

Abstract
The øX174 DNA pilot protein H forms an oligomeric DNA-translocating tube during penetration. However, monomers are incorporated into 12 pentameric assembly intermediates, which become the capsid's icosahedral vertices. The protein's N terminus, a predicted transmembrane helix, is not represented in the crystal structure. To investigate its functions, a series of absolute and conditional lethal mutations were generated. The absolute lethal proteins, a deletion and a triple substitution, were efficiently incorporated into virus-like particles lacking infectivity. The conditional lethal mutants, bearing cold-sensitive (cs) and temperature-sensitive (ts) point mutations, were more amenable to further analyses. Viable particles containing the mutant protein can be generated at the permissive temperature and subsequently analyzed at the restrictive temperature. The characterized cs defect directly affected host cell attachment. In contrast, ts defects were manifested during morphogenesis. Particles synthesized at permissive temperature were indistinguishable from wild-type particles in their ability to recognize host cells and deliver DNA. One mutation conferred an atypical ts synthesis phenotype. Although the mutant protein was efficiently incorporated into virus-like particles at elevated temperature, the progeny appeared to be kinetically trapped in a temperature-independent, uninfectious state. Thus, substitutions in the N terminus can lead to H protein misincorporation, albeit at wild-type levels, and subsequently affect particle function. All mutants exhibited recessive phenotypes, i.e., rescued by the presence of the wild-type H protein. Thus, mixed H protein oligomers are functional during DNA delivery. Recessive and dominant phenotypes may temporally approximate H protein functions, occurring before or after oligomerization has gone to completion.
INTRODUCTION
Prokaryotic viruses must transport their genomes across bacterial cell walls to gain access to the host cell cytoplasm. Many double-stranded DNA (dsDNA) bacteriophages utilize a tail to perform this function, whereas tailless phages generally rely on host-encoded channels or organelles, e.g., plasmid-encoded receptor complexes or pili (1–4). The øX174 capsid displays strict icosahedral symmetry (5); however, 10 H proteins form a 170-Å-long α-helical barrel (H-tube) with dimensions and physical properties ideal for genome translocation (6). Although the X-ray model is an oligomeric tube, the H proteins are stowed as monomers during procapsid assembly, a reaction that has been reconstituted in vitro (7). Upon cell contact, H proteins interact and a fully formed tube emerges from the capsid. After DNA delivery, the conduit appears to dissociate within the cell wall (6). Thus, the newly discovered øX174 phage tail is very ephemeral. The structure of the first 145 and last 50 amino acids are not represented in the crystal structure. Presumably, regions of the protein will need to traverse membranes, and protein H is known to interact with host cell lipopolysaccharide (LPS) (8, 9).
Previous biochemical and genetic analyses have focused on the α-helical barrel (10, 11), which resides in the C terminus. The overexpression of the modified gene encoding this region inhibits øX174 penetration. Presumably, the truncated H proteins interfere with the DNA-piloting function of the wild-type proteins within the infecting virion. Both ends of the H protein contain potential transmembrane domains, which could anchor the assembled structure into the inner and outer cell membranes. In this model, the tubes could span the periplasmic space or cell wall adhesion sites to transport the viral genome into the host's cytoplasm. The helical transmembrane prediction for the first 18 to 20 N-terminal amino acids is particularly strong (12). To elucidate the functions of the N terminus, a series of deletion and missense mutations were generated and the resulting mutants were characterized. All mutant proteins were assembled into virus-like particles at seemingly wild-type levels. While some defects directly affected host cell recognition, others appeared to kinetically trap particles during assembly or in a fully assembled noninfectious conformation. Thus, protein H can be misincorporated, albeit at wild-type levels, in conformations affecting other functions.
MATERIALS AND METHODS
Phage plating, media, buffers, stock preparation, attachment assays, eclipse assays, infected cell extracts, rate zonal sedimentation analyses, and SDS-PAGE.
Media, buffers, plating and liquid culture stock preparations (13), and protocols for attachments and eclipse assays (14) have been previously described. The protocols for generating infected cell extracts, rate zonal sedimentation analyses, and SDS-PAGE are identical to those described in a previous publication (15). To isolate particles from multistep growth infections, 2 liters of early exponential cells were infected at a multiplicity of infection (MOI) of 10−4 input phage/cell at 37°C. After lysis, assembled capsids (virions, degraded procapsids, products assembled off pathways) are removed by incubating lysates at 4°C for 12 h. Assembled capsids, regardless of infectivity and DNA content, attach to cellular debris (13), which is concentrated by centrifugation. To elute particles from membranes, pellets were resuspended in 8.0 ml 50 mM Na2B407–3.0 mM EDTA and shaken for 18 h at 4°C. Cellular debris was removed by centrifugation, and the supernatant was placed atop CsCl step gradients as previously described (13). Material with a density between 1.45 and 1.35 g/cm3, which would contain both virions and empty capsids, is concentrated and analyzed by rate zonal sucrose gradient sedimentation.
Bacterial strains and plasmids.
The Escherichia coli C strains C122, BAF5 (supE), BAF8 (supF), and BAF30 and the plasmid used to express the wild-type H gene (pøXH) have been described previously (10, 13). The C900 strain contains the host slyD mutation, which confers resistance to E protein-mediated lysis (16).
