

Abstract
Late in adenovirus assembly, the viral protease (AVP) becomes activated and cleaves multiple copies of three capsid and three core proteins. Proteolytic maturation is an absolute requirement to render the viral particle infectious. We show here that the L1 52/55k protein, which is present in empty capsids but not in mature virions and is required for genome packaging, is the seventh substrate for AVP. A new estimate on its copy number indicates that there are about 50 molecules of the L1 52/55k protein in the immature virus particle. Using a quasi-in vivo situation, i.e., the addition of recombinant AVP to mildly disrupted immature virus particles, we show that cleavage of L1 52/55k is DNA dependent, as is the cleavage of the other viral precursor proteins, and occurs at multiple sites, many not conforming to AVP consensus cleavage sites. Proteolytic processing of L1 52/55k disrupts its interactions with other capsid and core proteins, providing a mechanism for its removal during viral maturation. Our results support a model in which the role of L1 52/55k protein during assembly consists in tethering the viral core to the icosahedral shell and in which maturation proceeds simultaneously with packaging, before the viral particle is sealed.
INTRODUCTION
Adenovirus morphogenesis ends with a maturation step comprising proteolytic cleavage of several capsid and core precursor proteins. Without these cleavages, the immature particle lacks infectivity because of its inability to uncoat (1–3). Maturation primes the viral particle for stepwise uncoating by facilitating penton release and by loosening the condensed genome and its attachment to the icosahedral shell (4, 5). Proteolytic processing is carried out by the adenovirus protease (AVP; or L3 23K protein) (6). In human adenovirus type 2 (HAdV-2), AVP recognizes (M/I/L)XGX-G and (M/I/L)XGG-X sequence motifs (7, 8). These sequences are present in the precursor proteins pIIIa, pVI, and pVIII in the icosahedral shell, as well as in the DNA-binding polypeptides pVII, pre-μ, and the terminal protein. These six precursor proteins have been shown to be substrates for AVP. Another potential substrate, because it contains an AVP consensus sequence motif, is polypeptide L1 52/55k.
The L1 52/55k protein in HAdV-2 is 415 residues in length, with an AVP consensus cleavage site at the 351-352 position (LAGT-G). Although the molecular mass of L1 52/55k calculated from its sequence is 47 kDa, the protein was named by its electrophoretic mobility; it moved as a doublet due to two different phosphorylation states (9). L1 52/55k is part of the genome packaging machinery, together with polypeptides IIIa, IVa2, L4 33k, and L4 22k (10–15). An L1 52/55k deletion construct produces only empty capsids (10), and a thermosensitive mutation in the L1 52/55k C-terminal region (ts369; 333-EL-336 to 333-GP-336) causes partial packaging (16). L1 52/55k binds to the viral packaging sequence in vivo and to the putative packaging ATPase IVa2 in vitro (17–19). L1 52/55k contributes to the specificity of packaging, possibly via an interaction with capsid protein IIIa (12, 20). Furthermore, L1 52/55k has been reported to bind nonspecifically to DNA and to interact with pVII and its mature form VII in infected cells (21). The interaction with DNA may not be direct, because L1 52/55k does not bind to the DNA packaging sequence in vitro (17–19).
L1 52/55k has been considered a putative scaffolding protein because it is present in empty particles in its full-length form but is absent from the mature virion (9). It is not a bona fide scaffolding protein, however, since capsids of apparently the same size and composition as empty wild-type particles assemble in its absence (10). Fully packaged, immature particles produced by the HAdV-2 thermosensitive mutant ts1 at nonpermissive conditions contain precursor versions of all AVP targets because the virus particles do not contain AVP (3). They have been reported to also contain 2 copies of full-length L1 52/55k, compared to ∼4 copies in partially packaged capsids and 50 in empty capsids (9). Bands corresponding to molecular masses of 40 and 34 kDa are also recognized by antibodies generated against L1 52/55k in empty or partially packaged capsids (9, 10, 22). Notice that the bands described as 40 and 34 kDa in reference 9 are described as 47 and 40 kDa in reference 22. In the present study, we use the nomenclature of Hasson et al. (9) to avoid confusion with the calculated molecular mass of the full-length L1 52/55k protein (47 kDa). The 34-kDa protein and some smaller protein species are also revealed in overloaded Western blots of mature particles. The 40-kDa band would correspond to the expected product of proteolysis by AVP at the consensus cleavage site. Both the full-length and 40-kDa bands disappear and are replaced by higher-molecular-mass species in electrophoresis performed in the absence of β-mercaptoethanol, suggesting the formation of disulfide-linked homodimers mediated by the only Cys residue (Cys24) in L1 52/55k (9, 22). The 34-kDa band has been proposed to originate by an additional cleavage at the N terminus, because it does not react with antibodies against either residues 9 to 22 (9-22) or 402-415 in L1 52/55k, and its electrophoretic mobility is not sensitive to nonreducing conditions, a finding consistent with the absence of Cys24 (9, 22). However, the actual processing of L1 52/55k by AVP has not been experimentally observed, nor is the origin of the smaller bands recognized by anti L1 52/55k antibodies known. Furthermore, it is not known what possible role the cleavage by AVP might have in the ejection of the L1 52/55k protein and its fragments from the particle upon packaging of the genome.
AVP has a unique mode of action. It is synthesized as an inactive enzyme (23). It uses two cofactors for maximal enzyme activity: the 11-amino-acid peptide pVIc, derived from the C terminus of the polypeptide VI precursor pVI, and the viral DNA. AVP is activated when pVI slides on DNA into AVP bound to the same DNA (24). AVP, partially activated by being bound to DNA, cleaves pVI at its N terminus and then at its C terminus. Cleavage at the C terminus releases pVIc, which then binds to the AVP that cut it out. The AVP-pVIc complex then slides along DNA, processing the virus precursor proteins also bound to the DNA (25). These experiments were done with fully disrupted ts1 particles after heating at 60°C (26). When immature ts1 particles are incubated at 47°C for 10 min, they release pentons and peripentonal hexons but retain the spherical capsid arrangement (27). The viral genome remains inside the “whiffle ball” particle in a highly condensed form attached to the shell (4). Since these mildly disrupted particles have a structural organization close to that of the intact immature capsid, while presenting openings that allow recombinant AVP access to its substrates bound to the viral DNA, we now use them as an experimental system to follow the action of AVP in detail in a more “quasi-in vivo” situation. These assays reveal that L1 52/55k is a previously unrecognized substrate for AVP and provide new insights into its role during viral maturation.
MATERIALS AND METHODS
Materials.
The gene for AVP was cloned and expressed in Escherichia coli, and the protein was purified as described previously (26). Immature virus was obtained by propagating the HAdV-2 ts1 mutant in HeLa cells at 39.5°C as described previously (27). Particles were purified by equilibrium centrifugation in CsCl gradients, desalted on a 10DC column (Bio-Rad), and stored in 20 mM HEPES (pH 7.8)–150 mM NaCl plus 10% glycerol at −70°C at a final concentration of 1013 viral particles (vp)/ml. An E1 deletion HAdV-5 vector (Ad5GL) was used as a mature, structurally wild-type virus control (28). The following antibodies were used for immunoassays: rat polyclonal anti-pVII (29), mouse anti-V serum (30), rabbit anti-VIII N-terminal fragment (provided by U. F. Greber), rabbit anti-pVI (31), rabbit anti-L1 55/52k (17), and rabbit anti-HAdV-5 fiber knob (32).
Analysis of AVP function in disrupted ts1 virus.
Immature ts1 virus at a concentration of 1.6 × 1012 vp/ml was mildly disrupted by heating at 47°C for 10 min and then incubated with 0.25 μM AVP at 37°C for the indicated times. Reactions were carried out in 10 mM Tris-HCl (pH 7.4), 20 mM NaCl, and 10 mM EDTA and stopped by adding electrophoresis loading buffer. This is a suboptimal condition for enzyme activity (better nearer pH 8) (26) and for one-dimensional diffusion on DNA (better nearer pH 6) (25), but it allowed both sliding and enzyme activity to occur at a rate that best revealed details of the maturation process.
