

ABSTRACT
Bovine ephemeral fever virus (BEFV) is an arthropod-borne rhabdovirus that causes a debilitating disease of cattle in Africa, Asia, and Australia; however, its global geodynamics are poorly understood. An evolutionary analysis of G gene (envelope glycoprotein) ectodomain sequences of 97 BEFV isolates collected from Australia during 1956 to 2012 revealed that all have a single common ancestor and are phylogenetically distinct from BEFV sampled in other geographical regions. The age of the Australian clade is estimated to be between 56 and 65 years, suggesting that BEFV has entered the continent on few occasions since it was first reported in 1936 and that the 1955-1956 epizootic was the source of all currently circulating viruses. Notably, the Australian clade has evolved as a single genetic lineage across the continent and at a high evolutionary rate of ∼10−3 nucleotide substitutions/site/year. Screening of 66 isolates using monoclonal antibodies indicated that neutralizing antigenic sites G1, G2, and G4 have been relatively stable, although variations in site G3a/b defined four antigenic subtypes. A shift in an epitope at site G3a, which occurred in the mid-1970s, was strongly associated with a K218R substitution. Similarly, a shift at site G3b was associated primarily with substitutions at residues 215, 220, and 223, which map to the tip of the spike on the prefusion form of the G protein. Finally, we propose that positive selection on residue 215 was due to cross-reacting neutralizing antibody to Kimberley virus (KIMV).
IMPORTANCE
INTRODUCTION
Bovine ephemeral fever virus (BEFV) is an arthropod-borne rhabdovirus that is classified as the type species of the genus Ephemerovirus. The 14.9-kb negative-sense single-stranded RNA (ssRNA) genome encodes the 5 canonical rhabdovirus structural proteins (N, P, M, G, and L), as well as a nonstructural glycoprotein (GNS) and several small accessory proteins of unknown function (α1, α2, α3, β, and γ) in additional open reading frames (ORFs) located between the G and L genes (1). The virus is an important pathogen of cattle and water buffaloes and is enzootic in ruminant populations in tropical and subtropical regions of Africa, the Middle-East, Asia, and Australia. Bovine ephemeral fever (BEF), or 3-day sickness, also occurs seasonally in adjacent temperate zones, often with significant impacts on production. BEFV has been isolated from mosquitoes and biting midges (Culicoides spp.), but most data suggest that mosquitoes are the major vector (2).
Although several serologically related viruses have been isolated from cattle and insects in Africa and Australia (3, 4), BEFV occurs as a single cross-neutralizing serotype worldwide (5–10). The neutralizing immune response is induced by the envelope glycoprotein (G), which protects against experimental challenge in cattle (11). Neutralizing antigenic determinants are located in four independent antigenic sites (G1-G4) in the G protein ectodomain (12, 13). G1 is a linear site in the C-terminal region of the ectodomain, G2 is a nonlinear conformational site that appears to lie in the fusion domain adjacent to two highly conserved cysteine residues, and G3 is a complex conformational site composed of two elements (G3a and G3b) that lie in the cysteine-rich “head” of the G protein in a region that appears to form the receptor-binding pleckstrin homology (PH) domain (4, 14, 15). The location of antigenic site G4 has not yet been determined.
Previous studies of the molecular epidemiology of BEFV in east Asia and the Middle-East, using the amino acid sequence of the G protein ectodomain, have identified four genetic lineages comprising isolates that cluster chronologically and geographically (10, 16, 17). In Asia, genotype I comprises isolates sampled from epizootics in Taiwan in 1984 and Japan in 1988 to 1989, genotype II comprises isolates from Taiwan during 1996 to 2004 and Japan during 2001 to 2004, and genotype III is represented by the 1966 Japanese (Yamaguchi) vaccine strain. Isolates sampled from Turkey in 2000 and Israel in 2008 have been shown to form a separate lineage, and all of these lineages are distinct from Australian BEFV isolates (1, 10, 17). Indeed, multiple amino acid substitutions in antigenic sites G1 and G3, including two commonly occurring mutations that introduce potential N-glycosylation sites, distinguish the Australian and east Asian lineages (10).
To better understand the molecular evolution and epidemiology of BEFV, we studied viral isolates collected from cattle and insects at various locations across northern and eastern Australia during the period 1956 to 2012. Evolutionary analysis was conducted on nucleotide sequences of the G protein ectodomain, and the antigenic profiles of a selection of isolates were examined using a panel of monoclonal antibodies to identify shifts in the major neutralization sites, which we mapped to a 3-dimensional structural model of the G protein. The data indicate that BEFV has evolved progressively as a single clade across the continent, suggesting that introductions from Asia have been rare and that there is regular displacement of existing strains, likely through continual north-south viral traffic.
MATERIALS AND METHODS
Source and cultivation of viruses.
Details of the source species and the date and location of sample collection for all BEFV isolates and BEFV-infected tissue samples used in this study are shown in Table S1 in the supplemental material.
Source of antibodies.
The panel of BEFV antibodies used in this study is shown in Table 1. The panel included mouse monoclonal antibodies directed at all major antigenic sites (G1, G2, G3a, G3b, and G4) and nonneutralizing mouse monoclonal antibody 3A2. Preparation of the mouse ascitic fluids and characterization of the antibodies have been described previously (12). BEFV polyclonal mouse ascitic fluid and tissue culture medium were used as positive and negative controls, respectively.
TABLE 1.
Antibodies used for analysis of BEFV isolates
| Antibody | Designationa | Antigenic site | Isotype | Neutralization prototype strain (BB7721) |
|---|---|---|---|---|
| 17B1 | A | G1 | IgG2a | + |
| 13A3 | B | G1 | IgG3 | + |
| 15B5 | C | G2 | IgG1 | + |
| 12A5 | D | G2 | IgG2a | + |
| 8B6 | E | G3a | IgG2a | + |
| 8D3 | F | G3b | IgG2a | + |
| 16A6 | G | G3b | IgG2a | + |
| 5A5 | H | G4 | IgG1 | + |
| 3A2 | I | NDb | IgG2b | − |
| Polyclonal | J | NAc | ND | + |
| Culture fluid | K | NA | NA | − |
As depicted in Fig. 3.
ND, not determined; nonneutralizing monoclonal antibody.
NA, not applicable.
Neutralization tests.
Microneutralization tests were conducted in C6/36 cells in 96-well plates by using an immunofluorescence assay to detect viral infection (13). Briefly, viruses were grown in the presence of each monoclonal antibody, a mouse polyclonal anti-BEFV ascitic fluid, or uninfected tissue culture medium (negative control). After 6 days of incubation at 28°C, the presence or absence of neutralization was detected using indirect immunofluorescence.
RNA extraction and RT-PCR amplification and sequencing.
