

Abstract
The host eIF4F translation initiation complex plays a critical role the translation of capped mRNAs. Although human cytomegalovirus (HCMV) infection increases the abundance and activity of the host eIF4F complex, the requirement for eIF4F components in HCMV replication and mRNA translation has not been directly tested. In this study, we found that decreasing the abundance or activity of eIF4F from the start of infection inhibits HCMV replication. However, as infection progresses, viral mRNA translation and replication becomes increasingly resistant to eIF4F inhibition. During the late stage of infection the association of representative immediate-early, early, and late mRNAs with polysomes was not affected by eIF4F disruption. In contrast, eIF4F inhibition decreased the translation of representative host eIF4F-dependent mRNAs during the late stage of infection. A global analysis of the translation efficiency of HCMV mRNAs during the late stage of infection found that eIF4F disruption had a minimal impact on the association of HCMV mRNAs with polysomes but significantly diminished the translation efficiency of eIF4F-dependent host transcripts. Together, our data show that the translation of host eIF4F-dependent mRNAs remains dependent on eIF4F activity during HCMV infection. However, during the late stage of infection the translation efficiency of viral mRNAs does not correlate with the abundance or activity of the host eIF4F complex.
INTRODUCTION
As obligate intracellular parasites, viruses are reliant on cellular processes for their replication. At a minimum, viruses require host ribosomes to synthesize viral proteins. While host cells have evolved to limit mRNA translation during infection, viruses have evolved to limit host control of protein synthesis in infected cells to maximize viral protein expression. Thus, the interaction of viral mRNAs with the host translation machinery represents a fundamental aspect of the host-pathogen interface.
The recruitment of a ribosome to host mRNAs occurs through an ordered assembly of translation factors on the 5′ terminus of the message. In particular, the eIF4F complex is a critical host translation initiation complex required for the efficient recruitment of ribosomes to mRNAs containing a 7-methylguanosine cap (m7G cap) on their 5′ terminal nucleotide (1–3). Most cellular messages are capped (4), and therefore the eIF4F complex is thought to be required for the translation of the majority of cellular mRNAs. The three components of eIF4F each play specialized roles in translation initiation (5). The eIF4E protein nucleates the assembly of the eIF4F complex by binding to the m7G mRNA cap (6). eIF4E recruits the eIF4G scaffold protein, which in turn recruits the eIF4A RNA helicase to complete the eIF4F complex. The eIF4A helicase stimulates translation initiation by resolving secondary structures in the 5′ untranslated region (5′UTR) of mRNAs, thereby facilitating ribosomal scanning to the initiating codon (7, 8). Recruitment of the eIF4F complex to the m7G cap is a rate-limiting step in the initiation of mRNA translation, and reducing the amount of eIF4F complex results in a global decrease in protein synthesis.
Herpesviruses do not encode obvious homologs of eIF4F subunits, and herpesvirus mRNAs are thought to be translated in a cap-dependent manner, although cap-independent translation has been described for a limited number of herpesvirus mRNAs (9–12). This is based in part on studies showing that human cytomegalovirus (HCMV) infection induces the accumulation of eIF4F subunits (13). In addition, HCMV infection activates signaling pathways that stimulate eIF4F complex formation. For example, HCMV activates the mTOR kinase (13–16). Active mTOR facilitates eIF4F complex formation by phosphorylating and antagonizing the translation repressor 4EBP-1, which prevents the formation of eIF4F on the mRNA cap (17, 18). HCMV also stimulates the ERK and MEK kinase cascades, resulting in the phosphorylation of eIF4E by the Mnk1/2 kinases (13). In sum, these events ensure that the eIF4F complex remains active during infection despite the induction of a cellular stress response.
These findings suggest that HCMV infection increases eIF4F activity to stimulate the translation of viral mRNAs. However, several recent studies have suggested a more complicated role for eIF4F during herpesvirus infection (19). For example, while inhibiting the mTOR kinase from the start of HCMV infection disrupts the eIF4F complex and limits virus replication, some HCMV mRNAs continue to be efficiently translated (16). As infection progresses, both total protein synthesis and virus replication become increasingly resistant to the effects of mTOR inhibitors, despite significant disruption of the eIF4F complex (19). This suggests that mTOR has additional eIF4F-independent roles in virus replication, perhaps in the metabolic remodeling of HCMV-infected cells (20). These data suggest that the eIF4F complex may not be required for viral mRNA translation during the later stages of infection. However, the requirement for eIF4F activity for HCMV protein synthesis has not been directly assessed.
In this study, we measured the impact of eIF4F inhibition on host and viral mRNA translation during HCMV infection. Inhibiting eIF4F at the time of infection limits progression through the viral lytic cycle despite the efficient expression of HCMV immediate-early proteins. After the onset of viral DNA replication both viral protein synthesis and replication became increasingly resistant to eIF4F inhibition. We found that disrupting the eIF4F complex inhibited the association of host mRNAs with polysomes and consequently limited host protein synthesis during HCMV infection. In contrast, eIF4F inhibition had a minimal effect on the synthesis of representative viral proteins from each kinetic class and did not affect the association of the corresponding viral mRNAs with polysomes. Global analysis of the translation efficiency of viral transcripts revealed robust translation of HCMV mRNAs despite a significant reduction in eIF4F abundance. Our data suggest a differential requirement for eIF4F activity for the translation of host and viral mRNAs during the late stage of infection. Although host mRNAs continue to require eIF4F for their translation during infection, the association of viral mRNAs with ribosomes is insensitive to changes in eIF4F abundance or activity.
MATERIALS AND METHODS
Cells, viruses, and inhibitors.
Primary human foreskin fibroblasts (HFFs) were passaged in Dulbecco modified Eagle medium (DMEM; Sigma) containing 10% newborn calf serum and used between passages 7 and 14. Unless otherwise indicated, the cells were seeded at confluence and then serum starved for 48 h prior to infection. HCMV infections were performed with the BADinGFP strain (ADGFP) (21), which contains the green fluorescent protein (GFP) open reading frame under the control of the simian virus 40 promoter inserted in the nonessential UL21.5 locus. HCMV infections were performed at a multiplicity of infection (MOI) of three in serum-free medium unless otherwise noted. Cell-free HCMV was titered by the 50% tissue culture infectious dose (TCID50) method on primary human fibroblasts. The eIF4AI/II inhibitor hippuristanol (22) was generously provided by Jerry Pelletier (McGill University). Cycloheximide, actinomycin D, and phosphonoacetic acid (PAA), all obtained from Sigma, were used at final concentrations of 100, 5, and 200 μg/ml, respectively. Unless otherwise noted, Torin1 (Tocris) was used at a final concentration of 250 nM.
Western blot analysis of host and viral proteins.
Cell pellets were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) containing protease inhibitors (Complete EDTA-free; Roche). Cells were incubated on ice in RIPA buffer for 15 min and then cleared of debris by centrifugation for 10 min at 14,000 × g. The protein concentration of each sample was determined using the Bradford reagent. Equal amounts of protein from each sample were resolved on 10% polyacrylamide gels, transferred to nitrocellulose membranes (Whatman), and then blocked for at least 1 h in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and 5% nonfat milk. Primary mouse monoclonal antibodies were diluted in TBS-T containing 1% bovine serum albumin (BSA) and incubated with the membrane for 1 h at room temperature. Rabbit polyclonal antibodies were incubated overnight at 4°C in TBS-T containing 5% BSA. After being washed with TBS-T, blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The blots were again washed in TBS-T, and proteins were visualized by treatment with enhanced chemiluminescence reagent (Amersham or Advansta) and exposure to film. The following antibodies and dilutions were used in the present study: IE1 (1:100), UL44 (1:100), pp28 (1:100), tubulin (Sigma, 1:5,000), rpS6 (1:1,000), phospho-rpS6 (1:2,000), eIF4A (1:1,000), eIF4E (1:1,000), eIF4G (1:1,000), 4EBP1 (1:1,000) from Cell Signaling, hsp90 (Enzo,1:1,000), and RACK1 (Santa Cruz, 1:1,000).