The clone containing the three A or I→D substitutions at residues 6A, 9I, and 12A within the putative transmembrane domain was generated as previously described (10), with the exception that a mutagenic 5′ primer was used to amplify the gene by PCR. This primer also introduces an NcoI site, which contains an ATG start codon. The resulting fragment was digested with NcoI and XhoI, the site introduced by the 3′ primer, and ligated into pSE420 digested with the same enzymes. The DNA lacking the first 18 codons was generated in an identical manner with the exception that the 5′ primer introduced an NcoI site spanning codons 17 to 19.
øX174 mutants and second-site reversion analyses.
All am(H) mutants were generated by site-directed mutagenesis following previously published protocols (13, 17). All in vitro DNA synthesis reaction products were transfected into a host expressing a cloned wild-type H gene. Phages from the resulting plaques were screened by stabbing them into lawns seeded with C122 and BAF30 pøXH. Putative amber mutants were identified by a complementation-dependent phenotype. Second-site revertants were obtained by plating am(H) mutants on restrictive Su+ hosts. Phages from the resulting plaques were stabbed into lawns seeded with Su+ and Su− hosts. Second-site revertants were distinguished from am+ revertants by the retention of the amber phenotype and confirmed by a direct DNA sequence analysis.
The missense ts(H)G3Q and cs(H)G24Q mutants and the H-charged mutant, which contains D codons in place of alanine codons 6 and 12 and isoleucine codon 9, were generated by site-directed mutagenesis. Phage from the resulting plaques were screened by stabbing them into lawns seeded with C122 incubated 28°C, 37°C, and 42°C and with BAF30 pøXH incubated at 33°C. Putative mutants were identified by temperature-restricted growth or a complementation-dependent phenotype. Only one defective phenotype was recovered per mutagenesis, cold sensitive (cs) or temperature sensitive (ts). The sp(H)G24Y mutant was recovered by reverting the am phenotype at 33°C and screening for small-plaque phenotypes.
Multiple rounds of site-directed mutagenesis were conducted to generate the deletion mutation directly in the viral genome. In the first round of mutagenesis, an NcoI site at the 3′ end of the deletion was introduced along with an amber mutation, which served as the marker for the screen. Afterwards, the amber mutation was reverted. This genome then served as the template for the second round of mutagenesis, which introduced an NcoI at the 5′ end of the deletion. Again, an amber mutation, which resided between the two NcoI sites, was introduced for screening the mutagenized progeny. After reverting the amber mutation, replicative-form DNA was generated as previously described (18). The DNA was digested with NcoI, ligated, and transfected in cells expressing a cloned wild-type H gene. Mutants were identified by a complementation-dependent phenotype and verified by a direct sequence analysis.
RESULTS
Deletion and disruption of the putative transmembrane helix result in the production of noninfectious virus-like particles.
To investigate the requirement of the putative transmembrane domain during infection, two altered genes were cloned. The resulting protein sequences are given in Fig. 1. One protein contains a deletion of the first 18 amino acids (Δ18), whereas the second protein contains three A or I→D substitutions at residues 6A, 9I, and 12A (H-charged). A typical α-helix contains seven residues per two helical turns. Thus, the placement of aspartic acid residues at positions 1, 4, and 7 within a heptad sequence is likely to create steric hindrances as well as alter the charge. Bioinformatic predictions (12) supported these assertions. Expression of the cloned genes was assayed for the ability to complement a øX174 am(H) mutant. The genes neither complemented the amber mutant nor inhibited wild-type plaque formation, suggesting a recessive lethal phenotype. Thus, the mutations were engineered directly into the øX174 genome. Mutants were recovered in cells expressing a wild-type H gene, which was essential for plaque formation at all assayed temperatures (28 to 42°C).
FIG 1.

N-terminal amino acid sequences of the wild-type and H protein mutants.
To determine the molecular nature of the recessive lethal phenotype, extracts were prepared from mutant infected cells and analyzed for the presence of virus-like particles. As can be seen in Fig. 2A, material sedimenting at 114S was readily detected in all extracts. The protein composition was investigated by SDS-PAGE (Fig. 2C). Gels were silver stained to enhance the H protein band. The mutant proteins were readily detected in virus-like particles isolated from Δ18 and H-charged infected cells at levels comparable to those of the wild-type control. The specific infectivity (PFU/A280) of the mutant particles was calculated (Table 1). Mutant particles exhibited a specific infectivity approximately 2 orders of magnitude below that of the wild-type control. Wild-type revertants constitute the background signal in these specific infectivity assays and serve as a valuable internal S value marker. However, specific infectivity is strictly defined as PFU/A280, regardless of the genotype of the plaque-forming particle. Thus, the actual specific infectivity of the mutant particles is most likely lower than the assay suggests.
FIG 2.