Total RNA was extracted from infected cell culture supernatants using the RNeasy minikit (Qiagen, Germany) per the manufacturer's protocol. cDNA was synthesized by adding 5 to 10 μl RNA to 200 ng random hexamer primer (Promega, USA) and 20 nmol deoxynucleoside triphosphate (dNTP) mix (Promega, USA), heating at 65°C for 5 min, and then cooling on ice for at least 1 min. To this reaction mixture was added 4 μl of 5 × Superscript III first-strand buffer (Life Technologies, USA), 2 μl of dithiothreitol (DTT; 100 mM; Life Technologies, USA), 1 μl of RNase OUT (40 U/μl; Life Technologies, USA), and 1 μl of Superscript III reverse transcriptase (200U/μl; Life Technologies, USA). The reaction was mixed gently and then incubated at 50°C for 1 h and 70°C for 15 min, then it was cooled to 4°C. cDNA products were either placed on ice and used immediately or stored at −20°C until required. For each sample, 4 μl cDNA was used as the template to amplify the glycoprotein gene by PCR using primers BEFV G-All-F and BEFV G-All-R and/or BEFV M-262-F and BEFV GNS-262-R (primer sequences are available on request). The PCR mixture was 10 μl 5 × LongAmp reaction buffer (NEB, USA), 1.5 μl dNTP mix (10 mM), 1 μl each primer (20 μM), 2 μl LongAmp enzyme (NEB, USA), and nuclease-free water to a total volume of 50 μl. Amplification was performed under the following conditions: 1 cycle at 94°C for 30 s; 35 cycles of 94°C for 10 s, 50°C for 30 s, and 65°C for 2 min; 1 cycle at 65°C for 10 min; and then cooling to 4°C. Amplification products were separated on 1.5% agarose gels containing 0.01% Sybersafe, and bands were excised and purified using the QiaQuick gel extraction kit (Qiagen, Germany) per the manufacturer's protocol. The eluate was quantified on a Qubit (Life Technologies, USA) using the Quant-IT double-stranded DNA HS assay kit (Life Technologies, USA). Sequencing was performed using routine Sanger sequencing using the BigDye Terminator v3.1 kit (Life Technologies, USA) and the ABI PRISM 3130xl genetic analyzer (Life Technologies, USA) according to the manufacturer's protocols.
Evolutionary analysis.
Two BEFV G gene ectodomain data sets were compiled for evolutionary analysis: (i) a global data set (149 sequences, 1,527 nt) comprising the four genetic lineages of BEFV, including isolates from Australia, Japan, Taiwan, Turkey, and Israel, sampled between 1966 and 2012, which allowed us to place the genetic diversity of BEFV observed in Australia in a wider geographical context; and (ii) an Australian BEFV data set (97 sequences, 1,527 nt) comprising viruses sampled between 1956 and 2012, which allowed exploration of viral evolution in Australia in greater detail (although the scale of the sampling precluded a detailed phylogeographic analysis). Because few gaps needed to be introduced into the alignment, this could easily be achieved by eye.
Phylogenetic analysis on both data sets was undertaken using the maximum likelihood (ML) method, available within the PhyML package (18), and the Bayesian Markov Chain Monte Carlo (MCMC) method, available in the BEAST package (version 1.7.5) (19), incorporating information on the time (year) of sampling of each strain and displayed as the maximum clade credibility (MCC) tree. For the former analysis, we employed the GTR+Γ model of nucleotide substitution as well as SPR branch swapping. An ML bootstrap analysis was undertaken using 1,000 replicate trees again estimated under the GTR+Γ substitution model but with NNI branch swapping. For the BEAST analysis, we utilized (i) a GTR model of nucleotide substitution with a different substitution rate of each codon position, (ii) a relaxed (uncorrelated lognormal) molecular clock, and (iii) Bayesian skyline, time-aware Gaussian Markov random field (GMRF) Bayesian skyride and constant population size coalescent priors. In all cases, the MCMC chain was run for 100 million generations (with 10% burn-in), and convergence was achieved for all parameters. Statistical uncertainty was reflected in values for the 95% highest posterior density (HPD), and support for individual groupings on the MCC tree is reflected in posterior probability values. The BEAST analysis also enabled us to estimate rates of nucleotide substitution per site and the time to the most recent common ancestor of the tree (tMRCA). To determine whether there was sufficient temporal structure in the data to accurately infer evolutionary rates, and as an additional rate estimate without assuming a coalescent process, we plotted root-to-tip genetic distances determined from the ML tree against year of sampling using the Path-O-Gen program (http://tree.bio.ed.ac.uk/software/pathogen/).
To estimate the selection pressures acting on the BEFV G protein, we inferred the relative numbers of nonsynonymous (dN) and synonymous (dS) nucleotide substitutions per site (in which dN > dS is indicative of adaptive evolution) using a variety of methods implemented with the Datamonkey interface of the HyPhy package (http://www.datamonkey.org) (20, 21): SLAC (single likelihood ancestor counting), FEL (fixed-effects likelihood), FUBAR (a fast, unconstrained Bayesian approximation for inferring selection), and MEME (mixed-effects model of episodic selection). To examine whether episodic diversifying selection has occurred on individual branches of the Australian BEFV phylogeny, we utilized the branch-site REL (random effects likelihood) method also available in HyPhy. Only sites with P values of <0.1 or with posterior probability values greater than 0.95 provided statistically viable evidence for positive selection under these analytical criteria.
Structural modeling.
Three-dimensional models of the G protein were generated by homology modeling using Phyre2. Automatic template selection assigned the postfusion structure (PDB code 2CMZ) for which the fold departs significantly from the neutral pH form of the G protein that is anticipated at the surface of the infectious particle. Thus, we also produced a model using the prefusion structure of vesicular stomatitis virus G protein (VSV G) as the template (22) (PDB code 2J6J) using the Phyre2 one-to-one threading with a global alignment method. The trimer was generated by superposition of the BEFV G model with the three chains of the VSV structure (PDB code 2J6J). Figures were prepared using PyMOL.
Nucleotide sequence accession numbers.
The nucleotide sequences generated here have been deposited in GenBank under accession numbers KF679404 to KF679496.
RESULTS
Nucleotide sequence and evolutionary analysis of BEFV isolates.
Our phylogenetic analysis of 149 G gene sequences from a diverse array of BEFV sequences (i.e., the global data set), including 97 from Australia collected from cattle and insects over a 57-year span (1956 to 2012), revealed that the Australian viruses form a monophyletic group distinct from the east Asian and Middle-East lineages. As reported elsewhere (23), several isolates from Turkey in 2012 clustered with the east Asian lineage and were most closely related to viruses isolated in China in 2011. The monophyly of the Australian viruses is indicative of a single entry into the continent followed by sustained transmission over the sampling period (Fig. 1). The Australian phylogeny is also notable for its ladder-like structure, in which a single dominant lineage links viruses sampled from multiple time points and which, in the case of influenza A virus, is compatible with strongly immune-driven evolution (24).
FIG 1.

ML tree of 149 sequences of the G gene ectodomain of BEFV representing the known global genetic diversity in this virus. The geographic locations of the different viral lineages are marked, clearly showing the common ancestry of the Australian viruses (Fig. 2 provides a more detailed analysis of the Australian viruses). Bootstrap values are shown for key nodes, and all horizontal branch lengths are drawn according to a scale of nucleotide substitutions per site. The tree is midpoint rooted for purposes of clarity only.