To assess the role of DNA in L1 52/55k processing by AVP, viral DNA was removed from the samples as follows. Viruses were either mildly or completely disrupted by heating at 47 or 60°C, respectively, for 10 min and then incubated overnight at 37°C with 50 μg of DNase I (Sigma, catalog no. D5025)/ml in 10 mM Tris-HCl (pH 8.2)–5 mM MgCl2. The DNase was inactivated by adding 10 mM EDTA. After 30 min, 0.25 μM AVP was added, and the samples were incubated at 37°C for 24 h. In the indicated cases, purified ts1 DNA was added after DNase inactivation at a final concentration of 50 ng/ml. For DNA isolation, 25 × 1010 ts1 viral particles were treated with proteinase K at a final concentration of 400 μg/ml, and the DNA was extracted by phenol-chloroform precipitation.
Protein electrophoresis and Western blot analysis.
To separate viral proteins, samples were boiled in Laemmli loading buffer and subjected to electrophoresis under denaturing conditions (SDS-PAGE) in either 15% or 4 to 20% (Bio-Rad mini-Protean TGX) acrylamide gels. For Western blot analysis, proteins resolved via SDS-PAGE were transferred to polyvinylidene difluoride membranes and probed with the required antibodies. Bound antibodies were detected with the corresponding secondary antibody conjugated to alkaline phosphatase (Sigma), and the membranes were developed using an alkaline phosphatase conjugate substrate kit (Bio-Rad Laboratories, Hercules, CA) as recommended by the manufacturer.
Protein identification by mass spectrometry (MS).
Protein bands observed in SDS-PAGE were carefully excised from Coomassie blue-stained 4 to 20% acrylamide gradient gels and subjected to in-gel trypsin digestion as described previously (33). The gel pieces were swollen in a digestion buffer containing 50 mM NH4HCO3 and 12.5 μg/ml of trypsin (modified porcine trypsin, sequencing grade; Promega, Madison, WI) in an ice bath. After 30 min, the supernatant was removed, and 20 μl of 50 mM NH4HCO3 was added to the gel pieces. Digestion was allowed to proceed at 37°C overnight. The reaction was stopped by adding a mixture of 50% acetonitrile (ACN) and 0.5% trifluoroacetic acid (TFA). The extracted peptides were dried by speed-vacuum centrifugation and resuspended in 4 μl of matrix-assisted laser desorption ionization (MALDI) solution (30% ACN plus 15% isopropanol plus 0.5% TFA). Then, 20% of each peptide mixture was deposited onto a 384-well OptiTOF plate (Applied Biosystems, Framingham, MA) and allowed to dry at room temperature. A 0.8-μl aliquot of matrix solution (3 mg of α-cyano-4-hydroxycinnamic acid/ml in MALDI solution) was then added, and the sample was allowed to dry at room temperature.
Samples were automatically analyzed in an ABI 4800 MALDI time-of-flight (TOF)/TOF mass spectrometer (ABSciex, Framingham, MA) working in positive-ion reflector mode. The ion acceleration voltage was 25 kV for MS acquisition and 2 kV for tandem MS (MS/MS). Peptide mass fingerprinting and MS/MS fragment ion spectra were smoothed and corrected to zero baseline using routines embedded in the ABI 4000 Series Explorer Software v3.6. Internal and external calibration allowed us to achieve a typical mass measurement accuracy of <25 ppm. To submit the combined peptide mass fingerprinting and MS/MS data to MASCOT software v.2.1 (Matrix Science, London, United Kingdom), GPS Explorer v4.9 was used.
Negative-staining electron microscopy.
Virus samples were heated at 47°C for 10 min and then incubated with either AVP 0.25 μM in 10 mM Tris-HCl (pH 7.4), 20 mM NaCl, and 10 mM EDTA or only buffer for the indicated times. Proteolysis was stopped by adding NaCl to a final concentration of 150 mM. Samples were adsorbed for 5 min onto glow-discharged collodion/carbon-coated copper electron microscopy grids. The grids were transferred to a 2% uranyl acetate drop for negative staining, dried, and examined using a JEOL JEM 1011 transmission electron microscope.
Quantification of L1 52/55k copy number in ts1 particles.
For semiquantitative determination of the amount of L1 52/55k protein in different viral populations, identical quantities of purified wild-type or ts1 capsids (estimated by absorbance at 260 nm) were electrophoresed and silver stained, or probed with anti-V or anti-L1 52/55k antibodies for Western blot analysis as described above. Band intensities in the silver-stained gel images were measured with ImageJ (34). The area below each peak in the intensity plot was measured and corrected for the background intensity measured in a nearby area. Since in denaturing gel electrophoresis the full-length L1 52/55k protein runs at the same position as polypeptide V, to estimate the copy number of L1 52/55k in full ts1 viral capsids, we compared the intensity ratio between bands corresponding to polypeptides pVII (VII in the wild type) and V, or between IX and V, in ts1 and wild-type virions. Given that Western blot assays indicated that L1 52/55k was not present in the wild type (see Fig. 5), an estimation of the L1 52/55k content was derived from the excess intensity in the ts1 polypeptide V band.
FIG 5.

Estimation of L1 52/55k copy number. Serial dilutions of wild-type (wt) or ts1 virus samples were analyzed by SDS-PAGE and band densitometry, as well as by Western blotting for L1 52/55k or polypeptide V, as indicated. The intensity ratio between the bands labeled in the SDS-PAGE panel was used for the estimation, as explained in the text.
Southern blotting.
Detection of viral DNA by Southern blotting was performed according to standard methods (35). Briefly, DNA was extracted from purified ts1 virus by proteinase K-SDS digestion, followed by phenol-chloroform extraction. Purified DNA was electrophoresed on 1% agarose gels, transferred to Hybond nylon filters, and probed with DraIII-digested ts1 DNA labeled with alkaline phosphatase in conjunction with chemiluminescent detection with CDP-Star (Amersham, catalog no. RPN3680).
Far-Western blot.
Different fragments of L1 52/55k were obtained by AVP cleavage in disrupted ts1 capsids, followed by SDS-PAGE and extraction from the gel as follows. Immature ts1 virus particles at a concentration of 1.6 × 1012 vp/ml were mildly disrupted by heating at 47°C for 10 min and then incubated with 0.25 μM AVP at 37°C in 10 mM Tris-HCl (pH 8.2)–1 mM EDTA for 24 h. The sample was electrophoresed under denaturing conditions (SDS-PAGE) in 4 to 20% acrylamide gradient gels. Unstained bands corresponding to L1 52/55k proteolysis products were excised by using as a reference the stained first and last lanes of the gel. The gel pieces were washed three times, maintained in elution buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1 mM EDTA; pH 7.5) in a rotary shaker at 30°C overnight, and then centrifuged at 10,000 × g for 10 min.
Proteins present in the supernatant were labeled using two methods. In the first method, proteins were incubated overnight with rabbit anti-L1 52/55k in a rotary shaker at 4°C and cross-linked by adding 0.5% formaldehyde. Protein-antibody complexes were separated from free antibody by Sephacryl S-200HR gel filtration chromatography. In the second method, proteins were incubated with sulfo-NHS-biotin overnight, and the reaction was stopped by adding 10 mM Tris (pH 7.4). The excess biotin reagent was removed by using a desalting column.
Samples of mildly disrupted ts1 virus, digested or not digested with AVP, were electrophoresed and transferred to a nitrocellulose membrane as described for Western blots. For renaturation, the membranes were washed and incubated overnight in refolding buffer TBS-T (100 mM Tris-HCl [pH 7.6], 150 mM NaCl, 0.01% Tween 20), with 10% glycerol and 5 mM 2-mercaptoethanol. The membranes were blocked with 0.05% Tween 20 in phosphate-buffered saline (PBS) for 2 h and then with 1% (wt/vol) bovine serum albumin in PBS for another 2 h, followed by incubation with the labeled proteins overnight at 4°C with agitation. Finally, the membranes were incubated with either secondary antibody or streptavidin-horseradish peroxidase for 1 h, and the bound probes were detected by using an enhanced chemiluminescence method.
L1 52/55k release assay.
Immature ts1 particles at a concentration of 1.6 × 1012 vp/ml were mildly disrupted by heating at 47°C for 10 min and then incubated overnight at 37°C with either 0.25 μM AVP in 10 mM Tris-HCl (pH 8.2)–10 mM NaCl–10 mM EDTA or buffer alone. The samples were centrifuged at 20,200 × g for 60 min at 4°C. The supernatant was collected and concentrated using a speed-vacuum concentrator, and the sediment was dissolved in 10 mM Tris-HCl (pH 8.2), 10 mM NaCl, and 10 mM EDTA before being analyzed by SDS-PAGE, Western blotting, or native electrophoresis in agarose gels.