Metabolic labeling of HFFs.
At the indicated times postinfection, the medium was removed and replaced with serum-free DMEM lacking methionine and cysteine (Sigma). After a 15-min incubation, 125 μCi of 35S-labeled methionine-cysteine (EasyTag Express protein labeling mix; Perkin-Elmer) was added directly to each well. In experiments measuring the total protein synthesis the cells were labeled for 30 min. Cells were labeled for 1 h where the synthesis of specific proteins was measured. The medium was then removed, and the cells were washed three times with ice-cold phosphate-buffered saline (PBS). Cells were pelleted and stored at −80°C until analyzed. Where the effect of inhibitors was tested, the inhibitors were included in both the methionine-cysteine-free incubation and labeling periods.
Analysis of total protein synthesis.
35S-labeled cells were lysed in 100 μl of RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate) containing protease inhibitors (complete EDTA-free protease inhibitor; Roche). The protein concentration of the lysate was then determined by the Bradford assay. Fifteen microliters of cell extract was mixed with 0.1 ml of 1 mg of BSA/ml containing 0.02% (wt/vol) sodium azide (NaN3). One milliliter of 20% trichloroacetic acid (TCA) was added to the sample, and the mixture was vortexed and incubated on ice for 30 min. The precipitate was vacuum filtered onto 2.5-cm glass microfiber filters (Whatman). The filters were washed twice with 20% TCA, once with 100% ethanol, and allowed to air dry for 30 min. The amount of radioactivity retained on the filters was measured using a scintillation counter. The amount of precipitated radioactivity in each sample was normalized to the total amount of protein in the sample. In some cases, metabolically labeled proteins were visualized by resolving 10 μg of protein on SDS–10% PAGE gels. Gels were dried for 1 h and then exposed to film to visualize radiolabeled proteins.
Quantification of viral DNA.
HCMV DNA accumulation was measured essentially as described previously (23). Briefly, HFFs were seeded at confluence in six well plates, and then serum starved for 48 h prior to infection. The cells were infected at an MOI of 0.05, and inhibitors were added after removal of the inoculum. Control infected cells were treated with dimethyl sulfoxide vehicle. At 96 h postinfection (hpi), the cells were scraped from the dishes, pelleted, and frozen at −80°C. Frozen cell pellets were resuspended in 100 μl of lysis buffer (RIPA buffer) and digested overnight with proteinase K (10 mg/ml). The lysates were extracted with phenol-chloroform, and the DNA was precipitated with isopropanol. The DNA pellet was resuspended in distilled water and quantified using a NanoDrop spectrometer. Portions (500 ng) of DNA were analyzed by quantitative real-time PCR (qPCR) exactly as described previously (16) using primers specific for the HCMV major immediate-early promoter (MIEP) (see Table S1 in the supplemental material). The number of HCMV genomes in each sample was determined by comparing the results to a series of DNA standards containing from 108 to 101 HCMV genomes. To control for variations in pipetting, the results were normalized to the total amount of DNA in each sample by qPCR using primers specific for GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
Polysome isolation.
Polysomes were isolated by centrifugation through 10 to 50% linear sucrose gradients. Linear sucrose gradients were prepared by pouring sucrose step gradients and allowing them to equilibrate overnight at 4°C. Step gradients consisted of 10, 20, 30, 40, and 50% sucrose steps, each prepared in polysome gradient buffer (20 mM Tris-HCl [pH 7.4], 140 mM KCl, 5 mM MgCl2) containing 100 μg of cycloheximide/ml. Confluent serum-starved fibroblasts were infected at an MOI of 3 in serum-free media. At 72 h postinfection, the cells were treated with Torin1 or left untreated. After another 16 h, the cells were incubated in serum-free media containing cycloheximide for 10 min at 37°C. The cells were washed three times in PBS containing cycloheximide and scraped from the dishes and pelleted (5 min at 3,000 × g). The cell pellets were resuspended in polysome gradient buffer containing cycloheximide, 1 mM dithiothreitol, and 1% Triton X-100. The cells were swollen on ice for 10 min and then disrupted by five passes through a 27-gauge needle. Nuclei were removed by centrifugation for 5 min at 2,500 × g. The resulting supernatant was cleared of insoluble debris by centrifugation for 10 min at 13,000 × g. The resulting supernatant was layered onto the sucrose gradients and spun in an ultracentrifuge (Becton-Dickinson) for 2 h at 32,500 rpm in an SW41 swinging bucket rotor. The centrifuge brake was disabled to avoid disrupting the gradients during deceleration. After centrifugation, the gradients were manually fractionated from the top into 14 750-μl fractions. The bottom fraction of the gradient (fraction 15) containing any pelleted debris, was discarded. RNA was extracted from one third of each gradient fraction and resolved on 2% nondenaturing agarose gels to visualize rRNA. We routinely monitored the efficiency of fractionation by performing Western blots for the nuclear protein lamin A/C and cytoplasmic protein tubulin (data not shown). In each experiment, the efficiency of mTOR inhibition was measured by performing Western blots on whole-cell extracts using antibodies specific for either phosphorylated or total ribosomal protein S6 (rpS6).
Quantification of viral mRNA abundance.
The abundance of specific mRNAs in total cellular RNA was quantified by reverse transcriptase real-time PCR (qRT-PCR) as previously described (24). Briefly, frozen cell pellets were resuspended in TRIzol (Invitrogen). The mixture was extracted with chloroform, and the RNA was precipitated with isopropanol. RNA pellets were resuspended in RNase-free water, and RNA was quantified using a NanoDrop spectrometer. Two micrograms of RNA was reverse transcribed with a high-capacity reverse transcription kit (ABI) using random hexamers as primers. Two microliters of the resulting cDNA was mixed with gene-specific primers and SYBR green master mix and amplified in a Roche LightCycler 4800 using the following cycling parameters: 95°C for 5 min and then 40 cycles consisting of 95°C for 30 s and 60°C for 30 s. The amount of viral transcript in each sample was determined by using the ΔΔCT method, with GAPDH as the reference sample.
To quantify the abundance of mRNAs in sucrose gradient fractions, RNA was extracted from one-third of each gradient fraction. An equal volume of RNA from each fraction was reverse transcribed, and an equal volume of the cDNA reaction was analyzed by real-time PCR analysis as described above. The abundance of each transcript was determined by comparing the threshold values for each sample to a standard curve specific for each primer set. For viral transcripts, the standard curve consisted of a range of HCMV BAC DNA concentrations ranging from 108 to 101 copies. For host transcripts, the PCR product of each primer pair was cloned into the pGEM-T vector. The threshold values obtained from a series of DNA standards containing from 108 to 101 copies was used to determine the abundance of each transcript in each fraction. The total amount of each transcript in the gradient was determined by summing the copy number of the transcript across all fractions. The percentage of the mRNA present in each fraction of the gradient was then graphed to demonstrate the relative distribution of a transcript across the gradient. Primer sequences for both host and viral genes are listed in Table S1 in the supplemental material.
shRNA-mediated depletion of host mRNAs.
Lentivirus shRNA expression constructs targeting eIF4A1 were obtained from the University of North Carolina Lentivirus Core Facility. Lentivirus stocks were prepared by transfecting 293T cells with lentivirus shRNA construct together with packaging vectors. Cell-free supernatants were harvested 3 days posttransfection and filtered through a 0.45-μm-pore-size filter. One day prior to transduction, the HFFs were seeded at confluence into six-well plates. The cells were transduced with lentivirus overnight in the presence of 5 μg of Polybrene/ml. The next morning, the medium was removed, and the cells were washed with PBS. Serum-free media was then added to the wells, and the cells were incubated for an additional 48 h prior to infection with HCMV as described above.
