

Abstract
The vesicular stomatitis virus (VSV) RNA-dependent RNA polymerase consists of two viral proteins; the large (L) protein is the main catalytic subunit, and the phosphoprotein (P) is an essential cofactor for polymerase function. The P protein interacts with the L protein and the N-RNA template, thus connecting the polymerase to the template. P protein also binds to free N protein to maintain it in a soluble, encapsidation-competent form. Previously, five sites of phosphorylation were identified on the P protein and these sites were reported to be differentially important for mRNA synthesis or genomic replication. The previous studies were carried out by biochemical analysis of portions of the authentic viral P protein or by analysis of bacterium-expressed, exogenously phosphorylated P protein by mutagenesis. However, there has been no systematic biochemical search for phosphorylation sites on authentic, virus-expressed P protein. In this study, we analyzed the P protein isolated from VSV-infected cells for sites of phosphorylation by mass spectrometry. We report the identification of Tyr14 as a previously unidentified phosphorylation site of VSV P and show that it is essential for viral transcription and replication. However, our mass spectral analysis failed to observe the phosphorylation of previously reported C-terminal residues Ser226 and Ser227 and mutagenic analyses did not demonstrate a role for these sites in RNA synthesis.
INTRODUCTION
Nonsegmented, negative-strand RNA viruses encode their own RNA-dependent RNA polymerase (RdRp) to carry out the RNA synthetic activities of mRNA transcription and genome replication. The RdRp is composed of two multifunctional proteins, the large (L) protein and the phosphoprotein (P) (1). The L protein is the main catalytic subunit, harboring the RdRp, capping, methyltransferase, and polyadenylation activities (2–6). The P protein is the cofactor, essential for the formation of an active polymerase complex. The template for RNA synthesis is the negative-sense RNA genome encapsidated by oligomers of the viral nucleocapsid (N) protein (7, 8). The L protein gains access to the N-RNA template via the P protein, which interacts with the L protein and the oligomers of N protein simultaneously (9–12). The P protein also functions to maintain N protein in a soluble, encapsidation-competent form by acting as an N-specific chaperone (13–15). The vesicular stomatitis virus (VSV) P protein is divided into three domains, an N-terminal domain (PNTD), a central domain (PCD), and a C-terminal domain (PCTD) (Fig. 1) (10, 16–18). The PNTD has been shown to bind to both the L and N proteins. Upon binding to L protein, it causes a conformational rearrangement of L protein and increases the processivity of the polymerase (12, 19). The PNTD also binds free N protein (N°), acting as a chaperone protein (13, 14, 17). The PCTD contacts two adjacent N monomers of the N-RNA template and, along with PNTD binding of L, serves as a bridge between the polymerase and RNA template (9, 12). The PCD is a homodimerization domain (16). Recent data suggest that structured regions of P are separated by intrinsically disordered regions (IDRs) (18, 20).
FIG 1.

(A) Schematic representation of the organization of the VSV P protein domain in light of recent structural studies (16–18). The P protein has been divided into three domains, an N-terminal domain (PNTD), an autonomously folded central domain (PCD), and a negatively charged C-terminal domain (PCTD). Previously reported phosphorylation sites and surrounding residues are shown. (B) Protein profile of VSV-infected cells. BHK 21 cells either mock infected or infected with VSV were metabolically labeled with [35S]methionine-cysteine. Cell lysates were resolved by low-bis SDS-PAGE (see Materials and Methods). (C) P protein was isolated from VSV-infected BHK 21 cells by immunoaffinity purification (see Materials and Methods). N protein was copurified with P as shown. (D) The identities of the P and N proteins were determined by Western blotting analyses. The values between the blots are molecular sizes in kilodaltons.
The VSV P protein becomes phosphorylated during the viral life cycle. Recombinant P protein isolated from Escherichia coli and thus devoid of any phosphorylation was unable to support in vitro transcription, indicating that P protein phosphorylation is important for viral RNA synthesis (21, 22). P protein isolated by DEAE cellulose column chromatography, either from virions or from infected cells, was reported to have different phosphorylated forms (23). These differentially phosphorylated forms varied in the ability to support in vitro transcription (23, 24). Doublet bands of P proteins were observed when resolved through urea-SDS-polyacrylamide gels, and these two forms of the P protein were further divided into subspecies when subjected to isoelectric focusing (23, 25).
The specific sites of phosphorylation of the VSV P protein were analyzed initially by chemical and enzymatic cleavage, which indicated that most of the phosphate acceptor sites were in the PNTD (26, 27). Hsu and Kingsbury (28) localized these phosphorylation sites between amino acids 35 and 75 of P protein of the VSV Indiana (PInd) serotype. Residues Ser60, Thr62, and Ser64 in this region were subsequently identified as phosphorylation sites in PInd by incubating exogenous casein kinase II with bacterium-expressed mutant P protein; however, phosphorylation at residue 62 varied with the type of assay and the enzyme source (29). A separate study reported constitutive phosphorylation at all three sites (30). These phosphorylation sites in the PNTD were reported to be important for the homodimerization of the protein and its interaction with L (31–34). Mutation of residues Ser60, Thr62, and Ser64 individually or in pairs inhibited the ability of the polymerase to transcribe subgenomic templates in a transfection assay by 50 to 98%, and mutation of all three sites to alanine inhibited transcription by 98%; however, genome RNA replication was largely unaffected (35).
In the PCTD, residues Ser226 and Ser227 were identified as phosphorylation sites in PIND by in vitro phosphorylation of bacterium-expressed mutant P protein. Phosphorylation was abrogated if both residues 226 and 227 were changed to alanine but unaffected if either site was altered individually (29). In contrast to the N-terminal phosphorylation sites, mutation of these sites was reported to affect viral genome replication but not transcription (36). These studies led to the proposal that phosphorylation of the VSV P protein had a regulatory role in viral transcription and replication.
Despite the aforementioned efforts to characterize the different phosphorylation sites of P protein, to date, there has not been a systematic biochemical search for the phosphorylation sites of the authentic, virus-expressed P protein. Therefore, in the present study, we used mass spectrometry to analyze the phosphorylation state of the P protein isolated from VSV-infected cells. Our analysis revealed a previously unidentified phosphorylation site in the N-terminal region of the P protein that we show is required for the P protein to be able to support viral mRNA transcription and genome replication. Further, our analysis has revealed differences from previous reports of the C-terminal phosphorylation sites in the P protein, as did our analysis of their biological relevance in viral RNA synthesis.
MATERIALS AND METHODS
Viral infection and isolation of P protein for mass spectral analysis.
BHK-21 cells in 60-mm dishes were infected with VSV at a multiplicity of infection (MOI) of 30. Virus particles were allowed to adsorb for 1 h, after which the inoculum was aspirated and replaced with 1 ml of Dulbecco's modified Eagle's medium plus 2% fetal bovine serum. At 3.5 or 7.5 h postinfection, cells were incubated with a phosphatase inhibitor cocktail (1 mM sodium orthovanadate, 5 mM sodium β-glycerophosphate, 1 μM microcystin-LR) for 30 min, and then a cytosolic extract was prepared in lysis buffer (1% NP-40, 0.4% deoxycholate [DOC], 66 mM EDTA, 10 mM Tris-HCl, pH 7.4) also containing phosphatase inhibitor and protease inhibitor cocktails. P protein was purified from this cytosolic extract with protein A-Sepharose (Sigma) beads that had been cross-linked with a saturating amount of VSV-monospecific polyclonal anti-P antibody with dimethyl pimelimidate (Thermo Scientific). The cytosolic extract was incubated with the anti-P antibody–protein A–Sepharose beads overnight at 4°C. After washing, protein was eluted in 0.5% SDS and resolved by 10% SDS-PAGE (77:1 acrylamide-bisacrylamide) (37). Alternatively, in a separate set of experiments, VSV-infected BHK-21 cells were harvested directly into 1× Laemmli sample buffer at 3.5 or 7.5 h postinfection and the proteins were resolved on low-bis 10% SDS-polyacrylamide gels (77:1 acrylamide-bisacrylamide). The P protein bands were excised from both gels, subjected to in-gel tryptic digestion, and processed for mass spectrometry.
Mass spectrometry.