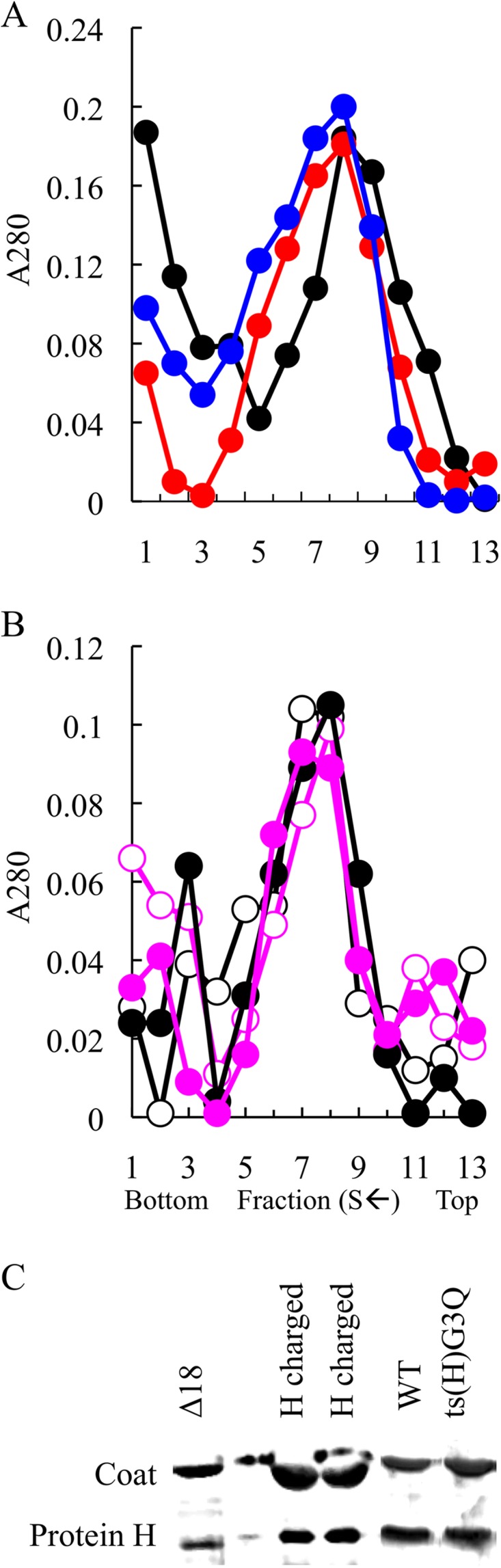
Particle isolation and H protein content of wild-type, Δ18, H-charged, and ts(H)G3Q infections. Gradients were fractionated from the bottom of the tube. Thus, S value decreases with higher fraction numbers. Particles, regardless of infectivity, were detected by UV spectroscopy. (A) Wild-type (black) particles and H-charged (blue) and Δ18 (red) mutants. (B) Wild type (black) and ts(H)G3Q (magenta) synthesized at 33°C (open circles) and 42°C (filled circles). (C) Silver-stained gels of the peak fractions.
TABLE 1.
Specific infectivitya of wild-type and mutant H particles
| Strain | Corresponding figure(s)b | Temp of synthesis (°C)c | PFU/A280 | Specific infectivity normalized to WT control |
|---|---|---|---|---|
| Wild type | 2A | 33° | 1.0 × 1012 | 1.0 |
| ΔH18 | 2A | 33 | 1.0 × 1010 | 1.0 × 10−2 |
| H-charged | 2A | 33 | 1.8 × 1010 | 1.8 × 10−2 |
| Wild type | 4C | 28 | 8.0 × 1011 | 1.0 |
| 2B and 3A | 33 | 1.9 × 1012 | 1.0 | |
| 4C | 37 | 8.2 × 1011 | 1.0 | |
| 3B | 37 | 5.0 × 1011 | 1.0 | |
| 2B and 3A | 42 | 7.7 × 1011 | 1.0 | |
| ts(H)G3Q mutant | 2B | 33 | 1.7 × 1012 | 0.8 |
| 2B | 42 | 3.5 × 109 | 4.5 × 10−3 | |
| ts(H)G8Y mutant | ||||
| 114S | 3A | 33 | 1.4 × 1012 | 0.7 |
| 114S | 3B | 37 | 1.2 × 1012 | 2.2 |
| 60-70S | 3B | 37 | 7.5 × 109 | 1.5 × 10−2 |
| cs(H)G24Q mutant | 4C | 28 | 2.7 × 1011 | 0.4 |
| 4C | 37 | 7.0 × 1011 | 0.9 |
Specific infectivity was determined by determining the titer of the peak fraction and dividing it by the A280 value depicted in the sedimentation profile. Values were normalized to the wild-type control, which was purified at the same temperature and the same time as the mutant being investigated. As specific infectivity assays are essentially plaque assays, the mutant and corresponding wild-type assays were conducted at the same time and with the same cells.
The figure contains the sedimentation profile from which the specific infectivity was calculated.
Temperature at which the particles were synthesized. Specific infectivity assays were conducted at permissive temperatures.
Initial characterization of nonsense and missense mutations in the 5′ end of the H gene.
Unlike nonviable deletion mutants, missense mutants with conditional lethal phenotypes can be more amenable to further analyses. Viable particles containing the mutant protein can be generated at the permissive temperature and subsequently analyzed at the restrictive temperature. To isolate a suitable set of conditional lethal mutants, amber mutations were first generated in glycine codons 3, 7, 8, 15, 16, 23, and 24. Mutants were screened for growth defects on informational suppressing strains (Su+) that insert either glutamine or tyrosine. For those amber mutants that displayed a tight lethal or conditional lethal phenotype on a Su+ host, the amber codons were replaced with either tyrosine or glutamine codons. The missense mutants used in further studies are listed in Table 2.
TABLE 2.