Analysis of cap structures on viral mRNAs.
Polysome-associated mRNAs were isolated from untreated of Torin1-treated, infected cells. The RNAs were treated with calf-intestinal phosphatase to remove 5′ phosphates from uncapped RNAs and then treated with TAP to remove the m7G cap. An RNA oligonucleotide was then ligated to the decapped mRNA. All reagents were from an RLM-RACE kit (Ambion) essentially as described in the product manual. The RNA was then reverse transcribed and PCR amplified using primers specific for the viral sequence and the RNA oligonucleotide. The omission of TAP should prevent ligation of the RNA oligonucleotide if the mRNA contains a 5′ m7G cap, and therefore amplification of the cDNA would be prevented. In some samples the TAP digestion step was omitted to test for the presence of the mRNA cap structure.
Microarray analysis of translation efficiency.
Polysome-associated and total RNA were isolated as described above. Sample quantity and quality was verified by using an Agilent 2100 bioanalyzer. Total RNA and polysome RNA was labeled with Cy3 or Cy5, respectively, using the Agilent LIQA two-color labeling protocol according to the manufacturer's instructions. Samples were hybridized to a custom microarray developed on the Agilent Human GE 4x44K v2 Microarray platform. In addition to probes specific for the whole human genome, the microarray contained oligonucleotide probes specific for all of the annotated genes of the WT Merlin HCMV genome (NCBI accession no. NC_006273). Microarray hybridization, washing, and signal intensity analysis were performed according to the manufacturer's directions. The data were collected using the Feature Extraction software. The data analysis was carried out using Partek Genomics Suite software. Only probes showing positive signal in all samples were subjected to further analysis. To determine the translation efficiency of a given gene, the signal intensity from the polysomes RNA was divided by that in the total RNA. To determine the relative change in translation efficiency in the presence of Torin1, the translation efficiency of mRNA in Torin1-treated cells was divided by that in untreated cells. Statistically significant changes in translation efficiency in the presence of Torin1 were calculated using a Student paired two-tailed t test. The experiment was performed three times using RNAs derived from three independent experiments.
RESULTS
To test the requirement for eIF4F activity for the translation of viral mRNAs, we measured the effect of a specific inhibitor of the eIF4A RNA helicase on viral replication and protein synthesis. Hippuristanol is an ATP-competitive inhibitor that binds directly to the active site of eIF4AI and eIF4AII and inhibits their RNA helicase activity (22, 25). Importantly, hippuristanol is a specific inhibitor of eIF4AI and eIF4AII, and it does not affect the activity of other RNA helicases (25). Therefore, unlike other eIF4F inhibitors that target signaling pathways that control eIF4F integrity, hippuristanol directly targets the enzymatic activity of the eIF4F complex. Consistent with the requirement of eIF4A activity for the translation of most cellular mRNAs, hippuristanol inhibits protein synthesis in mammalian cells (22, 25). We found that 100 nM hippuristanol potently inhibited the total rate of protein synthesis in uninfected cells (Fig. 1A) without affecting cell viability (Fig. 1B) or mTOR signaling (Fig. 1C). We therefore used hippuristanol treatment to determine the requirement for eIF4A activity for the translation of viral mRNAs during HCMV infection.
FIG 1.
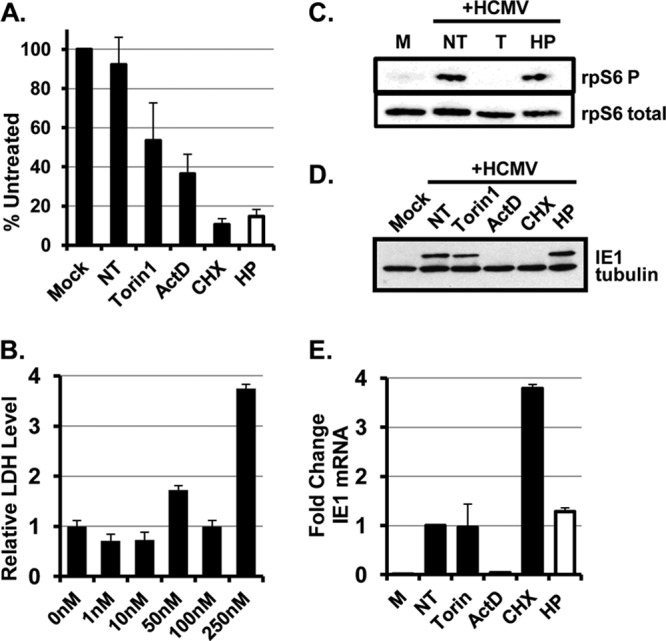
An HCMV immediate-early protein is efficiently translated in the presence of an eIF4A inhibitor. (A) HFFs were infected with HCMV (MOI of 3) and treated with vehicle (NT), Torin1 (T; 250 nM), actinomycin D (ActD; 10 μg/ml), cycloheximide (CHX; 100 μg/ml), or hippuristanol (HP; 100 nM) when the inoculum was removed. The rate of incorporation of radiolabeled amino acids into acid-insoluble protein during the final 30 min of the infection was measured (n = 3). Values are expressed as a percentage of the untreated, uninfected control. (B) Uninfected HFFs were treated with the indicated concentrations of hippuristanol for 5 days. Cell viability was determined by the LDH assay (n = 2). (C) Cells were mock infected (M) or infected and treated as for panel B. Cells were harvested at 6 h postinfection (hpi), and the levels of phosphorylated rpS6 and total rpS6 were determined by Western blotting (n = 2). (D) Same as for panel C, except the IE1 and tubulin levels were measured by Western blotting (n = 3). (E) Same as for panel D, except that RNA was harvested at 6 hpi, and IE1 mRNA abundance was measured by qRT-PCR. The abundance of IE1 mRNA in untreated cells was set to 1 (n = 3).
We first determined the effect of hippuristanol on HCMV immediate-early (IE) protein expression. Cells were serum starved for 48 h and then infected with HCMV and treated with hippuristanol at the time the inoculum was removed. Serum starvation was included to better assess the stimulatory effects of HCMV infection on eIF4F activity and to allow us to compare our results to previously published studies. The expression of HCMV immediate-early protein IE1 was then measured by Western blotting at 6 h postinfection. As expected, cycloheximide, an inhibitor of peptide elongation, prevented IE1 protein expression (Fig. 1D). Consistent with our previous studies (16), the mTOR inhibitor Torin1 did not limit IE1 protein expression. Surprisingly, hippuristanol treatment also had no effect on IE1 protein levels despite limiting total protein synthesis to a similar degree as cycloheximide. A potential explanation for this result could be that inhibiting eIF4A helicase activity resulted in a compensatory increase in IE1 mRNA transcription. However, qRT-PCR analysis of IE1 mRNA levels showed that similar amounts of the IE1 mRNA were present in untreated and hippuristanol-treated cells (Fig. 1E). Since similar amounts of IE1 mRNA and protein were made when eIF4A was inhibited, this result demonstrates that inhibiting eIF4A helicase activity does not decrease the efficiency of IE1 mRNA translation despite a global reduction in the rate of protein synthesis.