Each trypsinized sample was pressure loaded onto a self-prepared 100-μm-inside-diameter (i.d.) fused-silica column (Polymicro Technologies, Phoenix, AZ) packed with irregular (5 to 15 μm, 120 Å) reverse-phase phenyl resin (YMC, Kyoto, Japan) and then connected to a 75-μm-i.d. PicoFrit fused-silica column (New Objective, Woburn, MA) that had a prefritted 10-μm tip and had been self-packed with regular (5 μm, 120 Å) reverse-phase phenyl resin (YMC, Kyoto, Japan). Nanoflow electrospray ionization (ESI) was performed in the positive-ion mode with a 2.0-kV spray voltage applied to peptides that were eluted at a flow rate of about 200 nl/min from a high-performance liquid chromatography (HPLC) gradient of 0 to 70% solvent B in 105 min, where solvent A was 0.1 M acetic acid and solvent B was 80% acetonitrile in 0.1 M acetic acid.
Briefly, the Thermo LTQ-XL ion trap mass spectrometer (Thermo, San Jose, CA) was operated in the data-dependent mode with an Agilent 1100 HPLC system split to nanoflow. The acquisition duty cycle consisted of an initial MS1 centroid scan with a mass range of 300 to 1,800 m/z for all experiments. The five most abundant ions were sequentially selected for a Zoom MS1 scan acquired in profile with a width of 10 m/z centered on the precursor ion. Each Zoom MS1 scan was followed by a MS2 collision-induced dissociation (CID) spectrum of that same precursor. After repetition for each of the top five precursor ions, the cycle was repeated. The duty cycle for this data acquisition cycle of 11 mass spectral scans was about 2.9 s, on average. Further details are provided in reference 38.
Mass spectral data analysis.
Data sets were first processed with a custom Perl script, dubbed MAZIE, that accurately determines the charge and monoisotopic mass for each MS2 scan precursor ion by analyzing the preceding Zoom MS1 scan (39). The MAZIE script then generates a concatenated DTA file used for searching with the OMSSA engine (40). MAZIE is distributed under the Creative Commons License and is available, together with its dependencies, at http://faculty.virginia.edu/templeton. A composite protein database was created that contained both the entire Syrian golden hamster (taxonomy id 10036) and the VSV strain Indiana (taxonomy id 11277) protein library as extracted from the NCBI nr database (ftp://ftp.ncbi.nih.gov/blast/db/, downloaded on 17 August 2012) with the blastdbcmd command (ftp://ftp.ncbi.nih.gov/blast/executables/blast+/, version 2.2.25). Reversed sequences of all of the proteins were generated by an in-house Perl script and appended to the database so that a decoy search strategy could be used. Using this database, the MS2 data were searched as a tryptic digest by using parameters that were optimized as described previously (38, 39). Briefly, the masses of both the precursor and fragment ions were treated as monoisotopic with m/z tolerances of 0.3 and 0.5 Da, respectively; two missed cleavages were allowed, and variable modifications of carbamidomethylation (+57) of cysteine; oxidation (+16) and sulfonation (+32) of methionine; deamidation (+1) of asparagine and glutamine; and phosphorylation (+80) of serine, threonine, and tyrosine were permitted. A false-discovery rate was calculated by tabulating the OMSSA search results that identified natural (forward) protein sequences, representing potential real sequence matches, together with those that identified reversed protein sequences that are, by definition, false matches (41). All of the mass spectral data were compiled and stored in a MySQL database that is accessed and presented via a PHP Web-based user interface, WiMS-AMP, that will be presented in a future publication. All putative phosphorylated peptides were manually inspected to verify their authenticity, regardless of the OMSSA E value.
Plasmid construction and mutagenesis.
The plasmid containing the VSV genes for N, P, and L under the control of a T7 promoter and the ΔBgl-22 subgenomic replicon have been described previously (42, 43). Mutations were engineered into the gene for P by using the QuikChange (Stratagene) site-directed mutagenesis protocol. For each mutant protein, the entire P-encoding gene was sequenced independently three times to confirm that the correct mutations had been cloned.
Protein labeling and immunoprecipitation.
The synthesis of wild-type (WT) or mutant P protein was examined by transfection of 1.25 μg of a plasmid encoding the respective protein into BHK-21 cells that had been preinfected with the vaccinia virus T7 recombinant, vTF7-3 (44) with Lipofectin (Invitrogen) (45). At 16 h posttransfection, the transfected cells were starved in medium lacking methionine and cysteine for 30 min and subsequently exposed to 50 μCi/ml [35S]methionine-cysteine (Translabel; PerkinElmer) for 30 min. Cytoplasmic extracts were prepared with lysis buffer (1% NP-40, 0.4% DOC, 66 mM EDTA, 10 mM Tris-HCl, pH 7.4) and then incubated overnight with monospecific polyclonal anti-P antibody. The proteins were immunoprecipitated with protein A-Sepharose beads (Sigma) for 1 h and then washed with wash buffer (1% NP-40, 0.5% DOC, 0.1% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 7.4). Subsequently, the proteins were eluted in 1× Laemmli buffer and resolved by 10% SDS-PAGE (77:1 acrylamide-bisacrylamide). The gels were fixed, dried, and exposed to phosphoscreens.
To test the ability of the mutant P proteins to interact with N protein, BHK-21 cells were cotransfected with 4 and 1.25 μg of plasmids encoding the N and P proteins, respectively. The proteins were labeled with Translabel, and N-P interaction was assayed by immunoprecipitation with monospecific polyclonal anti-P antibody as described above.
Analysis of viral RNA synthesis.
For analysis of RNA synthesis, BHK-21 cells infected with the vTF7-3 recombinant vaccinia virus were transfected with plasmids that encode a VSV subgenomic replicon, ΔBgl-22 (4 μg), and the plasmids encoding the VSV N (4.5 μg), P (1.25 μg), and L (0.75 μg) proteins (45). The P plasmids containing mutations at the phosphorylation sites were substituted for the WT P plasmid as indicated in Results. Viral RNA synthesis was assayed by direct metabolic labeling as described previously (43, 45). Briefly, at 16 h posttransfection, cells were treated with 10 μg/ml of actinomycin D-mannitol (Sigma-Aldrich, St. Louis, MO) and subsequently labeled for 5 h in medium containing 50 μCi/ml of [3H]uridine (PerkinElmer, Waltham, MA) at 37°C. Cells were harvested in lysis buffer (1% NP-40, 0.4% desoxycholate, 66 mM EDTA, 10 mM Tris-HCl, pH 7.4), and the nuclei were removed by centrifugation. RNA was purified from cytoplasmic extracts by phenol-chloroform extraction and ethanol precipitation. The extracted RNA was electrophoresed through a 1.75% agarose-urea gel (pH 3) and visualized by fluorography (43).
The effect of P protein mutations on RNA synthesis of authentic defective interfering (DI) particle templates was analyzed by coinfecting BHK-21 cells with vTF7-3 recombinant vaccinia virus and VSV DI particles. Following an hour-long adsorption period, the cells were transfected with plasmids encoding the WT or mutant P protein and the WT N and L proteins. Cells were incubated at 37°C for 5 h, treated with actinomycin D-mannitol (10 μg/ml) for 30 min, and subsequently exposed to [3H]uridine (33 μCi/ml) for 5 h at 37°C. Cytoplasmic extracts were harvested, and RNA was resolved by 1.75% agarose-urea gel (pH 3) electrophoresis and detected by fluorography (43).
RESULTS
VSV P protein contains a previously unidentified phosphorylation site in the N-terminal region.
To examine the VSV P protein for sites of phosphorylation by mass spectrometry, P protein was isolated from virus-infected BHK-21 cells by immunoaffinity purification. Figure 1B shows the electrophoretic profile of the proteins from uninfected and VSV-infected cells metabolically labeled with [35S]methionine-cysteine. Because of the efficient shutoff of host protein synthesis, the five major VSV proteins were resolved as discrete bands on 10% SDS-polyacrylamide gels (77:1 acrylamide-bisacrylamide) (Fig. 1B). Figure 1C shows the electrophoretic profile of proteins isolated by immunoprecipitation with protein A-Sepharose beads cross-linked with anti-P antibody. The P protein interacts with the N protein, and thus, the N protein coprecipitates with it. The identities of the two bands as the N and P proteins were confirmed by Western blotting with the corresponding monospecific antibodies (Fig. 1D).
