

Abstract
Hepatitis C virus (HCV) predominantly infects human hepatocytes, although extrahepatic virus reservoirs are being discussed. Infection of cells is initiated via cell-free and direct cell-to-cell transmission routes. Cell type-specific determinants of HCV entry and RNA replication have been reported. Moreover, several host factors required for synthesis and secretion of lipoproteins from liver cells, in part expressed in tissue-specific fashion, have been implicated in HCV assembly. However, the minimal cell type-specific requirements for HCV assembly have remained elusive. Here we report that production of HCV trans-complemented particles (HCVTCP) from nonliver cells depends on ectopic expression of apolipoprotein E (ApoE). For efficient virus production by full-length HCV genomes, microRNA 122 (miR-122)-mediated enhancement of RNA replication is additionally required. Typical properties of cell culture-grown HCV (HCVcc) particles from ApoE-expressing nonliver cells are comparable to those of virions derived from human hepatoma cells, although specific infectivity of virions is modestly reduced. Thus, apolipoprotein B (ApoB), microsomal triglyceride transfer protein (MTTP), and apolipoprotein C1 (ApoC1), previously implicated in HCV assembly, are dispensable for production of infectious HCV. In the absence of ApoE, release of core protein from infected cells is reduced, and production of extracellular as well as intracellular infectivity is ablated. Since envelopment of capsids was not impaired, we conclude that ApoE acts after capsid envelopment but prior to secretion of infectious HCV. Remarkably, the lack of ApoE also abrogated direct HCV cell-to-cell transmission. These findings highlight ApoE as a host factor codetermining HCV tissue tropism due to its involvement in a late assembly step and viral cell-to-cell transmission.
INTRODUCTION
Currently, around 160 million people are chronically infected with hepatitis C virus (HCV) worldwide (1). A prophylactic vaccine is not available, but direct-acting antivirals (DAA) have recently been approved for treatment (2). However, the novel triple therapy including pegylated alpha interferon (PEG-IFN-α), ribavirin, and one of two available protease inhibitors is associated with side effects and is not licensed for all viral genotypes. A detailed understanding of the viral life cycle and the roles of specific viral and host factors may reveal novel targets for antiviral therapy and thus help to improve existing therapeutic options.
Chronic HCV infection is a leading cause of liver disease, including hepatitis, liver cirrhosis, and hepatocellular carcinoma (3). It is also associated with numerous extrahepatic manifestations, such as cryoglobulenimia and neuronal disorders (reviewed in reference 4). While hepatocytes are thought to be the primary site of HCV replication, a number of studies have highlighted possible extrahepatic sites of replication, including peripheral blood mononuclear cells and cells of neuronal origin (reviewed in references 5 and 6). These observations suggest that viral interactions with these cells could modulate the pathophysiology of HCV infections. Therefore, dissection of HCV tropism and host factor usage may help to delineate the etiology and mechanisms of extrahepatic manifestations associated with hepatitis C.
Tissue tropism of HCV is likely determined by specific host factor requirements for cell entry, translation, RNA replication, and virus assembly. Regarding HCV cell entry, it has been shown previously that expression of scavenger receptor class B type I (SCARB1), CD81, and the tight-junction proteins claudin-1 (CLDN1) and occludin (OCLN) renders human nonliver cells and nonhuman cells susceptible to HCV entry (7). Moreover, the functional relevance of each of these factors for HCV infection of human liver cells is supported by ample experimental evidence from different laboratories (8). Interestingly, CLDN6 and CLDN9 were shown to functionally substitute for a lack of CLDN1 in human nonliver cells (9–11). Therefore, these factors together with either CLDN1, -6, or -9 apparently make up the minimal requirements to render cells permissive for HCV entry. Notably, however, several additional host factor are known to modulate HCV entry, including, for instance, glycosaminoglycans (12, 13), the transferrin receptor (14), specific receptor tyrosine kinases (15), and the cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) (16), which could further contribute to HCV cell tropism (summarized in reference 8). Moreover, the low-density lipoprotein receptor is involved in HCV infection, possibly also by way of its physiological function, which may facilitate genome replication upon cell entry (17–19).
HCV RNA replication is generally low in most cultured cells and limited to only a few viral consensus genomes (20). Nevertheless, HCV RNA replication has been observed in several human cell lines originating from different tissues (e.g., Huh-7 and HepG2 [liver], 293 [embryonic kidney], Caco-2 [intestine], and HeLa [cervix]) and even in nonhuman cells (e.g., mouse hepatoma cell lines AML12, Hepa 1-6, and Hep56.1D and murine embryonic fibroblasts) (20). These results suggest that the basic host factors required for HCV translation/RNA replication are expressed in various human cells and conserved between humans and mice. However, the replication efficiency varies greatly between these cells, and it is generally highest in the human hepatocarcinoma cell line Huh-7 and its subclones (20). Yet, which specific host factors determine these large differences and how this reflects HCV tissue tropism are poorly understood. Nevertheless, the present experimental evidence indicates an important role of microRNA 122 (miR-122) in liver-specific HCV RNA translation/replication (21). In fact, several studies have shown that ectopic expression of the liver-specific miR-122 dramatically increases HCV replication in various cell lines, including human nonliver cells and mouse cells (22–24).
HCV particles display an unusually variable and generally low buoyant density, which is due to the tight interaction of HCV particles with lipoproteins (25–27). Congruently, assessment of the composition of serum-derived HCV revealed the presence of several apolipoproteins, including apolipoprotein AI (ApoAI), ApoB, ApoC1, and ApoE (26–29). In parallel, several studies have highlighted the involvement of numerous host factors for production of infectious HCV particles (summarized in reference 30). In part, these proteins display tissue-specific expression patterns, suggesting that availability of assembly cofactors may codetermine HCV tissue tropism. Notably, several studies reported the importance of host proteins involved in the synthesis and secretion of very-low-density lipoproteins (VLDL), a typical function of liver cells, for production of infectious HCV. Specifically, ApoB, microsomal triglyceride transfer protein (MTTP), ApoE, and ApoC1 have been shown to be relevant for production of infectious HCV (31–35). However, the minimal requirements for HCV assembly in nonliver cells and how liver-specific cofactors contribute to particle production and virus properties are incompletely understood. Therefore, in this study, we aimed at defining the minimal set of host factors required for virus production of infectious HCV in human nonliver cells and at determining the role of lipoproteins in HCV assembly and cell-to-cell transmission.
MATERIALS AND METHODS
Results of at least 3 independent biological replicates and the mean value for these replicates are shown in every figure, unless otherwise indicated.
Constructs.
The genotype 2a/2a chimera Jc1 (36), the construct harboring a deletion in p7 (53), the Renilla luciferase reporter virus JcR2A (37), the selectable subgenomic replicon with and without the adaptive mutation H2476L in NS5B (38), the packaging construct expressing core to NS2 (39), and the plasmid harboring miR-122 (40) have been described previously. The plasmids pWPI ApoE puro, pWPI ApoE BLR, and pWPI MTTP BLR were constructed using standard PCR-based cloning methods and were verified by sequencing. Detailed cloning strategies are available upon request.
Cell culture and cell lines.
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (PAA Laboratories GmbH) (complete DMEM). If required, blasticidin (Invivogen), puromycin (Sigma), or Geneticin (G418) (Life Technologies) was added for selection.
For Huh-7.5 and HeLa cells harboring a selectable subgenomic replicon, cells were cultured with 0.75 mg/ml and 1 mg/ml of Geneticin, respectively. Medium was replaced twice a week during selection.
