

Abstract
Hypervariable region 1 (HVR1) of envelope protein 2 (E2) of hepatitis C virus (HCV) serves important yet undefined roles in the viral life cycle. We previously showed that the viability of HVR1-deleted JFH1-based recombinants with Core-NS2 of H77 (H77ΔHVR1, genotype 1a) and S52 (S52ΔHVR1, genotype 3a) in Huh7.5 cells was rescued by E2 substitutions N476D/S733F and an E1 substitution, A369V, respectively; HVR1-deleted J6 (J6ΔHVR1, genotype 2a) was fully viable. In single-cycle production assays, where HCV RNA was transfected into entry-deficient Huh7-derived S29 cells with low CD81 expression, we found no effect of HVR1 deletion on replication or particle release for H77 and S52. HCV pseudoparticle assays in Huh7.5 cells showed that HVR1 deletion decreased entry by 20- to 100-fold for H77, J6, and S52; N476D/S733F restored entry for H77ΔHVR1, while A369V further impaired S52ΔHVR1 entry. We investigated receptor usage by antibody blocking and receptor silencing in Huh7.5 cells, followed by inoculation of parental and HVR1-deleted HCV recombinants. Compared to parental viruses, scavenger receptor class B type I (SR-BI) dependency was decreased for H77ΔHVR1/N476D/S733F, H77N476D/S733F, S52ΔHVR1/A369V, and S52A369V, but not for J6ΔHVR1. Low-density lipoprotein receptor (LDLr) dependency was decreased for HVR1-deleted viruses, but not for H77N476D/S733F and S52A369V. Soluble LDLr neutralization revealed strong inhibition of parental HCV but limited effect against HVR1-deleted viruses. Apolipoprotein E (ApoE)-specific HCV neutralization was similar for H77, J6, and S52 viruses with and without HVR1. In conclusion, HVR1 and HVR1-related adaptive envelope mutations appeared to be involved in LDLr and SR-BI dependency, respectively. Also, LDLr served ApoE-independent but HVR1-dependent functions in HCV entry.
INTRODUCTION
Approximately 180 million people worldwide are chronically infected with hepatitis C virus (HCV) with an increased risk of developing liver cirrhosis and hepatocellular carcinoma (1). HCV is an enveloped positive-strand RNA virus of the family Flaviviridae with a 9.6-kb genome consisting of 5′ and 3′ untranslated regions (UTRs) flanking an open reading frame (ORF) that encodes a single polyprotein. This polyprotein is processed into structural proteins (Core and envelope proteins E1 and E2), p7, and six nonstructural proteins (NS2 to NS5B) (2). HCV is a highly diverse virus, and isolates are divided into seven major genotypes, most containing multiple subtypes and differing by ∼30% and ∼20%, respectively, at the nucleotide and amino acid levels (2). Previous studies have shown genotype or isolate differences when analyzing HCV neutralization and in reverse genetics studies of HCV proteins (3–5). This highlights the importance of including several isolates, preferably of diverse genotypes, in functional studies.
While the process of HCV entry into the human hepatocyte remains incompletely understood, it is known to be a complex multistep process involving several receptors acting at (i) initial attachment, (ii) cell surface transport, and (iii) cellular uptake and infection initiation (6). Both the low-density lipoprotein receptor (LDLr) and scavenger receptor class B type I (SR-BI) are believed to be involved in early interactions between the cell and the virion, possibly priming conformational changes that allow further interactions with the late-stage receptor CD81 or entry factors Claudin I and Occludin (7–10). Apparently, E2 interacts directly with CD81, and it has recently been suggested that CD81 and Claudin I are endocytosed with the virus particle in a clathrin-dependent manner (11, 12). The initial cell interactions have been proposed to occur through the association of the virus with apolipoproteins B and especially E (ApoB and ApoE) (13–16). ApoE has been implicated in virus attachment to the host cell (17) by interaction with heparan sulfate proteoglycans (HSPGs) (18), whereas others have found recombinant E1 and E2 to interact directly with liver-derived HSPGs (19). However, a recent study demonstrated that virus-associated ApoE is responsible for interactions mediating attachment between the cell-associated HSPG syndecan 1 and HCV (20). In addition, there is indirect evidence suggesting that ApoE is responsible for HCV interactions with LDLr (14, 21). However, a recent study showed that HCV internalization through LDLr does not lead to infection of the cell, suggesting that the ApoE-LDLr interaction might not mediate productive uptake of HCV (22). Thus, LDLr might primarily mediate cell attachment, possibly through an interaction with virus-associated ApoE (23). SR-BI has also been reported to interact with ApoE on the surface of the HCV particle and to interact with the E2 protein motif hypervariable region 1 (HVR1) (16, 24, 25). The latter finding was supported by the loss of SR-BI dependency of an HVR1-deleted genotype 2a virus, Jc1 (26). HVR1-deleted viruses have been shown to be infectious in both the chimpanzee and the human liver chimeric mouse model (3, 27), but so far, only a few studies have addressed how the deletion might affect the HCV life cycle.
In this study, we first analyzed which step of the HCV life cycle was affected by HVR1 deletion and the adaptive mutations acquired by HVR1-deleted viruses. Using antibody blocking and receptor silencing, we explored the lipoprotein receptor dependency of parental and HVR1-deleted HCV. Interestingly, HVR1 deletion conferred decreased dependency on the LDLr, while decreased SR-BI dependency seemed to be linked to HVR1-related envelope mutations required to rescue the infectivity of some HVR1-deleted viruses. Finally, we found LDLr to be important at the entry step of the HCV life cycle and showed that the interaction between HCV and the LDLr might not require virus-associated ApoE or ApoB.
MATERIALS AND METHODS
Plasmids.
We used JFH1-based HCV recombinants with Core-NS2 of isolate H77C, J6CF, or S52 and UTRs and NS3-NS5B of JFH1, which we and others had previously developed (3, 28–30). All mutations were annotated based on the H77 reference sequence (GenBank accession number AF009606). Spread of H77/JFH1 and S52/JFH1 in cell culture was previously shown to depend on coding mutations, although not in the envelope proteins. Both parental and HVR1-deleted H77/JFH1 constructs had the mutation pair T2701C/A4081T (p7/NS3). Parental S52/JFH1 had the mutation pair T2701G/A4533C (p7/NS3), and the HVR1-deleted S52/JFH1 had the mutation pair T2701G/T7155C (p7/NS5A). All three JFH1-based recombinants are referred to by the isolate of Core-NS2, i.e., H77, J6, and S52. We previously found that HVR1-deleted H77 required envelope substitutions (E1/E2, H261R/Q444R, or E2/E2, N476D/S733F) for efficient spread in culture; HVR1-deleted H77 viruses with N476D/S733F are abbreviated H77ΔHVR1/N476D/S733F (3). HVR1-deleted S52 required a single envelope substitution (E1, A369V) and is abbreviated S52ΔHVR1/A369V, whereas J6 did not require adaptation and is abbreviated J6ΔHVR1 (3). For HCV pseudoparticle (HCVpp) studies, a plasmid encoding murine leukemia virus (MLV) Gag-Pol and another plasmid encoding firefly luciferase were used (31). The expression plasmids encoding E1/E2 from H77, J6, and S52 have been previously described (phCMV-7a and phCMV ires) (31–33). The deletion of HVR1 and the introduction of the adaptive mutations in the plasmids containing E1/E2 from H77, J6, or S52 were performed using standard molecular cloning techniques. A plasmid maxiprep (Qiagen) was obtained for each construct, and the HCV sequence was confirmed (Macrogen Inc., Seoul, Korea).
Antibodies.
Antibodies used for blocking studies of the HCV receptors were monoclonal anti-CD81 (JS81; BD Pharmingen) with the isotype control antibody 553447; monoclonal anti-SR-BI C16-71 and control antibody D (34); polyclonal anti-LDLr (AF2148; R&D Systems) with control antibody AB108C; and monoclonal anti-LDLr antibodies 5G2, 6E2, and 3D8, kindly provided as mouse ascites fluid by Robert Milne (35). Purification of LDLr-specific antibodies and isotype-matched controls from 500 μl of mouse ascites fluid was performed using standard protein G column purification with absorbance measurements at 280 nm for tracking flowthrough and antibody elution. Briefly, a 5-ml protein G-Sepharose 4 fast flow (Pharmacia) was prepared in phosphate-buffered saline (PBS). The ascites fluid was spun for 10 min at 14,000× relative centrifugal force (RCF), and the supernatant was applied to the column, which was then washed with PBS. The antibody was then eluted in alkaline PBS (pH > 12) and immediately adjusted to pH 7 using HCl. Finally, the antibody was dialyzed twice overnight at 4°C in PBS with 0.02% sodium azide. This amount of azide in the antibody stocks had been previously found to have no effect in HCV infection assays. For neutralization studies, we used monoclonal antibody 1D7 against ApoE, which was a kind gift from Robert Milne (36), and E2-specific antibody AR3A, which was a kind gift from Mansun Law (37). Monoclonal antibody 9E10, used in immunostaining of NS5A in infected cells, was a kind gift from Charles Rice (28). Antibodies used in Western blotting were anti-CD81 (5A6; sc-23962; Santa Cruz), anti-SR-BI (EP1556Y; Abcam), anti-LDLr (20R-LR002; Fitzgerald Industries), anti-β-actin (C4; sc-47778; Santa Cruz), anti-E1 (A4 [38]), anti-E2 (H52), and anti-MLV-Gag. The last two antibodies were kind gifts from Jean Dubuisson (38, 39). Horseradish peroxidase-conjugated secondary antibodies were goat anti-mouse (32430; Thermo Scientific) and anti-rabbit (32460; Thermo Scientific).
Cell culturing.
Huh7.5 human hepatoma cells, Huh7-derived S29 cells (with low CD81 expression), and 293T human embryo kidney (HEK) cells were grown in Dulbecco's modified essential medium supplemented with 10% fetal bovine serum (FBS) and antibiotics. Culturing, transfections, and infections of cells were done as described previously (29, 40), and infected cultures were evaluated every 2 or 3 days by HCV-specific immunostaining (29).
S29 cell transfection.