To determine the phosphorylation sites in the VSV P protein, the P protein was excised from a Coomassie-stained gel (Fig. 1C), subjected to in-gel tryptic digestion, and analyzed by mass spectrometry. Using a nanoflow HPLC gradient coupled with ESI-MS2 as described in Materials and Methods, 66% of the P protein amino acid sequence was observed from 20 unique peptides (Table 1). As shown in Fig. 2A, the region encompassing amino acids 51 to 109 lacks a canonical trypsin recognition sequence, thereby producing a 59-amino-acid peptide (representing ∼22% of the P protein sequence) with an average mass of 6,629 Da. The peptide was resistant to mass spectral observation for two main reasons. First, the peptide would have to have at least a +4 charge state [(6,629 + 4)/4 = 1,658 m/z] to fall within the upper end of the MS1 full scan (1,800 m/z). Because of the high number of negatively charged residues (11 Asp and 8 Glu residues) and the low number of positively charged residues (1 Lys, 0 Arg, and 0 His residues) contained within the peptide, the probability of its existence in a +4 charge state is low. Second, even if the peptide did exist in a +4 charge state, the CID fragmentation that was used is inherently ineffective in breaking it apart because it has many more than 25 residues. Since the previously reported phosphorylation sites at Ser60, Thr62, and Ser64 reside in this region, their phosphorylation status could not be verified through this analysis. However, as described below, we confirmed the biological importance of these residues in viral transcription by independent mutagenic analysis.
TABLE 1.
Coverage of predicted tryptic peptides for VSV strain Indiana protein P
| Ending amino acid | Sequencea | Length (amino acids) | Avg mass (Da) | Scan count |
|---|---|---|---|---|
| 6 | MDNLTK | 6 | 720.84 | 0 |
| 8 | VR | 2 | 273.34 | 0 |
| 12 | EYLK | 4 | 551.64 | 0 |
| 16 | SYSR | 4 | 511.54 | 0 |
| 31 | SYSRLDQAVGEIDEIEAQR | 19 | 2,259.33 | 9 |
| 31 | LDQAVGEIDEIEAQR | 15 | 1,685.81 | 126 |
| 34 | LDQAVGEIDEIEAQREAK | 18 | 2,014.20 | 41 |
| 34 | AEK | 3 | 346.38 | 0 |
| 109 | SNYELFQEDGVEEHTRPSYFQAADDSDTESEPEIEDNQGLYVPDPEAEQVEGFIQGPLDDYADEDVDVVFTSDWK | 75 | 8,563.86 | 0 |
| 120 | QPELESDEHGK | 11 | 1,268.30 | 7 |
| 123 | TLR | 3 | 388.47 | 0 |
| 135 | LTLPEGLSGEQK | 12 | 1,271.43 | 61 |
| 143 | LTLPEGLSGEQKSQWLLTIK | 20 | 2,241.64 | 3 |
| 143 | SQWLLTIK | 8 | 988.20 | 50 |
| 150 | AVVQSAK | 7 | 701.82 | 0 |
| 169 | HWNLAECTFEASGEGVIIK | 19 | 2,104.37 | 52 |
| 170 | K | 1 | 146.19 | 0 |
| 171 | R | 1 | 174.20 | 0 |
| 171 | RQITPDVYK | 9 | 1,119.30 | 11 |
| 179 | QITPDVYK | 8 | 963.10 | 11 |
| 202 | VTPVMNTHPYQSEAVSDVWSLSK | 23 | 2,575.88 | 208 |
| 210 | TSMTFQPK | 8 | 939.09 | 36 |
| 211 | TSMTFQPKK | 9 | 1,067.29 | 3 |
| 211 | K | 1 | 146.19 | 0 |
| 228 | KASLQPLTISLDELFSSR | 18 | 2,005.32 | 46 |
| 228 | ASLQPLTISLDELFSSR | 17 | 1,877.13 | 19 |
| 239 | ASLQPLTISLDELFSSRGEFISVGGNGR | 28 | 2,951.31 | 2 |
| 239 | GEFISVGGNGR | 11 | 1,092.18 | 46 |
| 243 | MSHK | 4 | 501.60 | 0 |
| 251 | MSHKEAILLGLR | 12 | 1,367.69 | 10 |
| 251 | EAILLGLR | 8 | 884.09 | 36 |
| 253 | YK | 2 | 309.37 | 0 |
| 254 | K | 1 | 146.19 | 0 |
| 260 | KLYNQAR | 7 | 892.04 | 11 |
| 260 | LYNQAR | 6 | 763.85 | 14 |
| 262 | VK | 2 | 245.32 | 0 |
Boldface type indicates an amino acid in the peptide sequence that is likely to be positively charged during mass spectral acquisition.
FIG 2.

Mass spectral sequence coverage and identification of a novel phosphorylation site in the VSV P protein: (A) Sequence of the VSV P protein where the underlined amino acids indicate those that were observed by mass spectral analysis. The small bold s/y (Ser13/Tyr14) represents the newly identified phosphorylation site with the slash indicating that, on the basis of the MS2 CID fragmentation spectra displayed in panel B (see Fig. S1 in the supplemental material), it cannot be determined if the single phosphorylation is located on Ser13 or Tyr14. The small bold and italic t and s letters (Ser60, Thr62, Ser64, Ser226, and Ser227) represent the phosphorylation sites that have been reported previously. (B) MS2 CID fragmentation pattern of the s13/y/SRLDQAVGEIDEIEAQR31 peptide containing the newly identified phosphorylation site. The peptide sequence with its theoretical b and y ions is located above the spectra, with the ions that were used for identification denoted with colored highlights in the sequence and corresponding labels in the spectrum. Though it is ambiguous from the two fragmentation spectra whether the phosphorylation site is at Ser13 or Tyr14, the ion fragment masses listed above the spectra are for the phosphorylation to be at Tyr14 for the sake of simplicity of the display.
The mass spectral results revealed two new findings concerning the phosphorylation status of the VSV P protein. First, a previously unreported phosphorylation site in the N-terminal region of the P protein was identified. As noted in Fig. 2B, the MS2 CID fragmentation of the +3 charge state of the s13/y/sRLDQAVGEIDEIEAQR31 peptide was observed, where the slash between the bold small s and y indicates that a single phosphate group is located on residue Ser13, Tyr14, or Ser15. Although the lack of fragmentation ions from the peptide N terminus prevented the identification of the specific location of the phosphorylated residue, the spectra unambiguously showed that the peptide was phosphorylated on one of those residues. Furthermore, the E9YLKs/ySR16 tryptic peptide was also analyzed and the corresponding fragmentation narrowed the potential phosphorylation site to either Ser13 or Tyr14 (see Fig. S1 in the supplemental material). Notably, both phosphopeptides, E9YLKs/ySR16 and s13/y/sRLDQAVGEIDEIEAQR31, contain an unused tryptic cleavage site at either Lys12 or Arg16 and the nonphosphorylated forms of these extended peptides were not observed. This is likely a direct consequence of the negative charge associated with the nearby phosphate group damping the trypsin cleavage efficiency at these sites, as has been observed previously (46).
Second, the mass spectral analysis failed to detect phosphorylation of the proposed C-terminal phosphorylation sites Ser226 and Ser227 (29). The nonphosphorylated A212SLQPLTISLDELFSSR228 tryptic peptide was observed routinely across repeated analyses of different biological samples harvested either in the presence of phosphatase inhibitors or directly into denaturing agents. This result was surprising, as the use of these sites as a substrate for phosphorylation was reported previously; however, as mentioned previously, these observations relied on the in vitro phosphorylation of bacterium-expressed P protein (29), which may explain the disparity in the findings.
Phosphorylation of the P protein at Ser226 or Ser227 is below the limit of detection.
Because of the confounding effects of sample complexity, ion suppression, and ionization efficiency of a particular peptide, the lack of observation of a particular peptide within a mass spectral acquisition, whether phosphorylated or not, does not demonstrate that it does not exist within the sample. Therefore, to more fully explore the phosphorylation state of Ser226 and Ser227, a synthetic phosphopeptide, Ser226p, was synthesized to mimic the tryptic peptide that encompasses the residues, A212SLQPLTISLDELFsSR228, as well as an extended version, A212SLQPLTISLDELFsSRGEFISVGGNGR239, that takes into account the potential of an unused trypsin cleavage site as a result of the phosphorylation. The elution time, the charge state profile of the precursor ion, and the MS2 CID fragmentation pattern (see Fig. S2 and S3 in the supplemental material) of both of these peptides were then determined with the same HPLC column and gradient. The characteristics of these peptides should be relatively insensitive to whether or not the phosphate is located on Ser226 or Ser227. By acquiring data from P protein samples both without (Fig. 3B) and with (Fig. 3C) the addition of these synthetic phosphopeptides at a known concentration, the relative level of phosphorylation at Ser226 or Ser227 in the biologically relevant sample could be determined. As displayed in Fig. 3B and C, the ion chromatograms of the +3 charge state of these phosphopeptides that were obtained during these experiments demonstrated that the phosphorylation level at Ser226 and Ser227 in the biological sample was below the 1% level, if present at all.
FIG 3.