Lentiviral gene transfer was performed as described previously (41). The plasmids pCMV-ΔR8.74 (42), pcz-VSV-G (43), and derivatives of pWPI (carrying the genes of interest and in addition either the blasticidin S deaminase gene of Aspergillus terreus or a puromycin N-acetyltransferase gene) (44) or pLenti (encoding miR-122) (40) were transfected at a ratio of 3:1:3 into 293T cells. Lentiviral particles were harvested at 48 h posttransfection. Transduced target cells were then selected with the corresponding antibiotic (blasticidin, 5 μg/ml; puromycin, 2 μg/ml; or Geneticin, 0.75 mg/ml). Expression of transduced constructs was confirmed by Western blotting, enzyme-linked immunosorbent assay (ELISA), or reverse transcription-PCR (RT-PCR).
In vitro transcription, electroporation, and virus preparation.
In vitro transcripts were generated as described previously (45). Briefly, MluI-linearized plasmid DNA was extracted with phenol and chloroform. Next, in vitro transcription was performed, and RNA was extracted with acidic phenol and chloroform. RNA concentration and integrity were determined by photometry and agarose gel electrophoresis, respectively. For electroporation, single-cell suspensions were washed with phosphate-buffered saline (PBS) and resuspended at 1.5 × 107 cells/ml in Cytomix (46) containing 2 mM ATP and 5 mM glutathione and were electroporated with 5 μg RNA. Cells were immediately transferred to 10 ml of complete DMEM and seeded in a 10-cm dish, or cells transferred to 16 ml of complete DMEM and 2 ml of the cell suspension was seeded per well of a 6-well plate.
Virus infection assays.
For standard infection assays with Renilla luciferase reporter viruses, Huh-7.5 cells were seeded at a density of 8 × 104 cells per well of a 12-well plate at 24 h prior to infection with filtered cell culture fluids. Infection was carried out for 4 h and stopped by addition of medium. Luciferase expression was quantified at 48 h postinfection by lysis in passive lysis buffer (Promega) and measured by addition of coelenterazine (P.J.K. GmbH).
HCV titers were determined as published recently (47). The 50% tissue culture infectious dose (TCID50) was calculated based on the methods described by Spearman and Kärber (48, 49).
For quantification of intracellular infectivity and core amounts at 48 h posttransfection, cells were washed with PBS, scraped, and centrifuged for 5 min at 1,000 × g at 4°C. Pellets were resuspended in 500 μl complete DMEM and subjected to five cycles of freezing and thawing in liquid nitrogen. Subsequently, cells were centrifuged at 10,000 × g for 10 min at 4°C to remove cell debris.
Virus neutralization.
For neutralization experiments, Huh-7.5 cells or Huh-7 Lunet N#3 hCD81-FLuc cells (J. Gentzsch and T. Pietschmann, unpublished data) were seeded at 104 cells per well in 96-well dishes at 1 day before infection. Viral supernatants from Huh-7.5 or 293T/mir-122/ApoE cells were diluted so as to normalize for the infectious titers (as determined by luciferase assay) and mixed with given amounts of antibody before infection of the cells (40 μl per well). The cell culture medium was replaced after 3 h and the luciferase activity was measured after 72 h as described above.
Antibodies and inhibitors.
The CD81-specific 5A6 antibody was purchased from Santa Cruz. The SCARB1-specific antibody was purchased from Novogen. M. Law kindly provided the AR4A antibody (50), and the ITX inhibitor was a kind gift from F. Wong-Staal. The IgG1 control isotype antibody was directed against a neuronal antigen and kindly provided by C. Erck. Total immunoglobulins derived from HCV (genotype 2a)-positive patient sera were purified using the MabTrap kit from GE Healthcare. Protein content was determined by the Bradford assay, and purity was controlled by Coomassie blue staining of an SDS gel. Sera from HCV-negative donors were purified as controls. Patient sera were obtained in the context of routine diagnostic workup. All patients gave written informed consent in accordance with local ethical committees.
Density gradient centrifugation.
To separate viruses according to buoyant density, filtered supernatants of HCV-transfected cells were concentrated with Centricon Plus-70 centrifugal filter units (Merck Millipore) according to the manufacturer's instructions. Subsequently, density gradient centrifugation was performed as described recently (45). Briefly, 1 ml of virus stock was mixed with 2 ml of a 60% iodixanol solution and layered under an iodixanol gradient (0 to 30%). Gradients were centrifuged at 154,000 × g in a TH641 swing-out rotor at 4°C using a Sorvall WX80 centrifuge. After 18 h, fractions of 1 ml were collected from bottom to top. For each fraction, the infectivity was determined by limiting dilution, and the density was measured via refractometry.
Oleic acid stimulation.
For oleic acid stimulation, the medium of cells was changed at 30 h postelectroporation and replaced by medium containing a final concentration of 360 μM oleic acid solubilized in a solution of bovine serum albumin (BSA) in PBS or only BSA in PBS (final concentration, 0.03 mg/ml). Incubation was carried out overnight on a shaker.
Proteolytic digestion protection assay.
Proteolytic digestion protection assays were performed essentially as published previously (51, 52). Briefly, cells transfected with wild-type (wt) Jc1 or a mutant harboring a deletion in p7 (Δp7half) (53) were seeded on poly-l-lysine-coated 10-cm dishes, scraped 48 h later in 255 μl proteinase K buffer (50 mM Tris-HCl [pH 8.0], 10 mM CaCl2, 1 mM dithiothreitol [DTT]) and subjected to five cycles of freezing and thawing. Subsequently, one-third of lysates was left untreated, one-third was incubated with 50 μg/ml proteinase K (Roche, Mannheim, Germany), and the third part was lysed with 5% Triton X-100 prior to proteinase K treatment. After 1 h of incubation on ice, the reaction was terminated by addition of phenylmethylsulfonyl fluoride (PMSF) (AppliChem) at a final concentration of 10 mM and additional incubation of samples on ice for 10 min. For quantification by core ELISA, samples were heated to 95°C for 10 min. After cooling, a protease inhibitor cocktail (Roche, Mannheim, Germany) was added.
SDS-PAGE and Western blotting.
For Western blotting, cells were scraped in SDS-PAGE sample buffer, boiled for 10 min at 95°C, and loaded onto a 12% SDS gel. After electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane with a semidry blotter. The membrane was blocked with 5% milk in PBS containing 0.5% Tween. NS5A (9E10, 1:1,000) (54), core (C7-50, 1:1,000) (55), ApoE (Calbiochem, 1:1,000), and actin (Sigma,1:1,000) were detected with specific antibodies and a secondary antibody coupled to horseradish peroxidase (Sigma-Aldrich, 1:20,000). The antibody signal was detected with the ECL Prime detection reagent (GE Healthcare).
Quantitative detection of viral and cellular RNAs.
RNA was isolated from cells by using Nucleo Spin RNAII kit (Macherey-Nagel) as recommended by the manufacturer. Levels of mature miR-122 and U6 were determined by using a TaqMan MicroRNA assay (Applied Biosystems). HCV-specific RNA and GAPDH (glyceraldehyde-3-phosphate dehydrogenase)-specific mRNA were determined as described previously (52). For detection of mRNA levels of ApoB, specific primers (S-ApoB, 5′-TGAACACCAACTTCTTCCACGAG-3′; A-ApoB, 5′-GTTCTCAATGAGAGGTGGGATCAC-3′) and an ApoB-specific probe (6FAM-CTGGAGGCTCATGTTGCCCTAAAAGC-BBQ; TIB Molbiol) were used. This combination detects both ApoB100 and a shortened version, ApoB48. ApoE mRNA levels were detected with the primers S-ApoE (5′-CAGGAACTGAGGGCGCT-3′) and A-ApoE (5′-GAGCCGCTTACGCAGCTT-3′) and a specific probe (5′-6FAM-CCCGCAGCTCCTCGGTGCTC-BBQ-3′; TIB Molbiol). For ApoC1, the primers S-ApoC1 (5′-AGCTGAAGGAGTTTGGAAACACA-3′) and A-ApoC1 (5′-AGAGGCCCCTCCTGGGAT-3′) and the probe 5′-6FAM-TGGAGGACAAGGCTCGGGAACTC-BBQ-3′ (TIB Molbiol) were utilized. MTTP mRNA levels were detected with the primers S-MTTP (5′-CAGAGAGGAGAGAAGAGCATCTTCAA-3′) and A-MTTP (5′-CTCATTGGTGGTTCCAGAGCTTA-3′) and a specific probe (5′-6FAM-AGGTCTTTGCAGAGCTTCCAAGTTTTCCT-BBQ-3′). A one-step RT-PCR LightCycler 480 RNA Master Hydrolysis Probe kit (Roche) was used for the reactions.