Plating efficiencya of missense mutants
| Mutationb | Plating efficiency at indicated temp |
||||
|---|---|---|---|---|---|
| No plasmid |
pøXH |
||||
| 28°C | 33°C | 42°C | 28°C | 42°C | |
| ts(H)G3Q | 1.0 | 1.0 | 10−4 | 1.0 | 1.0 |
| ts(H)G8Y | 10−1 | 1.0 | 10−6 | 1.0 | 1.0 |
| cs(H)G24Q | 10−6 | 1.0 | 1.0 | 10−6 | 1.0 |
Plating efficiency is defined as titer on mutant with no exogenous H gene expression/titer on host expressing a cloned wild-type H gene at 33°C.
All mutant stocks were grown in the absence of exogenous H gene expression. Thus, plaque formation assays the ability of the mutant protein to infect.
To characterize the missense strains, mutants were grown in the absence of the exogenously expressed, wild-type H gene. Thus, mutant particles contain only the missense protein. The exogenous expression of a wild-type H gene rescued the temperature-sensitive (ts) phenotype of the ts(H)G3Q and ts(H)G8Y mutants on the level of plaque formation. Both plates and cells were prewarmed to the restrictive temperature, 42°C. As the particles in the plaque assay initially contain only the mutant H proteins, rescue at elevated temperatures indicates that mutant genomes were able to penetrate cells and that the temperature-sensitive defect most likely affects an aspect of protein folding and/or particle morphogenesis. This hypothesis was tested by examining particles synthesized at 42°C.
A temperature-sensitive missense mutation that kinetically traps particles in a noninfectious conformation.
Extracts were prepared from lysis-resistant cells infected with the wild type or the ts(H)G3Q mutant at 33°C and 42°C. After the removal of cellular debris, the soluble extracts were analyzed by rate zonal sedimentation. As can be seen in Fig. 2B, particles sedimenting at 114S, the S value of the mature virion, were recovered from all infections. Mutant particle yields were roughly equivalent to wild-type yields, suggesting no gross changes in assembled particle solubility. The specific infectivity (PFU/A280) was determined. For these experiments, the titers of virions were determined at the permissive temperature. When synthesized at 33°C, the specific infectivity of the ts(H)G3Q 114S material was comparable to that of the wild-type control (Table 1). In contrast, the particles synthesized at 42°C were 3 orders of magnitude less infectious. These plaques did not arise from wild-type revertants, which was assessed in a 42°C plaque assay. The ts+ reversion frequency was 2 orders of magnitude lower than the specific infectivity determined at 33°C.
The proteins within the peak fractions were examined by SDS-PAGE. Again, gels were silver stained to enhance the protein H bands. Protein H was detected in mutant particles at seemingly wild-type levels (Fig. 2C), suggesting that the mutation does lead to gross alterations in H protein solubility that would remove it from the assembly pathway. The A260/A280 ratios for the wild-type and mutant samples were 1.28 and 1.3, respectively, indicating that the mutant fraction did not contain less nucleic acid. Moreover, empty capsids sediment at a considerably slower speed (70S) and were not present in the gradient at levels greater than the wild-type control. These data indicate that the mutation confers a temperature-sensitive synthesis phenotype, which kinetically traps particles in a noninfectious state.
A temperature-sensitive missense mutation that may confer a DNA-packaging defect.
Unlike the ts(H)G3Q mutant, the ts(H)G8Y mutant did not produce assembled particles at the restrictive temperature. However, particles formed at 33°C displayed a specific infectivity comparable to that of the wild-type control (Fig. 3A and Table 1). Although SDS-PAGE analysis of mutant-infected whole-cell lysates indicated that viral protein levels were equivalent to the wild-type control, particles composed of viral proteins were sparse in gradients that typically detect them within the 50S to 130S or the 4S to 50S ranges (data not shown). This suggests the formation of off-pathway product that is either insoluble or lost using the standard protocols to generate extracts.
FIG 3.

Particle isolation and protein and DNA content of ts(H)G8Y and wild-type infections. (A) Wild type (black) and ts(H)G8Y mutant (magenta) synthesized at 33°C (open circles) and 42°C (filled circles). Gradients were fractionated from the bottom of the tube. Thus, S value decreases with higher fraction numbers. Particles, regardless of infectivity, were detected by UV spectroscopy. (B) Particles isolated from multiround growth cultures of the wild type (black) and the ts(H)G8Y mutant (magenta) at 37°C. (C) Protein content. Fractions 7, 9, 11, 13, 15, and 17 depicted, used to generate the sedimentation profile in panel B, were analyzed by SDS-PAGE. Fraction 7 represents the virion peak, and later fractions contain slower-sedimenting material. (D) DNA content of particles pooled from fractions 7 to 12 and 13 to 17. Abbreviations: RF, purified ds replicative-form DNA; ss, purified single-stranded genomic DNA.
In an effort to identify the off-pathway product, an alternate protocol was devised. The protocol allowed for multiple rounds of growth at a less-than-optimal temperature, but not at entirely restrictive temperature. Briefly, 2 liters of early exponential lysis-sensitive cells were infected at an MOI of 10−4 at 37°C. After lysis, assembled capsids (virions, degraded procapsids, and assembled-off-pathway products) were concentrated as described in Materials and Methods. The capsids were then purified by buoyant density sedimentation (CsCl). Material with a density between 1.35 and 1.45 g/cm3, which would contain both virions and empty capsids, was then further analyzed by rate zonal sedimentation. The results of this experiment are depicted in Fig. 3B.