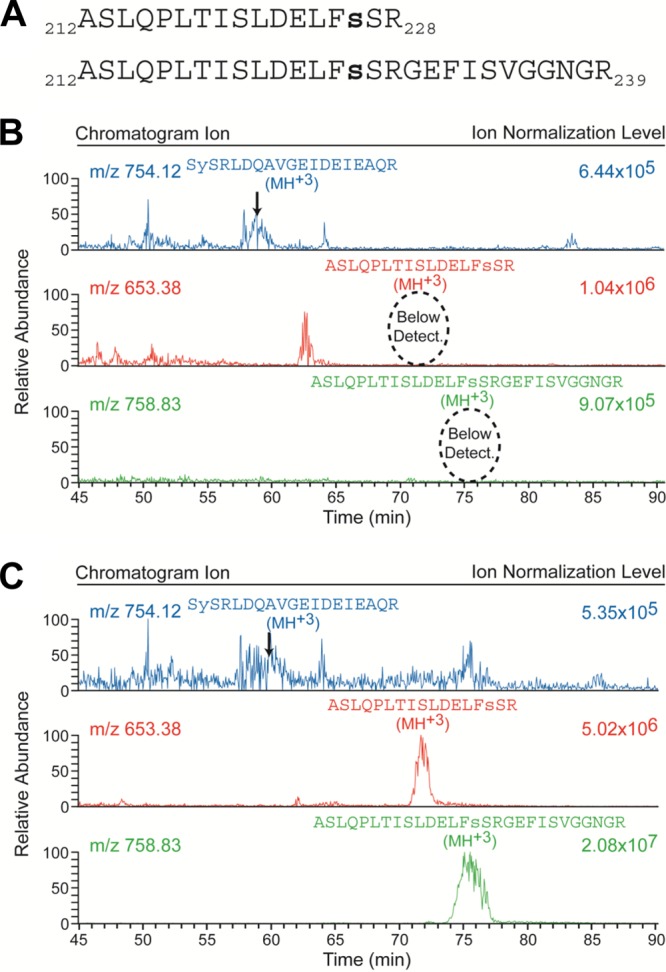
Phosphorylation at Ser226 and Ser227 is below the limit of detection. (A) Sequences of the two synthetic phosphopeptides representing the tryptic (residues 212 through 228) and extended (residues 212 through 239 with missed cleavage at R228) peptides of the VSV P protein that encompass the putative Ser226 and Ser227 phosphorylation sites. Both of these synthetic peptides were phosphorylated at residue Ser226, as indicated by the small bold letter s. (B) Chromatograms obtained from a biological sample without the addition of the two synthetic phosphopeptides. The top chromatogram presents the m/z = 754.12 ion that represents the +3 charge state of the newly identified Ser13/Tyr14 phosphopeptide; the indicated elution peak was verified via inspection of its corresponding MS2 CID spectrum as illustrated in Fig. 2B. The bottom two chromatograms present the m/z = 653.38 and 758.83 ions that represent the +3 charge state of the tryptic and extended synthetic phosphopeptides, respectively (as displayed in panel A), occurring in the biological sample. (C) The corresponding chromatograms obtained from a biological replicate of the sample represented in panel B but with 2 pmol of each of the two synthetic phosphopeptides in panel A added. A comparison of the corresponding bottom two chromatograms in panels B and C reveals that phosphorylated Ser226 and Ser227 are below the limit of detection in the biological sample and thus exist in, at most, <1% of the total abundance of P protein. Note that the individual chromatograms are each scaled independently by the indicated ion normalization level.
Phosphorylation site mutant forms of P and their effect upon N-P interaction.
The mass spectral analysis revealed a previously unidentified phosphorylation site in the N-terminal region of the VSV P protein; however, it could not discriminate whether the modification was at Ser13 or Tyr14. Additionally, the analysis failed to find evidence of the presence of the C-terminal Ser226 and Ser227 phosphorylations that had been reported previously (29). In order to evaluate the biological significance of these observations, we introduced different combinations of mutations into a cDNA of WT VSV P protein at each of the previously reported phosphoreceptor residues, as well as at the newly identified phosphorylation site(s), Ser13 and/or Tyr14. The various mutations introduced into the P protein are diagrammed in Fig. 4A. These can be divided into three groups. The first four include mutations of residues Ser13, Tyr14, and Ser15 introduced either individually or collectively. As mentioned, phosphorylation of these residues has not been reported previously, making their potential importance to P protein function unknown. The second group contains mutations at the previously reported phosphorylation sites of Ser60, Thr62, and Ser64, in the combinations shown in Fig. 4A. The third group consists of a mutant P protein with both Ser226 and Ser227 mutated to alanine. Since the mass spectrometric analysis failed to detect phosphorylation on these two residues, we wanted to reexamine the effect upon viral RNA replication of mutating these residues as reported previously (36). The effects of the different mutations on P protein expression were examined by transfection of the WT or mutant plasmids into cells as described previously (47). As shown in Fig. 4B, the expression levels of all of the mutant proteins were comparable to that of the WT P protein.
FIG 4.

Construction and expression of P proteins containing amino acid substitutions. (A) Schematic representation of different substitutions of potential P protein phosphorylation sites. Newly identified and previously reported phosphorylation sites were replaced with alanine residues individually or in combinations as shown. (B) WT and mutant P proteins were expressed under the control of the T7 promoter in BHK21 cells infected with vTF7-3 prior to transfection. Proteins were metabolically labeled with [35S]methionine-cysteine and then immunoprecipitated (IP) with monospecific polyclonal P antibody.
A major function of P protein resides in its interaction with N protein to maintain it in an encapsidation-competent form. Therefore, we examined whether the mutant P proteins were altered in the ability to interact with N protein by coexpressing either WT or mutant P proteins with N protein in BHK-21 cells transfected with plasmids encoding these proteins. Transfected-cell proteins were metabolically labeled with Translabel and subjected to immunoprecipitation with an anti-P antibody as described in Materials and Methods. As shown in Fig. 5A (top), the N protein coprecipitated well with WT P and all of the mutant P proteins, except P226A/227A. Significantly less N protein coprecipitated with P226A/227A than with WT P. Interestingly, consistently more N protein coprecipitated with the P14A mutant protein than with WT P.
FIG 5.
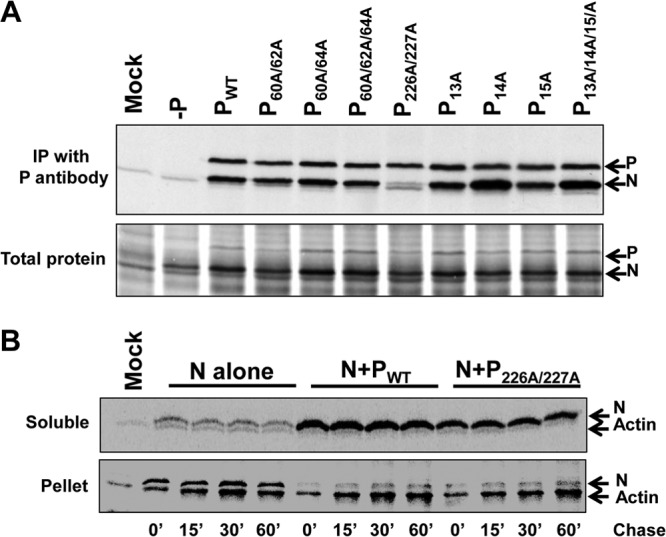
Interaction of amino acid substitution-containing P proteins with N protein. (A) WT P protein and P proteins with amino acid substitutions were coexpressed with N protein in BHK21 cells infected with vTF7-3 prior to transfection. Proteins were metabolically labeled with [35S]methionine-cysteine and then immunoprecipitated (IP) with monospecific polyclonal anti-P antibody. Either immunoprecipitates (top) or total protein samples (bottom) were resolved by SDS-PAGE, followed by autoradiography. (B) Pulse-chase analysis. N protein was expressed in vTF7-3-infected BHK21 cells either alone or with WT P or P226/227A. At 16 h posttransfection, proteins were metabolically labeled with [35S]methionine-cysteine by a 15-min (15′) pulse and then exposed to medium containing excess unlabeled methionine and cysteine for the indicated time periods. Proteins were immunoprecipitated from the soluble fractions with anti-N antibody and resolved by SDS-PAGE (top). Pellet fractions were dissolved in 1× Laemmli sample buffer and resolved by SDS-PAGE (bottom).