Fluorescence microscopy.
Cells were reseeded at 24 h postelectroporation on poly-l-lysine-coated glass coverslips at a density of 5 × 105 cells per well of a 24-well plate. At 6 h after seeding, cells were incubated with oleic acid (360 μM final concentration) or carrier alone overnight. The next morning, cells were fixed with 3% paraformaldehyde (PFA) and stained for lipid droplets (LDs); a solution of 1% Oil Red O (Sigma-Aldrich) in isopropanol was diluted freshly in water (6 parts Oil Red O and 4 parts water), filtered, and used as published recently (56). Cell nuclei were visualized with DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) (Invitrogen), and core protein was detected by immunofluorescence staining (C7-50, 1:1,000). Finally, cells were mounted on glass slides with Fluoromount (Southern Biotech). Cells were imaged with an inversed confocal laser-scanning microscope (Olympus Fluoview 1000), using an oil lens at a magnification of ×100. Further numerical zoom was applied to the acquisition of 293T cell fluorescence. The three channels (blue, green, and red) were read in a sequential acquisition mode, with an average of 3 frames for each picture (Kalman n = 3).
ELISA.
Amounts of human ApoE and human ApoB100 (Mabtech, Nacka Strand, Sweden) and human albumin (Bethyl, Montgomery, TX) in the cell culture fluid were determined by specific ELISAs according to the manufacturer's instructions. Core amounts were quantified with a diagnostic kit (Architect Anti-HCV; Abbott).
Cell-to-cell spread assay.
To measure HCV infection by direct cell-to-cell transmission between adjacent cells, an agarose overlay assay was performed as described recently (57). Briefly, transmission of HCV from HCV-positive donor cells to HCV-negative target cells in the coculture was assessed. Huh-7.5 target cells were transduced with the pTRIP-tagRFP-NLS-IPS reporter construct recently described by Jones and colleagues (58). As donor cells, 293T cells expressing miR-122 and ApoE or the empty vector and Huh-7.5 cells were used. These cells were transfected with Jc1 RNA. After transfection, the cells were washed 3 times with PBS to remove free RNA. When 293T cells were used as donor cells, they were mixed with target cells at a ratio of 4:1 and 2 × 105 cells were seeded per well into a 24-well plate. When Huh-7.5 cells were used as donor cells, a donor-to-target ratio of 1:1 was used and 8 × 104 cells were seeded per well into a 24-well plate. The cell mixture was overlaid with medium containing 1% SeaPlaque agarose (Lonza) and 20 mg/ml of the neutralizing AR4A antibody (50) to prevent virus spread through the cell-free route. After 72 h, nuclear localization of the tagRFP reporter, which results from cleavage by the HCV NS3-4A protease and thus indicates spread of HCV to the target cells, was monitored through fluorescence microscopy. Additionally, NS5A expression was detected by immunofluorescence staining. The number of spreading events was quantified by determining the number of cells in which the red fluorescent protein (RFP) reporter translocated into the nucleus among all counted cells. For each condition in each experiment, between 1,000 and 2,300 cells were counted.
Statistical analysis.
Statistical analyses were performed using Welch′s 2-sample t test or a paired t test. P values of <0.1 were considered marginally significant (*), P values of <0.05 were considered statistically significant (**), and P values of <0.01 were considered highly significant (***).
RESULTS
Ectopic expression of ApoE is necessary and sufficient for production of infectious trans-complemented HCV particles in HeLa cells.
To identify liver-specific host factors crucial for assembly and release of infectious HCV particles, we aimed at determining the host factor requirements for reconstituting HCV production in human nonhepatic cells. Given the generally modest HCV RNA replication efficiency in nonliver cells, which may limit virus production from full-length HCV RNAs, we made use of the recently described HCV trans-complementation system (41). In this model, subgenomic HCV replicons, which display a more robust RNA replication than full-length HCV RNAs, are packaged into single-round infectious particles if the remaining structural proteins are provided in trans. Thus, HeLa cells (cervical carcinoma) and, as a control, the highly HCV-permissive Huh-7.5 cells (hepatocarcinoma) were transfected with an assembly-defective neomycin-selectable subgenomic HCV-JFH1 replicon which lacks core-, E1-, E2-, p7-, and NS2-coding sequences and carries the adaptive mutation H2476L in NS5B (38). After several passages under selection pressure, the remaining viral factors were introduced into the cells via lentiviral gene transfer of a packaging construct which carries a second resistance gene (Fig. 1A).
FIG 1.

Ectopic expression of Apo E rescues production of infectious HCV in HeLa cells. (A) Schematic depiction of the constructs used for trans-complementation. The replicon (Rep.) harboring a neomycin resistance gene and the adaptive mutation H2476L is shown at the top, and the lentiviral vector (C-NS2) is shown at the bottom. The arrows indicate which gene products were detected in panel B to confirm expression of both constructs in Huh-7.5 and HeLa cells. (B) Protein expression of the given cell lysates was determined using Western blotting and antibodies against NS5A, core, and actin. +, ectopic expression of the constructs; −, only endogenous levels are present. (C) Cells were seeded in 6-well dishes, and 48 h later, total RNA was extracted to determine the amount of HCV RNA and mature miR-122 via RT-PCR. Shown are values normalized to amounts of GAPDH mRNA or U6 RNA, respectively. n.d., not detected. (D) Amounts of secreted ApoE, ApoB, and albumin were determined by collecting culture supernatants at 48 h postseeding and using specific ELISAs. The dashed line shows the detection limit of the assays. (E) HCVTCP titers in the supernatants of the different cell lines were determined at 48 h postseeding using a limiting-dilution assay.
Protein expression of the replicon as well as from the packaging construct was analyzed by Western blotting, thus confirming stable replication of the selectable HCV replicon and comparable production of the HCV structural proteins in both cellular backgrounds (Fig. 1B). Notably, NS5A protein expression was consistently lower in HeLa cells than in the Huh-7.5 cells, indicating somewhat lower HCV RNA replication in the former cells. Analysis of intracellular HCV RNA levels by quantitative RT-PCR confirmed this and revealed ca. 2-fold-lower steady-state levels of HCV RNA in the HeLa cell lines than in the Huh-7.5 cells (Fig. 1C). The lower replication efficiency of HCV in HeLa cells may at least in part be attributable to the lack of endogenous miR-122 expression, which acts as an enhancer of HCV RNA replication (21) (Fig. 1C).
Even though all viral factors were present in HeLa and Huh-7.5 cells, release of infectious HCV trans-complemented particles (HCVTCP) was observed only from Huh-7.5 cells (Fig. 1E). Therefore, we speculated that liver-specific host factors may be lacking in HeLa cells, thus precluding virus production. To test this hypothesis, we first determined expression of ApoB and ApoE, since both proteins have been implicated in production of infectious HCV and because they are expressed in a tissue-specific fashion. Albumin, which is also specifically secreted from liver cells but does not act on HCV assembly, was included as control. As depicted in Fig. 1D and the RT-PCR analysis shown in Fig. 2A, HeLa cells had undetectable protein expression of both ApoE and ApoB and expressed more than 100-fold- and 10,000-fold-lower levels of the respective mRNAs. Given these findings, we next explored whether ectopic expression of ApoE would reconstitute production of HCVTCP in the HeLa cell background. Thus, HeLa cells harboring the replicon as well as the packaging construct were additionally transduced to express ApoE. Thereafter, ApoE was readily detectable in the cell lysates by Western blotting (Fig. 1B) and was also secreted into the supernatant (Fig. 1D). Importantly, ectopic expression of ApoE in these HeLa cells efficiently rescued HCV production, yielding infectious HCVTCP titers of ca. 1 × 104 TCID50/ml, which is only 2- to 3-fold lower than the peak titer observed in the highly permissive human liver cell line Huh-7.5 (Fig. 1E). Hence, reconstitution of ApoE expression was necessary and sufficient to permit efficient production of infectious HCVTCP in HeLa cells.