For each recombinant to be tested, 2 wells in a 6-well plate (Nunc) were seeded with 400,000 S29 cells/well, along with either 1 well in a 24-well plate seeded with 100,000 S29 cells/well or an additional 6-well plate seeded with 400,000 S29 cells/well. All were incubated overnight. The following day, we prepared 100- to 150-μl in vitro transcription mixtures for use in transfection, as previously described (29). The cells were washed and transfected in Opti-MEM for 4 h with 5 μl Lipofectamine 2000 using 40 μl in vitro transcription mixture for the 6-well plates and 10 μl for the 24-well plates. S29 cells from the 24-well or the additional 6-well plates were trypsinized and then centrifuged at 1,000× RCF for 5 min at 4°C, washed once in cold PBS, and spun down again at 1,000× RCF for 5 min at 4°C. The cells were then lysed using 100 μl (for the 24-well plates) or 400 μl (for the 6-well plates) of ice-cold RIPA buffer containing protease inhibitor cocktail III (Calbiochem). The samples were cleared by centrifugation at 20,000× RCF at 4°C for 12 min, and the supernatant was transferred to a fresh tube for measurement of Core levels using either the Ortho HCV Antigen ELISA (Ortho Clinical Diagnostics) or Architect HCV Core antigen test (Abbott). Core enzyme-linked immunosorbent assays (ELISAs) were performed according to the manufacturer's instructions. Cells in 6-well plates were trypsinized 48 h and 72 h posttransfection, and 1/4 of the cells, corresponding to 100,000 plated cells, were prepared for Core ELISA as described above. Core values were normalized to the 4-h value from the 24-well plate, representing transfection efficiency. S29 cell culture supernatants were taken prior to cell trypsinization at 48 h and 72 h. Core values were measured as described above. The remaining 3/4 of the cells following trypsinization of 6-well plates at 48 h and 72 h were resuspended in 100 μl of complete medium and subjected to 4 or 5 freeze-thaw cycles to release intracellular infectious particles. Cellular debris was removed by two centrifugations at 1,500× RCF at 4°C for 5 min, and samples were titrated for HCV infectivity as previously described at a minimum dilution of 1:50 (41). Cell culture supernatants were infectivity titrated as described previously (41).
Cell-to-cell spread assays.
Huh7.5 cells were transfected with HCV RNA as previously described (29). The following day, the cells were trypsinized and mixed with naive trypsinized Huh7.5 cells prior to seeding 12,000 Huh7.5 cells in 100 μl per well in 96-well poly-d-lysine (PDL) plates (Nunc). Six wells per plate for each virus condition were seeded, along with 12 wells per plate with only naive Huh7.5 cells to estimate background staining on each plate. A ratio of transfected to naive Huh7.5 cells of 1:150 was used for cells plated in standard medium, and a ratio of 1:30 was used for cells plated in standard medium supplemented with 10 μg/ml of the cross-genotype-reactive HCV neutralizing antibody AR3A (37). This concentration of AR3A represented at least 500 times the 50% inhibitory concentration (IC50) for the tested HVR1-deleted viruses (data not shown). Six replicates of cell mixtures from each virus recombinant were plated in the absence or presence of AR3A for fixation at three time points: 24 h, 48 h, and 72 h postplating. At these time points postplating, 50 μl of supernatant was removed from each well of one plate for infectivity titration (as described above) in order to ensure the efficacy of the neutralizing antibody. No infectious particles were found in the antibody-treated supernatants for any of the viruses tested. The 50-μl volume was replaced with either standard medium (for the wells without antibody) or standard medium supplemented with 10 μg/ml AR3A (for wells with antibody). At 24 h, 48 h, and 72 h, respectively, a replicate plate was fixed in methanol, and all plates were subsequently stained for infection using the NS5A-specific antibody 9E10 (28). The number of single infected cells, as well as the number of focus-forming units (FFU) (clusters of single cells), was automatically determined as previously described (41). The FFU size was also automatically calculated by the BioSpot software (Cellular Technology Ltd.). The data were analyzed using GraphPad Prism v4.03.
RNA interference.
Huh7.5 cells (300,000 per well) were plated in 6-well plates and incubated overnight. The following day, the cells were transfected with small interfering RNAs (siRNAs) (Qiagen) specifically targeting CD81, SR-BI, or LDLr mRNA; with AllStars negative-control siRNA at a concentration identical to that of the specific siRNAs of the assay (Qiagen); or without siRNA (mock transfection) using Lipofectamine 2000 as described previously (29). For SR-BI, three specific siRNAs (HS_SCARB1_4, HS_SCARB1_6, and HS_SCARB1_9; Qiagen) were transfected independently at a final concentration of 5 nM. For CD81 (HS_CD81_6 and HS_CD81_7, Qiagen) and LDLr (HS_LDLr_2, HS_LDLr_4, and HS_LDLr_5; Qiagen), siRNAs were transfected at a final concentration of 25 nM. Twenty-four hours posttransfection, the cells were trypsinized, replated at 300,000 cells/well, and incubated overnight. The following day, to ensure robust silencing of the targeted protein, the cells were transfected a second time, as described above. Twenty-four hours following the second transfection, the cells were trypsinized and seeded into a 6-well plate with 400,000 cells/well and into a 96-well plate at 10,000 cells/well. The next day, 96-well plates were incubated with the indicated virus with and without HVR1 for 3 h prior to washing once with PBS and adding fresh medium. Forty-eight hours after infection, the number of HCV FFU/well in the 96-well plates was visualized by methanol fixation and NS5A-specific immunostaining with 9E10 antibody as described previously (28). The cells plated in 6-well plates were harvested at the time of infection in cold RIPA buffer (Thermo Scientific) supplemented with protease inhibitor (Calbiochem) to estimate the silencing efficiency of the targeted receptor by Western blotting.
Western blotting for estimating RNA interference efficiency.
Cell lysates were stored overnight at −80°C and cleared by centrifugation at 20,800× RCF for 15 min at 4°C. The total protein concentration of the cell lysate was determined by bicinchoninic acid assay (BCA; Pierce). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a 10% or 12% precast Novex Bis-Tris polyacrylamide gel (Invitrogen) in the presence of reducing agent (for CD81, the gel was run without reducing agent). After separation, the proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Hybond-P membrane; 0.45-μm pore size; GE Healthcare-Amersham) using an XCell SureLock Mini-Cell (Invitrogen) and incubated overnight at 4°C with specific primary antibodies. Then, the membranes were incubated for 1 h at room temperature with secondary horseradish peroxidase-conjugated antibody [stabilized, peroxidase-conjugated goat anti-mouse or anti-rabbit IgG(H+L); Pierce]. Proteins were revealed by enhanced chemiluminescence detection (SuperSignal West Femto Maximum Sensitivity Substrate; Pierce). Protein expression was quantified using ImageJ. The receptor signal was normalized to the β-actin signal and related to negative-control siRNA for calculation of the percent receptor downregulation.
Assessment of cell viability.
To ensure the viability of Huh7.5 cells that had been transfected twice with siRNA at the time of viral infection, cell proliferation was assessed by carboxyfluorescein diacetate succinimidyl ester (CFSE) staining and 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. In an independent experiment, 24 h prior to the first siRNA transfection, Huh7.5 cells were trypsinized, washed twice in PBS, and resuspended at 106 cells/ml in PBS before incubation with 5 μM CFSE (CellTrace CFSE Cell Proliferation; Molecular Probes) for 5 min at room temperature. CFSE-stained cells were washed twice in PBS supplemented with 5% FBS, and 450,000 cells were plated in 6-well plates. Then, the CFSE-stained cells were transfected twice with siRNAs as described above. Twenty-four hours after the second siRNA transfection, 400,000 cells were plated in duplicate in 6-well plates, and for MTS assays (Promega), 6,000, 8,000, and 10,000 cells per well were plated in three replicates in 96-well plates. The following day, the cells in the 6-well plate were harvested as described above for assessment of the silencing efficacy of the targeted receptor by Western blotting. The cells from the replicate wells of the other 6-well plate were trypsinized, washed twice in PBS, and fixed for 15 min at room temperature (10× CellFix; BD Pharmingen) before analyses of CFSE-stained cells by flow cytometry (Calibur; BD Pharmingen). CFSE-stained cells treated daily with 60 ng/ml of colchicine (KaryoMax Colcemid solution in PBS; Invitrogen) were used as a negative control for cell proliferation. Unlike the cells treated with colchicine, no significant differences between the proliferation rates of the nontransfected, mock-transfected, and siRNA-transfected CFSE-stained cells were observed. The MTS assay was conducted as recommended by the manufacturer. No significant change in the slope of the MTS signal between cells plated at 6,000, 8,000, and 10,000 per well for the differently treated cells was observed, showing that the specific silencing of CD81, SR-BI, or LDLr did not affect the cellular metabolic activity of transfected cells.
HCVpp generation and infection assays.
Generation of HCVpp was done as previously described (40) with the following minor modifications. To generate each independent batch of HCVpp, HEK 293T cells were seeded in 10-cm dishes or 6-well plates and transfected after 24 h. In infection assays, we used either fresh supernatants or supernatants stored at 4°C overnight. For statistical analysis of luciferase measurements, data were analyzed from three independent infection assays conducted with three different batches of HCVpp. For each infection assay, luciferase measurements were performed in 4 to 8 replicates. Incorporation of MLV-Gag and E1 and E2 (if possible; see below) into pseudoparticles was analyzed on material from 20% sucrose-pelleted supernatants as previously described (40).
HCVpp Western blotting of MLV-Gag and HCV E1 and E2.
For Western blots of MLV-Gag and HCV E1 and E2, 5 μg of sucrose-pelleted HCVpps was added to the gel for SDS-PAGE and transferred to PVDF membranes as described above. The proteins were visualized by overnight incubation at 4°C with specific antibodies (A4 anti-E1, H52 anti-E2, and anti-MLV-Gag), followed by washing and 1 h of incubation with secondary immunoglobulin-conjugated peroxidase antibody (ECLO Anti-mouse IgG; horseradish peroxidase-linked whole antibody; GE Healthcare-Amersham) and revealed by enhanced chemiluminescence detection (Signal West Femto Maximum Sensitivity Substrate; Pierce). As we were not able to acquire antibodies recognizing E1 and E2 of isolates J6 and S52, we were unable to perform envelope protein Western blots for these HCVpps.