RESULTS
Processing of proteins in mildly disrupted ts1 particles by AVP.
Immature ts1 particles (containing all protein precursors) were partially disrupted by heating at 47°C for 10 min and incubated with recombinant AVP as described in Materials and Methods. In the reactions, there were approximately 94 to 100 AVP molecules per viral particle, a ratio similar to that encountered in vivo (50 AVP/particle) (36). Processing of the viral proteins was followed by denaturing electrophoresis and by Western blotting to identify the precursors and their cleavage products.
Five minutes after the addition of AVP, processing of the precursors pVI and pVII was observed (Fig. 1A and B). For pVI, processing started with the appearance of the intermediate product corresponding to residues 33 to 250 (Fig. 1B), previously observed in in vitro assays using purified recombinant AVP and pVI (24). The mature protein, VI, appeared at t = 30 min, and became the dominant form after 1 h of incubation, although some precursor was still present in the sample until t = 5 h. For pVII, precursor and mature forms coexisted for the first hour of AVP treatment, after which only the mature form was observed. It was not possible to determine when cleavage of pVIII started, because the available antibodies did not reveal its cleavage products. Similarly to pVII, pVIII was completely processed after 1 h of AVP incubation, judging by the disappearance of the precursor band in Western blots. For controls, treating intact ts1 virus with AVP produced no cleavages, even after 6 h of incubation (Fig. 1E); neither did heat-disrupted ts1 viruses incubated in the absence of AVP show any proteolytic degradation (see Fig. 3B).
FIG 1.

Time course of AVP activity on mildly disrupted ts1 virus. (A) A 15% acrylamide SDS-PAGE shows changes in the viral proteins upon incubation with AVP for the times indicated. (B) Western blot assays to follow proteolytic processing of pVI, pVII, pVIII, and L1 52/55k, as indicated. (C) Time course analysis of AVP cleavages in a 4 to 20% gradient gel. Double stars indicate bands generated upon incubation with AVP that could correspond to L1 52/55k cleavage products. (D) Western blot for L1 52/55k protein on a 4 to 20% gradient gel reveals more AVP-generated proteolysis products. L1 52/55k full-length precursor (FL) and fragment products (F1 to F5) are indicated in panels B and D. (E) Control experiment. No proteolytic processing is observed in intact ts1 virions in the presence of AVP, even after 6 h of incubation. This observation also indicates that the virus particle is sealed, at least to a protein the size of AVP.
FIG 3.

DNA requirement for L1 52/55k cleavage by AVP. (A) DNA present in the preparations. DNA was extracted from either intact or heat disrupted ts1 virus particles after DNase treatment and revealed by Southern blotting with a viral genome probe. Lane 1, intact virus; lane 2, virus disrupted at 47°C; lane 3, virus disrupted at 60°C. (B) SDS-PAGE (top) and Western blot for L1 52/55k (bottom) showing dependence on DNA of AVP activity. Lane 1, control wild-type virus; lane 2, control, intact untreated ts1 virus; lane 3, ts1 virus disrupted at 47°C, treated with DNase, and then incubated with AVP; lane 4, ts1 virus disrupted at 60°C, treated with DNase, and DNase inactivated, and purified DNA was added back before incubation with AVP; lane 5, ts1 virus disrupted at 60°C and incubated with AVP; lane 6, ts1 virus disrupted at 60°C, treated with DNase, and incubated with AVP; lane 7, ts1 virus disrupted at 60°C and incubated with buffer in the absence of AVP.
L1 52/55k is a potential substrate for AVP, since it contains an AVP consensus cleavage sequence. Therefore, we probed disrupted ts1 virus for this protein (Fig. 1B). Western blot analysis using serum against L1 52/55k revealed the presence of a band corresponding to the full-length protein in ts1 virus. After the addition of AVP, two more bands appeared beginning 15 to 30 min later, a finding consistent with a 40-kDa doublet previously observed (9, 22). We named the bands in this doublet F1a and F1b (fragments 1a and b). A 40-kDa species is expected if the protein is cleaved at the consensus site. Consistent with this hypothesis, it has previously been shown that the 40-kDa doublet is not recognized by an antibody directed against the C-terminal region (residues 402 to 405) of L1 52/55k (22). Here, we show directly that indeed the expected cleavage is produced by AVP. An additional band of 34-kDa apparent molecular mass, which we call F2, also appeared in the Western blots. This 34-kDa band recognized by anti-L1 52/55k antibodies had also previously been observed (9, 22), but this is the first direct proof that it originates from the proteolytic action of AVP. Furthermore, in gradient SDS-PAGE (Fig. 1C), at least three other bands recognized by the anti-L1 52/55k serum were observed upon incubation with AVP (Fig. 1D): two with apparent molecular masses larger than 30 kDa (F3 and F4) and one of 18 kDa (F5). The presence of these bands hints at cleavage at nonconsensus sites. Interestingly, even after long incubations (t ≥ 5 h), a considerable amount of L1 52/55k in a variety of forms, from full length to 18 kDa, was observed, although the dominant form seemed to be the 40-kDa doublet bands. This is in contrast with the absence of L1 52/55k bands in wild-type, mature particles, suggesting that during maturation in vivo the processed forms of the L52/55k protein are somehow expelled, as opposed to them being completely degraded inside the nascent virion.
AVP cleaves L1 52/55k at noncanonical sites.
To further investigate the nature of the detected L1 52/55k species, we scanned the HAdV-2 L1 52/55k sequence (UniProt ID P03262) for consensus AVP cleavage motifs using the PATTINPROT server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_pattinprot.html). There are three important positions in the AVP consensus cleavage sequences: P4, P2, and P1-P−1 (Table 1). When the search was restricted to 100% compliance to either of the two previously reported AVP recognition patterns (7, 8), only the consensus cleavage at residues Thr351-Gly352 was found, as expected. However, a search with relaxed pattern similarity constraints yielded 13 more possible cleavage sites (Table 1). According to the PATTINPROT similarity criteria, six sites at residues 36-37, 66-67, 353-354, 382-383, 384-385, and 398-399 had 75% similarity to the consensus patterns, with a wobble in the P4 position. The other seven predicted sites had a lower similarity level (62%). Six of these (residues 65-66, 71-72, 124-125, 275-276, 276-277, 300-301, and 380-381) had a wobble in the P2 position, while the remaining site (residues 297-301) had a wobble in the P1-P−1 position. Positioning these sites in the protein sequence (Fig. 2A) revealed that the 14 possible cleavages were clustered in two regions at the N terminus, three regions at the C terminus, and three more regions at the central part of the polypeptide chain.
TABLE 1.
Potential AVP cleavage sites in L1 52/55k predicted by PATTINPROT
| Parameter | Sequencea |
|---|---|
| Position relative to cleavage site (--) | P4P3P2P1--P−1 |
| AVP consensus cleavage sequences | (M/I/L)XGX--G |
| (M/I/L)XGG--X | |
| Major cleavage sequence in the L1 52/55k protein | LAGT--G |
| Wobble in the P4 position | ASGG--A |
| EEGE--G | |
| GTGS--G | |
| AAGA--G | |
| GAGP--G | |
| NVGG--V | |
| Wobble in the P2 position | LEEG--E |
| LARL--G | |
| LRHG--L | |
| MLSL--G | |
| LSLG--K | |
| LEAA--G | |
| Wobble in the P1-P−1 position | IEGF--Y |
Discrepancies from the consensus patterns are indicated in boldface.
FIG 2.