In the wild-type sedimentation profile, there is one prominent peak, which may contain a shoulder. In contrast, the mutant sedimentation profile contains two distinct peaks. The proteins within the fractions spanning the peaks were analyzed by SDS-PAGE (Fig. 3C). The three viral structural proteins were present in the slower-sedimenting material, along with some host cell proteins. The identity of these host cell proteins is not known, nor is it known if they are physically associated with capsids or are contained in a host cell complex that shares a similar S value (∼60S to 70S). Scaffolding proteins were not detected in the mutant particles; however, the internal scaffolding protein was detected in the wild-type fractions in which degraded procapsids are often recovered.
The A260/A280 ratios of the two mutant fraction peaks indicated that the material sedimenting between 60S and 70S was associated with more nucleic acids than 114S material. These values were 1.7 (60S to 70S) and 1.4 (114S). To investigate this more thoroughly, fractions 6 to 11 and 12 to 17 were pooled and the DNA was extracted. As can be seen in Fig. 3D, the DNA associated with the particles in fractions 7 to 12 was primarily single-stranded DNA (ssDNA) genomes, confirming that this material is primarily infectious virions. The specific infectivity of the mutant particles was comparable to that of the wild-type particles (Table 1). Two species of DNAs were extracted from material in fractions 12 to 17 from the ts(H)G8Y gradient. One species migrates like ssDNA by agarose gel electrophoresis. The other species migrates as relaxed double-stranded replicative-form DNA. Although very little DNA was detected in this region from the wild-type gradient, an overexposed image demonstrates that both species of DNAs are present. Thus, the particles within this region of the gradient may be off-pathway products formed during DNA packaging, which appear to be augmented in mutant infections. If this hypothesis is correct, the host cell proteins, once identified, should be involved in DNA metabolism, and electron microscopy should show partially filled intact capsids with external double-stranded DNA.
The cold-sensitive (cs) missense mutant exhibits defects in host cell attachment.
Unlike what was seen with the mutants with ts phenotypes, exogenous expression of a cloned wild-type H gene could not rescue the cs(H)G24Q mutant, which had been grown at the permissive temperature without exogenous wild-type H gene expression. This suggested that the mutant H protein affects an early process in the infection life cycle, one that prevents the delivery of DNA. However, if first passed through cells expressing the wild-type H gene at permissive temperature, it formed small plaques at 28°C in cells expressing the wild-type gene. The plating efficiency was 0.5 (Table 3). If the wild-type gene was expressed neither during synthesis nor during plating, the efficiency of plaque formation fell more than 2 orders of magnitude. Thus, the wild-type H protein within the infecting virions along with its subsequent production within the infected cells rescued plaque formation. These data indicate that the G24Q substitution is both necessary and sufficient to confer the defective phenotype.
TABLE 3.
Rescue of cs(H)G24Q mutant by permissive synthesis with cloned wild-type gene expression
| Wild-type cloned H gene expression in: |
Plaque assay temp (°C) | Plating efficiencyc | |
|---|---|---|---|
| Synthesisa | Plaque assayb | ||
| No | No | 37 | 1.0 |
| No | 28 | <6.0 × 10−3 | |
| Yes | 28 | <6.0 × 10−3 | |
| Yes | No | 37 | 1.0 |
| No | 28 | <1.0 × 10−2 | |
| Yes | 28 | 0.5 | |
Indicates whether a cloned wild-type (WT) H gene was expressed during particle synthesis at the permissive 37°C temperature.
Indicates whether a cloned wild-type H gene was expressed for the plating assay.
Plating efficiency was defined as the assay titer/permissive titer ratio. The permissive titer of each lysates was assigned a value of 1.0.
The attachment kinetics of the mutant and wild-type particles were investigated. Wild-type and mutant particles were first generated at the permissive 37°C temperatures and then assayed for host cell attachment at both 37°C and 28°C. Lysis-resistant cells were used to reduce background that could arise from the release of progeny. At set time intervals after mixing, samples were centrifuged to remove attached virions and the titers of supernatants were determined for unattached phages (Fig. 4A). At both temperatures, the titer of unattached wild-type particles falls several orders of magnitude by the earliest assayed time point. In contrast, the cs(H)G24Q mutant exhibited attachment defects at both permissive and restrictive temperatures. The defect was particularly pronounced at 28°C. To determine whether attachment defects were associated with other mutations at this site, a small-plaque (sp)-forming revertant of am(H)G24 mutant was recovered, sp(H)G24Y mutant. It also exhibited attachment defects, displaying attachment efficiencies similar to that of the cs(H)G24Q mutant at 37°C (Fig. 4A). The sp(H)G24Y mutant was not analyzed at 28°C.
FIG 4.

Attachment kinetics and eclipse kinetics of virion production of the wild type and mutants at the Q24 site. (A) Attachment kinetics of the wild type (black) and cs(H)G24Q mutant (blue) at 37°C (open circle) and 28°C (filled circle) and of sp(H)G24Y mutant (magenta) kinetics at 37°C (this mutant was not analyzed at 28°C). (B) Eclipse kinetics of preattached wild-type (black) and cs(H)G24Q (blue) particles at 28°C. (C) Virions isolated from wild-type (black)- and cs(H)G24Q mutant (blue)-infected cells at 37°C (filled circles) and 28°C (open circles). Gradients were fractionated from the bottom of the tube. Thus, S value decreases with higher fraction numbers. Particles, regardless of infectivity, were detected by UV spectroscopy.