We next determined the effect of inhibiting eIF4A at the time of infection on the progression of the HCMV lytic cycle. As shown in Fig. 2A, the IE1 protein was expressed throughout the HCMV lytic cycle in the presence of hippuristanol. The expression of the HCMV early protein UL44 (pUL44) was delayed and reduced by hippuristanol treatment; however, pUL44 continued to increase in abundance as infection progressed. In contrast, the expression of pp28 was significantly diminished in hippuristanol treated cells at all times postinfection. We investigated the defect in pp28 expression in more detail by determining whether the decrease in pp28 protein levels was due to decreased transcription or translation of the pp28 mRNA. Quantitative real-time PCR analysis found that pp28 mRNA levels were reduced when hippuristanol was added to the cultures at the time of infection (Fig. 2B). Since transcription of HCMV late mRNAs is dependent on viral DNA replication, we hypothesized that the defect in pp28 transcription might be due to a requirement for eIF4A activity for efficient viral DNA accumulation. Infected cells were treated with hippuristanol at the time of infection, and viral DNA was isolated and quantified by real-time PCR at 96 h postinfection. As a control, we also measured the level of viral DNA in cells treated with PAA, a well-described inhibitor of HCMV DNA replication. Hippuristanol and PAA similarly reduced viral DNA accumulation by approximately 2 orders of magnitude (Fig. 2C). Therefore, the reduction in pp28 expression when hippuristanol is added at infection is due to a requirement for eIF4A activity for viral DNA replication.
FIG 2.
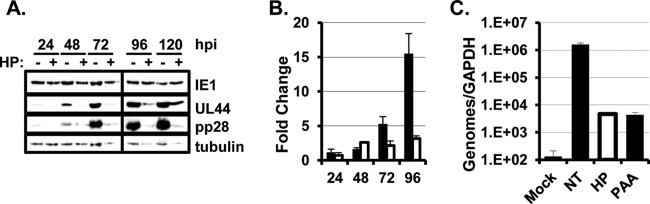
Hippuristanol inhibits progression through the HCMV lytic cycle. (A) Serum-starved confluent HFFs were infected with HCMV at an MOI of 3. Cells were left untreated or treated with hippuristanol (HP; 100 nM) when the inoculum was removed. Cells were harvested at 24-h intervals, and viral protein expression was measured by Western blotting (n = 3). (B) Cells were infected and treated as in panel A, and pp28 mRNA abundance was determined by qRT-PCR (n = 3). The fold change in pp28 mRNA levels relative to the 24-h time point is shown. Bars: ■, untreated samples; □, HP treated samples. (C) Serum-starved confluent HFFs were infected at an MOI of 0.05. Hippuristanol (HP; 100 nM) or phosphonoacetic acid (PAA; 200 μg/ml) was added at the time of infection. At 96 hpi, viral DNA was quantified by qPCR. The results were normalized to the abundance of GAPDH DNA in the sample to control for variations in loading (n = 3).
Consistent with the effect of hippuristanol on viral DNA accumulation and late gene transcription, hippuristanol potently inhibited the production of cell-free progeny virus when added at the time of infection (Fig. 3A). The effects of hippuristanol were dose dependent (Fig. 3B) consistent with the specific inhibition of eIF4A in a nontoxic manner. We used shRNA-mediated depletion of eIF4A1 as an additional approach to confirm the phenotypes observed following hippuristanol treatment. Similar to hippuristanol treatment, the HCMV IE protein IE1 and the early protein pUL44 were expressed in eIF4A1-depleted cells, although pUL44 expression was delayed and reduced (Fig. 3C). Limiting eIF4A1 expression prior to infection also decreased the yield of cell-free virus by ∼150-fold (Fig. 3D). Similar results were obtained using a separate eIF4A-specific shRNA (data not shown). These data confirm the results obtained in hippuristanol-treated cells and support the conclusion that eIF4A activity is required at the onset of infection for progression through the HCMV lytic cycle.
FIG 3.

Inhibiting eIF4AI at the time of infection limits HCMV replication. (A) Serum-starved confluent HFFs were infected with HCMV at an MOI of 3. Cells were left untreated (■) or treated with 100 nM hippuristanol (□) at the time of infection. Cell-free virus was quantified by the TCID50 method (n = 3). (B) Cells were infected as described for panel A. The indicated concentrations of hippuristanol were added at the time of infection. The amount of cell-free virus supernatants at 120 hpi was determined by the TCID50 method (n = 2). (C) Cells expressing control (−) or eIF4AI (+) specific shRNA were infected with HCMV at an MOI of 3. Viral protein expression was measured as in Fig. 2A (lanes marked with −, scrambled shRNA; eIF4AI KD, eIF4AI shRNA; n = 3). (D) Same as in panel C, except the amount of cell-free virus in the supernatant was quantified by the TCID50 assay (n = 3). Closed bars, scrambled shRNA; open bars, eIF4AI shRNA.
We next performed experiments to address the temporal requirement for eIF4A activity in HCMV mRNA translation and virus replication. Cells were infected with HCMV, and then hippuristanol was added to the infected cells for a 24-h period beginning at different times postinfection (e.g., from 24 to 48 h, 48 to 72 h, etc.). For comparison, vehicle-treated cells were harvested at the time of hippuristanol addition, and at the end of the 24-h treatment period. We first measured the rate of incorporation of radiolabeled amino acids into protein in the presence of hippuristanol of different times postinfection. Hippuristanol inhibited total protein synthesis in both uninfected and infected cells by >70% (Fig. 4A). We also visualized proteins made during the final 30 min of the hippuristanol treatment on acrylamide gels by autoradiography. Interestingly, in HCMV-infected cells a subset of proteins continued to be efficiently synthesized in the presence of hippuristanol during the later stages of infection (Fig. 4A). These proteins were only observed in infected cells, suggesting that they were viral proteins.
FIG 4.

HCMV replication and protein synthesis becomes increasingly resistant to inhibition by hippuristanol as infection progresses. (A) HFFs were left uninfected (Mock) or infected with HCMV at an MOI of 3. Cultures were treated with 100 nM hippuristanol for the indicated 24-h period. Nascent proteins were metabolically labeled during the final 30 min of drug treatment and visualized by autoradiography (n = 3). (B) Cells were infected and treated as in panel A. Cells were harvested at the indicated times, and viral protein expression was measured by Western blotting (n = 3). (C) Cells were infected as in panel A. At 80 hpi, the cells were treated with either hippuristanol or cycloheximide. Nascent proteins were metabolically labeled during the final hour of the drug treatments, and the indicated viral proteins were immunoprecipitated from the lysates and visualized by autoradiography (n = 2). (D) HFFs were infected as in panel A. At the indicated times, hippuristanol (HP; 100 nM) was added to the cultures. Cell-free virus present at the end of the 24-h hippuristanol treatment (□) was quantified by the TCID50 assay. Supernatants from untreated cultures (NT; ■) were harvested, and titers were determined at the indicated times (n = 3).
Using the same treatment protocol, we next evaluated the effect of hippuristanol on viral protein levels. Adding hippuristanol at any time following infection did not affect levels of the IE1 protein (Fig. 4B). Levels of the early UL44 protein increased between 24 and 48 h in the presence of hippuristanol, although to a lower level than in untreated cells. The addition of hippuristanol at later times after infection did not affect the levels of pUL44. The late protein pp28 was first detected at 48 h in untreated cells; however, pp28 was undetectable in cells treated with hippuristanol between 24 and 48 h. Hippuristanol also limited the accumulation of pp28 between 48 and 72 h postinfection. Beyond 72 h, hippuristanol had minimal effect on the steady-state levels of pp28 protein. As a more direct measure of the rate of viral protein synthesis, we measured the incorporation of radiolabeled amino acids into specific viral proteins during the final hour of hippuristanol treatment. Representative immediate-early, early, and late proteins were immunoprecipitated from the labeled cell lysates and visualized by autoradiography. The results show that the HCMV IE1 and UL44 proteins were efficiently synthesized in the presence of hippuristanol (Fig. 4C). Hippuristanol had a modest effect on the synthesis of the pp28 protein; however, nascent pp28 protein was clearly detectable. In contrast, cycloheximide completely inhibited nascent protein synthesis (Fig. 4C). These results suggest that viral protein synthesis is largely independent of eIF4A activity during the late stage of HCMV infection.