Cleavage pattern of L1 52/55k by AVP. (A) Location in the L1 52/55k sequence of the predicted AVP cleavage motifs and of the actual peptides identified by MS/MS in the different fragments generated by AVP cleavage. The possible cleavage sites predicted by PATTINPROT are indicated by vertical red lines. A thick, continuous line indicates the consensus site. Thick dashed lines indicate sites with 75% similarity to the consensus pattern. Thin dashed lines indicate sites with 62% similarity to the consensus pattern. The peptides identified for each fragment are indicated by horizontal lines below the sequence in different colors: blue for F1, orange for F2, green for F3, and magenta for F4. The single Cys residue is highlighted in green and the two phosphorylated Ser residues in light blue. (B) Schematics picturing the L1 52/55k sequence and cleavage products observed in this work. The box at the top represents the full-length sequence with some relevant motifs or regions indicated as follows: pink, domain required for genome packaging (39); orange, IVa2 interacting domain (12); green, Cys residue involved in homodimer formation; blue, phosphorylated Ser residues; and dark red, residues involved in the ts369 mutation (16). The fragment assignments and molecular masses are indicated in the center of each box. Initial and final positions in the L1 52/55k sequence are indicated at the left and right of each box. Asterisks indicate fragments identified by MS/MS.
The possible L1 52/55k proteolytic products according to the PATTINPROT prediction of AVP consensus and nonconsensus cleavage sites are shown in Table 2. Interestingly, there is a substantial correspondence between the sizes of the predicted fragments and the bands recognized by anti-L1 52/55k antibodies in Western blots. To further assess this point, these bands were extracted from gels and analyzed by MS. The MS analysis confirmed that the F1, F2, F3, and F4 bands were derived from L1 52/55k. It was not possible to determine the identity of the F5 band. The peptide fingerprint obtained for each of the identified bands (Fig. 2A) further supported their identity as fragments derived from nonconsensus cleavage sites. Altogether, our analyses indicate the following cleavage scheme for L1 52/55k (Fig. 2B): first, and most frequently, the C-terminal region is cleaved. The consensus cleavage would produce a 40.3-kDa fragment, corresponding to the lower band in the F1 doublet. Cleavage at either the 382 or the 398 position (Table 2) would explain the upper band F1a in the doublet, although there is no MS confirmation for this hypothesis. Next, the 36 N-terminal residues are removed to produce F2 (36.3 kDa), in agreement with previous results showing that this band did not appear in Western blots with antibodies against either the C-terminal or the N-terminal regions of L1 52/55k and that its electrophoretic mobility was not dependent on the presence of reducing agents, indicating that the single Cys residue at position 24 (Fig. 2) was absent (9, 22). Additional cleavages identified here, at the 66 and 124 positions, give rise to F3 (33.0 kDa) and F4 (26.1 kDa). Finally, removal of the C-terminal 275-351 or 300-351 stretch could produce the smallest fragment observed, F5. It is intriguing that F5, whose identity could not be confirmed by MS, consistently appears at early times during AVP incubation (Fig. 1C and D).
TABLE 2.
Possible L1 52/55k fragments derived from AVP cleavage at the PATTINPROT predicted sites and their correspondence to bands observed in Western blots after in vitro processing of mildly disrupted ts1 virus by AVPa
| Fragment | Calculated molecular mass (kDa) | pI | Possible correspondence to Western blot banda |
|---|---|---|---|
| 1-415 (full length) | 46.9 | 5.67 | Full length |
| 1-398 | 44.9 | 7.24 | F1a |
| 1-382 | 43.5 | 6.89 | F1a |
| 1-351 (consensus) | 40.3 | 7.92 | F1b* |
| 36-351 | 36.3 | 6.66 | F2* |
| 66-351 | 33.0 | 8.77 | F3* |
| 124-351 | 26.1 | 6.74 | F4* |
| 124-300 | 20.2 | 5.55 | F5 |
| 124-275 | 17.2 | 5.04 | F5 |
| 275-351 | 8.9 | 9.91 | Not observed |
The isoelectric point value for each fragment is indicated. An asterisk (*) indicates identification by mass spectrometry.
Our results are consistent with L1 52/55k being cleaved at eight positions, seven of them not conforming to the previously determined consensus sequence pattern (7, 8). Other authors have previously detected maturation cleavages in adenovirus where the P4 residue in the pattern was Gln or Asn, instead of Met, Leu, or Ile (37). We report here one cleavage site with Asn in the P4 position and other sites with Ala, Gly, and possibly even a charged residue, Glu (Fig. 2A). The successive cleavages of L1 52/55k by AVP result in fragments with drastic differences in their isoelectric points (Table 2), as well as in the removal of sequence motifs or regions relevant for protein function (Fig. 2B). These include the only cysteine residue in the polypeptide chain, the two phosphorylation sites (38), part of the IVa2 binding domain (12), part of the region required for interaction with the viral packaging sequence (39), and the region containing residues mutated in the thermosensitive mutant ts369 (16).
Cleavage of L1 52/55k by AVP requires the presence of dsDNA.
We have previously shown that the presence of double-stranded DNA (dsDNA) is required for both activation of AVP and for cleavage of its substrates by AVP-pVIc complexes, when assayed in ts1 virus particles completely disrupted by heating at 60°C (24, 25). To determine whether this is also the case for L1 52/55k, we analyzed by Western blotting its processing by AVP in the presence or absence of dsDNA (Fig. 3). As previously observed for other AVP substrates, when ts1 virus was completely disrupted and its dsDNA digested away by DNase treatment (Fig. 3A, lane 3, and Fig. 3B, lane 6), no proteolytic products, including any processed form of L52/55k, were observed after 24 h of incubation with AVP. When, after DNase treatment, the DNase was inactivated and purified ts1 DNA was added back to the reaction, full AVP activity was recovered, including processing of L52/55k (Fig. 3B, lanes 4 and 5). Therefore, cleavage of L1 52/55k by AVP is also dependent on dsDNA.
An intriguing behavior was observed when, instead of completely disrupting ts1 by heating at 60°C, we used milder disruption conditions, heating at 47°C. In this case, treatment with DNase did not completely degrade the dsDNA, but instead fragments of 200 to 300 bp remained in the solution, presumably protected by interactions with core proteins that are not disrupted in the whiffle ball particle (Fig. 3A, lane 2) (40). In the presence of these small dsDNA fragments, partial cleavage of L1 52/55k was observed, with only fragments F1 and F2 revealed in the Western blot (Fig. 3B, lane 3). That is, cleaving at internal positions in the polypeptide (interior to the 36- to 351-residue region) did not occur. Under these conditions, digestion of the pVI, pVII and pVIII precursors was also deficient or absent (Fig. 3B, SDS-PAGE). This result indicates that AVP function is hindered by the lack of long stretches of dsDNA or by less DNA being present. It has previously been shown that for AVP to perform its function, both protease and substrate have to be bound to the same DNA molecule (24, 25). Therefore, a possible reason for this hindrance is the lack of physical space in the short oligonucleotides to house both the activated AVP and its substrates, as well as other DNA-binding proteins. Another possible reason is that only those proteins with the lowest equilibrium dissociation constants for binding to DNA will be bound to the “small” amount of dsDNA left. In any case, the lack of minor, internal cleavages in L1 52/55k in a situation with limited access to dsDNA suggests that its interaction with the genome changes as a consequence of the major cleavages induced by AVP.
Cleavage of L1 52/55k impairs its interactions with other proteins in the viral particle.
In the assays presented above, L1 52/55k protein was cleaved at multiple sites by the viral protease, but extensive cleavage was not observed; rather, large L1 52/55k fragments (>17 kDa) were present even after several hours of incubation with AVP. However, no traces of L1 52/55k were found in the mature virion in a variety of experiments (Fig. 1B, 1D, and 3B; see also Fig. 5). Therefore, we sought to investigate how the limited proteolysis of L1 52/55k can result in complete removal of all fragments from the viral particle during in vivo assembly.
We have previously shown that in vivo maturation of adenovirus results in the removal of protein interactions that stabilize both capsid and core structures (4, 27). Here, we observe the same effect in our in vitro system. HAdV-2 ts1 preparations were imaged by negative-staining electron microscopy after heating at 47°C for 10 min and digestion with AVP for different periods. As expected, in the absence of AVP, ts1 capsids lost some capsomers but largely retained their icosahedral organization. In contrast, after 45 min of incubation with AVP, the structural integrity of the capsids was lost, and fragmented, flattened capsids were mainly observed, along with some unraveling cores (Fig. 4A). From this experiment, we conclude that in vitro processing by AVP in disrupted immature virus particles weakens interactions stabilizing the viral particle, similar to the effect of in vivo AVP processing during maturation. In addition, we observed that the multiple cleavages undergone by L1 52/55k result in fragments with different electrostatic properties (Table 2) and in the loss of relevant sequence domains (Fig. 2B). These observations suggest that AVP-induced cleavages in L1 52/55k may diminish its interactions with other viral proteins, or the viral DNA, facilitating its removal from viral particles during maturation.