As mentioned previously, the N-P interaction is crucial for maintaining N in a soluble form, necessary to support encapsidation of the nascent genomic and antigenomic RNA during replication. Therefore, we investigated whether the mutant protein P226A/227A had the ability to maintain N in soluble form by examining the solubility and stability of the N protein when expressed alone or in the presence of WT P or P226A/227A over time by pulse-chase analysis. Transfected cells were starved for methionine and cysteine for 30 min and subsequently exposed to Translabel for 15 min, followed by a chase with medium containing a 10-fold molar excess of unlabeled methionine and cysteine, for time periods of 0 to 60 min, as indicated in Fig. 5B. Soluble proteins were collected from the supernatant after centrifugation at 14,000 rpm for 30 min, followed by immunoprecipitation with a monoclonal anti-N antibody (Fig. 5B, top). The pellet fractions were solubilized in 1× Laemmli buffer and resolved on 10% SDS-polyacrylamide gels (Fig. 5B, bottom). As shown, the N protein, when expressed alone, formed insoluble aggregates that remained in the pellet fraction. However, when N was coexpressed with WT P protein, both proteins remained in the soluble supernatant fraction. The mutant P226A/227A protein also retained the ability to maintain the N protein in soluble form, and there was little change in the amount of soluble N protein throughout the time course, compared with that seen when WT P protein was used. This finding indicated that although mutation of Ser226 and Ser227 to alanine affected the level of N-P interaction, as assayed by coimmunoprecipitation, that level of interaction was sufficient to maintain N in soluble form.
Residue Tyr14 is required to support viral transcription, as well as genomic RNA replication.
Given previous reports showing that the phosphorylation state of the P protein can affect viral RNA synthesis (35, 36), we examined the role of the newly identified phosphorylatable residues at the extreme N terminus of the P protein on transcription and genomic replication. For completeness and because of the inability to find evidence of the phosphorylation of Ser226 and Ser 227, we reexamined the effect of mutation of these residues, as well as Ser60, Thr62, and Ser64, on viral transcription and replication. We examined RNA synthesis by using two different templates, a subgenomic replicon (ΔBgl-22) that carries out both genomic replication and the synthesis of a single mRNA (Fig. 6A) and a naturally occurring DI RNA particle that exclusively replicates genomic RNA (43, 45). BHK-21 cells were transfected with a plasmid encoding the subgenomic replicon and plasmids encoding the VSV N, L, and WT or mutant P proteins. RNA synthesis was analyzed by direct metabolic labeling with [3H]uridine in the presence of actinomycin D. RNA was extracted and resolved by acid agarose-urea gel electrophoresis (see Materials and Methods).
FIG 6.

RNA transcription and replication supported by P proteins containing amino acid substitutions. (A) Schematic representation of subgenomic replicon ΔBgl22, used as a template for viral RNA synthesis. This replicon contains a single noncoding transcriptional unit flanked by the 3′ and 5′ signals required for transcription and replication. The replicon generates a negative-sense RNA [(−)] that serves as the template for the transcription of a single mRNA and for the replication of a full-length antigenomic RNA [(+)] that, in turn, serves as the template to generate more genomic RNA. (B) RNA synthesis was examined by transfecting the plasmids encoding the replicon ΔBgl22, the N, and L proteins, and the WT and substituted P proteins into BHK21 cells infected with vTF7-3. [3H]uridine-labeled RNA products were resolved on acid agarose-urea gels (pH 3.0) and detected by fluorography. The replication products and the mRNAs are indicated. (C) Ability of mutant P proteins to support the replication of DI RNA particles. BHK21 cells were coinfected with vTF7-3 and DI particles and then transfected with plasmids encoding the N, L, and WT or amino acid-substituted P proteins. Labeled RNA products were analyzed as described above. The negative- and positive-sense DI genomic RNAs were separated as indicated. (D) P proteins with residue Tyr14 replaced with alanine, glutamate, aspartate, or phenylalanine as indicated were examined for the ability either to support viral RNA synthesis in the subgenomic replicon system (top) or to support genomic RNA template replication (Rep) by authentic DI particles (bottom) as described above.
The abilities of the WT and mutant P proteins to support viral mRNA transcription and genome RNA replication are shown in Fig. 6B. Mutation of Ser13 or Ser15 to alanine did not affect RNA synthesis, as both P13A and P15A supported mRNA transcription and RNA replication at levels comparable to those achieved with the WT P protein. However, P14A abrogated both mRNA and genomic RNA synthesis. This phenotype was verified further by analysis of the triple mutant protein P13A/14A/15A in the subgenomic replicon system, which also failed to support RNA synthesis. These data show that either residue Tyr14 or its phosphorylation is important to support viral RNA synthesis. Mutation of various combinations of the Ser60, Thr62, and Ser64 residues adversely affected viral transcription. All three mutant proteins supported transcription at less than 10% of the WT level. However, these mutations did not affect the ability of P to support viral RNA replication, as shown by the similar intensities of replication products in WT and P60A/62A, P60A/64A, and P60A/62A/64A mutant protein samples (Fig. 6B) and as confirmed by analysis (below) of the replication of the DI RNA template (Fig. 6C). These data corroborate previous reports (35) and confirm that phosphorylation at these sites is important for viral transcription but not genomic replication. In contrast, replacement of the serine residues at positions 226 and 227 with alanine had no effect on viral RNA synthesis. Mutant protein P226A/227A supported viral transcription and genomic replication at WT levels. This finding was surprising because a previous report indicated that these serine residues are important for viral RNA replication but not for transcription (29, 36, 48). As mentioned above, we were unable to detect evidence of phosphorylation of these residues by mass spectral analysis.
In light of this finding, we verified the results from the subgenomic replicon system by testing the abilities of the mutant P proteins to support the replication of authentic VSV DI particles. In the subgenomic replicon assay, the mRNAs are the major product of RNA synthesis, while replication products are relatively minor. The DI particle template exclusively replicates its genome, as the first four internal genes and the start of the L gene are deleted. In this assay, BHK-21 cells were coinfected with vTF7-3 and DI particles and then transfected with plasmids encoding the VSV N, L, and WT or mutant P proteins. At 5 h posttransfection, cells were exposed to [3H]uridine and metabolically labeled RNA products were extracted and analyzed as described in Materials and Methods. WT P protein supported the synthesis of both the genomic and antigenomic strands of RNA, which separate in the acid urea gel system on the basis of both size and base composition (Fig. 6C). The encapsidated, authentic DI particles already contain a very small amount of WT P protein. To account for any effect that this low level of P protein coming in on the purified DI template might have on the system, we included a negative control in which the plasmid encoding the P protein was omitted from the transfection. This control showed negligible activity compared to the transfection with WT P (Fig. 6C, lane −P). The importance of Tyr14 in supporting viral genomic RNA replication was confirmed, as the mutant proteins P14A and P13A/14A/15A did not support the replication of the DI genome. All of the other mutant proteins, including, P226A/227A, supported DI particle RNA replication at levels comparable to that of WT P protein. These data confirmed our finding that Ser226 and Ser227 are not essential to support viral RNA replication.
Replacement of Tyr14 with a negatively charged residue does not restore RNA synthetic activity.
Replacement of Tyr14 with alanine adversely affected viral RNA synthesis (Fig. 6B), indicating that either the tyrosine residue or its phosphorylation is important for P protein function. Negatively charged amino acids like glutamate or aspartate can be used to mimic the negative charge contributed by a phosphate group. To examine whether it is possible to restore the activity of P protein by the presence of negatively charged glutamate or aspartate residues at position 14, we introduced these mutations into the plasmid encoding the P protein and examined their ability to support RNA synthesis of a subgenomic replicon or DI RNA genome, as described above. As shown in Fig. 6C, the presence of a negatively charged residue at position 14 did not restore the ability of P protein to support RNA synthesis of either template tested (Fig. 6D). To investigate whether the overall structure of the tyrosine residue is important at position 14, we also examined the effect of a P protein with Tyr14 mutated to Phe on RNA synthesis. Similarly, this mutant P protein was also unable to support viral RNA synthesis (Fig. 6D). Together, these data indicate that neither the phenyl ring nor the negative charge alone is sufficient to restore the activity of the tyrosine/phosphotyrosine residue.
VSV P protein is phosphorylated at residue Tyr14.