To explore the evolution of the Australian viruses in more detail, we analyzed the 97 Australian sequences separately. Because our ML and Bayesian (MCC) trees exhibited very similar topologies, only the latter is shown here (Fig. 2; the ML tree is available from the authors on request). The Bayesian analysis is particularly notable for the strongly clock-like evolution exhibited by BEFV over the sampling period, as reflected in a coefficient of rate variation (CoV) approaching zero (95% HPD, 0.28 to 4.7 × 10−5). The existence of a strong molecular clock was also apparent in a regression analysis of root-to-tip genetic distance against time (year) of sampling (r2 = 0.98) on the ML phylogeny. Such clock-like evolutionary behavior enabled us to estimate a robust evolutionary rate for BEFV, at 0.9 to 1.3 × 10−3 nucleotide substitutions per site per year (substitutions/site/year), and a time to common ancestor of the tree at 56 to 65 years before present (1956 to 1947). The timing of other divergence events can be inferred from the MCC tree. Importantly, very similar estimates (i.e., strongly overlapping HPDs) of nucleotide substitution rate and tMRCA were obtained under the Bayesian skyline, skyride, and constant population size models, as well as using linear regression (rate, 0.81 × 10−3 substitutions/site/year; tMRCA, 65 years), strongly suggesting that they are robust.
FIG 2.

MCC tree of BEFV in Australia showing evolution in estimated real time (x axis). All sequences are color coded according to the region of origin: Northern Territory (12° to 15° S), olive green; northern-central Queensland (15° to 25° S), red; southern Queensland-northern New South Wales (25° to 30° S), brown; central-southern New South Wales (30° to 37° S), blue. A small clade of viruses sampled from Queensland is also indicated, as are those from insects (by the asterisk symbol): BEFV/CS42/AU/Beatrice Hill/1975 and BEFV/CS1622/AU/Peachester/1984 were taken from Anopheles bancrofti, while BEFV/CS1647/AU/Peachester/1984 was taken from Culicoides brevitarsis. The tree is automatically rooted on the assumption of a molecular clock, and tip times reflect the date of sampling. Node bars indicate the 95% HPD values for the height (i.e., age) of that node, and posterior probability values are shown for key nodes. The map inset indicates the geographic locations of the four sampled regions.
The MCC tree (obtained from the Bayesian skyline analysis) was also notable for the presence of a single dominant lineage (as shown in Fig. 1) with little sustained evolution of independent clades, such that there is a marked turnover of viral lineages with time. Indeed, on these data, the only evidence for the (short-term) persistence of a local clade is the presence of eight viruses sampled from Queensland (including four from Peachester) between 1979 and 1982 (Fig. 2). In addition, isolates clearly clustered temporally, and there was a close phylogenetic relationship between viruses isolated from distant geographic locations at similar times, highlighting the fluidity of BEFV spatial movement (Fig. 2; lineages are color coded by geographic region).
Analysis of selection pressures on the G protein ectodomain.
We used a variety of bioinformatic methods to determine the nature of the selection pressures acting on the BEFV G protein as it has evolved in Australia. Importantly, these results painted a relatively consistent picture of individual sites which might be subject to positive selection, suggesting that they are robust given these data. As is true of the vast majority of viral proteins, the overall dN/dS in the data, 0.26, indicates that most amino acid changes are deleterious and removed by purifying selection. However, within this background of negative selection, consistent evidence for adaptive evolution was found at amino acid sites 59 and 215, which were detected as positively selected in all four methods utilized here, usually with strong statistical support (P < 0.05; posterior probability, >0.95). There was also more tentative evidence for some form of adaptive evolution at sites 274, 277, and 459 (Table 2). No evidence of lineage-specific episodic diversifying selection was observed.
TABLE 2.
Putatively positively selected sites in the G protein of BEFV in Australia
| Amino acid site | Probability (P and PP values) determined bya: |
|||
|---|---|---|---|---|
| SLAC | FEL | FUBAR | MEME | |
| 59 | 0.041 | 0.084 | 0.967 | 0.016 |
| 215 | 0.038 | 0.024 | 0.995 | 0.026 |
| 274 | 0.953 | 0.036 | ||
| 277 | 0.095 | |||
| 459 | 0.953 | 0.087 | ||
SLAC, FEL, and MEME gave probability (P) values, while FUBAR produced posterior probability (PP) values.
Natural antigenic variation in neutralization sites.
A selection of 66 of the Australian BEFV isolates collected between 1956 and 1992 were tested for neutralization using a panel of monoclonal antibodies (MAbs) representing each of the recognized antigenic sites (Table 1). All isolates were neutralized by MAbs to antigenic sites G1 (17B1 and 13A3), G2 (15B5 and 12A5), and G4 (5A5), as well as one site G3b monoclonal antibody (16A6) and polyclonal anti-BEFV immune rabbit serum. As expected, nonneutralizing G protein monoclonal antibody 3A2 failed to neutralize any of the isolates. Neutralization by monoclonal antibodies 8B6 (site G3a) and 8D3 (site G3b) varied among the isolates, allowing the identification of four antigenic types (I to IV) based on the patterns of neutralization (Fig. 3). Twelve isolates (type I), including the prototype strain (BB7721), were neutralized by both MAbs. Of the remaining isolates, 36 were neutralized by 8D3 but not 8B6 (type II), eight were neutralized by 8B6 but not 8D3 (type III), and 10 isolates were not neutralized by either of these site G3 MAbs (type IV). Examination of the sites of isolation of the viruses in each category indicated no association with location. However, all viruses categorized as antigenic types I and III were isolated primarily up to 1974 to 1975, and all viruses categorized as antigenic types II and IV were primarily isolated from 1973 to 1974 onwards, indicating a shift in site G3a epitope 8B6 in the Australian episystem at approximately this time. There was no temporal association with the reactivity of epitope 8D3 for which isolates representing each of the two variants spanned the period 1968 to 1989.
FIG 3.

Antigenic profiles of a selection of Australian BEFV isolates (1956 to 1992) and associated amino acid substitutions. Virus neutralization was conducted in C6/36 cells using neutralizing monoclonal antibodies corresponding to antigenic sites G1, G2, G3a, G3b, and G4 (A to H), nonneutralizing monoclonal antibody (I), BEFV polyclonal rabbit antiserum (J), and uninfected cell culture fluid (K) (Table 1). Shaded boxes indicate positive neutralization as determined by the absence of BEFV-specific immunofluorescence. Amino acid substitutions mapping to the known antigenic sites are shown highlighting those associated with highly variable amino acids K215 and R218. Two of the isolates analyzed in this assay have not been sequenced (ns).
Mutations associated with the major antigenic subtypes.
An alignment was generated for the 509-amino-acid G protein ectodomain sequences of the set of 97 viruses used for evolutionary analysis. The set included 64 of the 66 Australian BEFV isolates (1956 to 1992) for which antigenic profiles were obtained and 33 other BEFV isolates collected from Queensland, the Northern Territory, and New South Wales during the period 1970 to 2012 (see Fig. S1 in the supplemental material). Sequence variation was detected in 100 sites across the ectodomain, including regions previously mapped to neutralizing antigenic sites G1, G2, and G3 (14).
Two conservative amino acid substitutions (R486K and K503R) were detected at the extreme ends of the region mapped to antigenic site G1 (I485-Q505). This linear site has been reported to have some local conformation and was represented in antigenic profiling experiments by MAbs 17B1 and 13A3, which target each end of the linear region (14). Interestingly, each of these MAbs solidly neutralized all BEFV isolates tested, indicating that the conservative K503R substitution did not change the neutralization phenotype, even though nonconservative substitutions at the same site have been reported in escape mutants selected using MAb 13A3 (14). Substitution R486K was first detected in 1984 and was present in all viruses isolated since, with the exception of EMAI66 isolated at Gloucester in NSW in 2011. The K503R substitution was first detected in CS967 isolated at Kairi in northeast Queensland in 1981 and was present in all viruses isolated since 1984, with the exception of CS1619 and CS1647, each isolated at Peachester in southeast Queensland in that same year.