HCV receptor blocking.
For receptor-blocking studies, we used antibodies against the receptors CD81 (monoclonal JS81), SR-BI (monoclonal C16-71), and LDLr (polyclonal AF2148 or monoclonal antibody 5G2, 6E2, or 3D8) and relevant control antibodies. A total of 6 × 103 Huh7.5 cells/well were plated on PDL 96-well plates (Nunc) and incubated overnight. The following day, a dilution series of the antibody was prepared and applied to the cells for 1 h at 37°C prior to the addition of 25 to 200 FFU of HCV. The cells were washed after 3 h of incubation with the virus and antibody or virus only, and following an additional 45 h of incubation, the cells were immunostained for NS5A (28). For HCV, the percent blocking was calculated by relating FFU counts to the mean FFU count of six replicates incubated with virus only. Blocking data were analyzed as variable-slope dose-response curves using GraphPad Prism v4.03.
Time-lapse LDLr blocking of HCV infection.
Huh7.5 cells were plated in PDL 96-well plates (Nunc) at 6 × 103 Huh7.5 cells/well. The following day, infection with the relevant viruses and blocking of the LDLr with 10 μg/ml of the polyclonal LDLr-specific antibody AF2148 was carried out. For each virus used in the infection, there were 4 wells blocked preinoculation by incubation with antibody for 1 h prior to inoculation. After washing with PBS, fresh medium containing 10 μg/ml of LDLr-specific antibodies was added to the cells. Four wells were blocked postinoculation by incubation with antibody following 4 h of virus inoculation. Additionally, we included 6 wells with virus only that were used for normalization. The cells were fixed after a total infection time of 48 h, and the staining was carried out as described above for HCV receptor blocking.
Neutralization of HCV infection.
Huh7.5 cells were plated in PDL 96-well plates (Nunc) at 6 × 103 Huh7.5 cells/well. The following day, a dilution series of either soluble LDLr, ApoE-specific antibody (1D7), or E2-specific antibody (AR3A) was prepared and incubated in triplicate or quadruplicate with the tested viruses for 1 h at 37°C, along with relevant antibody isotype controls. Next, the receptor-virus or antibody-virus mixtures, along with 6 replicates of virus only, were added to the 96-well plates and incubated for 3 h prior to washing and addition of fresh medium. Following a total infection time of 48 h, the cells were fixed and stained for HCV NS5A using 9E10 antibody (28). The data were normalized to the 6 replicates of virus only and analyzed using four-parameter curve fitting in GraphPad Prism v4.03.
HCV RNA immunoprecipitation.
Using the ApoE-specific antibody 1D7 and relevant isotype-matched control antibody, we washed 50 μl of magnetic-bead slurry (immunoprecipitation kit; Dynabeads Protein G; 100.070D; Invitrogen) and incubated it on a shaker with 5 μg of the indicated antibody in 50 μl for 20 min at room temperature. The beads were subsequently washed two times and incubated with 106 HCV RNA copies of the indicated virus with and without HVR1 in 500 μl of complete medium on a shaker for 1 h at room temperature. The beads were washed three times in 200 μl of washing buffer prior to elution in the lysis buffer using a QIAamp MinElute Virus Vacuum kit (57714; Qiagen), along with blanks and a dilution series of an internal sample with a known HCV RNA titer. Afterward, the beads were removed by spinning for 5 min at 1,400× RCF. The extraction of viral RNA was carried out according to the manufacturer's instructions and eluted in 22 μl of elution buffer; 8 μl of the eluted fractions was then used in a LightCycler for reverse transcription-quantitative PCR (RT-qPCR) using primers and protocols that have been described previously (29). The results are shown as the total HCV RNA amount in each fraction.
Measuring DiI-labeled LDL association with Huh7.5 cells by flow cytometry.
Huh7.5 cells were plated at 60,000/well in 24-well plates and incubated overnight. The following day, the cells were switched to medium supplemented with 10% lipoprotein-deficient serum and incubated for 48 h. The cells were then incubated for 10 min with either 20 μg/ml of the polyclonal antibody AF2148 and controls; 50 μg/ml of the monoclonal antibody 5G2, 6E2, or 3D8 and controls; or without antibody (for positive and negative controls). Subsequently DiI (1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate)-labeled LDL particles (L-3482; Life Technologies) were added at 10 μg/ml to all but the negative-control wells. After 4 h, the cells were washed once with PBS and harvested using trypsin-EDTA. The cells were washed three times in ice-cold PBS with spinning at 1,400× RCF. The DiI signal from bound and internalized LDL was measured using flow cytometry on 10,000 gated cells.
RESULTS
HVR1 deletion and HVR1-related adaptive envelope mutations did not affect viral replication/translation or assembly/release.
We have previously shown that H77ΔHVR1 depended on envelope mutation pair H261R/Q444R or N476D/S733F for efficient spread in Huh7.5 cells (3). Similarly, S52ΔHVR1 depended on the E1 mutation A369V, whereas J6ΔHVR1 did not depend on adaptive mutations for spread in culture (3). To investigate whether the adaptive envelope mutations in HVR1-deleted viruses could affect replication/translation or assembly/release, we conducted two independent single-cycle production assays in HCV entry-impaired S29 cells characterized by low CD81 expression (42).
We transfected S29 cells with HCV RNA transcripts of relevant H77 and S52 recombinants and performed intra- and extracellular Core measurements (Fig. 1A and B). HVR1 deletion and the adaptive envelope mutations (N476D and S733F, either singly or in combination, for H77, and A369V for S52) did not affect the production of intracellular Core (Fig. 1A) or the release of extracellular Core (Fig. 1B). This suggested that HVR1 deletion and the adaptive envelope mutations had no significant impact on replication through release of HCV particles, and we therefore did not perform assays to investigate the effects on these viral processes in further detail. However, the intracellular HCV infectivity was reduced for both H77ΔHVR1 and S52ΔHVR1 and restored by the introduction of the indicated envelope mutations (Fig. 1C). For H77ΔHVR1, the extracellular infectivity titer was similarly decreased, suggesting that HVR1 deletion lowered the entry capacity of the viral particles (Fig. 1D). Similar findings were obtained upon S29 cell analysis of intra- and extracellular Core and infectivity for the parental and HVR1-deleted H77 recombinant with the adaptive envelope mutations H261R and Q444R (data not shown). Interestingly, the extracellular infectivity of S52ΔHVR1 was comparable to that of S52 and also to that of recombinants harboring the A369V mutation (Fig. 1D). We confirmed the identity of S52ΔHVR1 by RNA extraction and direct sequencing of the structural Core, E1, and E2 genes in the extracellular samples from 48 h posttransfection. Furthermore, we inoculated Huh7.5 cells at a multiplicity of infection (MOI) of ∼0.006 FFU/cell using the same 48-h extracellular samples of S52ΔHVR1 and S52ΔHVR1/A369V. The number of infected cells at day 1 postinfection was similar, as observed by HCV-specific immunostaining, thus confirming comparable infectivity titers in the transfection supernatants. However, S52ΔHVR1/A369V was able to spread and infected the entire Huh7.5 cell culture 20 days postinfection, whereas no infected cells were observed at this time point for S52ΔHVR1 (data not shown). This confirmed the inability of S52ΔHVR1 to spread effectively in Huh7.5 cells, as previously reported (3). Overall, the data from the S29 cell transfections suggested that HVR1 deletion and the identified adaptive envelope mutations did not affect virus replication/translation or assembly/release. Therefore, we concluded that the envelope mutations adapting HVR1-deleted H77 for viral spread in Huh7.5 might increase the entry efficiency of H77, whereas the role of the E1 mutation A369V for S52 was less clear.
FIG 1.

HCV RNA transfection of S29 cells with low CD81 expression suggested that HVR1 deletion and HVR1-related adaptive envelope mutations had no effect on HCV replication/translation and assembly/release. The boxes on the x axes indicate which Core-NS2 JFH1-based recombinant served as the backbone for the indicated mutations, and Parental signifies the unmodified recombinant. A J6/JFH1 recombinant with the replication-deficient GND mutation was included as a negative control. Cells were plated and transfected with the indicated HCV RNA as described in Materials and Methods to allow quantification of intra- and extracellular Core at 4 h, 48 h, and 72 h posttransfection, as well as intra- and extracellular infectivity titers. (A) Protein was harvested from the S29 cells at 4 h, 48 h, and 72 h posttransfection and analyzed by Core ELISA. The Core values were normalized to the 4-h value, representing transfection efficiency. (B) S29 cell culture supernatants were collected at 48 h and 72 h posttransfection. Core values were measured directly on the supernatants by Core ELISA. (C) S29 cells were subjected to freeze-thaw cycles and cleared of cellular debris by centrifugation prior to HCV infectivity titration in triplicate to obtain intracellular infectious particles at 48 h and 72 h posttransfection. (D) S29 cell culture supernatants were HCV infectivity titrated in triplicate. The entire single-cycle production assay was repeated, and the values represent the means of the two independent experiments performed as described above, with error bars representing standard errors of the mean (SEM). #, values below the cutoff.
HVR1 deletion significantly decreased the infectivity of HCVpp in Huh7.5 cells, and adaptive envelope mutations restored the infectivity of HCVpp H77ΔHVR1, but not HCVpp S52ΔHVR1.