The studies described above indicate that the tyrosine at position 14 and/or its phosphorylation are required for P protein function in supporting viral RNA synthesis. Because mass spectral analysis could not discriminate whether the phosphorylated residue was Ser13 or Tyr14, we tested whether a phosphotyrosine-specific antibody could recognize the P protein from VSV-infected cells in an effort to determine the exact residue of phosphorylation. VSV-infected BHK21 cell lysates, either untreated or treated with calf intestine alkaline phosphatase, were resolved by SDS-PAGE and Western blot assays were carried out with phosphotyrosine antibody (clone4G10; Millipore) (Fig. 7A, lanes 1 to 3). The same membrane was then stripped and reblotted in succession with a polyclonal monospecific anti-P antibody (Fig. 7A, lanes 4 to 6) or a polyclonal monospecific anti-N antibody (Fig. 7A, lanes 7 to 9). The data in Fig. 7A (antiphosphotyrosine blot) show a significant change in the pattern of tyrosine-phosphorylated proteins (compare lanes 1 and 2) in BHK21 cells in response to VSV infection. A band migrating at the 50-kDa position, present only in the infected-cell lysate, was detected by both the antiphosphotyrosine and the anti-P antibodies upon respective blotting of the membrane (lanes 2 and 5, respectively). Treatment with calf intestine alkaline phosphatase removed the recognition of the P protein by the phosphotyrosine-specific antibody (lanes 3 and 6). Blotting of the membrane with anti-N protein antibody confirmed that the N protein was not recognized by the antiphosphotyrosine antibody, showing the antibody's specificity. These data show that the P protein, and not the N protein, is recognized by the antiphosphotyrosine antibody and that this recognition is eliminated by alkaline phosphatase treatment. These data, together with the mass spectral results, all support the conclusion that residue Tyr14 of the VSV P protein is phosphorylated.
FIG 7.

The VSV P protein contains a phosphotyrosine residue (A) BHK 21 cells either uninfected or infected with VSV (MOI, 1.0) were harvested at 16 h of postinfection. Uninfected cell extract (lanes 1, 4, and 7) and infected-cell extracts either untreated (lanes 2, 5, and 8) or treated with calf intestine alkaline phosphatase (CIP) (lanes 3, 6, and 9) were resolved by 10% SDS-PAGE and blotted to a PVDF membrane. The same membrane was then probed with phosphotyrosine antibody (lanes 1 to 3), stripped, and probed with polyclonal anti-P antibody (lanes 4 to 6) and then polyclonal anti-N antibody (lanes 7 to 9). The values to the left are molecular sizes in kilodaltons. (B) Phosphoprotein sequences from 27 different strains of 11 different vesiculoviruses (listed in the ViPR database) were aligned by using MUSCLE software. The tyrosine residue, indicated by the arrow, is conserved across all of them. The sequences aligned are those of Chandipura virus (CHPV1 and CHPV2; UniProt Knowledgebase [UniProtKB] accession no. of P proteins, E3T3B5 and E3T3A5, respectively; strains CIN 0327 and CIN 0451), Cocal virus (COCV; UniProtKB accession no. of P protein, B3FRK7; strain Indiana 2), Jurona virus (JURV; UniProtKB accession no. of P protein, I1SV83; strain unknown [accession no. HM566194]), Maraba virus (MARAV; UniProtKB accession no. of P protein, F8SPF1; strain unknown [accession no. HQ660076]), Perinet virus (PERV; UniProtKB accession no. of P protein, I1SV88; strain unknown [accession no. HM566195]), Pike fry rhabdovirus (PFRV; UniProtKB accession no. of P protein, C3VM12; strain F4), Spring viremia of carp virus (SVCV-1-5; UniProtKB accession no. of P protein, A8VJA2, Q91DS2, Q91DS5, A0FCR4, and Q1KK77; strain BJ0505-2), Fijan reference strains ATCC VR-1390 and ATCC VR-1390 (A1 and A2), Vesicular stomatitis Alagoas virus (VSAV; UniProtKB accession no. of P protein, B3FRL2; strain Indiana 3), VSV strain Indiana (VSIV-1 to -4; UniProtKB accession no. of P proteins, Q8B0I3, Q8B0H3, P03520], and Q8B0H8; strains 98COE, 94GUB, unknown [accession no. J02428], and 85CLB), Vesicular stomatitis New Jersey virus (VSNJV-1 to -9; UniProtKB accession no. of P proteins, I7CGJ1, I7CGJ9, I7CGL3, I7CGM8, I7CGK3, I7CGL8, I7CGJ5, I7CGL3, and I7CGM3; strains NJ0783NCP, NJ92CRB, NJ1184HDB, NJ95COB, NJ0703CRB6, NJ89GAS, NJ0185PNB, NJ92CLB, and NJ0405NME), Yug Bogdanovac virus (YBV; UniProtKB accession no. of P protein, K4FFQ0; strain unknown [accession no. JF911700]).
We next examined the conservation of the Tyr14 residue among the P proteins of different vesiculoviruses. Interestingly, P is one of the most divergent genes among different vesiculoviruses. We aligned 27 different P protein sequences from 11 different vesiculoviruses listed in the ViPR database. The alignment (Fig. 7B) showed that only two residues were completely conserved among different P proteins, one of which is Tyr14. This conservation throughout the genera, along with our previous results, strongly suggests that the Tyr14 residue is critically important for the biological function of vesiculovirus phosphoproteins.
DISCUSSION
By mass spectrometric analysis, we identified a previously unreported phosphorylation site in the N-terminal domain of the VSV P protein. CID fragmentation of an N-terminal peptide encompassing amino acids 13 to 34 of the P protein showed the presence of a single phosphorylation on residue 13, 14, or 15. Analysis of a shorter fragment of the phosphopeptide narrowed the site of phosphorylation to residue 13 or 14, although the exact residue could not be identified. Subsequent studies conclusively identified the phosphorylated residue as Tyr14 by immunoblotting of the P protein with a phosphotyrosine-specific antibody and demonstrating that the recognition was eliminated by alkaline phosphatase treatment.
Functional analysis of P proteins containing individual alanine substitutions at residues Ser13, Tyr14, and Ser15 showed that the alteration of Tyr14 abrogated the ability of the P protein to support viral transcription, as well as replication, while, in contrast, replacement of residue Ser13 or Ser15 had no detectable effect on RNA synthesis (Fig. 6B). The triple mutant protein P13A/14A/15A showed the same RNA synthesis phenotype as P14A. This observation was strengthened when mutant proteins were examined for the ability to support the replication of naturally occurring VSV DI particles; neither P14A nor P13A/14A/15A supported DI RNA replication. These data confirmed the importance of Tyr14 in viral RNA synthesis. To investigate whether the structure of tyrosine is important for P protein functionality or whether a negative charge can mimic phosphorylation of this site, we constructed three additional mutant forms of P protein, P14D, P14E, and P14F. None of these mutant P proteins was capable of supporting viral RNA synthesis. In this regard, it should be noted that the structures of glutamate and aspartate are significantly different from that of phosphotyrosine, a possible reason why these residues failed to mimic the role of a phosphotyrosine residue. Interestingly, comparison of P sequences from different vesiculoviruses showed that the Tyr14 residue is conserved across all vesiculoviruses (Fig. 7B), which adds weight to the potential significance of this residue and its phosphorylation in the viral life cycle.
While our mass spectrometric analysis identified a new, biologically important phosphorylation site in P, it also failed to confirm the previously reported phosphorylation sites within the C-terminal domain of the P protein (29). Through mass spectral analysis, we identified the peptide containing the C-terminal Ser226 and Ser227 residues; however, phosphorylation was not detected at these sites. We used synthetic peptides phosphorylated at residue Ser226 to determine the sensitivity of detection of our method. Coanalysis of the biological samples of P that included these synthetic peptides showed that the phosphorylation level at Ser226 and Ser227 must be below 1% of the total protein population, if present at all. Since these phosphorylation sites were reported to be phosphatase sensitive, we repeated our analyses under a variety of harvest conditions. The P protein was isolated in the presence of phosphatase inhibitors or directly into 1× Laemmli sample buffer, where the high percentage of denaturing agents should have inactivated the phosphatase activity present in the cell extract; similar results were obtained when the proteins were isolated by both methods. The major difference between the studies reported here and previous reports lies in the source of the P protein; we used P protein isolated from virus-infected cells, whereas much of the previous work used bacterium-expressed P that was subsequently phosphorylated enzymatically (29).