We also determined the temporal requirement for eIF4A activity for the production of HCMV progeny virus. In marked contrast to the results obtained when eIF4A was inhibited at the start of infection, hippuristanol had minimal impact on the accumulation of cell-free virus during the later stages of infection (Fig. 4D). In control cells the amount of cell-free virus increased by approximately 2 orders of magnitude between 48 and 72 h postinfection. Hippuristanol treatment had a <5-fold effect on virus replication during the same time frame. Virus titers increased an additional 10-fold between 72 and 96 h postinfection in untreated cells. Hippuristanol treatment had no negative effect on virus replication during this time and in fact slightly increased the yield of cell-free virus. These data show that HCMV replication becomes increasingly resistant to eIF4A inhibition as infection progresses.
As part of the eIF4F complex, the eIF4A helicase resolves secondary structures in the 5′UTR of mRNAs that might otherwise impede ribosome scanning (7, 8). A possible explanation for the translation of viral mRNAs when eIF4A is inhibited could be that viral mRNAs lack extensive secondary structure in their 5′UTRs and therefore do not require the eIF4A helicase activity for their mRNA translation. However, assembly of the eIF4F complex might still be required to recruit 40S ribosomal subunits to viral mRNAs, since the eIF4G subunit bridges interactions between eIF4F and the 43S preinitiation complex. We therefore determined the impact of eIF4F disruption on HCMV mRNA translation. We used the mTOR inhibitor Torin1, a specific ATP-competitive inhibitor of mTOR kinase activity, to decrease eIF4F abundance in infected cells (26). We focused on the late stage of infection since our data from hippuristanol-treated cells suggested a minimal requirement for eIF4F activity for the synthesis of viral proteins during this stage of infection. Torin1 treatment decreased the amount of eIF4G that copurified with m7G-Sepharose while conversely increasing the binding of the translational repressor 4EBP1 (Fig. 5A), demonstrating that Torin1 disrupted the eIF4F complex during the late stage of infection. Torin1 also limited nascent protein synthesis in both infected and uninfected cells. Similar to the results in hippuristanol-treated cells, the synthesis of a subset of infected cell specific proteins appeared to be less affected by Torin1 (Fig. 5B). We also found that polysome formation in infected cells was resistant to the effects of Torin1, while Torin1 treatment diminished polysomes abundance in uninfected cells (Fig. 5C). Our results show that a subset of mRNAs efficiently recruits ribosomes in infected cells despite significant disruption of the eIF4F complex.
FIG 5.
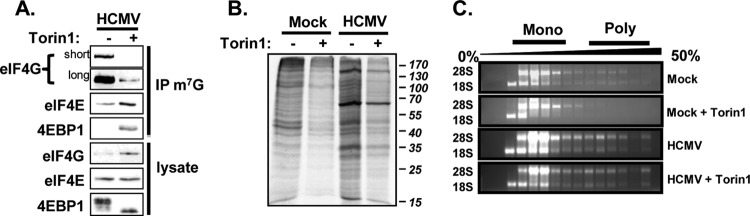
Effect of eIF4F disruption on protein synthesis in HCMV-infected cells during the late stage of infection. (A) At 80 hpi (MOI of 3), the cells were treated with Torin1 (250 nM) for 16 h or left untreated. The integrity of the eIF4F complex was measured using m7G Sepharose affinity purification. (B) Same as in panel A, except nascent proteins were metabolically labeled during the final 30 min of Torin1 treatment and visualized by autoradiography (C) Same as for panel A, except cytoplasmic lysates were resolved through 10 to 50% sucrose gradients to separate ribosomal subunits, monosomes, and polysomes. The location of ribosomes in the gradient was monitored by visualizing rRNA isolated from each fraction on agarose gels.
We next determined the effect of eIF4F disruption on the translation of host mRNAs during the late stage of infection. Cytoplasmic extracts from control or Torin1-treated infected cells were resolved through sucrose density gradients to separate ribosomal subunits, monosomes, and polysomes. The relative abundance of specific host mRNAs in each gradient fraction was measured by qRT-PCR. If the recruitment of ribosomes to the mRNA is dependent on the abundance of eIF4F complex, then Torin1 treatment should result in a shift of mRNAs from the polysomes-containing fractions to the lighter, monosome-containing fractions of the gradient. Conversely, if ribosomes bind the mRNA equally as well when eIF4F is disrupted, the distribution of the mRNA in the gradient should be unaffected by Torin1. Importantly, this assay only measures the distribution of cytoplasmic mRNAs across the gradient, allowing us to normalize for any potential effects of the mTOR inhibitor on mRNA transcription, processing, or nuclear export.
Using this approach, we determined the extent of polysome association for three host mRNAs previously shown to require the eIF4F complex for their efficient translation (27, 28). Figure 6A shows that the host HSP90 and GNB2L1 transcripts are efficiently translated in infected cells as determined by their relative abundance in polysome-containing fractions (fractions 11 to 14). Although the rps20 mRNA is less efficiently translated than either of the above, rps20 transcripts were clearly present in polysomes. Treating infected cells with Torin1 decreased the association of each of the host mRNAs with polysomes, with a corresponding increase in the amount of each mRNA in the monosome-containing fractions (Fig. 6A). Torin1 decreased the steady-state levels of the HSP90 and GNB2L1 proteins (Fig. 6B) demonstrating that changes in polysomes association are reflective of changes in protein abundance. We also measured the rate of incorporation of radiolabeled amino acids into nascent HSP90 and GNB2L1 protein during the final hour of Torin1 treatment. Fewer radiolabeled amino acids were incorporated into nascent HSP90 or GNB2L1 proteins in the presence of Torin1, confirming that their translation was inhibited (Fig. 6C). These results show that the degree of eIF4F disruption achieved with Torin1 is sufficient to inhibit the translation of eIF4F-dependent mRNAs. We conclude that host eIF4F-dependent mRNAs continue to require eIF4F for their translation during HCMV infection.
FIG 6.
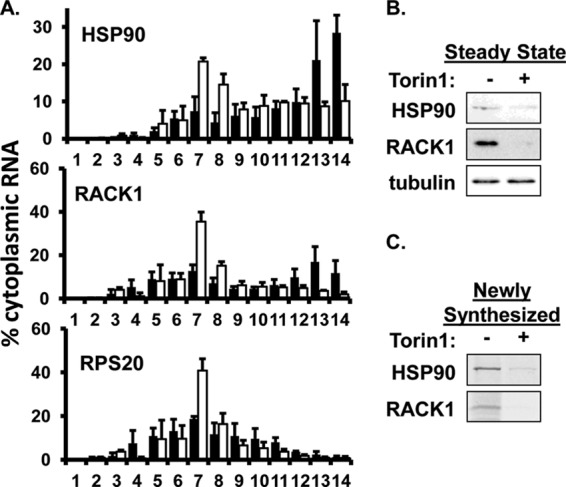
eIF4F disruption limits the association of host mRNAs with polysomes. (A) HFFs were infected and treated as in Fig. 5. At 96 hpi, cytoplasmic extracts were resolved through sucrose density gradients. The abundance of the indicated mRNAs in each gradient fraction was determined by qRT-PCR. The percentage of the total RNA in the gradient in each fraction is shown (■, untreated; □, Torin1 treated; n = 3). (B) Cells were treated as in panel A. Steady-state protein levels were measured by Western blotting. (n = 3). (C) Cells were treated as in panel A. Nascent proteins were metabolically labeled during the final 30 min of the assay. Immune complexes specific for the indicated proteins were visualized by autoradiography. The results of a representative experiment (n = 3) are shown in panels B and C.