Consistent with the antigenic profiling data, only a single amino acid variation occurred in the region previously mapped to antigenic site G2 (S166-W191), a conformational neutralization site that was predicted to be linked by a single disulfide bridge (14). This change (T170A) occurred in D901 from Kangaloon in NSW in 2010 and was not included in neutralization assays.
Notable sequence variations were detected in each of the three linear elements of conformational antigenic site G3, which spans much of the cysteine-rich region of the ectodomain. In the first element (residues 49 to 63), substitutions occurred at D57E in one isolate, H59P in seven isolates, and H59N in five isolates. The D57E substitution has been identified previously in a neutralization escape mutant selected with site G3a MAb 8B6 (14), and the naturally variant isolate (CS1188) also was not neutralized by this same MAb (Fig. 3). As discussed above, amino acid residue 59 was shown to be under positive selection. Two different substitutions occurred independently at this site in 12 viruses from several different sublineages isolated between 1970 and 2010. There was also a single amino acid deletion at residue 59 in one isolate and at residues 54, 60, and 61 in five other isolates. The significance of the deletion at residue 61 is unclear, but it occurred independently in viruses from several phylogenetically distinct sublineages. The second element of site G3 (residues 215 to 231) was relatively variable, with substitutions occurring in various isolates in 8 of the 17 amino acids, the most notable of which were at residues 215 and 218. There was a strong association between the R218K substitution and the loss of reactivity with site G3a MAb 8B6. The substitution occurred in each of the 46 viruses that failed to react with this MAb (types II and IV) and none of the 20 remaining viruses tested (types I and III). At residue 215, which dN/dS analysis indicated was likely to be subject to positive selection, there were 15 nonconservative substitutions (N, T, and E) and three deletions which occurred independently in various sublineages of viruses isolated over the 55-year period from 1956 to 2010. There was a partial correlation between loss of reactivity with site G3b MAb 8D3 (types III and IV) and substitutions at residue 215, along with substitutions at nearby residues 220 and 223 in this element of the G3 antigenic site (see Fig. S1 in the supplemental material). The third element of site G3 (residues 261 to 279) was characterized by substitutions in various isolates in 12 of the 19 amino acids identified in the domain. Residue 274, which dN/dS analysis indicated may also be under positive selection, displayed nonconservative substitutions (E and G) and a deletion in all except 11 of the 97 isolates.
Another region of the G protein (residues 455 to 482), located just upstream of linear antigenic site G1, was also variable, with substitutions in 12 of the 28 residues. Sequence variation was noted at residues 459 (9 substitutions and one deletion), 476 (5 substitutions), and 477 (8 substitutions and one deletion). This region has not previously been recognized as a neutralizing antigenic site. It may be an extension of the adjacent linear site G1 or could be antigenic site G4, which has been detected in competitive enzyme-linked immunosorbent assays (ELISAs) but has not yet been physically mapped to the sequence (13).
Structural model of the major antigenic sites.
A three-dimensional structural prediction of the BEFV G protein was generated by homology modeling using Phyre2 (Fig. 4). The template used for most analyses presented here corresponds to the prefusion form of VSV G expected at the surface of virions (22). The confidence rating of Phyre2 was 100%, supporting the validity of the global fold. However, given the relatively low sequence identity (22% over 408 aligned residues), local errors in the precise conformations of loops and side chains are to be anticipated. Inspection of disulfide bridges predicted by Walker and Kongsuwan (25) and subsequently adjusted following resolution of crystal structure of VSV G (4, 15) revealed that most are present, supporting the accuracy of the model for the low-resolution analyses presented here. Accordingly, using the notation introduced by Walker and Kongsuwan, disulfide bridges A (C39-C308), B (C79-C112), C (C88-C134), E (C204-C247), and G (C26-C393) have a correct geometry, while three other disulfide bonds, D (C172-C182), F (C242-C278), and I (C64-C211), were slightly disrupted. Cysteines C352 and C365 are not involved in a disulfide bridge in this model (Fig. 4A).
FIG 4.
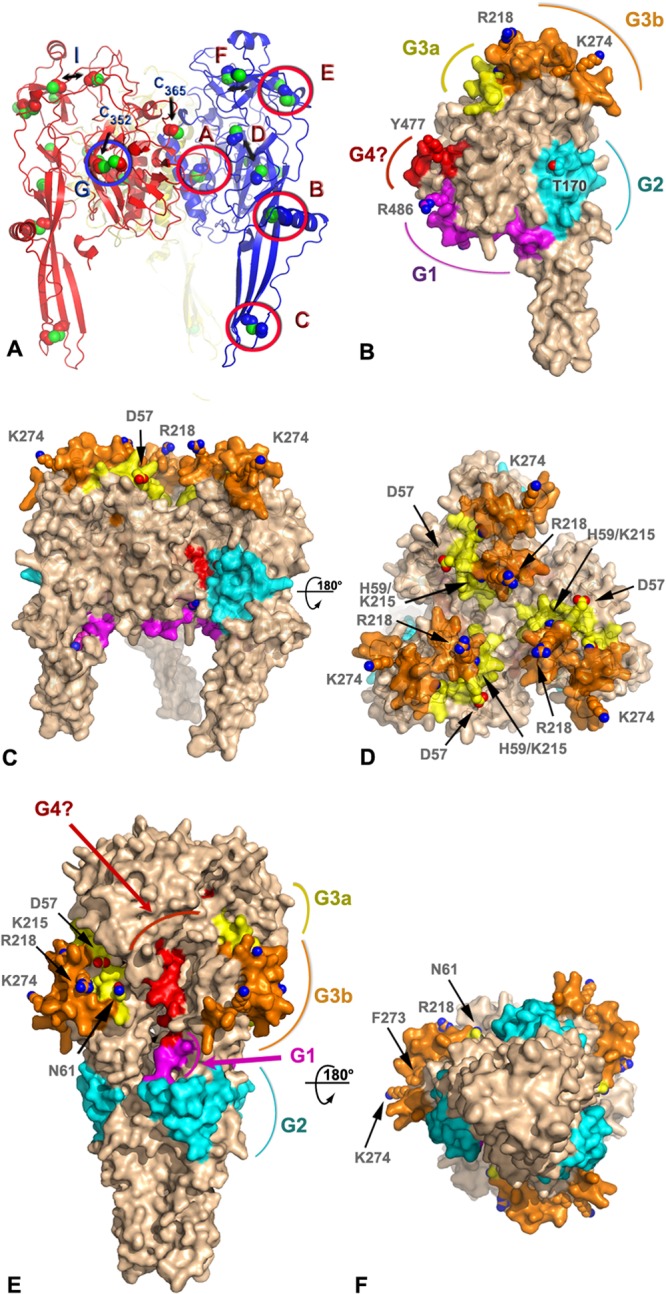
Model of antigenic sites on the prefusion and postfusion forms of the BEFV G trimer. (A) Ribbon representation of a homology model of BEFV G trimer derived by threading with the prefusion VSV G protein structure (PDB code 2J6J). The three subunits are shown in blue, red, and yellow with sulfur atoms in green. The side chains of the cysteine residues are shown as spheres. Disulfide bonds are labeled according to the nomenclature of Walker and Kongsuwan (25), and the residue number is indicated for the two free cysteines. Double arrows indicate cysteines that are expected to form disulfide bonds but are too far apart in the model. (B) Surface representation of a subunit of the BEFV G trimer approximately in the orientation of the subunit colored in blue in panel A. The antigenic sites G1, G2, G3a, and G3b and the putative G4 site are colored in magenta, cyan, yellow, orange, and red, respectively. The side chains of residues discussed in the text are shown as spheres and are labeled. The molecular surface around these residues has been omitted for clarity. (C and D) Orthogonal views of the BEFV G trimer in its prefusion conformation with the same representation and color scheme as that for panel B. The viral membrane is located at the bottom of the trimer in this orientation. (E and F) Orthogonal views of the BEFV G homology model derived by threading with the postfusion, trimeric structure of the VSV G protein (PDB code 2CMZ) with the same representation and color scheme as that for panel B.