FIG 4.

Effect of L1 52/55k cleavage in L1 52/55k associations with other components of the viral particle. (A) Electron microscopic images showing additional disruption of ts1 particles heated at 47°C upon incubation with AVP for 45 min. The inset shows an unraveling core. Scale bar, 200 nm. (B) Solubility of L1 52/55k and its cleavage products. Mildly disrupted ts1 virus, control or treated with AVP, was centrifuged. The input (I), supernatant (S), and pellet (P) were analyzed by native DNA electrophoresis, SDS-PAGE, and Western blotting with antibodies against fiber, L1 52/55k, or polypeptide V, as indicated. (C) Far-Western blot assays were performed to analyze interactions between L1 52/55k cleavage products and other proteins in the viral particle. The proteins in control or AVP-treated ts1 virus samples were separated by SDS-PAGE and probed with gel-purified L1 52/55k F1, F2, and F3 fragments, previously labeled with either antibodies against L1 52/55k or biotin, as indicated.
Further evidence supporting this hypothesis was obtained from experiments testing the solubility of L1 52/55k before and after incubation with AVP (Fig. 4B). When heat-disrupted ts1 was centrifuged, the supernatant contained vertex proteins (peripentonal hexons, penton, IIIa, fiber, and pVI), whereas cores (DNA, V, and pVII) and presumably large capsid fragments (hexons, pVI, pVIII, and IX) appeared in the sedimented material. Western blotting indicated that some L1 52/55k was released, together with the vertices, but the majority remained in the insoluble fraction. When the disrupted virus was treated with AVP before centrifugation, the distribution of most viral components between the supernatant and pellet remained essentially unaltered. However, all L1 52/55k fragments were solubilized, indicating that processing by AVP disrupts L1 52/55k interactions with the other proteins and also the viral genome.
Next, we addressed the interactions of the different L1 52/55K fragments with other viral proteins using far-Western blots (Fig. 4C). Disrupted ts1 virus incubated with AVP was subjected to SDS-PAGE, L1 52/55k fragments F1, F2, and F3 were extracted and purified from gels and labeled using either antibodies against L1 52/55k or biotin. The labeled L1 52/55k fragments were then used as probes against viral proteins from ts1, processed or unprocessed by AVP. In all cases, the most intense signal was obtained with bands corresponding to either full-length or processed L1 52/55k, indicating a strong tendency to form homo-oligomers. In addition, minor interactions were observed with hexon, penton base, pVII, and VII. Interestingly, bands for these minor interactions became fainter as the L1 52/55k cleavages became more extensive. Interaction with penton base was only observed with F1, the largest fragment analyzed, while for the smallest (F3), virtually only self-interaction was observed. This result indicates that the N-terminal region of L1 52/55k (residues 1 to 65) participates in interactions with hexon, penton base and pVII/VII, but it is not required for self-interaction, in spite of containing the single cysteine residue in the sequence. Binding of L1 52/55k to hexon and penton base had not been previously described. The present analysis did not reveal other known L1 52/55k binding partners, such as polypeptides IIIa and IVa2 (12, 19). The signal for IVa2 may be obscured by the signal for L1 52/55k itself, since their electrophoretic mobilities are similar. As for IIIa, it is possible that the far-Western blot refolding conditions did not produce the proper conformation to maintain the native interactions. Nevertheless, the experiments presented here indicate that in the viral particle, L1 52/55k maintains an extensive network of interactions, not only with itself, but also with both shell (hexon and penton) and core proteins (pVII and VII), as well as with the genome. This network is disrupted when L1 52/55k is processed by AVP, providing a mechanism for scaffold ejection during maturation.
Estimation of the L1 52/55k copy number in immature ts1 particles.
In the course of both the present (Fig. 1C) and previous work (24), we noticed that bands appearing upon incubation of ts1 with AVP, consistent with L1 52/55k cleavage products, were often visible by silver staining. This observation suggested the possibility that ts1 full particles might contain L1 52/55k in larger quantities than expected from the copy number of 2 calculated previously (9). Therefore, we decided to perform a new analysis of the L1 52/55k copy number in ts1 using gel band densitometry (Fig. 5). Taking into account that ts1 contains only full-length L1 52/55k protein, that wt virions do not contain L1 52/55k, and that full-length L1 52/55k and polypeptide V have similar electrophoretic mobilities (∼50 kDa), we estimated the amount of L1 52/55k in ts1 by comparing the density ratio between the 50-kDa band and other bands corresponding to proteins with known copy numbers and present in both viral species. The excess protein content in the ts1 50-kDa band was attributed to L1 52/55k (Table 3). These calculations indicated that immature particles contain more than 50 copies of L1 52/55k, an order of magnitude more than previously reported (9).
TABLE 3.
Estimation of L1 52/55k copy numbers in ts1 virusa
| Polypeptides | Mass ratioa |
Deduced L1 52/55k copy no. in ts1 virusb | |||
|---|---|---|---|---|---|
| Wild-type virus |
ts1 mutant virus |
||||
| Theoretical | Calculated | Theoretical | Calculated | ||
| VII/V | 2.50 | 2.64 ± 0.20 | 2.84 | 2.12 ± 0.09 | 53 ± 5 |
| IX/V | 0.53 | 0.51 ± 0.04 | 0.53 | 0.34 ± 0.03 | 55 ± 3 |
Theoretical mass ratios were determined from the corresponding protein molecular mass (M) and copy numbers (N) as follows. For the wild type, ratioVII/V = (MVII·NVII)/(MV·NV). For the ts1 mutant, ratiopVII/V = (MpVII·NpVII)/(MV·NV). In both, ratioIX/V = (MIX·NIX)/(MV·NV). The following values were used: MV = 41.0 kDa, MpVII = 22.0 kDa, MVII = 19.4 kDa, MIX = 14.3 kDa, NV = 157, NpVII = NVII = 833, and NIX = 240. Calculated mass ratios were determined from the optical densities (D) of gel bands as measured with ImageJ, for example: ratioVII/V = DVII/DV. The results report the averages ± the standard deviations from three independent electrophoresis assays.
Calculated by solving the following equations for NL1: ratiopVII/(V+L1) = DpVII/D(V+L1) = (MpVII·NpVII)/(MV·NV + ML1·NL1) and ratioIX/(V+L1) = DIX/D(V+L1) = (MIX·NIX)/(MV·NV + ML1·NL1), with ML1 = 47.0 kDa.
DISCUSSION
Adenovirus morphogenesis is a complex process involving a considerable number of players. The final components of the HAdV-2 ∼150-MDa mature particle include multiple copies of seven different proteins in the icosahedral shell (hexon, penton base, fiber, IIIa, VI, VIII, and IX) and three putative DNA condensing proteins bound to the genome in the core (V, VII, and μ) (41, 42). In addition, several other factors are required to assemble the fully infectious particle. The concerted action of at least five different proteins is required for successful genome packaging: IIIa (also a part of the icosahedral shell), IVa2, L4 33k, L4 22k, and L1 52/55k (10, 12–15, 20, 43). Finally, proteolytic maturation carried out by AVP, together with its cofactors (dsDNA and pVIC), is required to render the particle infectious, i.e., metastable and primed for uncoating (2, 4, 23).
We have followed here the processing of immature, fully packaged virus particles using recombinant AVP. We observed that in this quasi-in vivo system, processing of polypeptide VI proceeds in two steps, from the precursor pVI to the intermediate iVI to the mature form VI, as previously observed using purified, recombinant pVI (24). However, the most novel information provided by this work links the genome encapsidation and maturation processes by showing that L1 52/55k is the seventh substrate for AVP and pointing to the effect of the AVP-induced cleavages on its extrusion from the maturing particle.