We next determined the effect of eIF4F disruption on the translation of viral mRNAs. We found representative viral mRNAs from each kinetic class associated with polysomes as efficiently in Torin1 treated cells as in untreated cells (Fig. 7A). The steady-state levels of each viral protein were minimally affected by Torin1 treatment as determined by Western blotting (Fig. 7B). Similarly, the rate of nascent protein synthesis for each viral protein was unaffected by Torin1 treatment (Fig. 7C). A potential caveat of this approach stems from the fact that HCMV mRNAs are packaged into the tegument of virions. Therefore, the migration of viral mRNAs in the gradient could reflect their association with virions rather than polysomes. However, we found that the inclusion of EDTA in lysis buffer, which disrupts polysomes (29) (see also Fig. S1 in the supplemental material), shifted both the IE1 and UL99 mRNAs to lighter fractions of the gradient. Together, these data show that substantial disruption of the eIF4F complex does not inhibit the recruitment of ribosomes to HCMV mRNAs.
FIG 7.
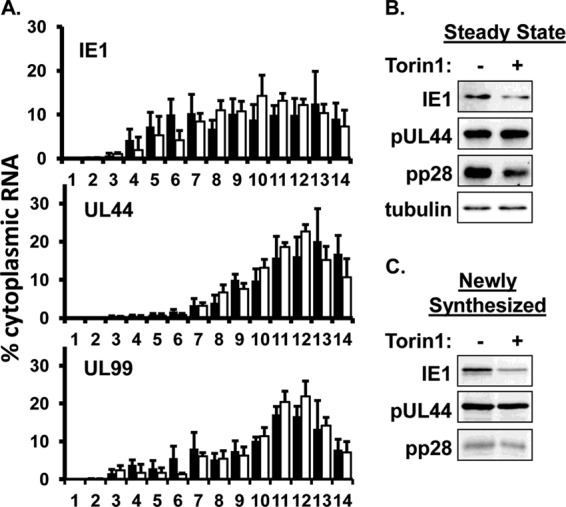
eIF4F disruption does not affect the association of viral mRNAs with polysomes late in infection. (A) HFFs were infected and treated as in Fig. 5. The distribution of viral mRNAs across a sucrose gradient was determined by qRT-PCR as in Fig. 6A (n = 3). (B) Cells were treated as in Fig. 6B. Steady-state protein levels were measured by Western blotting (n = 3). (C) Cells were treated as in Fig. 6C. Nascent proteins were metabolically labeled during the final 30 min of the assay. Immune complexes specific for the indicated proteins were visualized by autoradiography (n = 3). The results of a representative experiment (n = 3) are shown in panels B and C.
Several potential explanations existed for the differential effect of eIF4F disruption on host and viral mRNA translation. We considered the hypothesis that the viral mRNAs examined lacked sufficient 5′UTR structure to require eIF4F activity for their translation. In vitro studies have shown that mRNAs lacking stable secondary structure have a minimal requirement for eIF4F for their translation (30). The 5′UTR for each of the examined viral mRNAs has previously been mapped. In each case the free energy constraints of the viral 5′UTRs would predict that eIF4F was needed for their efficient translation (see Fig. S2A in the supplemental material). Another possibility was that HCMV mRNAs are not capped and therefore would not require the eIF4F complex for their translation. Surprisingly, we were unable to find any information concerning the presence of an m7G mRNA cap on either the UL44 or UL99 mRNAs. We therefore used tobacco acid pyrophosphatase (TAP)-mediated RNA ligation to test for the presence of an m7G mRNA cap on the viral mRNAs (see Materials and Methods). We found that polysome-associated mRNAs for both viral genes were capped and therefore have the potential to interact with eIF4F (see Fig. S2A).
Based on these results, we performed a global analysis of the effect of eIF4F disruption on the translation of host and viral mRNAs. For each mRNA we determined the translation efficiency, which is the ratio of mRNA abundance in the polysome compared to the total RNA sample. We then compared the translation efficiency for each mRNA in untreated cells and Torin1-treated cells. In these experiments, we used a custom microarray containing oligonucleotide probes specific for all annotated HCMV open reading frames, as well as 44,000 human genes. Table S1 in the supplemental material lists host mRNAs with statistically significant changes in their translation efficiency in the presence of Torin1. We identified more than 340 host mRNAs whose translation efficiency was decreased by >40% in Torin1-treated cells. The results of our analysis of host mRNAs were similar to those described previously (27, 28). For example, we found that Torin1 inhibited the translation of mRNAs encoding ribosomal proteins (Fig. 8A), which are translated in an eIF4F-dependent manner. We found that several host mRNAs previously shown to require eIF4F for their translation during HCMV infection were translated less efficiently in the presence of Torin1 (31).
FIG 8.
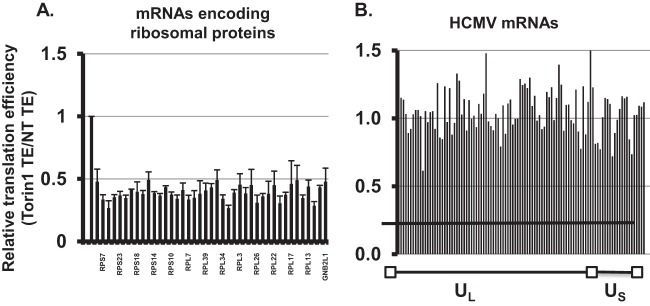
Global analysis of the effect of eIF4F disruption on the translation efficiency of host and viral mRNAs. (A) HFFs were infected and treated as in Fig. 5. Polysome-associated and total RNA were isolated from infected cells treated with Torin1 or left untreated. The translation efficiency of each mRNA was calculated by determining the ratio of the mRNA in the polysome and total fractions by microarray analysis. The graph shows the ratio of the translation efficiency (TE) in Torin1-treated cells to that in untreated cells. (n = 3) mRNAs translated as efficiently in both conditions have a value of 1. Values of <1 indicate that translation was inhibited in the presence of Torin1. (A) Relative translation efficiency of representative mRNAs encoding ribosomal proteins in Torin1-treated cells. (B) Relative translation efficiency of HCMV mRNAs in Torin1-treated cells (n = 3). The translation efficiencies of both host and viral mRNAs in untreated and Torin1-treated cells are given in Tables S1 and S2 in the supplemental material.
In contrast, the relative translation efficiency of HCMV mRNAs was for the most part similar in untreated and Torin1-treated cells (Fig. 8B and see Table S2 in the supplemental material). Torin1 treatment resulted in a statistically significant decrease in the translation of only one viral mRNA (UL17). In addition, the translation of four viral mRNAs (UL34, UL25, US1, and UL99) showed a statistically significant increase in translation efficiency in Torin1-treated cells. Torin1 treatment did not have a significant impact on the translation of the remaining 98 viral genes measured. The array data were confirmed by qRT-PCR for several viral mRNAs (see Fig. S3 in the supplemental material) using monosome and polysome fractions isolated from two additional experiments. No signal was obtained from either step when reverse transcriptase step was omitted, indicating the absence of DNA contamination (data not shown). These results further demonstrate that the Torin1 treatment used in these experiments is sufficient to inhibit the translation of host mRNAs that utilize the eIF4F complex. However, the translation of HCMV mRNAs as a group does not correlate with the abundance or activity of the eIF4F complex.
DISCUSSION
In this study, we measured the requirement for the host eIF4F translation initiation complex for the translation of host and viral mRNAs during HCMV infection. Our results demonstrate a differential requirement for eIF4F in the translation of host and viral mRNAs. We found that eIF4F is required at the start of infection for efficient progression through the viral lytic cycle. However, as infection progresses, both viral protein synthesis and replication become increasingly insensitive to inhibition or disruption of the eIF4F complex. In contrast, host eIF4F-dependent mRNAs continue to require eIF4F for their translation in infected cells. Our results therefore reveal a fundamental difference between host and HCMV mRNAs in their requirement for host translation factors.