When mapped onto the molecular surface of the BEFV G protein, antigenic sites G1, G2, G3a, and G3b form distinct patches mostly located on one face of the molecule (Fig. 4B), with the other face apparently silent antigenically (Fig. 4C). Site G1 is located just before the C-terminal stalk of the G protein at the end of the trimerization domain (DII). Site G2 is at the base of the fusion domain (DIV), while the tip of DIV that comprises the fusion loop does not present recognized antigenic sites. Site G3, the largest antigenic site, occupies most of the base of the PH domain (DIII) with a slight overlap at the beginning of DII (Fig. 4B). Finally, the variable region that may represent proposed site G4 (H455-E482) is found on the inner side of the lateral domain (DI) that faces a neighboring subunit in the trimer. This site is largely buried in our model of the trimer and is in direct contact with site G2 of the next subunit (Fig. 4C).
All recognized antigenic sites are exposed at the surface of the prefusion G trimer, except for the tentative site G4. However, in the context of the virion, site G1 faces the viral membrane; thus, it is inaccessible to antibody binding in the prefusion conformation modeled here. Site G2 is on the side of the tripod formed by the G trimer, and sites G3a/G3b are directly available to be recognized by neutralizing antibodies in the prefusion form.
Sites G3a and G3b are particularly exposed at the distal end of the spike compared to the viral membrane. Residues D59 and K215, which were shown to be likely subject to positive selection, are located on this surface, as are variable residues R218 and K274 (Fig. 4C and D). Their position at the tip of the spike also correlates well with the antigenic subtypes: types II and IV for residues R218 and K274 and types III and IV for residue K215. This suggests that the footprint of MAb 8B6 and 8D3 at least partially overlaps the ∼270-Å2 surface delimited by these four residues.
Site G4 is exposed at the surface of the BEFV G subunit but almost totally buried in the prefusion model of the trimer. This region of the protein is involved in much of the trimeric interactions. It would become partially exposed only in the postfusion structure (Fig. 4E and F) or in a monomeric form (Fig. 4B).
DISCUSSION
Although BEFV is widely distributed throughout many tropical and subtropical regions of the world, its insect vectors have not been clearly defined and the global geodynamics of infection are poorly understood. Here, we describe the evolution and phylogeography of BEFV in continental Australia, where the disease has been reported to be enzootic and seasonally epizootic for more than 75 years.
BEF was first reported in February 1936 when a major epizootic emerged in the Northern Territory. The widespread distribution of the first reports suggested that the virus had been in Australia for some time prior to its emergence, and it was assumed to have been introduced by infected insects from the Indonesian Archipelago (26). The epizootic swept 1,000 km westward into the Kimberley region of Western Australia, eastward into Queensland, and then progressively moved 3,000 km southward on a wide front through eastern states to reach Victoria in February 1937 (26). Similar sweeping wave-like epizootics occurred in 1955 to 1956 and then again in 1967 to 1968, 1970 to 1971, 1972 to 1974, and 1974 to 1976, coinciding with periods of extreme weather and heavy rainfall (27). Seroconversion rates of up to 90% were reported in cattle in eastern Australia during these wave-like epizootics, but in the intervening periods only sporadic seasonal outbreaks were reported, mostly in northern regions (27, 28). Following the 1974 to 1976 epizootic, the epidemiological pattern changed, with the disease becoming enzootic throughout eastern regions of Australia and as far south as the central coast of New South Wales, where a separate enzootic focus was reported to have developed (27, 29, 30). BEF has since occurred primarily as scattered regional disease epizootics interspersed with sporadic cases accompanying the summer rains and occasional widespread epizootics, such as those that have occurred since 2008 (30–32).
The phylogenetic data presented here indicate that all BEFV isolates sampled from Australia since 1956 form a single lineage that has evolved at a relatively constant rate across the continent. The estimated age of the Australia clade (56 to 65 years) suggests that no new BEFV genotype has entered the continent since the early 1950s. Indeed, unless the lineage responsible for the first reported outbreak in 1936 subsequently became extinct, there may have only been a single incursion of the virus since the introduction of cattle and water buffalo by European settlers during the 19th century. This contrasts with bluetongue virus (BTV), for which 10 different serotypes are enzootic in cattle in northern Australia and new serotypes and genotypes are regularly detected in sentinel herds (33). Indeed, modeling suggests that frequent incursions of BTV, which is transmitted by biting midges (Culicoides spp.), occur by wind-borne displacement of infected vectors across the Timor Sea from Indonesia and Timor Leste (34, 35). Although BEFV has been isolated from both mosquitoes and biting midges in Australia and Africa, it has been argued that its epidemiological pattern is not consistent with the distribution or seasonal abundance of culicoides and that the need for direct intravenous injection to initiate experimental infection of cattle implicates mosquitoes (capillary feeders) rather than midges (pool feeders) as the principal vectors (2, 36, 37). It has also been reported that the most abundant livestock-associated culicoides species in South Africa are refractory to oral infection with BEFV (38). As the virus is known to be enzootic in cattle in the Indonesian Archipelago (39, 40), the absence of multiple BEFV incursions into Australia also supports the view that mosquitoes, which may be less amenable to long-distance aerial displacement over water, are its major vectors.
The phylogeny of Australian BEFV isolates is also notable for its ladder-like structure, which is reminiscent of those obtained for the hemagglutinin (HA) gene of influenza A virus (24, 41, 42) and which is compatible with continuous positive selection (adaptive evolution), such that only a single (fittest) lineage survives between seasonal epizootics (see below). However, unlike influenza A virus, BEFV is relatively stable antigenically with a single known serotype worldwide, as evidenced by strong cross-neutralization by immune sera of isolates from Africa, Asia, and Australia (5–10) and the continuing effectiveness of vaccines developed using viruses isolated more than 50 years ago. BEFV neutralizing antibody is protective in cattle, and infection usually results in lifelong protective immunity (11). We have also shown here that all Australian isolates analyzed were neutralized by mouse polyclonal antiserum against the 1968 isolate (BB7721) and that epitopes representing neutralization sites G1, G2, and G4 have been stable antigenically since 1956. Despite this, antigenic variations have occurred in the major conformational neutralization site G3, and at least two amino acids that form part of this site (residues 59 and 215) were under putative positive selection. Several other ephemeroviruses are known to infect cattle in Australia, including Kimberley virus (KIMV), which has been isolated on multiple occasions from mosquitoes, midges, and healthy cattle and has a geographic distribution similar to that of BEFV (4, 43–45). It has also been shown that cross-reactive neutralizing antibodies develop in sentinel cattle infected naturally with BEFV or other ephemeroviruses (46), and BEFV neutralizing monoclonal antibody 8D3, which defines the highly variable epitope in neutralization site G3b, has been shown to cross-react with KIMV (13). Hence, it is possible that adaptive evolution of BEFV is driven by cross-reactive neutralizing antibodies to KIMV and/or other ephemeroviruses. Interestingly, KIMV is also present in Indonesia and China, and it has been shown recently that Malakal virus, which was isolated in Sudan in 1963, is a geographic variant of KIMV (47), suggesting that the evolutionary dynamics of BEFV and KIMV may be linked on a wider geographic scale.