We studied the effects of HVR1 deletion on HCV entry by using HCVpps presenting parental or HVR1-deleted H77, J6, and S52. For H77 HCVpps, deletion of HVR1 decreased infectivity 10- to 20-fold in three independent experiments (in Fig. 2A and B, a representative experiment for H77 HCVpp shows the entry efficiency of each HCVpp as measured in relative light units [RLU] and particle-incorporated proteins in Western blots of HCV E1, E2, and MLV-Gag). Insertion of the single adaptive envelope mutations H261R, Q444R, N476D, and S733F in H77 HCVpps partially restored infectivity. Insertion of a mutation pair (H261R/Q444R or N476D/S733F) restored infectivity to levels comparable to those of the parental H77 HCVpps (Fig. 2A to C), suggesting that these mutation pairs restored entry of HVR1-deleted H77 HCVpps. Neither the deletion of HVR1 nor the adaptive envelope mutations seemed to affect the incorporation of envelope protein 1 or 2 (Fig. 2B). Three independent experiments were performed in which the effects of HVR1 deletion on J6 and S52 HCVpps was also tested (Fig. 2C) (the presence of the MLV-Gag protein was confirmed for all recombinants). Surprisingly, for J6, deletion of HVR1 led to an ∼100-fold decrease in infectivity (Fig. 2C), which was in contrast to the retained viability of J6ΔHVR1 in the JFH1-based HCV model system. For HVR1-deleted S52 HCVpps, we observed a 10- to 20-fold decrease in infectivity. However, in contrast to H77 HCVpps, the adaptive effect of envelope mutations was not observed for HVR1-deleted S52, as the E1 mutation A369V impaired entry completely (Fig. 2C). Thus, we confirmed the role of the envelope mutations in increasing entry of HVR1-deleted H77 and found further evidence supporting a more complex role of the E1 mutation for HVR1-deleted S52.
FIG 2.

Deletion of HVR1 from H77, J6, and S52 HCVpps decreased infectivity, while HVR1-related adaptive envelope mutations restored infectivity of H77ΔHVR1, but not of S52ΔHVR1, HCVpps. 293T cells were transfected as described in Materials and Methods to produce HCVpps with the indicated HCV envelope proteins. The HCVpps were used for infection of Huh7.5 cells, and infection was assessed by measuring the RLU of luciferase activity. (A) Mean RLU from 8 replicates in Huh7.5 cells following infection with the indicated HCVpps. The error bars indicate standard deviations (SD). #, the luciferase signal was below the cutoff. (B) Western blot of MLV-Gag and HCV envelope proteins, E1 and E2, from supernatants used in the infection experiment in panel A. (C) For each set of data (H77, J6, and S52), mean RLU normalized to the parental virus from three HCVpp experiments carried out with three independent batches of HCVpps are shown. Each experiment contained 4 or 8 independent luciferase measurements. RLU values from infections of Huh7.5 cells are normalized to the parental envelope protein control. The error bars indicate SEM. #, luciferase activity was below the cutoff. RLU (background subtracted) for H77, J6, and S52 without envelope mutations varied from 8,703 to 91,947, 2,561 to 55,469, and 95,499 to 2,844,709, respectively, among the three experiments.
A369V increased both cell-free and cell-to-cell spread of S52ΔHVR1, whereas N476D/S733F increased only the cell-free spread of H77ΔHVR1.
As we were unable to clearly define the adaptive role of A369V with a single-cycle production assay using entry-impaired S29 cells and the HCVpp system, we assessed whether the mutation had an influence on cell-to-cell spread. We transfected Huh7.5 cells with in vitro-transcribed HCV RNA, mixed the transfected cells with naive Huh7.5 cells, and plated them in 96-well plates at nearly complete confluence to ensure immediate cell contact. To assess overall viral spread, including cell-free and cell-to-cell spread, we monitored the number of FFU 24, 48, and 72 h postplating in the absence of neutralizing antibody. At 24 h postplating, all cultures transfected with the different recombinants had 5 to 10 FFU/well. At 72 h, for J6ΔHVR1 and S52ΔHVR1/A369V, ∼120 FFU/well was recorded, while for H77ΔHVR1/N476D/S733F and S52ΔHVR1, ∼30 FFU/well was recorded. For H77ΔHVR1, the number of FFU per well stayed constant over time. Finally, the number of FFU per well for the negative-control recombinant, JFH1ΔE1E2, slowly decreased over time (Fig. 3A). In parallel with the above-mentioned cultures without antibody, we assessed cell-to-cell spread by plating cells in medium supplemented with neutralizing antibody, AR3A (37), at a concentration of at least 500 times the IC50 against HVR1-deleted virus isolates H77, J6, and S52 (data not shown). Supernatants were taken at 24, 48, and 72 h postplating and replaced either with fresh medium (for cultures to assess overall viral spread) (Fig. 3A) or with medium containing AR3A (for cultures to assess cell-to-cell spread) (Fig. 3B to D). Infectivity titration of the supernatants from all three time points revealed that no infectious particles were present in the AR3A-treated culture supernatants, whereas particles could be detected in the nontreated cultures, especially for J6ΔHVR1 and S52ΔHVR1/A369V (data not shown). In the wells treated with neutralizing AR3A, the number of FFU per well had remained constant at 30 to 60 FFU/well (Fig. 3B), indicating that cell-free spread was indeed ablated. Analyzing FFU size for AR3A-treated cultures, we found that the increase in FFU size at 72 h postplating was greatest for J6ΔHVR1 and S52ΔHVR1/A369V compared with the other virus recombinants (Fig. 3C). This correlated with an increase in HCV antigen-positive single cells per FFU (Fig. 3D). The data suggested that the mutation A369V increased cell-to-cell spread. However, compared to the observed increase in overall viral spread (Fig. 3A), the contribution of this effect to the overall adaptive effect of the mutation was probably minor.
FIG 3.

A369V increased both cell-free and cell-to-cell spread of S52ΔHVR1, whereas N476D/S733F increased only cell-free spread of H77ΔHVR1. Huh7.5 cells were transfected with HCV RNA of the indicated viral recombinants, including JFH1ΔE1E2 as a negative control. They were mixed with naive cells and plated at 12,000 cells/well in 96-well plates to ensure nearly 100% cell confluence. The number of FFU, size of the FFU, and number of single infected cells were automatically determined for the 6 replicates of each virus as described in Materials and Methods following HCV-specific staining after 24 h, 48 h, and 72 h. The error bars indicate SD. (A) Average numbers of FFU per well for wells not treated with AR3A antibody. (B) Average numbers of FFU per well for wells treated with AR3A antibody. (C) Average sizes of FFU in wells treated with AR3A. (D) Numbers of single infected cells per FFU in wells treated with AR3A.
Deletion of HVR1 and HVR1-related adaptive envelope mutations decreased HCV dependency on LDLr and SR-BI, respectively.
Previous reports had indicated a relationship between HVR1 and the lipoprotein receptor SR-BI in mediating HCV entry (9, 24, 26, 43). Investigation of this link using HVR1-deleted HCV has so far been done only for a single HCV genotype 2a isolate. In addition, the altered physiochemical properties observed for infectious HVR1-deleted viruses suggest decreased lipid association (3). This implied that HVR1 deletion might also affect interaction with another lipoprotein receptor, LDLr.
To compare the dependencies of parental and HVR1-deleted H77 (1a), J6 (2a), and S52 (3a) recombinants on CD81, SR-BI, and LDLr, we performed dose-response blocking of these three HCV receptors in Huh7.5 cells. The virus stocks used here and in subsequent experiments were sequenced to verify the isolate sequence of the envelope proteins. Blocking of HCV infection using antibodies targeting the nonlipoprotein receptor CD81 had similar effects on the genotype 1a, 2a, and 3a viruses with and without HVR1, as they displayed similar dose-response curves with complete inhibition of infection at the highest antibody concentrations (Fig. 4A). H77, J6, and S52 responded similarly to the blocking of HCV infection by targeting SR-BI. However, differences in the highest attainable blocking efficiency were observed, specifically, lower dependency for J6 (Fig. 4B). In contrast to the parental recombinants, the HVR1-deleted variants H77ΔHVR1/N476D/S733F and S52ΔHVR1/A369V both completely lost SR-BI dependency, while J6ΔHVR1 displayed dependency on SR-BI similar to that of J6 (Fig. 4B). The findings for J6 were verified in two independent experiments (data not shown). For J6 and J6ΔHVR1 the results differ from those of a previous study reporting that a JFH1-based HVR1-deleted J6 HCV recombinant (Jc1) lost dependency on SR-BI (26). We also observed that HVR1 deletion of H77, J6, and S52 conferred decreased dependency on LDLr in HCV infection; this effect was least pronounced for H77 (Fig. 4C).
FIG 4.

Blocking of HCV receptors on Huh7.5 cells revealed that HVR1-deleted viruses had decreased SR-BI and LDLr dependency. Huh7.5 cells were plated in 96-well plates. The following day, the blocking antibody or a relevant control antibody was incubated with the cells for 1 h at each concentration in three replicates, also including 6 wells without antibody. Infection was carried out by 3 h of incubation with the indicated viruses; the cells were washed once, and FFU were visualized after an additional 45 h by HCV-specific immunostaining. (A) Anti-CD81 antibody JS81 blocking of HCV infection against the indicated viruses. (B) Anti-SR-BI antibody C16-71 blocking of HCV infection against the indicated viruses. (C) Anti-LDLr antibody AF2148 blocking of HCV infection against the indicated viruses. Blocking data are shown as dose-response curves, with the means and SD of three replicates at the given antibody dilution normalized to 6 replicates of virus only. The open symbols represent data for the control antibody only tested at the highest antibody concentration. Four-parameter nonlinear curve regression was used to fit a curve to the data points (Graphpad Prism, v4.03). Z-tests were used to compare the differences in maximum attained blocking for each antibody. In the three bottom panels, the pairwise comparisons were made between viruses of the same isolate with and without the indicated envelope mutations. ***, statistical significance at a P value of <0.001; ns, not statistically significant.
To investigate the impact of the HVR1-related adaptive envelope mutations on receptor dependency, we first generated relevant stock viruses by transfecting Huh7.5 cells with H77N476D/S733F and S52A369V. Both viruses showed spread and infectivity titers similar to those of H77 and S52 (data not shown). No additional coding mutations were identified in the envelope proteins of first-passage viruses, while the presence of the engineered mutations was confirmed. Both CD81 and LDLr blocking sensitivities were unaltered by the introduction of the envelope mutations N476D/S733F in H77 and A369V in S52 (Fig. 4A and C, bottom). In contrast, introduction of these mutations significantly decreased SR-BI dependency (Fig. 4B, bottom).