We found that inhibiting or depleting the eIF4A subunit of the eIF4F complex from the start of infection inhibited progression through the HCMV lytic cycle. Specifically inhibition of eIF4A from the start of infection prevented viral DNA replication. These data are consistent with previous studies showing that inhibition of the mTOR kinase with Torin1 from the start of infection limits HCMV DNA accumulation and replication. Precisely how eIF4F contributes to viral DNA replication is currently unclear. Seven HCMV proteins are required for viral DNA replication in vitro (32). Perhaps one or more of these viral factors is dependent on eIF4F activity for their synthesis prior to viral DNA replication. This is consistent with our finding that pUL44 expression is delayed and reduced when eIF4A is depleted or inhibited at the start of infection despite the efficient expression on an IE protein. Alternatively, eIF4F activity might be required for the expression of a host protein needed for viral DNA replication or for the expression of viral DNA replication proteins. Recent studies have shown that eIF4F is required for the translation of several host metabolic enzymes (27). As HCMV infection remodels host metabolism (20, 33–36), perhaps the increased eIF4F abundance and activity in infected cells promotes the expression of metabolic enzymes needed for virus replication.
Of particular interest is our finding that eIF4A activity is not required for the efficient translation of viral mRNAs during the later stages of infection. One explanation could be that the viral mRNAs examined have minimal structure in their 5′UTRs. In this case, 40S ribosomal subunits would be capable of scanning viral 5′UTRs without a requirement for helicase activity. However, the 5′UTRs of the representative viral mRNAs examined in the present study have been defined and are predicted to have sufficient structure to impede ribosome scanning (see Fig. S2B in the supplemental material), suggesting that a helicase is required for their translation. While our data demonstrate a minimal requirement for eIF4A activity, perhaps another RNA helicase is required for the resolution of secondary structures in viral mRNAs. Human cells encode 64 potential RNA helicases, and HCMV itself encodes a putative RNA helicase (37). If indeed an alternative helicase contributes to viral mRNA translation, our data suggest that this helicase is directed specifically to viral mRNAs since the total protein synthesis remains significantly dependent on eIF4A activity in infected cells.
Perhaps the most surprising finding was that ribosomes efficiently associate with HCMV mRNAs despite significant disruption of the eIF4F complex. Early in infection expression of the HCMV immediate-early protein was resistant to eIF4A inhibition, while later in infection most mRNAs efficiently associated with ribosomes despite eIF4F disruption. In a stochastic model in which eIF4F binds to mRNA in a sequence-independent manner, one would expect that the translation of all mRNAs would be equally affected by disruption of the eIF4F complex. Our results demonstrating the differential effects of eIF4F inhibition on host and viral mRNAs argue against this model. However, if eIF4F preferentially associated with a subset of mRNAs in a sequence-specific manner, disruption of the eIF4F complex should preferentially affect the translation of that subset of mRNAs. In fact, two recent studies found that the eIF4F complex is most important for the translation of host mRNAs containing a tract of pyrimidines (TOP) or a pyrimidine-rich motif (PRTE) immediately adjacent to the 5′ mRNA cap (27, 28). Our microarray analysis of host mRNA translation closely matches the results of these previous studies. The translation of host mRNAs containing TOP motifs (e.g., ribosomal mRNAs) was preferentially inhibited by eIF4F disruption. Analysis of the 5′UTRs of the representative viral mRNAs studied herein did not reveal TOP or PRTE-like motifs, which is consistent with a minimal role for the eIF4F complex in their translation. Furthermore, examination of additional HCMV mRNAs with defined transcript structure did not reveal the presence of these motifs (unpublished observations). However, the 5′UTRs for the majority of HCMV mRNAs have not been defined. A more comprehensive understanding of HCMV message structure is needed to globally assess a role for cryptic TOP motifs in HCMV mRNA translation.
Our results raise the question of how ribosomes are recruited to HCMV mRNAs when eIF4F activity is inhibited. Although Torin1 treatment resulted in sufficient eIF4F disruption to limit host mRNA translation, some residual eIF4F complex was still present. Therefore, one explanation could be that a host or viral factor preferentially recruits the residual eIF4F complex to HCMV mRNAs. An example might be found in the herpes simplex virus (HSV) ICP6 protein, which stimulates eIF4F formation (38). Perhaps a viral protein specifically recruits any remaining eIF4F to viral mRNAs. Alternatively, a protein or protein complex expressed during infection might limit the association of the 4EBP1 translation repressor with viral mRNAs. To this end, the HCMV UL69 protein (pUL69) binds to m7G mRNA cap and associates with viral mRNAs. Although infection with wild-type HCMV limits 4EBP1 binding to the mRNA cap, 4EBP1 robustly binds m7G Sepharose in cells infected with a pUL69 mutant virus (39). Perhaps pUL69 specifically inhibits 4EBP1 binding to eIF4E-associated viral mRNAs, resulting in the preferential accumulation of eIF4F on viral messages. The interaction of pUL69 with poly(A)-binding protein (PABP) further suggests a role for pUL69 in viral mRNA translation. In uninfected cells, PABP promotes translation initiation via its interaction with eIF4F, thereby promoting the formation of the “closed loop” (40). In addition, PABP is required for efficient HCMV replication (41). pUL69 could bridge the association of PABP with the mRNA cap on viral transcripts when eIF4F is limiting. However, in both of the above scenarios some mechanism must exist to discriminate between host and viral messages, since the degree of eIF4F disruption obtained in our experiments was sufficient to limit the translation of host eIF4F-dependent mRNAs.
It is also possible that the recruitment of ribosomes to viral mRNAs does not require the eIF4F complex. Many viruses limit the abundance of the eIF4F complex while efficiently synthesizing viral proteins. For example, many RNA viruses encode proteases that cleave eIF4G and inactivate the eIF4F complex (42). Viruses that inactivate eIF4F often rely on an alternative suite of translation initiation factors (43). Host cells also utilize alternative translation initiation complexes, most notably the cap binding complex (CBC) (44). Like eIF4F, the CBC binds to the m7G cap and can recruit ribosomes to facilitate mRNA translation. Unlike eIF4F, the CBC does not require the eIF4E protein and is therefore insensitive to inhibition by 4EBP1 (45). Perhaps viral mRNAs utilize the CBC to recruit ribosomes to viral mRNAs when eIF4F activity is limiting. Equally plausible is the existence of an HCMV-encoded protein or protein complex that facilitates the recruitment of ribosomes to viral transcripts. For example, the N protein of bunyavirus is capable of replacing the entire eIF4F complex in the translation of viral mRNAs (46). If viral proteins contribute to HCMV mRNA translation, these factors would be excellent candidates for novel antiviral therapeutics, since they would specifically limit viral protein synthesis while leaving host protein synthesis intact. Clearly, additional studies are needed to further define the complement of viral and/or host factors that govern HCMV protein synthesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chad Cecil for contributions to this study. We thank members of the Moorman lab and the laboratory of Cary Moody for helpful discussions and insights. We also thank Steve Bachenheimer for critical reading of the manuscript and Blossom Damania, Dirk Dittmer, and Nancy Raab-Traub for making this work possible. We are grateful to Jerry Pelletier for his generous gift of hippuristanol.
This study was supported by grant 1R01AI103311-01 from the U.S. National Institutes of Health to N.J.M. and funds from the North Carolina University Cancer Research Fund to N.J.M. NIH/NINDS grant R01 NS067037 and NIH/NCI grant R01 CA134844 to D.A.M. also supported this work.