The evolutionary rate of BEFV documented here (∼10−3 substitutions/site/year) was relatively high. Although similarly high rates have previously been estimated for BEFV (48), these were based on very limited data with sparse sampling and not made with a robust Bayesian framework. Indeed, the rates estimated here are higher than those previously estimated for the rhabdovirus VSV (48, 49) and for most estimates of substitution rates in animal rabies virus (50, 51). In addition, they are particularly notable in the context that evolutionary rates are generally lower in vector-borne compared to directly transmitted RNA viruses (48, 52). Together, these data are consistent with the continual adaptive evolution of BEFV over a 50-year time span in Australia.
Even though BEFV is evolving rapidly, there is limited genetic diversity at individual time points, with a single lineage usually being dominant in each location (a pattern also noted in influenza virus). Evidence based on serosurveillance and patterns of disease indicates that BEFV is perennially enzootic in the northern tropical zone of the Northern Territory, and although annual and seasonal fluctuations in prevalence do occur, it is also enzootic throughout much of eastern Australia (1). Hence, the combination of phylogeny and epidemiology suggests that most adaptive evolution occurs in the perennial enzootic focus in the Northern Territory, resulting in a dominant lineage which emerges seasonally and sporadically into the eastern states of Queensland and New South Wales, where low seroconversion rates occur in winter and during periods of prolonged drought. Thus, although BEFV may overwinter in the east, the enzootic focus in the far north likely serves as the source of strains which are continually selected for optimum fitness and which regularly move southeastward with the onset of summer and autumn rains, displacing the scattered remnants of enzootic infection.
Similar epidemiological patterns in Australia have been reported previously for the mosquito-borne flaviviruses Murray Valley encephalitis virus (MVEV) and the Kunjin strain of West Nile virus (WNV), which emerge in southern regions from enzootic foci in the north and exist as single genotypes evolving uniformly in widely separated regions with no independent divergence (53, 54). In contrast, the alphavirus Ross River virus has evolved in Australia as separate genotypes in different geographic regions, and Sindbis virus has evolved as a series of temporally related genotypes (53). The evolutionary patterns of MVEV and WNV have been attributed to avian hosts which allow widespread and rapid dispersal from a northern enzootic focus, raising the interesting possibility that birds also are involved in the epidemiology of BEF.
Although the phylogenetic data provided no evidence for a major change in the epidemiological pattern in the mid-1970s, our antigenic analysis has shown that there was a shift in the G3a epitope represented by MAb 8B6 sometime during the period 1973 to 1975. This shift, which was suggested previously in a more limited antigenic analysis (13), was strongly associated with an R218K mutation that became fixed in all subsequent isolates. Although no temporal pattern was evident, there were also variations in the G3b epitope represented by MAb 8D3 associated primarily with amino acid substitutions at residue 215, which, along with residues 59 and possibly 274 in site G3, was shown to be under positive selection. The three-dimensional structural model of the BEFV G protein has confirmed the prominent surface location of sites G1, G2, G3a, and G3b and has allowed mapping of the variable-site G3 amino acids at the distal end of the spike formed by domain DIII. This complex site, which appears to adopt a surface topology similar to that of VSV neutralization sites A1 and A2 and rabies virus site II (22, 55, 56), would be exposed in the prefusion form of G and is likely to be associated with the receptor-binding domain. The variable amino acids defined by MAbs 8B6 and 8D3 appear to be particularly prominent at the tip of the spike.
The model also suggests that although neutralization site G1 would be exposed, it would face the viral envelope in both the pre- and postfusion forms of BEFV G. The variable region adjacent to site G1 (H455-E482) that may represent the unmapped antigenic site G4 was not predicted to be exposed on the surface of either form of the trimer, as it is located on the inner side of the lateral domain (DI) that faces a neighboring subunit. However, a characteristic of class III fusion proteins is that they undergo a transition between two distinct trimeric states during the fusion process. The topological arrangements of domains in the pre- and postfusion forms imply a mandatory dissociation of the trimer during fusion. Consistent with these structural constraints, a monomeric intermediate between the pre- and postfusion forms has been observed in vivo (57). These monomers may represent intermediates in the transition between the pre- and postfusion conformations. They have been identified in solution where the ectodomain of VSV G is primarily in a monomeric form as well as on the virions at both pH 7.5 and pH 6.7 (58). Thus, the equilibrium observed at the surface of the virion would expose both the region H455-E482, tentatively assigned to antigenic site G4, which is buried in the trimeric interface, and the linear neutralization site G1, which faces the viral membrane in the prefusion model. These observations suggest that neutralizing antibodies bind differentially to the pre- and postfusion forms of BEFV G, as observed previously for the rabies virus G protein (22, 59, 60).
Supplementary Material
ACKNOWLEDGMENTS
E.C.H. is supported by an NHMRC Australia Fellowship. F.C. is supported by an Australian Research Council Future Fellowship.
We thank T. D. St. George for his generous advice and assistance and Debbie Eagles (CSIRO Australian Animal Health Laboratory) for preparing the map inset in Fig. 2.
Footnotes
Published ahead of print 13 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02797-13.