The biological significance of the newly identified site and the previously reported phosphorylation sites was tested by replacement of the residues with alanine. We confirmed the importance of the N-terminal residues at positions 60, 62, and 64 in viral transcription and confirmed that they do not affect genomic replication (Fig. 6B and C), corroborating previous work (35). However, according to a previous report, replacement of both Ser226 and Ser227 with alanine was reported to affect genomic RNA replication (36). In contrast, we found that alteration of these residues to alanine had no effect on the levels of transcription or on genome replication when templated by a subgenomic replicon or an authentic DI RNA particle. Concerned that our template might be different from that used in the previous study, we further examined these mutant P proteins by using additional subgenomic replicons p8+, p8−, and p8+NP (45, 49, 50), which initiate RNA synthesis from cDNA by expressing an RNA of either positive or negative polarity, and found the same results (data not shown). Furthermore, we validated the sequences of our mutated P cDNA clones by three independent sequencing strategies. The major difference between the work carried out here and that done in the previous study was in the method of analysis. We examined RNA synthesis by direct metabolic labeling of RNA with [3H]uridine, followed by phenol extraction and gel analysis. In the previous study (36), RNA replication was analyzed by immunoprecipitation of encapsidated, replicated RNA prior to gel analysis. If alteration of residues Ser226 and Ser227 to alanine affected the conformation of the N binding site on P or the site of binding by the antibodies used in immunoprecipitation, this might possibly have affected detection.
Unphosphorylated P protein expressed in bacteria has been shown to be able to interact with N protein, suggesting that P interaction with N is unaffected by phosphorylation (7, 9). This was confirmed by our studies, as all of the N-terminal mutant P proteins we examined interacted with N protein, as determined by coimmunoprecipitation of levels of N protein similar to those obtained with WT P protein. In fact, the mutant proteins P14A and P13A/14A/15A coprecipitated greater amounts of N protein than did the WT protein. In contrast, P226A/227A interacted less well with N protein than did the WT P (Fig. 5A). Although these two serine residues reside in the C-terminal, N-interacting domain of P, they have not been indicated as points of contact with N by structural studies (9). However, it is possible that alteration of these residues can affect the conformation of the N binding site and indirectly affect contact between P and N. Additionally, this alteration of N-P interaction did not affect RNA synthesis, as shown by the data in Fig. 6, and we conclude that the level of N-P interaction that did occur was sufficient to support RNA synthesis. Further, this mutant P protein was competent to maintain N protein in soluble form, as shown by pulse-chase analysis (Fig. 5B). Maintenance of N in soluble form is one major role of P protein in supporting viral replication (13). The P226A/227A mutant protein could maintain most of the N protein in soluble form at levels similar to those of WT P (Fig. 5B). This finding is consistent with the fact that it supported viral replication as well as the WT P protein (Fig. 6). However, alteration of residues 226 and 227 simultaneously to alanine was reported previously to prevent the recovery of infectious virus from cDNA, indicating a role for these residues somewhere in the viral life cycle (48).
The finding that alteration of residue Tyr14 to alanine, phenylalanine, glutamate, or aspartate eliminated the ability of the P protein to support both transcription and replication indicates that either this Tyr residue or phosphorylation at this site affects a fundamental function of P with the polymerase. A recent deletion analysis showed that the extreme N terminus (amino acids 1 to 41) of P was not required for stimulation of the conformational change in L and its processivity (12). Furthermore, nonphosphorylated P produced in bacteria served to promote initiation of RNA synthesis and processivity of L (12, 51). These data together suggest that these steps in RNA synthesis do not require P phosphorylation. Phosphorylation of P was reported previously to be essential for interaction with L and template RNA (32, 33). However, recent findings showed that unphosphorylated P was competent to interact with both L and the N-RNA template (9, 12). Also, in vitro transcription reactions could be reconstituted with the N-RNA template isolated from detergent-disrupted virus particles and bacterium-expressed, unphosphorylated P protein (12). However, it is important to note that the N-RNA template purified from virions contains a small, residual amount of viral P protein (12, 52). It is possible that a small fraction of this residual P is phosphorylated, which might be sufficient to initiate RNA synthesis. As evident from the discussion above, a more detailed investigation of putative phosphorylation events is necessary to understand the relevance of phosphoregulation in the VSV life cycle.
VSV P protein and the phosphoprotein of rabies virus (18, 53, 54), another member of the Rhabdoviridae family, have some structural similarities that include IDRs. These regions may undergo transition between different structural states. The increased flexibility of the disordered regions could be advantageous to proteins that may recognize multiple target molecules with different affinities (55–57). As reported here, the functionally important residues (Tyr14, Ser60, Thr62, Ser64) of P lie in the N-terminal disordered domain. It has been predicted that intrinsic disorder in and around potential phosphorylation target sites is an essential common feature (58). In this context, it would be interesting to know if phosphorylation affects the structural disorder of P protein.
In summary, our results show that residue Tyr14, and likely its phosphorylation, is important for the VSV P protein to be functional in all viral RNA synthesis events, while phosphorylatable residues Ser60/Thr62/Ser64 are required to support transcription but do not affect genomic RNA replication. Further studies are necessary to understand whether phosphorylation is involved in determining the balance of transcription versus replication in the viral life cycle. In this regard, an investigation of the kinase responsible for phosphorylation of the newly identified site would be of interest, as it could be targeted to investigate effects on the viral life cycle and perhaps shed light on whether the site or its phosphorylation is important for VSV RNA synthesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant AI12464 to G.W.W. from the NIAID. Funding for the work done in the University of Virginia Pathology Mass Spectrometry Facility was provided through the National Cancer Institute (grant CA126101).
We thank our colleagues from the Wertz Lab and the Mehle Lab for constructive comments and Andrew Mehle and John Yin for generously providing key reagents.
Footnotes
Published ahead of print 20 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02384-13.
REFERENCES
- 1.Emerson SU, Wagner RR. 1972. Dissociation and reconstitution of the transcriptase and template activities of vesicular stomatitis B and T virions. J. Virol. 10:297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sleat DE, Banerjee AK. 1993. Transcriptional activity and mutational analysis of recombinant vesicular stomatitis virus RNA polymerase. J. Virol. 67:1334–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogino T, Banerjee AK. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 25:85–97. 10.1016/j.molcel.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 4.Li J, Fontaine-Rodriguez EC, Whelan SP. 2005. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol. 79:13373–13384. 10.1128/JVI.79.21.13373-13384.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J. Virol. 83:11043–11050. 10.1128/JVI.01426-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galloway SE, Richardson PE, Wertz GW. 2008. Analysis of a structural homology model of the 2′-O-ribose methyltransferase domain within the vesicular stomatitis virus L protein. Virology 382:69–82. 10.1016/j.virol.2008.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. 10.1126/science.1126953 [DOI] [PubMed] [Google Scholar]
- 8.Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363. 10.1126/science.1125280 [DOI] [PubMed] [Google Scholar]
- 9.Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. U. S. A. 106:11713–11718. 10.1073/pnas.0903228106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 84:5655–5659. 10.1073/pnas.84.16.5655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellon MG, Emerson SU. 1978. Rebinding of transcriptase components (L and NS proteins) to the nucleocapsid template of vesicular stomatitis virus. J. Virol. 27:560–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, Whelan SP. 2012. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 109:14628–14633. 10.1073/pnas.1209147109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howard M, Wertz G. 1989. Vesicular stomatitis virus RNA replication: a role for the NS protein. J. Gen. Virol. 70(Pt 10):2683–2694. 10.1099/0022-1317-70-10-2683 [DOI] [PubMed] [Google Scholar]