To further substantiate the receptor-blocking data, we performed gene silencing of CD81, SR-BI, and LDLr. The HCV permissiveness of Huh7.5 cells 48 h after the second siRNA transfection was assessed and related to the permissiveness of cells treated with negative-control siRNA (Fig. 5A, C, and E). The cell division rate and mitochondrial activity, assessed by CFSE cell proliferation assay and MTS assay, respectively, were not affected by efficient silencing of CD81, SR-BI, and LDLr (data not shown). Both CD81-specific siRNAs decreased CD81 protein expression by >90%, which completely inhibited HCV infection (Fig. 5A and B), confirming CD81 dependency of HCV infection. The three SR-BI-specific siRNAs, HS_SCARB1_4, HS_SCARB1_6, and HS_SCARB1_9, decreased SR-BI protein expression by 82%, 94%, and 92%, respectively (Fig. 5D). Silencing of SR-BI had a similar effect on HCV infection, as observed in the antibody-blocking experiments. Thus, H77ΔHVR1/N476D/S733F and S52ΔHVR1/A369V were only minimally affected by SR-BI silencing compared with H77 and S52, while J6 and J6ΔHVR1 were similarly inhibited (Fig. 5C). In addition, the lower SR-BI dependency of J6 than of H77 and S52, as observed in antibody-blocking experiments, was apparent for all three SR-BI-specific siRNAs. The three LDLr-specific siRNAs, HS_LDLr_2, HS_LDLr_4, and HS_LDLr_5, decreased LDLr protein expression by 47%, 65%, and 87%, respectively (Fig. 5F). In agreement with the data from the blocking experiments, all parental viruses were similarly inhibited by LDLr silencing (Fig. 5E). However, in this assay, HVR1 deletion did not influence the LDLr dependency of the recombinants, possibly due to the incomplete silencing of the receptor and the partial, although statistically significant, effects on entry observed in antibody-blocking experiments.
FIG 5.

siRNA-specific silencing of CD81, SR-BI, and LDLr reduced HCV permissiveness of Huh7.5 cells relative to cells transfected with the negative-control siRNA. Silencing was performed as described in Materials and Methods. Briefly, Huh7.5 cells were transfected twice with a 48-h interval using the indicated receptor-specific silencing RNAs. (A, C, and E) Infection of receptor-silenced cells was done in 96-well plates incubated with the indicated virus with and without HVR1 for 3 h prior to washing once with PBS and an additional 45 h of incubation in complete medium. The data points are means and SD of four replicates for the given siRNA normalized to the four negative-control siRNA replicates. Quantification of the amount of protein was done by chemiluminescence analysis of the Western blots, avoiding pixel saturation, and analyzed using ImageJ (B, D, and F). The degree of silencing was ascertained as described in Materials and Methods by Western blotting from cell lysates harvested at the time of infection. (B) CD81 was visualized using 5A6 antibody (Santa Cruz). (D) SR-BI was visualized using EP1556Y (Abcam). (F) LDLr was visualized using 20R-LR002 (Fitzgerald Industries). β-Actin was visualized using C4 (Santa Cruz). The order of the different cell treatments in the Western blots in panels B, D, and F mirrors the order of transfected siRNAs shown in panels A, C, and E, respectively. t tests were performed comparing permissiveness to HCV, with and without HVR1, of cells treated with specific siRNAs. ** and ***, statistical significance at P values of <0.01 and <0.001, respectively; ns, not statistically significant.
LDLr dependency of HCV was at the entry step of the viral life cycle.
It was recently reported that the LDLr might primarily be involved in the HCV life cycle at a later stage than viral entry (22). Therefore, we wanted to investigate if the decreased LDLr dependency of HVR1-deleted viruses was due to a decreased need to interact with the receptor during viral entry. Thus, we performed neutralization experiments in which HCV with and without HVR1 was incubated with different concentrations of soluble LDLr prior to infection of Huh7.5 cells. These results clearly showed that parental H77, J6, and S52 were efficiently neutralized by soluble LDLr. H77ΔHVR1/N476D/S733F was slightly affected at the highest tested soluble-LDLr concentration, while no neutralization was observed for J6ΔHVR1 and S52ΔHVR1/A369V (Fig. 6A). These results indicate the existence of a direct interaction between HVR1 and LDLr at the entry step. Furthermore, we conducted time-lapse antibody blocking of LDLr to assess the effects on HCV infection of receptor blocking prior to, or following, viral inoculation (Fig. 6B). We found that HCV infection was greatly diminished by preinoculation antibody blocking but only minimally affected if the antibody was added after the 4-h inoculation period (Fig. 6C). Overall, our data support an HVR1-dependent role for LDLr in viral entry.
FIG 6.
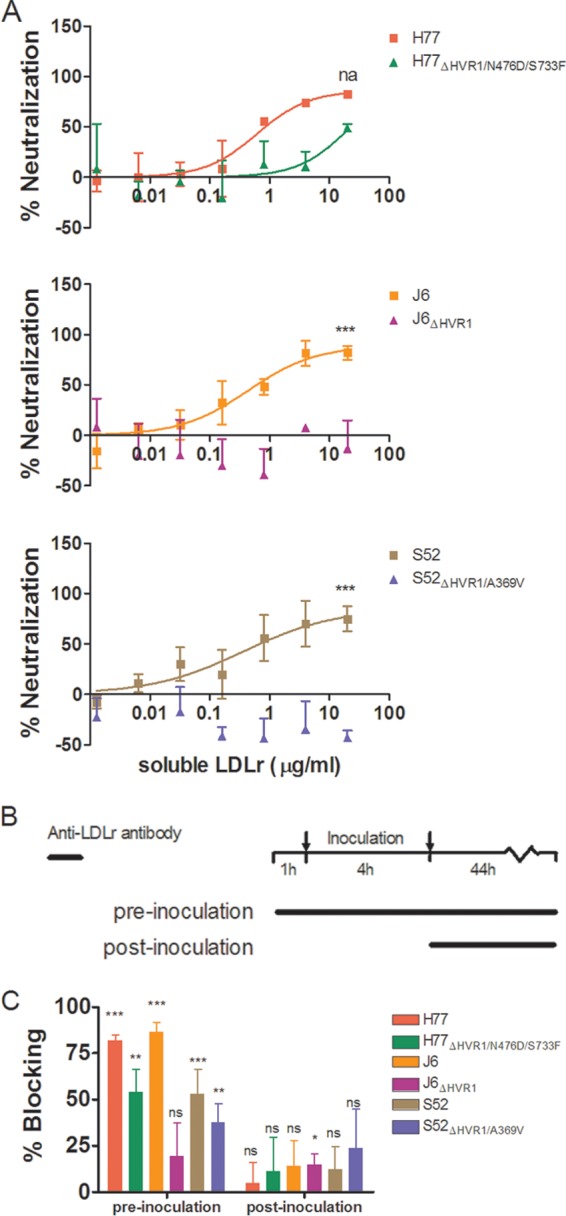
Hepatitis C virus depends on the LDLr during virus entry. (A) Huh7.5 cells were plated in 96-well plates. The following day, a dilution series of soluble LDLr was prepared and incubated in triplicate with the viruses shown at the indicated concentrations. Next, the receptor and virus and 6 wells of virus only were added to the 96-well plates and incubated for 3 h prior to washing and addition of fresh medium. Following 45 h of additional incubation, the cells were fixed and stained as described in Materials and Methods. The data were normalized to the 6 replicates of virus only and analyzed using four-parameter curve fitting in GraphPad Prism v4.03. The error bars represent SD. Z-tests were used to compare the differences in maximum attained neutralization. ***, statistical significance at a P value of <0.001; na, not applicable. (B) Schematic representation of the experimental setup for time-lapse blocking of the LDLr using polyclonal antibody AF2148. (C) Huh7.5 cells were plated in 96-well plates. The following day, a dose of 10 μg/ml of AF2148 was added to 4 wells for each tested virus either 1 h prior to the 4-h infection (and added again following the 4-h infection) or only after the 4-h infection. The data were normalized to 6 replicates of virus only, and the error bars represent SD. t tests were performed relative to virus only for pre- and postinoculation addition of blocking antibody. *, **, and ***, statistical significance at P values of < 0.05, 0.01, and 0.001, respectively; ns, not statistically significant.
Blocking of HCV infection by monoclonal antibodies targeting LDLr regions differently involved in ApoB and ApoE binding.
To further substantiate the HVR1-related differences in dependency on LDLr during viral infection and to better understand how the viral particle interacts with the receptor, we performed antibody-blocking experiments with polyclonal anti-LDLr antibodies and with monoclonal antibodies (MAbs) targeting three distinct regions of the LDLr ligand binding domain (LBD), consisting of seven semiconserved repeats (R1 to R7). MAb 5G2 (R5 specific) blocks both ApoE- and ApoB-dependent LDLr association, and 6E2 (R7 specific) specifically blocks ApoB-dependent association, while the antibody 3D8 (R1/2 specific) does not efficiently block interaction with either molecule (35). All three MAbs have been shown to have high affinity for the LDLr by surface plasmon resonance measurements (35). We confirmed the abilities of these antibodies to decrease LDLr-mediated uptake of fluorescently labeled LDL particles (Fig. 7A and data not shown). In HCV-blocking assays using a single high antibody concentration, we observed blocking using the polyclonal anti-LDLr antibody similar to that seen at the highest doses in the dose-response experiments described above (Fig. 4C and 7B). However, of the three MAbs, the most efficient blocking of HCV infection was observed with 3D8 (Fig. 7D), which does not block ApoE- or ApoB-dependent LDLr association. These results indicate that the interaction between HCV and LDLr probably did not depend on the presence of ApoE or ApoB on the surface of the virion. The high background blocking for S52ΔHVR1/A369V was evaluated in a repeat experiment for S52 and S52ΔHVR1/A369V in which we observed no effect of the isotype controls against either virus (data not shown). Since the experiment was carried out at a relatively high antibody concentration, we performed dose-response blocking as described above using MAb 3D8 (Fig. 8A to C). As expected, we observed dose-dependent blocking of infection of up to 75 to 85% for parental H77, J6, and S52 and reduced efficiency of blocking for the three HVR1-deleted variants (Fig. 8A to C). The relatively small differences in LDLr dependency between H77 and H77ΔHVR1/N476D/S733F may explain why this difference was not observed in the previous experiment using a single high concentration of 3D8 (Fig. 7D). Thus, our results suggest that the interaction between the LDLr and HCV does not depend on repeats in the LBD involved in ApoB and ApoE binding but possibly depends on R1/2.