FIG 2.

Ectopic expression of ApoE is necessary and sufficient to permit HCV assembly in several human nonliver cell lines. Huh-7.5, HeLa, 293T, 293, and Caco-2 cells were engineered to express HCV C-NS2 proteins and ApoE or the empty vector as a control. Subsequently, these cells were transiently transfected with a subgenomic HCV replicon. Analyses were carried out at 48 h postelectroporation. +, ectopic expression of the different constructs; −, only endogenous levels are present. The dashed lines in panels B and D show the detection limit. n.d., not detected. (A) Total cellular RNA was extracted, and mRNA levels of ApoE, ApoC1, MTTP, and ApoB were determined via mRNA-specific RT-PCR and normalized to GAPDH mRNA. Values were normalized to the corresponding mRNA abundance in Huh-7.5 C-NS2 cells. (B) Secretion of ApoE into the cell culture fluid of the given cell lines was quantified by specific ELISA. (C) Core, NS5A, and actin were detected in cell lysates by Western blotting. (D) Release of infectious HCVTCP from the different cell lines was quantified by a limiting-dilution assay.
Ectopic expression of ApoE rescues HCVTCP production in various human nonliver cell lines.
Next, we explored whether infectious HCV production is also arrested in other human nonliver cell lines and, if so, whether it is restored upon ectopic expression of ApoE alone. To this end, we created packaging cell lines expressing core to NS2 in the context of 293T and 293 (both derived from human embryonic kidney) and Caco-2 (colon carcinoma) cells. Moreover, ApoE or the empty expression vector was introduced into these cells by lentiviral gene transfer. Huh-7.5 and HeLa cells prepared in parallel served as controls. These cells were then analyzed for mRNA expression of different components of the VLDL pathway by using strictly mRNA-specific RT-PCR assays based on probe and primer sets across exon-intron boundaries (Fig. 2A). Using this approach, we detected comparably high levels of ApoE, ApoB, and ApoC1 mRNAs in Huh-7.5 and Caco-2 cells, while the abundance of MTTP mRNA was ca. 100-fold lower in Caco-2 cells than in Huh-7.5 cells (Fig. 2A). In contrast, HeLa, 293T, and 293 cells expressed very little mRNA of these VLDL components, with more than 1,000-fold lower ApoB and MTTP mRNA levels, ca. 100- to 1,000-fold-reduced ApoC1 mRNA levels, and at least 10-fold-lower ApoE mRNA levels than in Huh-7.5 cells. Ectopic overexpression of ApoE further increased mRNA abundance in Huh-7.5 and Caco-2 cells by ca. 10-fold and established mRNA levels of ApoE in HeLa, 293T, and 293 cells that were slightly higher than the endogenous level of ApoE mRNA expressed in Huh-7.5 cells (Fig. 2A). Consistent with this, ectopic expression of ApoE established robust ApoE expression in the latter nonliver cells and further elevated ApoE secretion from Caco-2 cells, which already released modest levels of this lipoprotein (Fig. 2B).
To assess HCVTCP production in the presence or absence of ApoE, these cell lines were transfected with a subgenomic JFH1 replicon. In contrast to the previous assay, however, selection was not carried out, and HCV protein expression as well as virus production was monitored at 48 h after transient transfection (Fig. 2C and D). Core protein expressed from the packaging construct was readily detected in all cell lines, whereas NS5A expressed from the replicon was detectable only in the transfected Huh-7.5 cells and was below the detection limit of our Western blot assay in all nonliver cells (Fig. 2C). These results suggested comparatively inefficient transient HCV RNA replication and/or translation in these nonliver cells. Regardless of this, ectopic expression of ApoE resulted in the production of infectious HCVTCP in all nonliver cell lines tested (Fig. 2D). Notably, virus titers obtained from nonliver cells were between 20- and 200-fold lower than those from Huh-7.5 cells, which is likely due to the reduced transient HCV replication in these cells. Moreover, among the nonliver cells, only Caco-2 cells produced infectious HCVTCP in the absence of ectopic expression of ApoE. Since among these tested nonliver cells only Caco-2 cells expressed endogenous ApoE, these results are consistent with strict ApoE dependence of HCV production. Thus, collectively these results highlight that in human nonliver cells that do not express endogenous ApoE, introduction of this host protein is necessary and sufficient for production of infectious HCVTCP.
Ectopic expression of miR-122 permits ApoE-dependent release of infectious HCV from replicating full-length HCV RNAs.
Next, we explored if wild-type HCV particles produced from transfected full-length HCV RNAs also are produced in these nonliver cells in an ApoE-dependent fashion. We focused our further analyses on 293T and HeLa cells, as both cell lines are easy to manipulate and allowed robust production of HCVTCP in the previous assay (Fig. 1E and 2D). Furthermore, in contrast to Caco-2 cells, neither HeLa nor 293T cells endogenously express ApoE (Fig. 2B). As miR-122 is an important liver-specific host factor facilitating efficient RNA replication, we introduced miR-122 into the nonliver cells and confirmed expression and processing by RT-PCR (Fig. 3).
FIG 3.
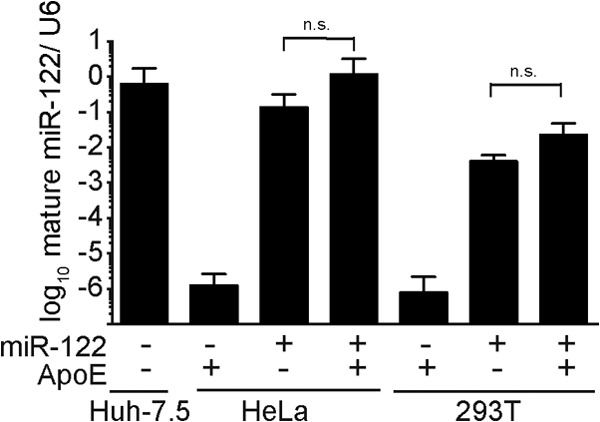
Abundance of mature miR-122 in different human cell lines. Levels of mature miR-122 in Huh-7.5, HeLa, and 293T cells were determined via specific RT-PCR and are shown relative to U6 RNA. +, indicates ectopic expression of miR-122; −, only endogenous levels are present; n.s., nonsignificant.
Naive HeLa and 293T cells expressed little endogenous miR-122. This low level of expression was increased by more than 100,000-fold and more than 1,000-fold due to lentiviral gene transfer, respectively. To analyze HCV RNA replication and particle release, these cells were transfected with a full-length Jc1 reporter virus RNA expressing a Renilla luciferase reporter gene (25). Thus, replication of the transfected RNA was determined by measuring luciferase activity in the transfected cells, and infectious virus production was quantified by inoculation of naive Huh-7.5 cells with the culture fluid of these cells and subsequent luciferase assays. In these assays, expression of ApoE alone did not rescue production of detectable infectious HCV from the full-length reporter virus genomes upon transfection of HeLa and 293T cells (Fig. 4A). This lack of virus production was, however, likely due to the poor RNA replication of the reporter virus RNA in these cells, as is evidenced by the low intracellular luciferase expression in the transfected cells (Fig. 4A, left panel). Ectopic expression of miR-122 dramatically increased HCV RNA replication in 293T cells and, as a consequence, also rescued virus production provided that ApoE was coexpressed (Fig. 4A). In the case of HeLa cells, ectopic expression of miR-122 only slightly increased RNA replication of the reporter virus genome. Therefore, likely due to very modest levels of viral RNA replication in these cells, we observed only very weak transduction of luciferase activity to the Huh-7.5 cells inoculated with culture fluid collected at 48 and 72 h posttransfection. Nevertheless, these results suggested that both HeLa and 293T cells produce infectious HCV reporter viruses in an miR-122- and ApoE-dependent fashion.