Footnotes
Published ahead of print 6 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02321-13.
REFERENCES
- 1.Brown-Luedi ML, Meyer LJ, Milburn SC, Yau PM, Corbett S, Hershey JW. 1982. Protein synthesis initiation factors from human HeLa cells and rabbit reticulocytes are similar: comparison of protein structure, activities, and immunochemical properties. Biochemistry 21:4202–4206. 10.1021/bi00261a002 [DOI] [PubMed] [Google Scholar]
- 2.Gingras AC, Raught B, Sonenberg N. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913–963. 10.1146/annurev.biochem.68.1.913 [DOI] [PubMed] [Google Scholar]
- 3.Grifo JA, Tahara SM, Morgan MA, Shatkin AJ, Merrick WC. 1983. New initiation factor activity required for globin mRNA translation. J. Biol. Chem. 258:5804–5810 [PubMed] [Google Scholar]
- 4.Muthukrishnan S, Both GW, Furuichi Y, Shatkin AJ. 1975. 5′-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255:33–37. 10.1038/255033a0 [DOI] [PubMed] [Google Scholar]
- 5.Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell. Biol. 11:113–127. 10.1038/nrm2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonenberg N, Rupprecht KM, Hecht SM, Shatkin AJ. 1979. Eukaryotic mRNA cap binding protein: purification by affinity chromatography on Sepharose-coupled m7GDP. Proc. Natl. Acad. Sci. U. S. A. 76:4345–4349. 10.1073/pnas.76.9.4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grifo JA, Tahara SM, Leis JP, Morgan MA, Shatkin AJ, Merrick WC. 1982. Characterization of eukaryotic initiation factor 4A, a protein involved in ATP-dependent binding of globin mRNA. J. Biol. Chem. 257:5246–5252 [PubMed] [Google Scholar]
- 8.Rozen F, Edery I, Meerovitch K, Dever TE, Merrick WC, Sonenberg N. 1990. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol. Cell. Biol. 10:1134–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieleski L, Talbot SJ. 2001. Kaposi's sarcoma-associated herpesvirus vCyclin open reading frame contains an internal ribosome entry site. J. Virol. 75:1864–1869. 10.1128/JVI.75.4.1864-1869.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grainger L, Cicchini L, Rak M, Petrucelli A, Fitzgerald KD, Semler BL, Goodrum F. 2010. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 84:9472–9486. 10.1128/JVI.00855-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundhoff A, Ganem D. 2001. Mechanisms governing expression of the v-FLIP gene of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:1857–1863. 10.1128/JVI.75.4.1857-1863.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimura K, Ueda K, Guwanan E, Sakakibara S, Do E, Osaki E, Yada K, Okuno T, Yamanishi K. 2004. A posttranscriptional regulator of Kaposi's sarcoma-associated herpesvirus interacts with RNA-binding protein PCBP1 and controls gene expression through the IRES. Virology 325:364–378. 10.1016/j.virol.2004.04.041 [DOI] [PubMed] [Google Scholar]
- 13.Walsh D, Perez C, Notary J, Mohr I. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057–8064. 10.1128/JVI.79.13.8057-8064.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. 2004. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 78:11030–11039. 10.1128/JVI.78.20.11030-11039.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253–262. 10.1016/j.chom.2008.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moorman NJ, Shenk T. 2010. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 84:5260–5269. 10.1128/JVI.02733-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. 1999. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13:1422–1437. 10.1101/gad.13.11.1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gingras AC, Raught B, Sonenberg N. 2001. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15:807–826. 10.1101/gad.887201 [DOI] [PubMed] [Google Scholar]
- 19.Clippinger AJ, Maguire TG, Alwine JC. 2011. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J. Virol. 85:3930–3939. 10.1128/JVI.01913-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spencer CM, Schafer XL, Moorman NJ, Munger J. 2011. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 85:5814–5824. 10.1128/JVI.02630-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang D, Bresnahan W, Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. U. S. A. 101:16642–16647. 10.1073/pnas.0407233101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. 2006. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2:213–220. 10.1038/nchembio776 [DOI] [PubMed] [Google Scholar]
- 23.Mitchell DP, Savaryn JP, Moorman NJ, Shenk T, Terhune SS. 2009. Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J. Virol. 83:10187–10197. 10.1128/JVI.00396-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84:7803–7814. 10.1128/JVI.00139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau ME, Vogt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, Wagner G, Pelletier J. 2008. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS One 3:e1583. 10.1371/journal.pone.0001583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284:8023–8032. 10.1074/jbc.M900301200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. 2012. The translational landscape of mTOR signaling steers cancer initiation and metastasis. Nature 485:55–61. 10.1038/nature10912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485:109–113. 10.1038/nature11083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnstein HR, Cox RA, Gould H, Potter H. 1965. A comparison of methods for the isolation and fractionation of reticulocyte ribosomes. Biochem. J. 96:500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. 2001. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 7:382–394. 10.1017/S135583820100108X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez C, McKinney C, Chulunbaatar U, Mohr I. 2011. Translational control of the abundance of cytoplasmic poly(A) binding protein in human cytomegalovirus-infected cells. J. Virol. 85:156–164. 10.1128/JVI.01778-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pari GS. 2008. Nuts and bolts of human cytomegalovirus lytic DNA replication. Curr. Top. Microbiol. Immunol. 325:153–166. 10.1007/978-3-540-77349-8_9 [DOI] [PubMed] [Google Scholar]
- 33.Chambers JW, Maguire TG, Alwine JC. 2010. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 84:1867–1873. 10.1128/JVI.02123-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McArdle J, Moorman NJ, Munger J. 2012. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 8:e1002502. 10.1371/journal.ppat.1002502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2:e132. 10.1371/journal.ppat.0020132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179–1186. 10.1038/nbt.1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umate P, Tuteja R, Tuteja N. 2010. Genome-wide analysis of helicase gene family from rice and Arabidopsis: a comparison with yeast and human. Plant Mol. Biol. 73:449–465. 10.1007/s11103-010-9632-5 [DOI] [PubMed] [Google Scholar]
- 38.Walsh D, Mohr I. 2006. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev. 20:461–472. 10.1101/gad.1375006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aoyagi M, Gaspar M, Shenk TE. 2010. Human cytomegalovirus UL69 protein facilitates translation by associating with the mRNA cap-binding complex and excluding 4EBP1. Proc. Natl. Acad. Sci. U. S. A. 107:2640–2645. 10.1073/pnas.0914856107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kahvejian A, Roy G, Sonenberg N. 2001. The mRNA closed-loop model: the function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harbor Symp. Quant. Biol. 66:293–300. 10.1101/sqb.2001.66.293 [DOI] [PubMed] [Google Scholar]
- 41.McKinney C, Perez C, Mohr I. 2012. Poly(A) binding protein abundance regulates eukaryotic translation initiation factor 4F assembly in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 109:5627–5632. 10.1073/pnas.1202829109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 9:860–875. 10.1038/nrmicro2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzgerald KD, Semler BL. 2009. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 1789:518–528. 10.1016/j.bbagrm.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maquat LE, Hwang J, Sato H, Tang Y. 2010. CBP80-promoted mRNP rearrangements during the pioneer round of translation, nonsense-mediated mRNA decay, and thereafter. Cold Spring Harbor Symp. Quant. Biol. 75:127–134. 10.1101/sqb.2010.75.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu SY, Lejeune F, Ranganathan AC, Maquat LE. 2004. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev. 18:745–754. 10.1101/gad.1170204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Panganiban AT, Mir MA. 2009. Bunyavirus N: eIF4F surrogate and cap-guardian. Cell Cycle 8:1332–1337. 10.4161/cc.8.9.8315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.