- 14.Chen M, Ogino T, Banerjee AK. 2007. Interaction of vesicular stomatitis virus P and N proteins: identification of two overlapping domains at the N terminus of P that are involved in N0-P complex formation and encapsidation of viral genome RNA. J. Virol. 81:13478–13485. 10.1128/JVI.01244-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mondal A, Roy A, Sarkar S, Mukherjee J, Ganguly T, Chattopadhyay D. 2012. Interaction of Chandipura virus N and P proteins: identification of two mutually exclusive domains of N involved in interaction with P. PLoS One 7:e34623. 10.1371/journal.pone.0034623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding H, Green TJ, Lu S, Luo M. 2006. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol. 80:2808–2814. 10.1128/JVI.80.6.2808-2814.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leyrat C, Jensen MR, Ribeiro EA, Gérard FC, Ruigrok RW, Blackledge M, Jamin M. 2011. The N(0)-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient α-helices. Protein Sci. 20:542–556. 10.1002/pro.587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leyrat C, Schneider R, Ribeiro EA, Yabukarski F, Yao M, Gérard FC, Jensen MR, Ruigrok RW, Blackledge M, Jamin M. 2012. Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J. Mol. Biol. 423:182–197. 10.1016/j.jmb.2012.07.003 [DOI] [PubMed] [Google Scholar]
- 19.Rahmeh AA, Schenk AD, Danek EI, Kranzusch PJ, Liang B, Walz T, Whelan SP. 2010. Molecular architecture of the vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:20075–20080. 10.1073/pnas.1013559107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leyrat C, Gérard FC, de Almeida Ribeiro E, Ivanov I, Ruigrok RW, Jamin M. 2010. Structural disorder in proteins of the rhabdoviridae replication complex. Protein Pept. Lett. 17:979–987. 10.2174/092986610791498939 [DOI] [PubMed] [Google Scholar]
- 21.Barik S, Banerjee AK. 1991. Cloning and expression of the vesicular stomatitis virus phosphoprotein gene in Escherichia coli: analysis of phosphorylation status versus transcriptional activity. J. Virol. 65:1719–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barik S, Banerjee AK. 1992. Phosphorylation by cellular casein kinase II is essential for transcriptional activity of vesicular stomatitis virus phosphoprotein P. Proc. Natl. Acad. Sci. U. S. A. 89:6570–6574. 10.1073/pnas.89.14.6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kingsford L, Emerson SU. 1980. Transcriptional activities of different phosphorylated species of NS protein purified from vesicular stomatitis virions and cytoplasm of infected cells. J. Virol. 33:1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masters PS, Banerjee AK. 1986. Phosphoprotein NS of vesicular stomatitis virus: phosphorylated states and transcriptional activities of intracellular and virion forms. Virology 154:259–270. 10.1016/0042-6822(86)90452-6 [DOI] [PubMed] [Google Scholar]
- 25.Hsu CH, Kingsbury DW. 1982. NS phosphoprotein of vesicular stomatitis virus: subspecies separated by electrophoresis and isoelectric focusing. J. Virol. 42:342–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bell JC, Prevec L. 1985. Phosphorylation sites on phosphoprotein NS of vesicular stomatitis virus. J. Virol. 54:697–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marnell LL, Summers DF. 1984. Characterization of the phosphorylated small enzyme subunit, NS, of the vesicular stomatitis virus RNA polymerase. J. Biol. Chem. 259:13518–13524 [PubMed] [Google Scholar]
- 28.Hsu CH, Kingsbury DW. 1985. Constitutively phosphorylated residues in the NS protein of vesicular stomatitis virus. J. Biol. Chem. 260:8990–8995 [PubMed] [Google Scholar]
- 29.Chen JL, Das T, Banerjee AK. 1997. Phosphorylated states of vesicular stomatitis virus P protein in vitro and in vivo. Virology 228:200–212. 10.1006/viro.1996.8401 [DOI] [PubMed] [Google Scholar]
- 30.Jackson RL, Spadafora D, Perrault J. 1995. Hierarchal constitutive phosphorylation of the vesicular stomatitis virus P protein and lack of effect on P1 to P2 conversion. Virology 214:189–197. 10.1006/viro.1995.9941 [DOI] [PubMed] [Google Scholar]
- 31.Das T, Gupta AK, Sims PW, Gelfand CA, Jentoft JE, Banerjee AK. 1995. Role of cellular casein kinase II in the function of the phosphoprotein (P) subunit of RNA polymerase of vesicular stomatitis virus. J. Biol. Chem. 270:24100–24107. 10.1074/jbc.270.41.24100 [DOI] [PubMed] [Google Scholar]
- 32.Gao Y, Lenard J. 1995. Multimerization and transcriptional activation of the phosphoprotein (P) of vesicular stomatitis virus by casein kinase-II. EMBO J. 14:1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y, Lenard J. 1995. Cooperative binding of multimeric phosphoprotein (P) of vesicular stomatitis virus to polymerase (L) and template: pathways of assembly. J. Virol. 69:7718–7723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spadafora D, Canter DM, Jackson RL, Perrault J. 1996. Constitutive phosphorylation of the vesicular stomatitis virus P protein modulates polymerase complex formation but is not essential for transcription or replication. J. Virol. 70:4538–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattnaik AK, Hwang L, Li T, Englund N, Mathur M, Das T, Banerjee AK. 1997. Phosphorylation within the amino-terminal acidic domain I of the phosphoprotein of vesicular stomatitis virus is required for transcription but not for replication. J. Virol. 71:8167–8175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang LN, Englund N, Das T, Banerjee AK, Pattnaik AK. 1999. Optimal replication activity of vesicular stomatitis virus RNA polymerase requires phosphorylation of a residue(s) at carboxy-terminal domain II of its accessory subunit, phosphoprotein P. J. Virol. 73:5613–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wertz GW, Moudy R, Ball LA. 2002. Adding genes to the RNA genome of vesicular stomatitis virus: positional effects on stability of expression. J. Virol. 76:7642–7650. 10.1128/JVI.76.15.7642-7650.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyons CE, Victor KG, Moshnikov SA, Bachmann LM, Baras AS, Dettmann KM, Cross JV, Templeton DJ. 2011. PICquant: a quantitative platform to measure differential peptide abundance using dual-isotopic labeling with 12C6- and 13C6-phenyl isocyanate. Anal. Chem. 83:856–865. 10.1021/ac102461e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Victor KG, Murgai M, Lyons CE, Templeton TA, Moshnikov SA, Templeton DJ. 2010. MAZIE: A mass and charge inference engine to enhance database searching of tandem mass spectra. J. Am. Soc. Mass Spectrom. 21:80–87. 10.1016/j.jasms.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang XY, Shi WY, Bryant SH. 2004. Open mass spectrometry search algorithm. J. Proteome Res. 3:958–964. 10.1021/pr0499491 [DOI] [PubMed] [Google Scholar]
- 41.Käll L, Storey JD, MacCoss MJ, Noble WS. 2008. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7:29–34. 10.1021/pr700600n [DOI] [PubMed] [Google Scholar]
- 42.Pattnaik AK, Wertz GW. 1990. Replication and amplification of defective interfering particle RNAs of vesicular stomatitis virus in cells expressing viral proteins from vectors containing cloned cDNAs. J. Virol. 64:2948–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pattnaik AK, Ball LA, LeGrone AW, Wertz GW. 1992. Infectious defective interfering particles of VSV from transcripts of a cDNA clone. Cell 69:1011–1020. 10.1016/0092-8674(92)90619-N [DOI] [PubMed] [Google Scholar]
- 44.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126. 10.1073/pnas.83.21.8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wertz GW, Whelan S, LeGrone A, Ball LA. 1994. Extent of terminal complementarity modulates the balance between transcription and replication of vesicular stomatitis virus RNA. Proc. Natl. Acad. Sci. U. S. A. 91:8587–8591. 10.1073/pnas.91.18.8587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siepen JA, Keevil EJ, Knight D, Hubbard SJ. 2007. Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics. J. Proteome Res. 6:399–408. 10.1021/pr060507u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harouaka D, Wertz GW. 2009. Mutations in the C-terminal loop of the nucleocapsid protein affect vesicular stomatitis virus RNA replication and transcription differentially. J. Virol. 83:11429–11439. 10.1128/JVI.00813-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das SC, Pattnaik AK. 2004. Phosphorylation of vesicular stomatitis virus phosphoprotein P is indispensable for virus growth. J. Virol. 78:6420–6430. 10.1128/JVI.78.12.6420-6430.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barr JN, Whelan SP, Wertz GW. 1997. Role of the intergenic dinucleotide in vesicular stomatitis virus RNA transcription. J. Virol. 71:1794–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barr JN, Whelan SP, Wertz GW. 1997. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J. Virol. 71:8718–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morin B, Rahmeh AA, Whelan SP. 2012. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31:1320–1329. 10.1038/emboj.2011.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emerson SU, Yu Y. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 15:1348–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerard FC, Ribeiro EeA, Albertini AA, Gutsche I, Zaccai G, Ruigrok RW, Jamin M. 2007. Unphosphorylated rhabdoviridae phosphoproteins form elongated dimers in solution. Biochemistry 46:10328–10338. 10.1021/bi7007799 [DOI] [PubMed] [Google Scholar]
- 54.Gerard FC, Ribeiro EeA, Leyrat C, Ivanov I, Blondel D, Longhi S, Ruigrok RW, Jamin M. 2009. Modular organization of rabies virus phosphoprotein. J. Mol. Biol. 388:978–996. 10.1016/j.jmb.2009.03.061 [DOI] [PubMed] [Google Scholar]
- 55.Lobley A, Swindells MB, Orengo CA, Jones DT. 2007. Inferring function using patterns of native disorder in proteins. PLoS Comput. Biol. 3:e162. 10.1371/journal.pcbi.0030162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. 1998. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac. Symp. Biocomput. 1998:473–484 [PubMed] [Google Scholar]
- 57.Radivojac P, Obradovic Z, Smith DK, Zhu G, Vucetic S, Brown CJ, Lawson JD, Dunker AK. 2004. Protein flexibility and intrinsic disorder. Protein Sci. 13:71–80. 10.1110/ps.03128904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. 2004. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32:1037–1049. 10.1093/nar/gkh253 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.