FIG 4.

Expression of ApoE and miR-122 is sufficient for production of full-length HCV particles from 293T and HeLa cells. +, ectopic expression of the indicated constructs; −, only endogenous levels are present. The dashed lines in panel A and the right panel of panel B show the detection limit of the assay. n.d., not detected. (A) The indicated cell lines were transfected with JcR2A HCV RNA, which encodes the Renilla luciferase reporter. HCV RNA replication was determined at 4 h, 24 h, 48 h, and 72 h postelectroporation by luciferase measurement and is expressed as relative light units (left). Additionally, cell-free culture fluids of the different cell lines were used to inoculate Huh-7.5 cells, and infectivity was determined by luciferase assay at 48 h postinoculation (right). (B) The same set of cells was transfected with wild-type Jc1 RNA, and at 48 h postelectroporation, the abundance of core protein in the cell lysates was determined by Western blotting (left). Actin served as a loading control. Release of infectious particles was determined by using a limiting-dilution assay (right).
To extend these findings, we transfected these cell lines with wt Jc1 RNA and measured HCV protein expression with a core-specific antibody (Fig. 4B). Accumulation of HCV core protein was clearly detectable in both HeLa and 293T cells upon introduction of miR-122, albeit to a much greater extent in the latter cells. This likely reflects the translation/replication-enhancing function of miR-122, which is apparently much more pronounced in the context of 293T cells than in that of HeLa cells (Fig. 4A and B). Importantly, in both cell lines ectopic expression of miR-122 and ApoE was necessary and sufficient to permit production of infectious HCV particles from the full-length Jc1 genome. Therefore, efficient HCV production by full-length HCV genomes in human nonliver cells depends on miR-122 replication/translation enhancement and expression of ApoE. As 293T/miR-122/ApoE cells were more permissive to HCV RNA replication and particle production than HeLa/miR-122/ApoE cells, we focused on these cells in our subsequent analyses.
293T cell-derived HCV particles display properties comparable to those of particles obtained from Huh-7.5 cells.
Serum-derived HCV was reported to associate with various lipoproteins, including ApoAI, -B, -C1, and -E (26, 27, 29), which is thought to influence particle properties with regard to density, neutralization by antibodies, and receptor interactions. Given that Huh-7.5 cells secrete both ApoE and ApoB and express ApoC1 mRNA, we hypothesized that HCV particles produced in these cells may carry a different spectrum of lipoproteins than those released from 293T/miR-122/ApoE cells (and thus in the absence of detectable ApoB and ApoC1). Since such differences in lipoprotein decoration of HCV particles may translate into different functional properties of released viruses, we conducted a series of virus neutralization assays using antibodies from HCV-positive sera and anti-CD81, anti-SCARB1, and anti-ApoE antibodies. Moreover, we explored the susceptibility of both particle types to inhibition by ITX5061 which prevents HCV infection by binding to SCARB1 (59) (Fig. 5). Finally, we compared the buoyant densities of these HCV particles by using density gradients (Fig. 6). Remarkably, we did not observe any striking differences between Huh-7.5 and 293T/miR-122/ApoE cell-derived HCV particles in these experiments. Therefore, we conclude that HCV particles produced in these cell types share fundamental properties, including density and susceptibility to receptor competitors and to patient-derived antibodies. However, we cannot exclude the existence of other compositional differences between these particle types. Our observation that the specific infectivity of viruses released from 293T/miR-122/ApoE cells is more than 5-fold lower than that of Huh-7.5 derived virus supports this notion (see Fig. 8B).
FIG 5.

Susceptibility of 293T-derived and Huh-7.5-derived HCV particles to neutralizing antibodies and entry inhibitors. Reporter viruses encoding a Renilla luciferase were produced in Huh-7.5 and in 293T/miR-122/ApoE cells. Cell culture supernatant was harvested at 48 h and 72 h posttransfection. For neutralization and inhibitor treatments, the virus stocks were normalized for Renilla counts prior to infection. Normalized values are shown. (A) Neutralization with patient-derived IgGs. Patient-derived IgGs were purified from HCV-positive donors. HCV-negative donors served as control. (B) Neutralization with anti-CD81 and anti-SCARB1 antibodies and control IgG. (C) Susceptibility to the SCARB1 inhibitor ITX5061; a nonfunctional compound (ITX7874) served as a control. (D) Neutralization with anti-ApoE antibodies.
FIG 6.
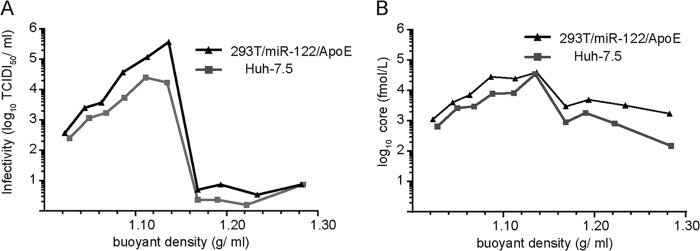
Buoyant densities of HCV particles derived from 293T/miR-122/ApoE and Huh-7.5 cells. Equilibrium density centrifugation was performed on concentrated cell culture supernatants. The density was determined by refractometry and is given on the x axis. A representative gradient is shown. (A) The infectivity in the different fractions of the gradient was quantified by a limiting-dilution assay. (B) Core amounts were determined by core-specific ELISA.
FIG 8.
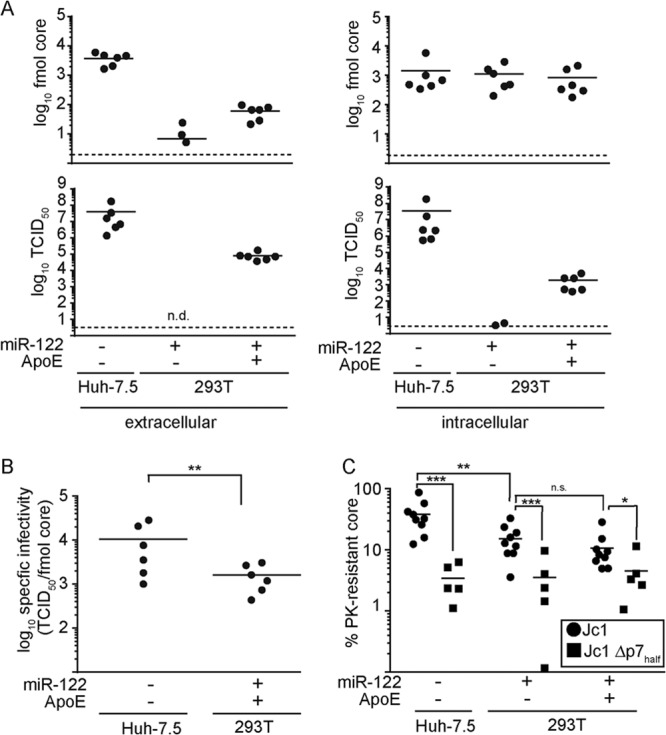
ApoE is required prior to particle release and after capsid envelopment. +, ectopic expression of the constructs; −, only endogenous levels are present. (A) At 48 h after transfection of Jc1 RNA into the different cell lines, viral titers and core protein were determined in the cell culture supernatants (left) as well as in freeze-thaw lysates (right). Shown are the total amount of core protein (upper panel)s and the viral titer (lower panels) per 10-cm culture dish. n.d., not detected. The dashed line represents the detection limit of the assay. (B) The specific infectivities of particles in the supernatant, from Huh-7.5 and 293T/miR-122/ApoE cells were calculated based on the data presented in panel A. (C) Cells were lysed by freezing and thawing at 48 h postelectroporation with Jc1 or Jc1 Δp7half RNA and subjected to proteinase K digestion. Lysates were divided into three parts: the first part was left untreated, the second was incubated with proteinase K, and the third was lysed with Triton prior to proteinase K-mediated digestion. The amounts of core protein in the different samples were determined by core-specific ELISA. Normalized values for proteinase K-treated samples are shown. For this, the core amounts in the samples treated with proteinase K and Triton were set as background and those in the untreated samples were set as 100%.
