenza A (H1N1) virus susceptibility to neuraminidase inhibitors in three enzyme activity inhibition assays. Antimicrob. Agents Chemother. 54:3671–3677. 10.1128/AAC.00581-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen JT, Hoopes JD, Le MH, Smee DF, Patick AK, Faix DJ, Blair PJ, de Jong MD, Prichard MN, Went GT. 2010. Triple combination of amantadine, ribavirin, and oseltamivir is highly active and synergistic against drug resistant influenza virus strains in vitro. PLoS One 5:e9332. 10.1371/journal.pone.0009332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen JT, Hoopes JD, Smee DF, Prichard MN, Driebe EM, Engelthaler DM, Le MH, Keim PS, Spence RP, Went GT. 2009. Triple combination of oseltamivir, amantadine, and ribavirin displays synergistic activity against multiple influenza virus strains in vitro. Antimicrob. Agents Chemother. 53:4115–4126. 10.1128/AAC.00476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prichard MN, Prichard LE, Shipman C., Jr 1993. Strategic design and three-dimensional analysis of antiviral drug combinations. Antimicrob. Agents Chemother. 37:540–545. 10.1128/AAC.37.3.540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prichard MN, Shipman C., Jr 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–205. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 49.Wang SY, Su CY, Lin M, Huang SY, Huang WI, Wang CC, Wu YT, Cheng TJ, Yu HM, Ren CT, Wu CY, Wong CH, Cheng YS. 2009. HA-pseudotyped retroviral vectors for influenza antagonist screening. J. Biomol. Screen. 14:294–302. 10.1177/1087057108330786 [DOI] [PubMed] [Google Scholar]
- 50.Sakabe S, Iwatsuki-Horimoto K, Horimoto T, Nidom CA, Le M, Takano R, Kubota-Koketsu R, Okuno Y, Ozawa M, Kawaoka Y. 2010. A cross-reactive neutralizing monoclonal antibody protects mice from H5N1 and pandemic (H1N1) 2009 virus infection. Antiviral Res. 88:249–255. 10.1016/j.antiviral.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corti D, Lanzavecchia A. 2013. Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 31:705–742. 10.1146/annurev-immunol-032712-095916 [DOI] [PubMed] [Google Scholar]
- 52.Corti D, Suguitan AL, Jr, Pinna D, Silacci C, Fernandez-Rodriguez BM, Vanzetta F, Santos C, Luke CJ, Torres-Velez FJ, Temperton NJ, Weiss RA, Sallusto F, Subbarao K, Lanzavecchia A. 2010. Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J. Clin. Invest. 120:1663–1673. 10.1172/JCI41902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. 10.1126/science.1171491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nabel GJ, Fauci AS. 2010. Induction of unnatural immunity: prospects for a broadly protective universal influenza vaccine. Nat. Med. 16:1389–1391. 10.1038/nm1210-1389 [DOI] [PubMed] [Google Scholar]
- 55.Wei CJ, Boyington JC, McTamney PM, Kong WP, Pearce MB, Xu L, Andersen H, Rao S, Tumpey TM, Yang ZY, Nabel GJ. 2010. Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 329:1060–1064. 10.1126/science.1192517 [DOI] [PubMed] [Google Scholar]
- 56.Dreyfus C, Ekiert DC, Wilson IA. 2013. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J. Virol. 87:7149–7154. 10.1128/JVI.02975-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sui J, Sheehan J, Hwang WC, Bankston LA, Burchett SK, Huang CY, Liddington RC, Beigel JH, Marasco WA. 2011. Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza A antibodies. Clin. Infect. Dis. 52:1003–1009. 10.1093/cid/cir121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leneva IA, Russell RJ, Boriskin YS, Hay AJ. 2009. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 81:132–140. 10.1016/j.antiviral.2008.10.009 [DOI] [PubMed] [Google Scholar]
- 59.Russell RJ, Gamblin SJ, Haire LF, Stevens DJ, Xiao B, Ha Y, Skehel JJ. 2004. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 325:287–296. 10.1016/j.virol.2004.04.040 [DOI] [PubMed] [Google Scholar]
- 60.Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, Skehel JJ. 2008. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 105:17736–17741. 10.1073/pnas.0807142105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Staschke KA, Hatch SD, Tang JC, Hornback WJ, Munroe JE, Colacino JM, Muesing MA. 1998. Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 248:264–274. 10.1006/viro.1998.9273 [DOI] [PubMed] [Google Scholar]
- 62.Yoshimoto J, Kakui M, Iwasaki H, Fujiwara T, Sugimoto H, Hattori N. 1999. Identification of a novel HA conformational change inhibitor of human influenza virus. Arch. Virol. 144:865–878. 10.1007/s007050050552 [DOI] [PubMed] [Google Scholar]
- 63.Yoshimoto J, Kakui M, Iwasaki H, Sugimoto H, Fujiwara T, Hattori N. 2000. Identification of amino acids of influenza virus HA responsible for resistance to a fusion inhibitor, stachyflin. Microbiol. Immunol. 44:677–685. 10.1111/j.1348-0421.2000.tb02549.x [DOI] [PubMed] [Google Scholar]