FIG 7.
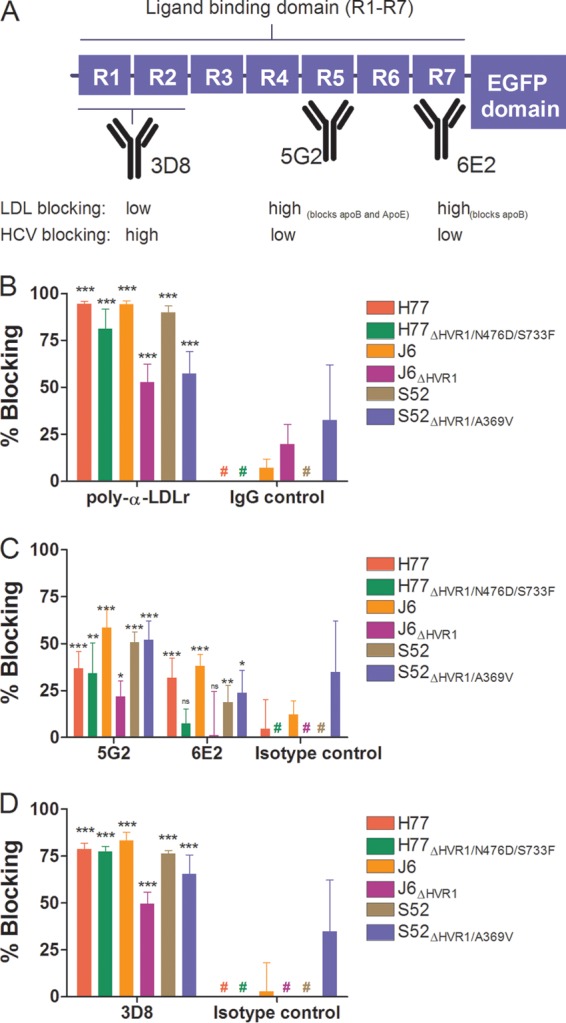
Monoclonal antibodies to three distinct regions of the LDLr ligand binding domain revealed that ApoB and ApoE are unlikely candidates for interaction between LDLr and HCV. (A) Schematic representation of LDLr with emphasis on the seven repeats (R1 to R7) of the ligand binding domain. The specificities of three MAbs are depicted, along with their abilities to decrease native LDL binding or HCV infection (cf. panels C and D). (B to D) Huh7.5 cells were plated in 96-well plates. The following day, the indicated LDLr blocking antibody (20 μg/ml of AF2148 [B], 50 μg/ml of 5G2 or 6E2 [C], or 50 μg/ml of 3D8 [D]) was added to the cells for 1 h at 37°C in 6 replicates, along with 6 replicates with only medium and 6 replicates with the relevant isotype controls (goat IgG, mouse IgG1, and mouse IgG2a). The indicated virus with and without HVR1 was added to the cells for 3 h of infection, and the cells were washed once with PBS and incubated for an additional 45 h in complete medium prior to HCV-specific staining. FFU counts at the given antibody concentrations were normalized to the mean FFU count of the 6 replicates of virus only. The data points are means of 6 replicates with SD. #, value below 0%. t tests were performed comparing the indicated LDLr-specific antibodies with virus only. *, **, and ***, statistical significance at P values of <0.05, 0.01, and 0.001, respectively; ns, not statistically significant.
FIG 8.
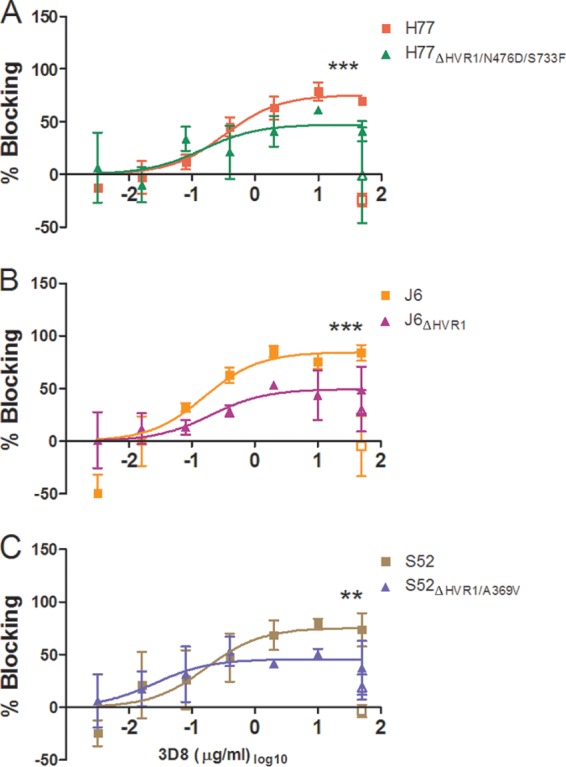
Dose-response blocking of H77, J6, and S52 with and without HVR1 with LDLr-specific monoclonal antibody 3D8. Huh7.5 cells were plated in 96-well plates. The following day, the indicated concentrations of blocking antibody or a single high dose of control antibody was incubated with the cells for 1 h at 37°C (each concentration in three replicates), including 6 wells without antibody. Following 3 h of infection, washing, and an additional 45 h of incubation, the plates were stained with 9E10 antibody for H77 and H77ΔHVR1/N476D/S733F (A), J6 and J6ΔHVR1 (B), or S52 and S52ΔHVR1/A369V (C). Blocking data are shown as dose-response curves with the means and SD of three replicates at the given antibody dilution normalized to 6 replicates of virus only. The open symbols represent data for the control antibody only tested at the highest antibody concentration. Four-parameter nonlinear curve regression was used to fit a curve to the data points (Graphpad Prism, v4.03). Z-tests were used to compare the differences in maximum attained blocking. ** and ***, statistical significance at P values of < 0.01 and <0.001, respectively.
HVR1 deletion did not alter sensitivity to ApoE-specific neutralization or efficiency of immunoprecipitation with ApoE antibodies.
Although previous reports suggested that ApoE association of HCV was involved in virus interactions with lipoprotein receptors SR-BI and LDLr (21, 44, 45), our data suggested this was not necessarily the case. To investigate this further, we examined if the decrease in the dependency of HVR1-deleted viruses on these receptors could be linked with a decrease in virus-associated ApoE. Therefore, we performed neutralization experiments using a MAb against ApoE known to block the receptor-binding site of the molecule (36). The neutralization experiment, performed as described above, showed 70 to 80% neutralization at the highest antibody concentration used for H77, J6, and S52 with and without HVR1 (Fig. 9A). We utilized ApoE-specific immunoprecipitation of HCV to investigate if neutralization was a direct effect of antibody binding to virus-associated ApoE and not merely of blocking important indirect functions of non-virus-associated ApoE in HCV infection. Using the same ApoE antibody bound to protein G-coupled beads and measuring the amount of bead-associated HCV RNA by RT-qPCR, we observed efficient particle pulldown with the six viruses with and without HVR1 (Fig. 9B), suggesting that ApoE was similarly associated with the virus irrespective of HVR1 deletion. Thus, our data support the idea that altered ApoE association of HVR1-deleted viruses does not explain the observed decrease in SR-BI or LDLr dependency.
FIG 9.
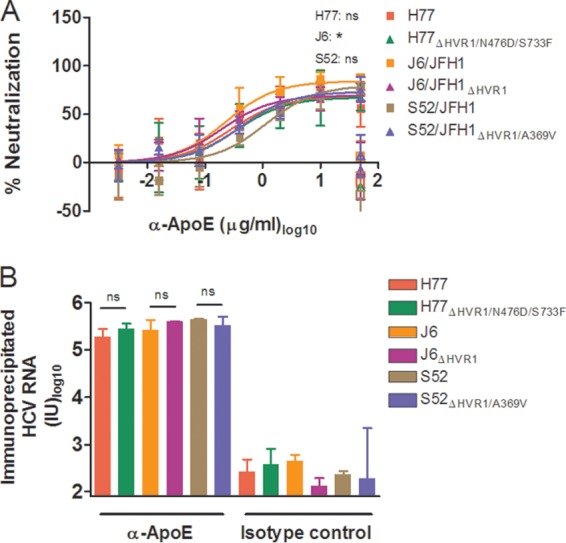
ApoE-specific neutralization and ApoE HCV particle association were similar for viruses with and without HVR1. (A) H77, J6, or S52 HCV with and without HVR1 was incubated for 1 h at 37°C in four replicates with anti-ApoE MAb (1D7) in the indicated dilution series, along with four replicates with 50 μg/ml of control antibody (mouse IgG1) and 6 replicates with medium only. The previous day, Huh7.5 cells were plated in 96-well plates. These cells were incubated with the above-mentioned virus or virus-antibody mixtures, washed after 3 h, and incubated for an additional 45 h with complete medium prior to HCV staining. FFU counts at the given antibody dilutions were normalized to the mean FFU count of the 6 replicates of virus only. The data points are means and SD of four replicates. The open symbols represent the control antibody only tested at the highest antibody concentration. Four-parameter nonlinear curve regression was used to fit the data points. Z-tests were used to compare the differences in maximum attained blocking for each antibody. Pairwise comparisons were made between viruses of the same isolate. *, statistical significance at a P value of <0.05; ns, not statistically significant. (B) ApoE-specific immunoprecipitation was carried out as described in Materials and Methods using ApoE-specific antibody (1D7) or relevant IgG1 mouse control antibody, and the amounts of immunoprecipitated HCV RNA were measured by RT-qPCR in duplicate on the eluted fractions in a LightCycler with primers and protocols that have been described previously (29). The results are shown as the total amount of HCV RNA in each sample (cutoff, 80 IU). The error bars represent SD. t tests were performed comparing HCV RNA immunoprecipitation for HCV with and without HVR1. ns, not statistically significant.