Determinants of HCV particle production in 293T cells.
Comparison of HCV titers released from Huh-7.5 and 293T/miR-122/ApoE cells transfected with Jc1 revealed that ca. 100-fold-lower peak infectivity was produced in the latter cells (Fig. 4). Since introduction of miR-122 had established comparatively robust HCV replication in the 293T background (Fig. 4), we hypothesized that the abundance/activity of assembly cofactors and/or the availability of cellular structures crucial for virus production may limit HCV assembly in 293T/miR-122/ApoE cells. Recently, lipid droplets were recognized as crucial cellular platforms for HCV assembly (60). As Oil Red O staining of lipid droplets of 293T/miR122 cells in the presence or absence of ApoE revealed modest numbers of relatively large lipid droplets compared with Huh-7.5 cells (Fig. 7A), we hypothesized that the low number of lipid droplets may limit production of infectious HCV in the 293T background. To test this, we supplied both cell lines with exogenous oleic acid in order to augment cellular lipid droplet size and number. However, this treatment neither allowed particle production in the absence of ApoE nor increased the virus titer from 293T/miR-122/ApoE cells (Fig. 7B), even though the lipid droplet content of the cells was greatly increased (Fig. 7A). Therefore, we conclude that augmentation of lipid droplets in 293T cells does not circumvent the ApoE dependence of HCV assembly and that endogenous levels of lipid droplets are unlikely to limit HCV production in these cells. Notably, core protein associated with the perimeter of lipid droplets in 293T cells in the absence of ApoE, ruling out the possibility that absence of ApoE precludes trafficking of core to these organelles (Fig. 7A).
FIG 7.

Lipid droplets and MTTP are not limiting for HCV assembly in 293T/miR-122/ApoE cells. +, ectopic expression; −, only endogenous levels are present. The dashed lines in panels B and D represent the detection limit of the assay. n.d., not detected. (A) The indicated cell lines were transfected with Jc1 RNA, and at 30 h postelectroporation, cells were stimulated with oleic acid (final concentration, 360 μM) or vehicle control overnight. Lipid droplets were stained with Oil Red O (red), core protein was detected by specific antibodies (green), and cell nuclei were visualized with DAPI (blue). Representative pictures are shown, and the scale bar corresponds to 10 μm. (B) The virus titer in the cell culture fluid was determined by using a limiting dilution assay. (C) The indicated cell lines were transfected with JcR2a HCV RNA. Total RNA was extracted at 48 h posttransfection, and mRNA levels of MTTP were determined in the different cell lines by RT-PCR. (D) HCV RNA replication in these cells was determined at 48 h posttransfection by luciferase measurement. Additionally, cell-free culture fluids were used to inoculate naive Huh-7.5 cells. Infectivity was determined by luciferase assay at 48 h postinoculation. Luciferase values are expressed as relative light units (RLU) per 6-well plate (for RNA replication) or per 12-well plate (for infectivity).
MTTP, which is important for lipid transfer to nascent ApoB in the context of VLDL production within liver cells, has been reported to contribute to HCV assembly (33). As MTTP mRNA levels in 293T cells were more than 1,000-fold lower than in Huh-7.5 cells (Fig. 2A), we reasoned that HCV particle production may be increased by ectopic MTTP expression. Hence, we introduced MTTP into 293T cells harboring miR-122 and ApoE or the empty vector. The abundance of MTTP mRNA was measured by quantitative RT-PCR (Fig. 7C). Even though levels of MTTP mRNA were robustly increased after lentiviral transduction, reaching levels comparable to those in Huh-7.5 cells, neither HCV RNA replication nor production of infectious particles was affected by expression of MTTP (Fig. 7D). Thus, the reduced efficiency of HCV particle production in the 293T background is either caused indirectly by somewhat lower RNA replication or due to other, as-yet-unknown host factors.
ApoE is required for a step prior to particle release and after capsid assembly.
Since 293T cells do not express endogenous ApoE (Fig. 2) and since ectopic ApoE expression restores production of infectious HCV, this cell system opens unique opportunities to explore the involvement of ApoE in HCV morphogenesis. Thus, to dissect the step of assembly and/or release at which ApoE was required, we first compared extra- and intracellular amounts of core protein and production of intra- and extracellular infectious particles for 293T/miR-122 cells with and without ApoE (Fig. 8A). Huh-7.5 cells transfected with Jc1 served as a control. After transfection of HCV into the 293T/miR-122 cells, core protein release into the culture fluid, a marker for release of virus particles, was ca. 10-fold increased by simultaneous coexpression of ApoE. In the absence of ApoE, core release was detected in only 3 out of 6 experiments and to a low level, which may reflect core content from cell debris in the supernatant. Extracellular infectivity was observed only in the presence of ApoE, whereas intracellular core protein levels were comparable between all three cell lines, excluding defects in translation and genome replication. Infectious intracellular HCV particles were produced in Huh-7.5 cells and 293T cells expressing ApoE. In the absence of ApoE, very low levels of infectivity could be detected in two out of six experiments, representing one or two foci in the lowest dilution of the titration. Collectively, these results indicate that in the absence of ApoE, noninfectious particles are not released and infectious particles do not accumulate intracellularly. In contrast, these results support the notion that ApoE is needed to allow assembly of infectious particles before particles could be secreted.
The current model of HCV assembly suggests that core protein traffics to lipid droplets and recruits viral NS5A and RNA, which then triggers assembly and budding of HCV capsids into intracellular membranes decorated with viral glycoproteins E1 and E2. Therefore, membrane envelopment of HCV core protein is an essential step during assembly of infectious HCV progeny. To test whether lack of ApoE arrests HCV assembly at a step prior to capsid envelopment, we analyzed the susceptibility of intracellular core protein to degradation by proteinase K (51, 52). In this experimental setup, the degree of resistance of core to proteolytic digestion, which is measured by a core-specific ELISA and Western blotting, reflects the amount of core protein that has acquired a membrane envelope. As it was shown recently that p7 is critical for capsid assembly and envelopment (51), a mutant HCV genome with partial deletion in p7 (Δp7half) was used as a control for arrested capsid envelopment. We observed that upon transfection with wt Jc1, ca. 40% of core was protected from proteinase K degradation in Huh-7.5 cells (Fig. 8C), while ca. 15% of core was protected in 293T cells, irrespective of whether ApoE was expressed. In all cell lines, transfection with Δp7half resulted in a reduced amount of enveloped core protein compared to wt Jc1 transfection, confirming that our assay is able to reveal defects in HCV capsid envelopment in both Huh-7.5 and 293T cells. Therefore, the presence or absence of ApoE does not influence the degree of capsid membrane envelopment. Collectively, these results suggest that the environment of 293T/miR-122 cells is slightly less conducive for HCV core protein envelopment and that the lack of ApoE likely ablates an HCV assembly step after envelopment of viral capsids.
ApoE is essential for direct cell-to-cell transmission of HCV.