Although host cell attachment was extremely inefficient at 28°C, relatively efficient attachment (∼95%) could be obtained by prolonged incubation at 16°C with 10× concentrated exponential-phase cells that had been resuspended in a starvation buffer, which lacks a carbon source. Thus, it was possible to conduct eclipse assays at the restrictive temperature. In these experiments, the cells and preattached bacteriophage are transferred into medium containing a carbon source at 28°C (t = 0). At set time points, samples are diluted into a borate-EDTA buffer, which releases uneclipsed viruses from cell membranes, and their titers are determined for surviving particles. As can be seen in Fig. 4B, the mutant particles were indistinguishable from those of the wild type in this assay. To determine if particles delivered DNA and produced progeny at the restrictive temperature, infections were allowed to continue for an additional 3 h. Prepared extracts were analyzed by rate-zonal sedimentation (Fig. 4C). Progeny was produced, and they exhibited a specific infectivity comparable to that of the wild-type control (Table 1). The results of these experiments suggest that the primary defect associated with the cs(H)G24Q mutant appears to be attachment.
Second-site suppressors of the mutant phenotype are intragenic and near the original mutations.
To determine whether viruses could adapt to mutant DNA pilot protein defects, a second-site genetic analysis was conducted. To facilitate the analysis, amber mutants were used to mark the site of the original mutation. The suppressors were selected by plating am(H)G3 and am(H)G24 mutants on the supE host, which inserts glutamine at amber codons during protein synthesis. The resulting plaques were stabbed into Su+ and Su− indicator lawns. Putative second-site suppressors were identified by the retention of the amber phenotype. The suppressors were located in gene H and restored viability in the Su+ hosts at all temperatures. Although the suppressors were isolated multiple times, only one stock of each mutant was used in the selections; thus, these do represent independent isolations. The suppressor of am(H)G24 conferred an A→V change at amino acid 17 in the H protein. Two genetically distinct 8-codon insertions were associated with the suppressors of am(H)G3. The insertions were located after alanine codon 10. The resulting 8-amino-acid insertions are depicted in Fig. 1. They are very hydrophobic and appear to duplicate part of the local protein sequence, suggesting that they may act by preserving the length of the predicted transmembrane helix. A similar analysis was conducted with the am(H)G8 mutant in the supF host, which inserts tyrosine at the amber site. However, no second-site suppressors were recovered.
DISCUSSION
As described in the introduction, 10 H protein monomers oligomerize to form an α-helical tube with dimensions long enough to span either the periplasmic space or membrane adhesion sites and wide enough to accommodate the genome (6). The hydrophobic nature of the N terminus suggests that it may interact with host cell membranes. More specifically, the first 20 amino acids are predicted to form a transmembrane helix. Similar to fusogenic peptides of animal viruses, this region is thought to be sequestered within the virion to prevent absorbing to bacteria in which øX174 cannot replicate. Upon some undefined signal, it would become exposed to facilitate genome translocation.
Recessive lethal phenotypes and the requirement of N-terminally mediated H-H protein interactions.
The possible function of the N terminus was investigated with a deletion mutation and a multiple mutant containing three A→D codons. The A→D substitutions would theoretically disrupt hydrophobicity and helical structure. When the mutant genes were expressed from the viral genome, a clone of the wild-type H protein efficiently complemented the lethal phenotypes. When expressed from a plasmid, they did not inhibit wild-type plaque formation. These data indicate that the mutant genes are recessive and suggest that a full complement of the wild-type protein is not required for viability. In the absence of the wild-type protein, the mutant proteins are incorporated into virus-like particles that lack infectivity.
Kinetic traps: the consequences of H protein misincorporation.
Unlike absolute lethal mutations, which must always be complemented with a wild-type gene, conditional lethality allows mutant particles to be generated at a permissive temperature. Thus, subsequent assays directly examine the effects of the missense proteins. Both ts mutants characterized in this study were rescued in cells expressing a cloned wild-type H gene at elevated temperatures, indicating that the ts defect most likely affected a postpenetration phenomenon. These data also demonstrate that the ts mutations are recessive.
Particles synthesized at the restrictive temperature in the absence of the wild-type protein were isolated and characterized. Compared to the wild-type control, an abundance of material sedimenting at ∼60S was isolated from extracts of ts(H)G8Y mutant-infected cells. The protein and DNA composition of this material is consistent with an off-pathway product arising from a DNA-packaging intermediate (19). Similar particles have been observed in earlier studies with am(H) and ts(H) strains (20). However, the locations of those mutations within the gene were not known and the H protein content of the particles was not characterized. These results could indicate that H protein plays a direct role in the packaging reaction, but a particle with similar characteristics was also isolated from wild-type-infected cells, albeit at a significantly smaller quantity. Thus, an indirect effect seems more plausible. The mutant H proteins may be incorporated incorrectly, in a conformation that hinders packaging efficiently and/or fidelity.