Because L1 52/55k is easily detected in HAdV light density particles (considered assembly intermediates) but is absent from the mature virion, it has been proposed to act as a scaffolding protein (9). Scaffolding elements are crucial in the assembly of large, complex capsids and are best understood in the dsDNA bacteriophage and the structurally related herpesvirus systems (44–46). Some of their functions include initiating assembly by nucleation of coat proteins, determining capsid size, and stabilizing labile assembly intermediates. Although structural knowledge of scaffolding proteins is scarce, studies on bacteriophage assembly indicate that these proteins have a tendency to form homo-oligomers, bind to major coat proteins and packaging machinery (47, 48), and show nonspecific dsDNA-binding activity (49). Once their functions have been fulfilled, scaffolding elements are released from the immature capsid before or during genome packaging. Release is often mediated by degradation by viral proteases and is thought to occur by extrusion through channels in the immature shell. In some bacteriophage, such as HK97, there are no separate scaffolding polypeptides. Instead, specific domains in the coat protein aid in assembly initiation and elongation and are proteolytically removed before packaging.
Adenovirus seems to use a dual scaffolding system, including both a detached polypeptide (L1 52/55k) and flexible regions of minor coat proteins that are removed during maturation (27, 41). We have previously shown that the immature ts1 particle is more stable than the mature one, largely due to a remarkable core compaction and the presence of strong capsid-core interactions mediated by the pIIIa, pVI, pVII, pVIII, and pre-μ precursors (4, 5, 27). In those studies, the presence of L1 52/55k was not considered, since a very low copy number (n = 2) had been reported in ts1 full particles (9). However, the results shown here indicate that there is a much larger quantity of L1 52/55k (n = 50) in ts1, implying that L1 52/55k may also contribute to the higher stability of the immature virus. We show here for the first time that L1 52/55k is proteolytically processed by AVP and that this processing facilitates its removal from the assembled particle by impairing interactions with other core and shell proteins and possibly with the dsDNA genome itself. Thus, cleavage of L1 52/55k must be considered part of the maturation process of adenovirus during which it is primed for uncoating.
Why our estimates for the copy number of the L1 52/55k protein in ts1 virus differ from previous reports is not clear. The highly dynamic and transitory nature of the L1 52/55k protein in the virus makes its copy number difficult to measure. Several potential problems could give rise to this disagreement. First, the amount of viral DNA per virion, as maturation proceeds, is variable. Second, in the growth of ts1 virus, slight, local variations in temperature could allow some AVP to be packaged that would decrease the amount of L1 52/55k protein. Finally, quantitating proteins by staining or antibody reactivity can give variable results. In the future, other methods for quantitating the copy number of L1 52/55k in ts1 particles may help to settle this discrepancy. For example, metabolic labeling by propagating the virus in the presence of 35S provided a very accurate stoichiometric analysis of the complete AdV mature particle (50). However, the occurrence of polypeptides IVa2, V, and L1 52/55k in a narrow range of electrophoretic mobility may complicate the procedure. An alternative method would be the use of quantitative MS (51). For these studies, representative tryptic peptides for the protein under study are synthesized, isotopically labeled, and mixed in known quantities with the viral sample. The MS signal intensities of the labeled peptides are compared to those of their unlabeled partners, thus serving as internal controls for estimation of the abundance of the corresponding unlabeled peptide in the virus. This methodology has also been used for AdV, where it helped to determine the copy number of the low-abundance polypeptide IVa2 (52).
Incubation with recombinant AVP produced L1 52/55k fragments different from those expected if the protein was cleaved at the single AVP consensus pattern in its sequence (7). The observation of cleavages at sites not fully conforming to the consensus was not that surprising, given that this phenomenon had already been observed (37, 53). The four empirically identified fragments shown by MS to be derived from the L1 52/55k protein were all derived by cleavage at the AVP consensus sequence near the C terminus, at position 351. Three of these fragments were also derived by a second cleavage, near the N terminus of L1 52/55k, at sites that resemble consensus cleavage sites (Fig. 2 and Table 1). In fact, all of the putative cleavage sites resemble AVP consensus cleavage sites; they all contain two of the three determinants most preferred by the proteinase. This observation argues that cleavage occurred by AVP and not by a contaminating E. coli protease in the AVP preparation. Consistent with this conclusion is that no cleavage by AVP occurred in disrupted virions treated with DNase (Fig. 3). No E. coli protease is known to have a DNA-dependent activity, whereas DNA is a cofactor for AVP activity (23–25). Since three of the four fragments were cleaved at the AVP consensus sequence, as well as a secondary site, it is possible that cleavage at the consensus sequence resulted in a conformational change of the protein, thereby exposing several secondary cleavage sites. Indeed, one could hypothesize that this first conformational change is the trigger for a cascade of cleavages required for release of L1 52/55k fragments from the nascent virion.
The picture emerging from this and other work highlights the similarities and the differences between adenovirus and bacteriophage/herpesvirus regarding assembly and maturation. Adenovirus scaffolding elements, whether peptides attached to minor coat proteins or polypeptide L1 52/55k, form part of an extensive network of interactions between the icosahedral shell and the viral core. Like the phage P22, ϕ29, or SPP1 scaffolding proteins (45), L1 52/55k has the ability to form homo-oligomers, either mediated by sulfur bridges using its single cysteine (9) or not (Fig. 4C). In addition, L1 52/55k interacts with coat and core proteins (Fig. 4C) (12, 21), as well as with other elements of the packaging machinery (17), including the genome itself (21). The latter property is a requisite for its functioning as an AVP substrate (Fig. 3) (25). L1 52/55k is cleaved at multiple sites, including several nonconsensus ones, by the viral protease (Fig. 2), but the protein is not completely degraded (Fig. 1). Cleavages impair some of its interactions, leading to release of L1 52/55k from the maturing particle, similar to the release mechanism reported for herpesvirus (46). The exact nature of the altered interactions is not known. However, it is noticeable that the different L1 52/55k fragments have widely different charge values (Table 1), suggesting possible changes in electrostatic interactions. Interestingly, in bacteriophage P22, electrostatic interactions between scaffold and coat proteins have been directly observed in high-resolution cryo-electron microscopic studies (47).
In adenovirus, the existence of light density, empty, or partially packaged capsids containing immature protein precursors, together with the similarities between polypeptide IVa2 and other packaging motors, point to a sequential packaging mechanism similar to that of bacteriophage, with the dsDNA being pumped into a preformed procapsid (13, 17, 43, 54–56). Unlike in bacteriophage, in adenovirus the scaffolding protein L1 52/55k does not seem to be required for capsid assembly or for incorporation of the packaging motor (10). Rather, L1 52/55k participates in bridging the viral core to the shell (Fig. 6). A possible role for this protein during packaging would then be to tether stably the partially packaged genome to the procapsid while DNA pumping is taking place. Alternatively, in a concerted assembly and packaging model, the role of L1 52/55k could be to recruit capsid proteins, or capsid fragments, onto condensing core precursors. In any case, since binding to DNA of both AVP and its substrates is required for proteolysis to occur (24, 25), scaffold maturation and release must occur during or after genome encapsidation.
FIG 6.
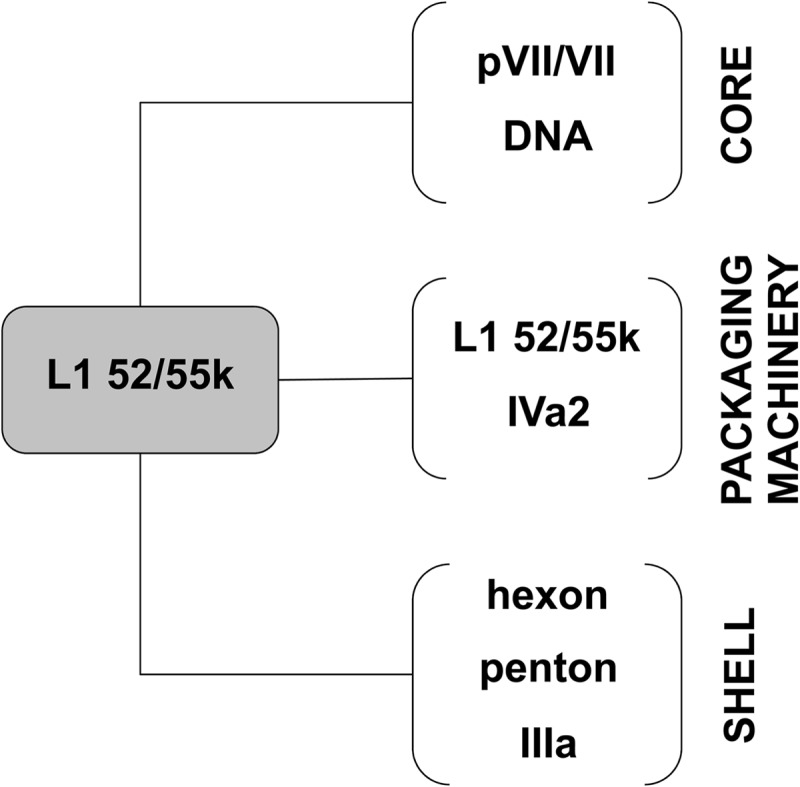
Diagram summarizing the multiple interactions established by L1 52/55k in the viral particle. The figure is based both in results presented here and in previously published studies (see the text for details).