HCV is transmitted to new target cells via cell-free as well as direct cell-to-cell transmission routes (61, 62). However, the determinants at the cellular and viral levels for cell-to-cell spread are not fully established. Therefore, we investigated if ApoE is required for direct cell-to-cell transmission using the 293T/miR122 cells and a recently described assay for cell-to-cell spread (57). Huh-7.5 cells, which readily sustain HCV cell-to-cell spread, served as a control. Specifically, 293T/miR-22 cells with or without ApoE or Huh-7.5 cells were transfected with Jc1 and served as “donor” cells that were cocultured under agarose overlay and in the presence of a neutralizing antibody (AR4A) (50) with Huh-7.5 “recipient” cells that had been transduced to express a tagRFP-NLS-IPS reporter protein (58). This reporter consists of a red fluorescent protein with nuclear localization signal that is fused to the C-terminal domain of beta interferon promoter stimulator 1 protein (IPS-1) including the NS3-4A cleavage site (63). Consequently, expression of HCV N3-4A causes cleavage of tagRFP-NLS-IPS and subsequent relocalization to the nucleus of tagRFP-NLS, which is a simple fluorescent marker for HCV infection (63). Interestingly, under these conditions we observed tagRFP-NLS nuclear staining in Huh-7.5 recipient cells cocultured with Huh-7.5 and also 293T/miR-122/ApoE donor cells but not when recipient cells were cocultured with 293T/miR-122 cells lacking ApoE expression (Fig. 9A). Quantification of the efficiency of cell-to-cell transmission did not show pronounced differences in the efficiency of cell-to-cell spread between transfected 293T/miR-122/ApoE and Huh7.5 cells (Fig. 9B). Therefore, these results suggest that HCV cell-to-cell spread depends on the presence of ApoE in the virus-producing cells.
FIG 9.
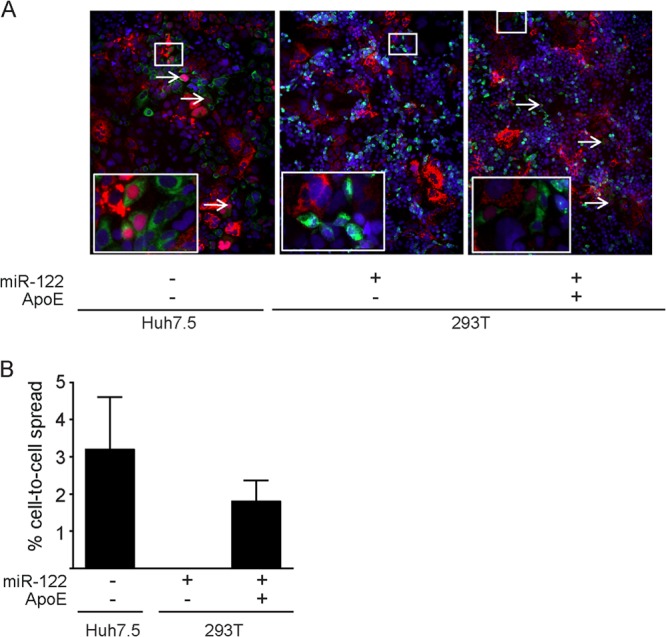
ApoE is required for HCV cell-to-cell transmission. 293T cells expressing miR-122 and ApoE or the empty vector and Huh-7.5 cells were transfected with Jc1 RNA. After electroporation, the cells were cocultured with Huh-7.5 cells harboring an RFP reporter construct that translocates into the nucleus upon cleavage by the HCV protease NS3-4A. Cell-free infection was blocked by agarose overlay and by addition of 20 mg/ml AR4A antibody. (A) After 72 h, cells were fixed with PFA, stained for NS5A, and analyzed by fluorescence microscopy. Representative images are shown. The inset is an enlarged version of a part of the picture. White arrows indicate cells where the RFP reporter translocated to the nucleus. (B) Cell-to-cell transmission of HCV was quantified by counting the total number of cells and the percentage of cells in which the RFP reporter translocated to the nucleus. Shown are averages from 3 experiments. In each experiment, between 1,000 and 2,000 cells were analyzed.
DISCUSSION
Chronic HCV infection is associated with liver disease and numerous extrahepatic manifestations. If these consequences are caused directly via interaction of HCV with extrahepatic tissues or indirectly due to deteriorating liver function is poorly understood. At the same time, host determinants of HCV tissue tropism are incompletely defined. In this study, we provide evidence that ApoE is the only cell type-specific host factor essential for production of infectious HCV particles. Moreover, our results suggest that ApoE functions during a late step of virus production subsequent to envelopment of viral capsids. Finally, we highlight that the function of ApoE during virus production is essential for viral cell-to-cell transmission. These findings imply that ApoE, which is expressed in a tissue-specific fashion, likely codetermines HCV tropism.
These conclusions, which have several interesting implications, are based on the following observations. First, ectopic expression of ApoE in the human cell lines HeLa, 293, and 293T rescued production of infectious HCV particles. Since 293T and HeLa cells are derived from different patients and tissues (kidney and cervix, respectively), these results suggest that other cellular assembly cofactors are broadly expressed. This notion is further supported by our observation that Caco-2 cells, a human colon carcinoma cell line that expressed endogenous ApoE, sustained assembly of infectious HCVTCP particles (Fig. 2). Given these findings, it is likely that human cells, provided they express ApoE, should be competent to produce infectious HCV progeny. In fact, Long et al. recently reported that infectious HCVTCP were produced in a mouse hepatocarcinoma cell line when human (or mouse) ApoE was introduced (39). Furthermore, we recently showed that in mouse hepatocyte-derived cell lines, ApoE also rescues production of HCV particles from a full-length genome (23). These findings confirm that with the exception of ApoE, HCV assembly cofactors are broadly expressed and, moreover, conserved between humans and mice.
Furthermore, our findings suggest that within infected humans, HCV should be able produce infectious progeny only from those cells and tissues that express ApoE. Considering that ApoE mRNA, for instance, is expressed in human liver, colon, and brain (based on the Tissue-Specific Gene Expression and Regulation database [TiGER]; http://bioinfo.wilmer.jhu.edu/tiger/), in principle these tissues may contribute to viremia. However, clearly these extrahepatic, ApoE-expressing tissues will produce virus only if they are permissive for HCV cell entry and at least low-level virus replication. Notably, Ploss and colleagues recently reported that CD81, SCARB1, CLDN1, and OCLN are the minimal cell type-specific host factors required to render nonliver cells as well as nonhuman cells permissive to HCV cell entry (7). Moreover, miR-122, a microRNA expressed predominantly in liver cells, has emerged as an important cell type-specific enhancer of translation/replication for HCV (22, 24). Collectively, these results indicate that those human cells that express CD81, SCARB1, CLDN1 (or CLDN6/9 which can substitute for CLDN1 [10, 11]), OCLN, miR-122, and ApoE should in principle be able to sustain the entire HCV replication cycle. In fact, while this paper was in preparation, Da Costa et al. reported that 293T cells engineered to express all the aforementioned factors were susceptible to HCV and produced viral progeny (64). Presently, it is unclear if any extrahepatic cells or tissues do express sufficient levels of these host factors to significantly contribute to viremia. Nevertheless, according to the TiGER expression database, human colon tissue, for instance, expresses all cell type-specific entry factors and ApoE. Moreover, we and others have observed low-level but measurable HCV replication in the absence of or with very little endogenous miR-122 expression. Clearly, more work involving primary cells and tissue samples is needed to elucidate whether extrahepatic tissues are relevant HCV replication sites or viral reservoirs. In principle, such sites may be relevant not only for extrahepatic disease manifestations but also for seeding reinfection after liver transplantation and possibly for initiation of viral relapse after antiviral therapy. With regard to the latter, a very low level of viral replication which could prevent immune detection and clearance and/or a lower level of drug exposure could increase chances of viral relapse from such extrahepatic tissues.