The mutant G3Q protein also appeared to kinetically trap particles, but after DNA packaging. Like the G8Y protein, it was incorporated, but into virion-like particles at levels comparable to that of the wild-type control. However, the particles synthesized at the restrictive temperature exhibited a significantly decreased specific infectivity when assayed at the permissive temperature. Unlike experiments conducted with absolute lethal mutations, specific infectivity values were not obfuscated by the presence of wild-type revertants. Viable particles retained the mutant phenotype. Thus, some particles retained the ability to infect. Again, these data indicate that mutant H proteins are misincorporated. Bacteriophages are generally synthesized in a high-energy state, the highly compacted DNA containing the potential energy to deliver genome to the subsequently infected cell (5, 21, 22). However, the requisite conformational change required for genome delivery most likely has a high activation energy, which confers stability to the packaged particle in the absence of the appropriate host. At restrictive temperatures, the G3Q protein may be incorporated in a conformation that elevates the activation energy, thus locking particles in an inactive state.
While the length of this helix may be critical for spanning a membrane, which typically requires 18 to 20 amino acids (23), mutant particles, if synthesized at the permissive temperature, exhibited near-wild-type attachment kinetics at elevated temperature. Thus, they are fully capable of delivering DNA under restrictive conditions. However, the particles produced at the restrictive temperature displayed decreased infectivity. This suggests that the length of the helix may be equally important during particle assembly. The genetic nature of the second-site suppressors supports this hypothesis. The suppressors conferred small duplications in the 5′ end of the gene, which results in the local insertions of hydrophobic amino acids, which may be required to pack the H proteins into virions to retain an infectious state.
A cold-sensitive missense mutation affects host cell attachment.
The only mutant that displayed a cs phenotype could not be rescued in cells expressing a cloned wild-type H gene at the restrictive temperature. This suggested that the mutant protein failed to mediate a requisite step for DNA delivery. Particles synthesized under permissive conditions displayed host cell attachment defects at both permissive and restrictive temperatures. The defect was particularly pronounced at the lower temperature. However, with prolonged incubation times, 95% attachment could be achieved. The attached particles exhibited no defects in DNA piloting or particle assembly at the restrictive temperature. The cs mutation lies outside the region predicted to encode the transmembrane helix and may indicate that a region of the H protein is exposed on the outer side of the virion or the conformation of the packaged H protein indirectly affects the virions outer structure.
The atomic structure of the H protein demonstrates that 10 copies form a 170-Å-long α-helical barrel with an internal diameter large enough to accommodate two antiparallel single-stranded DNA strands. This tube structure is clearly visible by tomography of infecting cells (6). The external surface of the øX174 virion is strictly icosahedral, containing no visible unique vertex. It is not known whether the H protein exits as a tube or partially formed tube. This could form a core that predestines one of the 12 vertices to be the site through which the tube must emerge. During early morphogenesis, the protein is monomeric in all assembly intermediates that contain a pentamer of the major coat protein, and these intermediates can be used to assemble capsids in vitro (7, 15, 24). During assembly, the external scaffolding protein organizes 12 pentameric intermediates into procapsids. Thus, at some time during the viral life cycle, protein H must transition from a monomeric, or vertex-associated, to an oligomeric state.
All of the mutations used in this study conferred a recessive phenotype. This indicates that mutations do not affect the oligomeric functions of the protein. It may also suggest that the initial infection stages are mediated by vertex-associated H protein monomers. In a simple model, one in which there is no predestined unique vertex from which the tube must emerge, each vertex, or a subset of vertices, would be functionally equivalent until interactions with the host cell select one of them for DNA delivery. The ts(H)G3Q protein may kinetically trap only the vertex with which it is most strongly associated. If wild-type protein is also present in the virion, other vertices would remain functional. A similar situation may occur with vertices containing the cs(H)G24Q protein: these vertices would be unable to attach to host cells, but vertices with the wild-type protein would retain function. In a more complex model, one in which a predestined unique vertex exists in the mature virion, the incorporation of the wild-type protein at this vertex would preselect it during particle maturation. The results presented here are consistent with either of these models. They do not prove or eliminate other mechanisms, which will require more-targeted and sophisticated biophysical and/or structural characterization.
ACKNOWLEDGMENTS
This research was supported by NSF grant MCB-0948399 and USDA-Hatch funds to the University of Arizona (B.A.F.).