REFERENCES
- 1.Walker PJ. 2005. Bovine ephemeral fever in Australia and the world. Curr. Top. Microbiol. Immunol. 292:57–80. 10.1007/3-540-27485-5_4 [DOI] [PubMed] [Google Scholar]
- 2.St George TD. 2009. Evidence that mosquitoes are the vectors of bovine ephemeral fever virus, p 161–164 In Ryan P, Aaskov J, Russell R. (ed), Arbovirus research in Australia, vol 10 QIMR, Coffs Harbour, Australia [Google Scholar]
- 3.Blasdell KR, Voysey R, Bulach D, Joubert DA, Tesh RB, Boyle DB, Walker PJ. 2012. Kotonkan and Obodhiang viruses: African ephemeroviruses with large and complex genomes. Virology 425:143–153. 10.1016/j.virol.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 4.Walker PJ, Blasdell KR, Joubert DA. 2012. Ephemeroviruses: arthropod-borne rhabdoviruses of ruminants, with large and complex genomes. In Dietzgen RG, Kuzman IV. (ed), Rhabdoviruses: molecular taxonomy, evolution, genomics, ecology, cytopathology and control. Horizon Scientific Press, Norwich, CT [Google Scholar]
- 5.Lecatsas G, Theodoridis A, Els HJ. 1969. Morphological variation in ephemeral fever virus strains. Onderstepoort J. Vet. Res. 36:325–326 [PubMed] [Google Scholar]
- 6.Snowdon WA. 1970. Bovine ephemeral fever: the reaction of cattle to different strains of ephemeral fever virus and the antigenic comparison of two strains of virus. Aust. Vet. J. 46:258–266. 10.1111/j.1751-0813.1970.tb15773.x [DOI] [PubMed] [Google Scholar]
- 7.Kemp GE, Mann ED, Tomori O, Fabiyi A, O'Connor E. 1973. Isolation of bovine ephemeral fever virus in Nigeria. Vet. Rec. 93:107–108 [DOI] [PubMed] [Google Scholar]
- 8.Tian FG, Jiang CL, Zakrzewski H, Davis SS. 1987. A comparison of a Chinese and an Australian strain of bovine ephemeral fever virus. Aust. Vet. J. 64:159. [DOI] [PubMed] [Google Scholar]
- 9.Inaba Y, Tanaka Y, Omori T, Matsumoto M. 1969. Serological relation between bovine epizootic fever and ephemeral fever. Jpn. J. Microbiol. 13:129–130. 10.1111/j.1348-0421.1969.tb00444.x [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Aizawa M, Takayoshi K, Kokuba T, Yanase T, Shirafuji H, Tsuda T, Yamakawa M. 2009. Phylogenetic relationships of the G gene sequence of bovine ephemeral fever virus isolated in Japan, Taiwan and Australia. Vet. Microbiol. 137:217–223. 10.1016/j.vetmic.2009.01.021 [DOI] [PubMed] [Google Scholar]
- 11.Uren MF, Walker PJ, Zakrzewski H, St George TD, Byrne KA. 1994. Effective vaccination of cattle using the virion G protein of bovine ephemeral fever virus as an antigen. Vaccine 12:845–850. 10.1016/0264-410X(94)90295-X [DOI] [PubMed] [Google Scholar]
- 12.Cybinski DH, Walker PJ, Byrne KA, Zakrzewski H. 1990. Mapping of antigenic sites on the bovine ephemeral fever virus glycoprotein using monoclonal antibodies. J. Gen. Virol. 71:2065–2072. 10.1099/0022-1317-71-9-2065 [DOI] [PubMed] [Google Scholar]
- 13.Cybinski DH, Davis SS, Zakrzewski H. 1992. Antigenic variation of the bovine ephemeral fever virus glycoprotein. Arch. Virol. 124:211–224. 10.1007/BF01309803 [DOI] [PubMed] [Google Scholar]
- 14.Kongsuwan K, Cybinski DH, Cooper J, Walker PJ. 1998. Location of neutralizing epitopes on the G protein of bovine ephemeral fever rhabdovirus. J. Gen. Virol. 79:2573–2581 [DOI] [PubMed] [Google Scholar]
- 15.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. 10.1126/science.1127683 [DOI] [PubMed] [Google Scholar]
- 16.Hsieh Y-C, Chen S-H, Chou CC, Ting L-J, Itakaru C, Wang F-I. 2005. Bovine ephemeral fever in Taiwan (2001–2002). J. Vet. Med. Sci. 67:411–416. 10.1292/jvms.67.411 [DOI] [PubMed] [Google Scholar]
- 17.Aziz-Boaron O, LKlausner Z, Shenkar J, Gafni O, Gelman B, David D, Klement E. 2012. Circulation of bovine ephemeral fever in the Middle-East–strong evidence for transmission by winds and animal transport. Vet. Microbiol. 158:300–307. 10.1016/j.vetmic.2012.03.003 [DOI] [PubMed] [Google Scholar]
- 18.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22:1208–1222. 10.1093/molbev/msi105 [DOI] [PubMed] [Google Scholar]
- 21.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. 2012. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848. 10.1126/science.1135710 [DOI] [PubMed] [Google Scholar]
- 23.Tonbak S, Berber E, Yoruk MD, Azkur AK, Pestil Z, Bulut H. 2013. A large-scale outbreak of bovine ephemeral fever in Turkey, 2012. J. Vet. Med. Sci. [Epub ahead of print.] https://www.jstage.jst.go.jp/article/jvms/advpub/0/advpub_13-0085/_article [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327–332. 10.1126/science.1090727 [DOI] [PubMed] [Google Scholar]
- 25.Walker PJ, Kongsuwan K. 1999. Deduced structural model for animal rhabdovirus glycoproteins. J. Gen. Virol. 80:1211–1220 [DOI] [PubMed] [Google Scholar]
- 26.Seddon HR. 1938. The spread of ephemeral fever (three-day sickness) in Australia 1936-37. Aust. Vet. J. 14:90–101. 10.1111/j.1751-0813.1938.tb04222.x [DOI] [Google Scholar]
- 27.St George TD, Standfast HA, Christie DG, Knott SG, Morgan IR. 1977. The epizootiology of bovine ephemeral fever in Australia and Papua-New Guinea. Aust. Vet. J. 53:17–28. 10.1111/j.1751-0813.1977.tb15812.x [DOI] [PubMed] [Google Scholar]
- 28.St George TD, Standfast HA, Armstrong JM, Christie DG, Irving MR, Knott SG, Rideout BL. 1973. A report on the progress of the 1972-73 epizootic of ephemeral fever–1 December 1972 to 30 April 1973. Aust. Vet. J. 49:441–442. 10.1111/j.1751-0813.1973.tb06858.x [DOI] [PubMed] [Google Scholar]
- 29.Kirkland PD. 1982. Bovine ephemeral fever in the Hunter Valley of New South Wales, 1972–1981, p 65–75 In St George TD, Kay BH. (ed), Arbovirus research in Australia, vol 3 CSIRO/QIMR, Brisbane, Australia [Google Scholar]
- 30.Uren MF, St George TD, Kirkland PD, Stranger RS, Murray MD. 1987. Epidemiology of bovine ephemeral fever in Australia 1981–1985. Aust. J. Biol. Sci. 40:125–136 [DOI] [PubMed] [Google Scholar]
- 31.Finlaison DS, Read AJ, Kirkland PD. 2010. An epizootic of bovine ephemeral fever in New South Wales in 2008 associated with long-distance dispersal of vectors. Aust. Vet. J. 88:301–306. 10.1111/j.1751-0813.2010.00596.x [DOI] [PubMed] [Google Scholar]
- 32.Uren MF, St George TD, Stranger RS. 1983. Epidemiology of ephemeral fever of cattle in Australia 1975-1981. Aust. J. Biol. Sci. 36:91–100 [DOI] [PubMed] [Google Scholar]
- 33.Boyle DB, Bulach DM, Amos-Ritchie R, Adams MM, Walker PJ, Weir R. 2012. Genomic sequences of Australian bluetongue virus prototype serotypes reveal global relationships and possible routes of entry into Aust. J. Virol. 86:6724–6731. 10.1128/JVI.00182-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eagles D, Deveson T, Walker PJ, Zalucki MP, Durr P. 2012. Evaluation of long-distance dispersal of Culicoides midges into northern Australia using a migration model. Med. Vet. Entomol. 26:334–340. 10.1111/j.1365-2915.2011.01005.x [DOI] [PubMed] [Google Scholar]