How are the large cleavage products from L1 52/55k ejected from the virus particle? The final products of L1 52/55k proteolytic processing are rather large polypeptides, ranging in size from 17 to 40 kDa. Cleavage and release of L1 52/55k is not a requirement for successful packaging, since immature virus particles containing full-length L1 52/55k and genome are readily assembled in the ts1 mutant. The immature capsids of bacteriophage and herpesvirus have large openings (ca. 4 by 2 nm), thought to be used as extruding conduits for the scaffold proteins (45). These channels disappear due to large conformational rearrangements occurring during maturation and packaging to produce a tightly closed shell. Such a mechanism does not seem to exist in adenovirus. Light density particles in CsCl gradients are thought to represent the adenovirus procapsid, but their structure has not been solved. However, negative-staining electron microscopic images (see, for example, those in reference 13) suggest that the gross organization of adenovirus light particles is highly similar to that of the mature virion. Empty and full capsids of the adenovirus-like bacteriophage PRD1 have practically identical structures, as do the adenovirus mature and immature, fully packaged viral particles (27, 57). Therefore, in adenovirus, building of the tightly knit immature shell must proceed in such a way that a large opening is present until the last stages of maturation, so that scaffold fragments can be released before building of the icosahedral shell is completed. Once core and capsid precursors are brought together, whether in a sequential or a concerted manner, the DNA-bound AVP can be activated by interaction with pVIC; maturation and packaging would proceed in a simultaneous fashion, and assembly would end by fitting a last capsid piece after complete release of scaffold fragments. What this last capsid piece would be, and whether the large opening matches the genome packaging channel, are questions that remain to be answered.
ACKNOWLEDGMENTS
This study was supported by grant BFU2010-16382 from the Ministerio de Economía y Competitividad of Spain (to C.S.M.) and by National Institutes of Health grants R01AI41599 (to W.F.M.) and GM037705 (to S.J.F.). A.J.P.-B. was a recipient of a Juan de la Cierva postdoctoral contract from the Ministerio de Ciencia e Innovación of Spain.
We gratefully acknowledge U. F. Greber (University of Zurich, Zurich, Switzerland), K. Nagata (University of Tsukuba, Tsukuba, Japan), R. Gerard (UT Southwestern Medical Center), and P. Ostapchuk and P. Hearing (Stony Brook University) for the gift of antibodies. We also thank Mario Ciordia, Ambra Lo Piano, Sylvia Ayora, and María López Sanz (CNB-CSIC) for help with mass spectrometry and Southern blot analyses.
Footnotes
Published ahead of print 13 November 2013
REFERENCES
- 1.Cotten M, Weber JM. 1995. The adenovirus protease is required for virus entry into host cells. Virology 213:494–502. 10.1006/viro.1995.0022 [DOI] [PubMed] [Google Scholar]
- 2.Greber UF, Webster P, Weber J, Helenius A. 1996. The role of the adenovirus protease on virus entry into cells. EMBO J. 15:1766–1777 [PMC free article] [PubMed] [Google Scholar]
- 3.Weber J. 1976. Genetic analysis of adenovirus type 2. III. Temperature sensitivity of processing viral proteins. J. Virol. 17:462–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pérez-Berná AJ, Ortega-Esteban A, Menéndez-Conejero R, Winkler DC, Menéndez M, Steven AC, Flint SJ, de Pablo PJ, San Martín C. 2012. The role of capsid maturation on adenovirus priming for sequential uncoating. J. Biol. Chem. 287:31582–31595. 10.1074/jbc.M112.389957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ortega-Esteban A, Perez-Berna AJ, Menendez-Conejero R, Flint SJ, San Martin C, de Pablo PJ. 2013. Monitoring dynamics of human adenovirus disassembly induced by mechanical fatigue. Sci. Rep. 3:1434. 10.1038/srep01434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster A, Kemp G. 1993. The active adenovirus protease is the intact L3 23K protein. J. Gen. Virol. 74(Pt 7):1415–1420. 10.1099/0022-1317-74-7-1415 [DOI] [PubMed] [Google Scholar]
- 7.Diouri M, Keyvani-Amineh H, Geoghegan KF, Weber JM. 1996. Cleavage efficiency by adenovirus protease is site-dependent. J. Biol. Chem. 271:32511–32514. 10.1074/jbc.271.51.32511 [DOI] [PubMed] [Google Scholar]
- 8.Webster A, Russell S, Talbot P, Russell WC, Kemp GD. 1989. Characterization of the adenovirus proteinase: substrate specificity. J. Gen. Virol. 70(Pt 12):3225–3234. 10.1099/0022-1317-70-12-3225 [DOI] [PubMed] [Google Scholar]
- 9.Hasson TB, Ornelles DA, Shenk T. 1992. Adenovirus L1 52- and 55-kilodalton proteins are present within assembling virions and colocalize with nuclear structures distinct from replication centers. J. Virol. 66:6133–6142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gustin KE, Imperiale MJ. 1998. Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J. Virol. 72:7860–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostapchuk P, Anderson ME, Chandrasekhar S, Hearing P. 2006. The L4 22-kilodalton protein plays a role in packaging of the adenovirus genome. J. Virol. 80:6973–6981. 10.1128/JVI.00123-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma HC, Hearing P. 2011. Adenovirus structural protein IIIa is involved in the serotype specificity of viral DNA packaging. J. Virol. 85:7849–7855. 10.1128/JVI.00467-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostapchuk P, Almond M, Hearing P. 2011. Characterization of empty adenovirus particles assembled in the absence of a functional adenovirus IVa2 protein. J. Virol. 85:5524–5531. 10.1128/JVI.02538-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu K, Guimet D, Hearing P. 2013. The adenovirus L4-33K protein regulates both late gene expression patterns and viral DNA packaging. J. Virol. 87:6739–6747. 10.1128/JVI.00652-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guimet D, Hearing P. 2013. The adenovirus L4-22K protein has distinct functions in the posttranscriptional regulation of gene expression and encapsidation of the viral genome. J. Virol. 87:7688–7699. 10.1128/JVI.00859-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasson TB, Soloway PD, Ornelles DA, Doerfler W, Shenk T. 1989. Adenovirus L1 52- and 55-kilodalton proteins are required for assembly of virions. J. Virol. 63:3612–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostapchuk P, Yang J, Auffarth E, Hearing P. 2005. Functional interaction of the adenovirus IVa2 protein with adenovirus type 5 packaging sequences. J. Virol. 79:2831–2838. 10.1128/JVI.79.5.2831-2838.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Romero P, Tyler RE, Abend JR, Dus M, Imperiale MJ. 2005. Analysis of the interaction of the adenovirus L1 52/55-kilodalton and IVa2 proteins with the packaging sequence in vivo and in vitro. J. Virol. 79:2366–2374. 10.1128/JVI.79.4.2366-2374.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gustin KE, Lutz P, Imperiale MJ. 1996. Interaction of the adenovirus L1 52/55-kilodalton protein with the IVa2 gene product during infection. J. Virol. 70:6463–6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wohl BP, Hearing P. 2008. Role for the L1-52/55K protein in the serotype specificity of adenovirus DNA packaging. J. Virol. 82:5089–5092. 10.1128/JVI.00040-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Arcos R. 2005. Interaction of the adenovirus major core protein precursor, pVII, with the viral DNA packaging machinery. Virology 334:194–202. 10.1016/j.virol.2005.01.048 [DOI] [PubMed] [Google Scholar]
- 22.Sutjipto S, Ravindran S, Cornell D, Liu YH, Horn M, Schluep T, Hutchins B, Vellekamp G. 2005. Characterization of empty capsids from a conditionally replicating adenovirus for gene therapy. Hum. Gene Ther. 16:109–125. 10.1089/hum.2005.16.109 [DOI] [PubMed] [Google Scholar]