DISCUSSION
The present study addresses the role of HVR1 in the HCV life cycle and focuses on the importance of the motif in mediating viral interactions with SR-BI and LDLr. These issues are highly relevant for understanding the complex entry process of HCV, which could prove to be a key element in the development of novel therapies. The study was conducted using cell culture-adapted JFH1-based Core-NS2 recombinant viruses of isolates H77 (1a), J6 (2a), and S52 (3a) (3, 28–30) and the corresponding Huh7.5 cell culture-viable HVR1-deleted viruses H77ΔHVR1/N476D/S733F, J6ΔHVR1, and S52ΔHVR1/A369V, as well as relevant HCVpps.
For H77, we found that HVR1 deletion decreased infectious HCV production in entry-deficient S29 cells (Fig. 1), while HCV intra- and extracellular Core levels were unaffected. Furthermore, HVR1 deletion significantly decreased HCVpp entry efficacy, indicating that viral entry was affected (Fig. 2). Interestingly, N476D/S733F rescued the infectivity of H77ΔHVR1 for both JFH1-based HCV and HCVpps. In contrast, the E1 mutation A369V, which adapted HVR1-deleted JFH1-based S52 in Huh7.5 cells, decreased the entry efficacy of S52 HCVpps in Huh7.5 cells. Furthermore, while A369V increased intracellular infectious-virus production of S52ΔHVR1 in entry-deficient S29 cells, no effect was observed for released extracellular infectious HCV (Fig. 1 and 2). However, our data confirmed our previous finding that A369V is important for viral spread in Huh7.5 cells (Fig. 3A).
We found that the mutation A369V slightly increased cell-to-cell spread of HVR1-deleted S52 (Fig. 3). However, it should be noted that in the same assay we observed greatly increased cell-free spread of S52ΔHVR1/A369V compared with S52ΔHVR1, suggesting that increased cell-to-cell spread did not entirely explain the increased infectivity of S52ΔHVR1/A369V observed in Huh7.5 cells. Furthermore, J6ΔHVR1 and S52ΔHVR1/A369V exhibited comparable levels of overall and cell-to-cell spread, suggesting that the increased cell-to-cell spread of S52ΔHVR1/A369V might simply be an indirect effect of an increase in overall infectivity. Others have developed more direct assays for measuring cell-to-cell spread (46, 47), and they would be relevant to further study this aspect of HCV infection and spread in future studies. It is important to recognize that differences between the current in vitro HCV culture model systems (i.e., Huh7.5 infectious culture systems, S29 single-cycle production assay, and HCVpps) likely exist, such as differences in E1-E2 interactions between JFH1-based HCV recombinants and HCVpps (48). However, HCVpp experiments and S29 HCV production assays of S52ΔHVR1 recombinants suggest that A369V might not simply increase entry of HCV in Huh7.5 cells. The fact that intracellular infectivity titers from S29 assays of S52ΔHVR1 are low whereas extracellular titers are high suggests that the infectivity of the particle might depend on cell-specific properties of relevance during assembly/release. It is therefore conceivable that differences between Huh7.5 cells and S29 cells render S52ΔHVR1 dependent on the A369V mutation for this process during assembly/release only in Huh7.5 cells. Such a mechanism would also fit with the observation that the mutation is not adaptive in the HCVpp model, in which the assembly process differs widely from viral production in the JFH1-based HCV system.
Previous findings, showing that HVR1-deleted viruses had increased density, suggest an alteration in the lipid composition of the viral particle, which could conceivably result in altered apolipoprotein association (3, 26). Furthermore, the HVR1-deleted Jc1 virus no longer required the lipoprotein receptor SR-BI for infection (26). Therefore, using receptor blocking and gene silencing, we investigated the role of HVR1 in dependency of infection on lipoprotein and the HCV receptors SR-BI and LDLr in comparison to dependency on the late-stage receptor CD81 (Fig. 4 and 5). We found that CD81 dependencies were similar for HCV genotypes 1 to 3 irrespective of HVR1 deletion, as shown by both antibody blocking and gene silencing. Analyzing SR-BI dependency for the three parental viruses, we observed small differences in their sensitivities to blocking antibodies. Furthermore, both H77ΔHVR1/N476D/S733F and S52ΔHVR1/A369V completely lost SR-BI dependency, as previously reported for HVR1-deleted Jc1 by Bankwitz et al. (26). However, no such loss was observed for J6ΔHVR1. This discrepancy is especially striking, since the only difference between J6 and Jc1 is the NS2 junction between the J6 and the JFH1 sequences. Analyzing the effects of the adaptive envelope mutations N476D/S733F and A369V on SR-BI dependency, we found that S52A369V completely lost SR-BI dependency, while it was greatly decreased for H77N476D/S733F (Fig. 4). Envelope mutations with similar effects on SR-BI dependency and neutralization susceptibility have been reported by others (49–51), suggesting that these mutations also might be linked to the role of HVR1 in entry. Overall, it seemed that dependency on both lipoprotein receptors, SR-BI and LDLr, was either indirectly or directly linked with the role of HVR1 in viral entry, whereas dependency on CD81 was not. Analyzing LDLr dependency, we found that all parental viruses could be blocked up to 90% using high concentrations of antibody, whereas HVR1 deletion consistently conferred decreased dependency on LDLr. Others have reported a decrease in HCV replication by antibodies against the LDLr (22). To address whether the observed effect on HCV infection by LDLr-specific antibodies was at the entry step, we performed soluble LDLr neutralization of HCV (Fig. 6A) and time-lapse antibody blocking of infection in which the antibody was added before or after infection of the cells (Fig. 6B and C). Both experiments indicated that LDLr was involved at the entry step of the viral life cycle and suggested that HVR1 might be directly involved in the interaction with the LDLr.
SR-BI and LDLr both interact with their native ligands through apolipoproteins (35, 52). LDLr interacts with ApoB on native LDL particles, while LDLr and SR-BI receptors can both interact with ApoE (52). ApoE has been shown to be present on the surface of the HCV particle (13), and the isoforms of ApoE with the highest affinity to LDLr also confer the greatest increase in HCV infectivity during assembly and release (14). In addition, buoyant density centrifugation of HCV particles revealed an association between the density of ApoE-containing HCV particles, HCV infectivity, and the LDLr requirement (21). However, we found that genotype 1 to 3 viruses with and without HVR1, irrespective of the decreased LDLr dependency of HVR1-deleted viruses, were comparably sensitive to neutralization with ApoE-specific antibodies, as well as to HCV immunoprecipitation with anti-ApoE antibodies (Fig. 9). The interaction of ApoE and ApoB with the seven semiconserved repeats in the LBD has been extensively studied (35). Using MAbs with similar affinities for the LDLr but targeting different repeats of the LBD of LDLr, we investigated whether the ApoE and/or ApoB binding sites on LDLr were involved in interactions with HCV. As expected, decrease in the uptake of the native ligand, LDL, was significant only with MAbs that specifically block the ApoB and ApoB/ApoE interactions with the receptor. The decreases in LDL uptake in Huh7.5 cells were similar to those in a previous report using LDLr-expressing fibroblasts (35). We did observe higher residual binding/uptake of LDL, but it might be due to Huh7.5 cells expressing other receptors able to bind LDL. Interestingly, we found that the antibody with the least effect on ApoB- and ApoE-related binding was the most effective in blocking HCV infection, indicating that ApoE was not the principal interacting partner between HCV and LDLr (Fig. 7). However, the observation that ApoE-specific neutralization and ApoE particle immunoprecipitation were similar for HCV recombinants no longer requiring SR-BI (i.e., H77ΔHVR1/N476D/S733F and S52ΔHVR1/A369V [Fig. 4B]) is not sufficient to conclude that virus-associated ApoE did not interact with SR-BI during viral entry. This is because antibodies binding to ApoE on the surface of the virion could bind and neutralize the virus even if ApoE no longer served a purpose in SR-BI binding. Also, recent studies have shown that the HCV interaction with SR-BI is probably multimodal (25, 53), which might explain why we observed differences in SR-BI dependency for HVR1-deleted viruses (Fig. 4B).
In summary, HVR1 deletion and HVR1 deletion adaptive envelope mutations did not affect the amount and trafficking of Core in the H77 viral life cycle from replication to release, whereas they had a significant impact on the infectivity of virus particles and thus viral entry. We speculate that the role of the A369V mutation for adapting HVR1-deleted S52 in Huh7.5 cells might be linked with increasing infectivity during the assembly/release stages of the viral life cycle. However, this conclusion must be tempered by recognizing the limitations of the model systems used to study the role of the mutation. In addition, HVR1 deletion conferred a consistent decrease in LDLr dependency during viral entry, whereas only HVR1-deleted viruses depending on additional envelope mutations had decreased SR-BI dependency. As it appeared that the genotype 1 to 3 viruses with and without HVR1 were similarly associated with ApoE, the presence or absence of this molecule was probably not the explanation for these observations. Furthermore, our studies indicated that ApoE was not likely to mediate interaction between HCV and LDLr (14, 21), as the repeats of the LBD of LDLr confirmed to be involved in ApoB-ApoE interactions were not greatly involved in the HCV-LDLr interaction. In combination with reports of the involvement of virion-associated ApoE in HCV cell attachment, our findings suggest that LDLr plays a critical role in HCV entry but is not important for attachment, as was also recently suggested by others (18). While our data confirm an important role for HVR1 in early receptor interactions with SR-BI and LDLr, but not for the late-stage receptor CD81, it would be of interest to conduct further studies investigating whether HVR1 might influence other late-stage receptor interactions. Our study suggests that HVR1 is primarily involved in viral entry and that it modulates LDLr dependency directly, while SR-BI dependency is, at least partly, indirectly modulated by the adaptive envelope mutations required for some HVR1-deleted viruses.