- 35.Eagles D, Walker PJ, Zalucki MP, Durr PA. 2013. Modelling spatio-temporal patterns of long-distance Culicoides dispersal into northern Australia. Preventive Vet. Med. 110:312–322. 10.1016/j.prevetmed.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 36.Kirkland PD. 1993. The epidemiology of bovine ephemeral fever in south-eastern Australia: evidence for a mosquito vector, p 33–37 In St George TD, Uren MF, Young PL, Hoffmann D. (ed), Bovine ephemeral fever and related rhabdoviruses. ACIAR, Canberra, Australia [Google Scholar]
- 37.St George TD. 1993. The natural history of ephemeral fever of cattle, p 13–19 In St George TD, Uren MF, Young PL, Hoffmann D. (ed), Bovine ephemeral fever and related rhabdoviruses. ACIAR, Canberra, Australia [Google Scholar]
- 38.Venter GJ, Hamblin C, Paweska JT. 2003. Determination of the oral susceptibility of South African livestock-associated biting midges, Culicoides species, to bovine ephemeral fever virus. Med. Vet. Entomol. 17:133–137. 10.1046/j.1365-2915.2003.00414.x [DOI] [PubMed] [Google Scholar]
- 39.Daniels PW, Sendow I, Soleha E, Hunt SNT, Bahri S. 1995. Australian-Indonesian collaboration in veterinary arbovirology–a review. Vet. Microbiol. 46:151–174. 10.1016/0378-1135(95)00080-T [DOI] [PubMed] [Google Scholar]
- 40.Daniels PW, Soleha E, Sendow I, Sukarsih 1993. Bovine ephemeral fever in Indonesia, p 41–44 In St George TD, Uren MF, Young PL, Hoffmann D. (ed), Bovine ephemeral fever and related rhabdoviruses. ACIAR, Canberra, Australia [Google Scholar]
- 41.Fitch WM, Leiter JM, Li XQ, Palese P. 1991. Positive Darwinian evolution in human influenza A viruses. Proc. Natl. Acad. Sci. U. S. A. 88:4270–4274. 10.1073/pnas.88.10.4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferguson NM, Galvani AP, Bush RM. 2003. Ecological and immunological determinants of influenza evolution. Nature 422:428–433. 10.1038/nature01509 [DOI] [PubMed] [Google Scholar]
- 43.Cybinski DH, Zakrzewski H. 1983. The isolation and preliminary characterization of a rhabdovirus in Australia related to bovine ephemeral fever virus. Vet. Microbiol. 8:221–235. 10.1016/0378-1135(83)90075-5 [DOI] [PubMed] [Google Scholar]
- 44.Liehne PFS, Anderson S, Stanley NF, Liehne CG, Wright AE, Chan KH, Leivers S, Britten DK, Hamilton NP. 1981. Isolation of Murray Valley encephalitis virus and other arboviruses in the Ord River Valley 1972-1976. Aust. J. Exp. Biol. Med. Sci. 59:347–356. 10.1038/icb.1981.29 [DOI] [PubMed] [Google Scholar]
- 45.Cybinski DH, Muller MJ. 1990. Isolation of arboviruses from cattle and insects at two sentinel sites in Queensland, Australia, 1979-85. Aust. J. Zool. 38:25–32. 10.1071/ZO9900025 [DOI] [Google Scholar]
- 46.Cybinski DH. 1987. Homologous and heterologous antibody reactions in sera from cattle naturally infected with bovine ephemeral fever group viruses. Vet. Microbiol. 13:1–9. 10.1016/0378-1135(87)90092-7 [DOI] [PubMed] [Google Scholar]
- 47.Blasdell KR, Voysey R, Bulach DM, Trinidad L, Tesh RB, Boyle DB, Walker PJ. 2012. Malakal virus from Africa and Kimberley virus from Australia are geographic variants of a widely distributed ephemerovirus. Virology 433:236–244. 10.1016/j.virol.2012.08.008 [DOI] [PubMed] [Google Scholar]
- 48.Hanada K, Suzuki Y, Gojobori T. 2004. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 21:1074–1080. 10.1093/molbev/msh109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jenkins GM, Rambaut A, Pybus OG, Holmes EC. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156–165. 10.1007/s00239-001-0064-3 [DOI] [PubMed] [Google Scholar]
- 50.Streicker DG, Lemey P, Velasco-Villa A, Rupprecht CE. 2012. Rates of viral evolution are linked to host geography in bat rabies. PLoS Pathog. 8:e1002720. 10.1371/journal.ppat.1002720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Talbi C, Holmes EC, de Benedictis P, Faye O, Nakoune E, Gamatie D, Diarra A, Elmamy BO, Sow A, Adjogoua EV, Sangare O, Dundon WG, Capua I, Sall AA, Bourhy H. 2009. Evolutionary history and dynamics of dog rabies virus in western and central Africa. J. Gen. Virol. 90:783–791. 10.1099/vir.0.007765-0 [DOI] [PubMed] [Google Scholar]
- 52.Woelk CH, Holmes EC. 2002. Reduced positive selection in vector-borne RNA viruses. Mol. Biol. Evol. 19:2333–2336. 10.1093/oxfordjournals.molbev.a004059 [DOI] [PubMed] [Google Scholar]
- 53.Mackenzie JS, Poidinger M, Lindsay MD, Hall RA, Sammels LM. 1995. Molecular epidemiology and evolution of mosquito-borne flaviviruses and alphaviruses enzootic in Australia. Virus Genes 11:225–237. 10.1007/BF01728662 [DOI] [PubMed] [Google Scholar]
- 54.Mackenzie JS, Broom AK. 1995. Australian X disease, Murray Valley encephalitis and the French connection. Vet. Microbiol. 46:79–90. 10.1016/0378-1135(95)00074-K [DOI] [PubMed] [Google Scholar]
- 55.Luo LH, Li Y, Snyder RM, Wagner RR. 1988. Point mutations in glycoprotein gene of vesicular stomatitis virus (New Jersey serotype) selected by resistance to neutralization by epitope-specific monoclonal antibodies. Virology 163:341–348. 10.1016/0042-6822(88)90274-7 [DOI] [PubMed] [Google Scholar]
- 56.Luo LZ, Li Y, Snyder RM, Wagner RR. 1990. Spontaneous mutations leading to antigenic variations in the glycoproteins of vesicular stomatitis virus field isolates. Virology 174:70–78. 10.1016/0042-6822(90)90055-V [DOI] [PubMed] [Google Scholar]
- 57.Zagouras P, Rose JK. 1993. Dynamic equilibrium between vesicular stomatitis virus glycoprotein monomers and trimers in the Golgi and at the cell surface. J. Virol. 67:7533–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albertini AA, Merigoux C, Libersou S, Madiona K, Bressanelli S, Roche S, Lepault J, Melki R, Vachette P, Gaudin Y. 2012. Characterization of monomeric intermediates during VSV glycoprotein structural transition. PLoS Pathog. 8:e1002556. 10.1371/journal.ppat.1002556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaudin Y, Raux H, Flamand A, Ruigrok RW. 1996. Identification of amino acids controlling the low-pH-induced conformational change of rabies virus glycoprotein. J. Virol. 70:7371–7378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaudin Y. 1997. Folding of rabies virus glycoprotein: epitope acquisition and interaction with endoplasmic reticulum chaperones. J. Virol. 71:3742–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.