- 23.Mangel WF, McGrath WJ, Toledo DL, Anderson CW. 1993. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 361:274–275. 10.1038/361274a0 [DOI] [PubMed] [Google Scholar]
- 24.Graziano V, Luo G, Blainey PC, Pérez-Berná AJ, McGrath WJ, Flint SJ, San Martín C, Xie XS, Mangel WF. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. II. Adenovirus proteinase is activated in an unusual one-dimensional biochemical reaction. J. Biol. Chem. 288:2068–2080. 10.1074/jbc.M112.407312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blainey PC, Graziano V, Pérez-Berná AJ, McGrath WJ, Flint SJ, San Martín C, Xie XS, Mangel WF. 2013. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. IV. Viral proteinase slides along DNA to locate and process its substrates. J. Biol. Chem. 288:2092–2102. 10.1074/jbc.M112.407460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mangel WF, Toledo DL, Brown MT, Martin JH, McGrath WJ. 1996. Characterization of three components of human adenovirus proteinase activity in vitro. J. Biol. Chem. 271:536–543. 10.1074/jbc.271.1.536 [DOI] [PubMed] [Google Scholar]
- 27.Pérez-Berná AJ, Marabini R, Scheres SHW, Menéndez-Conejero R, Dmitriev IP, Curiel DT, Mangel WF, Flint SJ, San Martín C. 2009. Structure and uncoating of immature adenovirus. J. Mol. Biol. 392:547–557. 10.1016/j.jmb.2009.06.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seki T, Dmitriev I, Kashentseva E, Takayama K, Rots M, Suzuki K, Curiel DT. 2002. Artificial extension of the adenovirus fiber shaft inhibits infectivity in coxsackievirus and adenovirus receptor-positive cell lines. J. Virol. 76:1100–1108. 10.1128/JVI.76.3.1100-1108.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haruki H, Gyurcsik B, Okuwaki M, Nagata K. 2003. Ternary complex formation between DNA-adenovirus core protein VII and TAF-Iβ/SET, an acidic molecular chaperone. FEBS Lett. 555:521–527. 10.1016/S0014-5793(03)01336-X [DOI] [PubMed] [Google Scholar]
- 30.Lunt R, Vayda ME, Young M, Flint SJ. 1988. Isolation and characterization of monoclonal antibodies against the adenovirus core proteins. Virology 164:275–279. 10.1016/0042-6822(88)90645-9 [DOI] [PubMed] [Google Scholar]
- 31.Burckhardt CJ, Suomalainen M, Schoenenberger P, Boucke K, Hemmi S, Greber UF. 2011. Drifting motions of the adenovirus receptor CAR and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 10:105–117. 10.1016/j.chom.2011.07.006 [DOI] [PubMed] [Google Scholar]
- 32.Henry LJ, Xia D, Wilke ME, Deisenhofer J, Gerard RD. 1994. Characterization of the knob domain of the adenovirus type 5 fiber protein expressed in Escherichia coli. J. Virol. 68:5239–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860 [DOI] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorincz AT, Lancaster WD, Temple GF. 1986. Cloning and characterization of the DNA of a new human papillomavirus from a woman with dysplasia of the uterine cervix. J. Virol. 58:225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown MT, McGrath WJ, Toledo DL, Mangel WF. 1996. Different modes of inhibition of human adenovirus proteinase, probably a cysteine proteinase, by bovine pancreatic trypsin inhibitor. FEBS Lett. 388:233–237. 10.1016/0014-5793(96)00569-8 [DOI] [PubMed] [Google Scholar]
- 37.Blanche F, Monegier B, Faucher D, Duchesne M, Audhuy F, Barbot A, Bouvier S, Daude G, Dubois H, Guillemin T, Maton L. 2001. Polypeptide composition of an adenovirus type 5 used in cancer gene therapy. J. Chromatogr. A 921:39–48. 10.1016/S0021-9673(01)00896-2 [DOI] [PubMed] [Google Scholar]
- 38.Bergstrom Lind S, Artemenko KA, Elfineh L, Zhao Y, Bergquist J, Pettersson U. 2012. The phosphoproteome of the adenovirus type 2 virion. Virology 433:253–261. 10.1016/j.virol.2012.08.012 [DOI] [PubMed] [Google Scholar]
- 39.Perez-Romero P, Gustin KE, Imperiale MJ. 2006. Dependence of the encapsidation function of the adenovirus L1 52/55-kilodalton protein on its ability to bind the packaging sequence. J. Virol. 80:1965–1971. 10.1128/JVI.80.4.1965-1971.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vayda ME, Flint SJ. 1987. Isolation and characterization of adenovirus core nucleoprotein subunits. J. Virol. 61:3335–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.San Martín C. 2012. Latest insights on adenovirus structure and assembly. Viruses 4:847–877. 10.3390/v4050847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giberson AN, Davidson AR, Parks RJ. 2012. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res. 40:2369–2376. 10.1093/nar/gkr1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W, Low JA, Christensen JB, Imperiale MJ. 2001. Role for the adenovirus IVa2 protein in packaging of viral DNA. J. Virol. 75:10446–10454. 10.1128/JVI.75.21.10446-10454.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson JE. 2010. Virus particle maturation: insights into elegantly programmed nanomachines. Curr. Opin. Struct. Biol. 20:210–216. 10.1016/j.sbi.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prevelige PE, Fane BA. 2012. Building the machines: scaffolding protein functions during bacteriophage morphogenesis. Adv. Exp. Med. Biol. 726:325–350. 10.1007/978-1-4614-0980-9_14 [DOI] [PubMed] [Google Scholar]
- 46.Brown JC, Newcomb WW. 2011. Herpesvirus capsid assembly: insights from structural analysis. Curr. Opin. Virol. 1:142–149. 10.1016/j.coviro.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen DH, Baker ML, Hryc CF, DiMaio F, Jakana J, Wu W, Dougherty M, Haase-Pettingell C, Schmid MF, Jiang W, Baker D, King JA, Chiu W. 2011. Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U. S. A. 108:1355–1360. 10.1073/pnas.1015739108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y, Parker MH, Weigele P, Casjens S, Prevelige PE, Jr, Krishna NR. 2000. Structure of the coat protein-binding domain of the scaffolding protein from a double-stranded DNA virus. J. Mol. Biol. 297:1195–1202. 10.1006/jmbi.2000.3620 [DOI] [PubMed] [Google Scholar]
- 49.Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BA, McMurray CT, Anderson DL, Rossmann MG. 2003. Bacteriophage phi29 scaffolding protein gp7 before and after prohead assembly. Nat. Struct. Biol. 10:572–576. 10.1038/nsb939 [DOI] [PubMed] [Google Scholar]
- 50.van Oostrum J, Burnett RM. 1985. Molecular composition of the adenovirus type-2 virion. J. Virol. 56:439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. 2003. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U. S. A. 100:6940–6945. 10.1073/pnas.0832254100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christensen JB, Ewing SG, Imperiale MJ. 2012. Identification and characterization of a DNA binding domain on the adenovirus IVa2 protein. Virology 433:124–130. 10.1016/j.virol.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruzindana-Umunyana A, Imbeault L, Weber JM. 2002. Substrate specificity of adenovirus protease. Virus Res. 89:41–52. 10.1016/S0168-1702(02)00111-9 [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Imperiale MJ. 2000. Interaction of the adenovirus IVa2 protein with viral packaging sequences. J. Virol. 74:2687–2693. 10.1128/JVI.74.6.2687-2693.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christensen JB, Byrd SA, Walker AK, Strahler JR, Andrews PC, Imperiale MJ. 2008. Presence of the adenovirus IVa2 protein at a single vertex of the mature virion. J. Virol. 82:9086–9093. 10.1128/JVI.01024-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ostapchuk P, Hearing P. 2008. Adenovirus IVa2 protein binds ATP. J. Virol. 82:10290–10294. 10.1128/JVI.00882-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.San Martín C, Huiskonen JT, Bamford JK, Butcher SJ, Fuller SD, Bamford DH, Burnett RM. 2002. Minor proteins, mobile arms and membrane-capsid interactions in the bacteriophage PRD1 capsid. Nat. Struct. Biol. 9:756–763. 10.1038/nsb837 [DOI] [PubMed] [Google Scholar]