ACKNOWLEDGMENTS
We are grateful to Lotte Mikkelsen (Copenhagen University Hospital, Hvidovre, Copenhagen, Denmark) for technical assistance; to Steen Ladelund (Copenhagen University Hospital, Hvidovre, Copenhagen, Denmark) for statistical advice; to Jens Ole Nielsen, Bjarne Ørskov Lindhardt, and Ove Andersen (Copenhagen University Hospital, Hvidovre, Copenhagen, Denmark) for their support of the project; to Mansun Law (Scripps Institute, USA), Robert Milne and Anna Toma (Ottawa Heart Institute, Canada), Charles Rice (Rockefeller University, USA), Robert Purcell and Suzanne Emerson (National Institutes of Health, USA), Jean Dubuisson (CNRS, France), François-Loïc Cosset (CNRS, France), and Takaji Wakita (National Institute of Infectious Diseases, Japan) for providing reagents; and to Henrik Krarup (Aalborg University Hospital, Aalborg, Denmark) for performing the Abbott HCV Core antigen analyses.
This study was supported by Ph.D. stipends from the Faculty of Health Sciences, University of Copenhagen (J.P. and S.B.N.S.); individual DFF-postdoctoral grants from the Danish Council for Independent Research, Medical Sciences (J.P. and S.R.); and research grants from the Lundbeck Foundation (J.P. and J.B.), the Danish Cancer Society (J.M.G. and J.B.), the Novo Nordisk Foundation (J.M.G. and J.B.), and the A. P. Møller and Chastine McKinney Møllers Medical Research Foundation (J.M.G. and J.B.).
Footnotes
Published ahead of print 20 November 2013
REFERENCES
- 1.Alter HJ, Seeff LB. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 20:17–35. 10.1055/s-2000-9505 [DOI] [PubMed] [Google Scholar]
- 2.Gottwein JM, Bukh J. 2008. Cutting the Gordian knot-development and biological relevance of hepatitis C virus cell culture systems. Adv. Virus Res. 71:51–133. 10.1016/S0065-3527(08)00002-X [DOI] [PubMed] [Google Scholar]
- 3.Prentoe J, Jensen TB, Meuleman P, Serre SB, Scheel TK, Leroux-Roels G, Gottwein JM, Bukh J. 2011. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 85:2224–2234. 10.1128/JVI.01594-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavillette D, Tarr AW, Voisset C, Donot P, Bartosch B, Bain C, Patel AH, Dubuisson J, Ball JK, Cosset FL. 2005. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 41:265–274. 10.1002/hep.20542 [DOI] [PubMed] [Google Scholar]
- 5.Scheel TK, Prentoe J, Carlsen TH, Mikkelsen LS, Gottwein JM, Bukh J. 2012. Analysis of functional differences between hepatitis C virus NS5A of genotypes 1–7 in infectious cell culture systems. PLoS Pathog. 8:e1002696. 10.1371/journal.ppat.1002696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith LW, Wilson GK, Fletcher NF, McKeating JA. 2012. Hepatitis C virus entry: beyond receptors. Rev. Med. Virol. 22:182–193. 10.1002/rmv.723 [DOI] [PubMed] [Google Scholar]
- 7.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 8.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cormier EG, Tsamis F, Kajumo F, Durso RJ, Gardner JP, Dragic T. 2004. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 101:7270–7274. 10.1073/pnas.0402253101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farquhar MJ, Hu K, Harris HJ, Davis C, Brimacombe CL, Fletcher SJ, Baumert TF, Rappoport JZ, Balfe P, McKeating JA. 2012. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 86:4305–4316. 10.1128/JVI.06996-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. 2008. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 82:8316–8329. 10.1128/JVI.00665-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793. 10.1128/JVI.01091-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 84:12048–12057. 10.1128/JVI.01063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cun W, Jiang J, Luo G. 2010. The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84:11532–11541. 10.1128/JVI.01021-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maillard P, Huby T, Andreo U, Moreau M, Chapman J, Budkowska A. 2006. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/ClaI is mediated by ApoB-containing lipoproteins. FASEB J. 20:735–737. 10.1096/fj.05-4728fje [DOI] [PubMed] [Google Scholar]
- 17.Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T. 2012. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 56:484–491. 10.1002/hep.25665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86:7256–7267. 10.1128/JVI.07222-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi F, Yamada S, Taguwa S, Kataoka C, Naito S, Hama Y, Tani H, Matsuura Y, Sugahara K. 2012. Specific interaction of the envelope glycoproteins E1 and E2 with liver heparan sulfate involved in the tissue tropismatic infection by hepatitis C virus. Glycoconj. J. 29:211–220. 10.1007/s10719-012-9388-z [DOI] [PubMed] [Google Scholar]
- 20.Shi Q, Jiang J, Luo G. 2013. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 87:6866–6875. 10.1128/JVI.03475-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen DM, Huang H, Ye J, Gale M., Jr 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. 10.1016/j.virol.2009.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J. 2012. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55:998–1007. 10.1002/hep.25501 [DOI] [PubMed] [Google Scholar]
- 23.Ploss A, Evans MJ. 2012. Hepatitis C virus host cell entry. Curr. Opin. Virol. 2:14–19. 10.1016/j.coviro.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, Pawlotsky JM, Lavillette D, Cosset FL. 2005. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 79:8217–8229. 10.1128/JVI.79.13.8217-8229.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dao Thi VL, Granier C, Zeisel MB, Guerin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, Cosset FL, Dreux M. 2012. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 287:31242–31257. 10.1074/jbc.M112.365924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY, Foung SK, Pecheur EI, Pietschmann T. 2010. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 84:5751–5763. 10.1128/JVI.02200-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forns X, Thimme R, Govindarajan S, Emerson SU, Purcell RH, Chisari FV, Bukh J. 2000. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. U. S. A. 97:13318–13323. 10.1073/pnas.230453597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 29.Gottwein JM, Scheel TK, Hoegh AM, Lademann JB, Eugen-Olsen J, Lisby G, Bukh J. 2007. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133:1614–1626. 10.1053/j.gastro.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 30.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U. S. A. 105:997–1002. 10.1073/pnas.0711044105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642. 10.1084/jem.20021756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meunier JC, Engle RE, Faulk K, Zhao M, Bartosch B, Alter H, Emerson SU, Cosset FL, Purcell RH, Bukh J. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. U. S. A. 102:4560–4565. 10.1073/pnas.0501275102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serre SB, Krarup HB, Bukh J, Gottwein JM. 2013. Identification of IFN-alpha induced envelope mutations of hepatitis C virus in vitro associated with increased viral fitness and interferon resistance. J. Virol. 87:12776–12793. 10.1128/JVI.00901-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meuleman P, Catanese MT, Verhoye L, Desombere I, Farhoudi A, Jones CT, Sheahan T, Grzyb K, Cortese R, Rice CM, Leroux-Roels G, Nicosia A. 2012. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 55:364–372. 10.1002/hep.24692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen AT, Hirama T, Chauhan V, Mackenzie R, Milne R. 2006. Binding characteristics of a panel of monoclonal antibodies against the ligand binding domain of the human LDLr. J. Lipid Res. 47:1399–1405. 10.1194/jlr.M600130-JLR200 [DOI] [PubMed] [Google Scholar]
- 36.Weisgraber KH, Innerarity TL, Harder KJ, Mahley RW, Milne RW, Marcel YL, Sparrow JT. 1983. The receptor-binding domain of human apolipoprotein E. Monoclonal antibody inhibition of binding. J. Biol. Chem. 258:12348–12354 [PubMed] [Google Scholar]
- 37.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- 38.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68:6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, Dubuisson J. 2011. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J. Virol. 85:1777–1792. 10.1128/JVI.02170-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlsen TH, Scheel TK, Ramirez S, Foung SK, Bukh J. 2013. Characterization of hepatitis C virus recombinants with chimeric E1/E2 envelope proteins and identification of single amino acids in the E2 stem region important for entry. J. Virol. 87:1385–1399. 10.1128/JVI.00684-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheel TK, Gottwein JM, Carlsen TH, Li YP, Jensen TB, Spengler U, Weis N, Bukh J. 2011. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J. Virol. 85:2891–2906. 10.1128/JVI.01605-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 105:4370–4375. 10.1073/pnas.0800422105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roccasecca R, Ansuini H, Vitelli A, Meola A, Scarselli E, Acali S, Pezzanera M, Ercole BB, McKeating J, Yagnik A, Lahm A, Tramontano A, Cortese R, Nicosia A. 2003. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 77:1856–1867. 10.1128/JVI.77.3.1856-1867.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A. 2010. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J. Virol. 84:34–43. 10.1128/JVI.02199-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazumdar B, Banerjee A, Meyer K, Ray R. 2011. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54:1149–1156. 10.1002/hep.24523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert TF, Tellinghuisen TL, Wong-Staal F, Balfe P, McKeating JA. 2011. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 85:596–605. 10.1128/JVI.01592-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. 10.1002/hep.21959 [DOI] [PubMed] [Google Scholar]
- 48.Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. 2010. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 84:10159–10168. 10.1128/JVI.01180-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koutsoudakis G, Dragun J, Perez-del-Pulgar S, Coto-Llerena M, Mensa L, Crespo G, Gonzalez P, Navasa M, Forns X. 2012. Interplay between basic residues of hepatitis C virus glycoprotein E2 with viral receptors, neutralizing antibodies and lipoproteins. PLoS One 7:e52651. 10.1371/journal.pone.0052651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tao W, Xu C, Ding Q, Li R, Xiang Y, Chung J, Zhong J. 2009. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology 395:67–76. 10.1016/j.virol.2009.09.006 [DOI] [PubMed] [Google Scholar]
- 51.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. 2008. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J. Virol. 82:12020–12029. 10.1128/JVI.01569-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Kan HY, Lavrentiadou S, Krieger M, Zannis V. 2002. Reconstituted discoidal ApoE-phospholipid particles are ligands for the scavenger receptor BI. The amino-terminal 1–165 domain of ApoE suffices for receptor binding. J. Biol. Chem. 277:21149–21157. 10.1074/jbc.M200658200 [DOI] [PubMed] [Google Scholar]
- 53.Zahid MN, Turek M, Xiao F, Dao TV, Guerin M, Fofana I, Bachellier P, Thompson J, Delang L, Neyts J, Bankwitz D, Pietschmann T, Dreux M, Cosset FL, Grunert F, Baumert TF, Zeisel MB. 2013. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 57:492–504. 10.1002/hep.26097 [DOI] [PubMed] [Google Scholar]