Footnotes
Published ahead of print 27 November 2013
REFERENCES
- 1.Grahn AM, Butcher SJ, Bamford JKH, Bamford DH. 2006. PRD1: dissecting the genome, structure and entry, p 161–170 In Calendar R. (ed), The bacteriophages, 2nd ed. Oxford Press, London, United Kingdom [Google Scholar]
- 2.Molineux IJ, Panja D. 2013. Popping the cork: mechanisms of phage genome ejection. Nat. Rev. Microbiol. 11:194–204. 10.1038/nrmicro2988 [DOI] [PubMed] [Google Scholar]
- 3.Russel M, Model P. 2006. Filamentous phage, p 146–160 In Calendar R. (ed), The bacteriophages, 2nd ed. Oxford Press, London, United Kingdom [Google Scholar]
- 4.Van Duin JT N. 2006. Single-stranded RNA phages, p 175–196 In Calendar R. (ed), The bacteriophages. Oxford Press, London, United Kingdom [Google Scholar]
- 5.McKenna R, Xia D, Willingmann P, Ilag LL, Krishnaswamy S, Rossmann MG, Olson NH, Baker TS, Incardona NL. 1992. Atomic structure of single-stranded DNA bacteriophage phi X174 and its functional implications. Nature 355:137–143. 10.1038/355137a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L, Young LN, Zhang X, Boudko SP, Fokine A, Zbornik E, Roznowski AP, Moulineux I, Rossmann MG, Fane BA. Icosahedral bacteriophage ΦX174 forms a tail for DNA transport. Nature, in press [DOI] [PubMed] [Google Scholar]
- 7.Cherwa JE, Jr, Organtini LJ, Ashley RE, Hafenstein SL, Fane BA. 2011. In vitro assembly of the ox174 procapsid from external scaffolding protein oligomers and early pentameric assembly intermediates. J. Mol. Biol. 412:387–396. 10.1016/j.jmb.2011.07.070 [DOI] [PubMed] [Google Scholar]
- 8.Inagaki M, Kawaura T, Wakashima H, Kato M, Nishikawa S, Kashimura N. 2003. Different contributions of the outer and inner R-core residues of lipopolysaccharide to the recognition by spike H and G proteins of bacteriophage phiX174. FEMS Microbiol. Lett. 226:221–227. 10.1016/S0378-1097(03)00601-3 [DOI] [PubMed] [Google Scholar]
- 9.Inagaki M, Tanaka A, Suzuki R, Wakashima H, Kawaura T, Karita S, Nishikawa S, Kashimura N. 2000. Characterization of the binding of spike H protein of bacteriophage phiX174 with receptor lipopolysaccharides. J. Biochem. 127:577–583. 10.1093/oxfordjournals.jbchem.a022643 [DOI] [PubMed] [Google Scholar]
- 10.Cherwa JE, Jr, Young LN, Fane BA. 2011. Uncoupling the functions of a multifunctional protein: the isolation of a DNA pilot protein mutant that affects particle morphogenesis. Virology 411:9–14. 10.1016/j.virol.2010.12.026 [DOI] [PubMed] [Google Scholar]
- 11.Ruboyianes MV, Chen M, Dubrava MS, Cherwa JE, Jr, Fane BA. 2009. The expression of N-terminal deletion DNA pilot proteins inhibits the early stages of phiX174 replication. J. Virol. 83:9952–9956. 10.1128/JVI.01077-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- 13.Fane BA, Hayashi M. 1991. Second-site suppressors of a cold-sensitive prohead accessory protein of bacteriophage phi X174. Genetics 128:663–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cherwa JE, Jr, Sanchez-Soria P, Wichman HA, Fane BA. 2009. Viral adaptation to an antiviral protein enhances the fitness level to above that of the uninhibited wild type. J. Virol. 83:11746–11750. 10.1128/JVI.01297-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherwa JE, Jr, Uchiyama A, Fane BA. 2008. Scaffolding proteins altered in the ability to perform a conformational switch confer dominant lethal assembly defects. J. Virol. 82:5774–5780. 10.1128/JVI.02758-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roof WD, Fang HQ, Young KD, Sun J, Young R. 1997. Mutational analysis of slyD, an Escherichia coli gene encoding a protein of the FKBP immunophilin family. Mol. Microbiol. 25:1031–1046. 10.1046/j.1365-2958.1997.5201884.x [DOI] [PubMed] [Google Scholar]
- 17.Fane BA, Shien S, Hayashi M. 1993. Second-site suppressors of a cold-sensitive external scaffolding protein of bacteriophage phi X174. Genetics 134:1003–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burch AD, Ta J, Fane BA. 1999. Cross-functional analysis of the Microviridae internal scaffolding protein. J. Mol. Biol. 286:95–104. 10.1006/jmbi.1998.2450 [DOI] [PubMed] [Google Scholar]
- 19.Fujisawa H, Hayashi M. 1976. Viral DNA-synthesizing intermediate complex isolated during assembly of bacteriophage phi X174. J. Virol. 19:409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spindler KR, Hayashi M. 1979. DNA synthesis in Escherichia coli cells infected with gene H mutants of bacteriophage phi X174. J. Virol. 29:973–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kostyuchenko VA, Leiman PG, Chipman PR, Kanamaru S, van Raaij MJ, Arisaka F, Mesyanzhinov VV, Rossmann MG. 2003. Three-dimensional structure of bacteriophage T4 baseplate. Nat. Struct. Biol. 10:688–693. 10.1038/nsb970 [DOI] [PubMed] [Google Scholar]
- 22.Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. 2004. The bacteriophage T4 DNA injection machine. Curr. Opin. Struct. Biol. 14:171–180. 10.1016/j.sbi.2004.02.001 [DOI] [PubMed] [Google Scholar]
- 23.Hildebrand PW, Preissner R, Frommel C. 2004. Structural features of transmembrane helices. FEBS Lett. 559:145–151. 10.1016/S0014-5793(04)00061-4 [DOI] [PubMed] [Google Scholar]
- 24.Cherwa JE, Jr, Fane BA. 2009. Complete virion assembly with scaffolding proteins altered in the ability to perform a critical conformational switch. J. Virol. 83:7391–7396. 10.1128/JVI.00479-09 [DOI] [PMC free article] [PubMed] [Google Scholar]