HCV has long been known to directly associate with human lipoproteins (27), and it has been postulated that this interaction may facilitate escape from neutralizing antibodies, and homing to liver cells by influencing viral interactions with cell entry factors. At least for virus-associated ApoE and -C1, functional relevance during HCV cell entry and membrane fusion has been reported (34, 65). Whether other lipoproteins also shape HCV particle properties and modulate cell entry as well as evasion from antibodies, however, is not resolved. In this work we used a number of functional assays to compare the properties of HCV particles released from Huh-7.5 cells and from 293T cells. Importantly, compared to Huh-7.5 cells, 293T cells express very little ApoB, ApoC1, and MTTP mRNA. Moreover, ApoB protein expression and secretion was readily detected in Huh-7.5 cells but was absent in 293T cells. Therefore, these cell types clearly provide a different environment for the interaction of HCV with lipoproteins. However, despite this, we noted comparable density profiles for Huh-7.5- and 293T-derived viruses. Moreover, we did not observe differences with regard to neutralization by patient-derived anti-HCV antibodies, as well as receptor-specific antibodies and a small-molecule inhibitor against SCARB1. While these findings suggest that expression of MTTP, ApoC1, and ApoB during HCV assembly is irrelevant for these tested viral functions, other interpretations are certainly possible. For instance, it is has been questioned that the host cell environment of Huh-7.5 cells fully recapitulates all aspects of lipoprotein secretion and in turn HCV association with lipoproteins (66). Moreover, Merz et al. were unable to detect ApoB within highly purified preparations of Huh-7.5-derived HCVcc particles (25), whereas serum-derived HCV is associated with ApoB (26, 27, 67). Thus, it is conceivable that Huh-7.5 cells, despite ApoC1, ApoB, ApoE, and MTTP expression, may not permit proper association of lipoproteins (e.g., ApoB) with released HCV particles. Consequently, also Huh-7.5-derived particles may only be decorated with a suboptimal lipoprotein coat. In this scenario, Huh-7.5 cells would be not well suited to study the relevance of additional HCV-associated lipoproteins. Of note, Lindenbach et al. observed that HCV particles derived from sera of humanized mice, which produce HCV from the engrafted primary human liver cells, display lower buoyant density and higher infectivity than Huh-7.5-derived viruses (68). In parallel, Podevin et al. observed that HCVcc particles collected from primary human hepatocytes in vitro display lower density than Huh-7.5-derived viruses (69). These two reports further support the notion that HCV particles originating from primary human hepatocytes may have different functional properties, possibly due to differential lipoprotein association, than those from Huh-7.5 cells. Notably, we observed a significant difference regarding the specific infectivity of 293T-derived and Huh-7.5 derived HCV particles, with the former being ca. 5-fold less infectious. Therefore, future work should explore and compare the compositions and the functional properties of HCV from 293T, Huh-7, and primary cells to find out which viral properties account for these differences. Moreover, the newly emerging HCV cell culture systems based on stem cell-derived hepatocyte-like cells may open novel opportunities to dissect the function of lipoproteins in the HCV replication cycle (70–72). In any case, the nonhepatic cell lines described in this study should be a useful model to reconstitute individual host factors involved in lipoprotein secretion and function to dissect their relevance for HCV.
Finally, we used our system, which permits conditional production of HCV upon expression of ApoE, to dissect the relevance of this host factor for the HCV assembly process. Our results reveal that ApoE is necessary for efficient release of HCV core protein from HCV-replicating 293T cells. Moreover, in the absence of ApoE, no infectious secreted particles were produced. The individual foci that were observed in the lysates of 293T/miR-122 cells in two of six experiments could be a low-level contamination of these cells. These findings indicate that lack of ApoE does not lead to accumulation of fully infectious intracellular particles or release of noninfectious particles. In contrast, an HCV assembly step prior to generation and secretion of infectious virus progeny is arrested. Interestingly, we observed trafficking of HCV core protein to lipid droplets in 293T cells lacking ApoE expression, confirming that core protein is deposited at this organelle independently of ApoE. Analysis of HCV capsid envelopment by protease K protection assay (51, 52) did not reveal gross differences between 293T cells with or without ApoE. However, compared with Huh-7.5 cells, capsid envelopment was slightly reduced. This finding may explain why virus titers released from 293T cells are lower than those from Huh-7.5 cells. Since overexpression of MTTP and supplementation of 293T cells with exogenous oleic acid did not stimulate virus production, we do not think that limited abundance or size of lipid droplets or absence of MTTP is responsible for this. Since absence of ApoE did not further decrease capsid envelopment, we conclude that ApoE functions in a late assembly step after HCV capsids have acquired a lipid envelope. This conclusion is supported by our observation that a defect in HCV capsid envelopment, as exerted by partial deletion of p7 (51), clearly further reduced the level of protease K-resistant and thus enveloped core protein, highlighting that our assay was able to detect defective capsid envelopment. More work is needed to find out how ApoE acts on enveloped capsids to render them infectious and to facilitate their secretion from infected cells. It is possible that ApoE acts on glycoprotein folding, which may be needed to assemble functional glycoprotein complexes and to permit secretion of enveloped particles. Moreover, given that ApoE is naturally loaded from intraluminal lipid droplets onto primordial lipoprotein complexes, it is possible that premature enveloped HCV capsids acquire ApoE during budding or their passage through the secretory compartment. Since absence of ApoE did ablate HCV cell-to-cell spread, we postulate that such late assembly steps are also essential for rendering HCV competent for this route of virus transmission. The novel infectious full-length HCV genomes with tetracysteine-tagged core protein (73, 74) could be an elegant system to track these late ApoE-dependent HCV assembly steps directly in living cells. Combined with the assembly-competent novel nonliver cell lines described in this report, these models may in the future reveal unique facets of the interaction between HCV, lipoproteins, and host cells.
ACKNOWLEDGMENTS
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 900, project A6) and by a grant from the Helmholtz Association (SO-024) to T.P. K.H. was supported by a stipend from the Hannover Biomedical Research School and the Centre for Infection Biology funded by the Excellence Initiative of the German Government.
We are very grateful to Takaji Wakita for the gift of the JFH1 isolate, to Charles Rice for Huh-7.5 cells, the 9E10 antibody, and the pTRIP-tagRFP-NLS-IPS reporter construct, to Ralf Bartenschlager for the plasmid encoding core to NS2, to Flossie Wong-Staal for ITX5061 and ITX7874, to Mansun Law for the AR4A antibody, and to Matthew Evans for the miR122 expression construct. We also thank all members of the Institute for Experimental Virology at Twincore for helpful comments and discussions of this work.
Twincore is a joint venture of Hannover Medical School and the Helmholtz-Centre for Infection Research.
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115. 10.1111/j.1469-0691.2010.03432.x [DOI] [PubMed] [Google Scholar]
- 2.Delang L, Neyts J, Vliegen I, Abrignani S, Neddermann P, De Francesco R. 2013. Hepatitis C virus-specific directly acting antiviral drugs. Curr. Top. Microbiol. Immunol. 369:289–320. 10.1007/978-3-642-27340-7_12 [DOI] [PubMed] [Google Scholar]
- 3.Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–S46. 10.1002/hep.1840360706 [DOI] [PubMed] [Google Scholar]
- 4.Yamane D, McGivern DR, Masaki T, Lemon SM. 2013. Liver injury and disease pathogenesis in chronic hepatitis C. Curr. Top. Microbiol. Immunol. 369:263–288. 10.1007/978-3-642-27340-7_11 [DOI] [PubMed] [Google Scholar]
- 5.Weissenborn K, Tryc AB, Heeren M, Worthmann H, Pflugrad H, Berding G, Bokemeyer M, Tillmann HL, Goldbecker A. 2009. Hepatitis C virus infection and the brain. Metab. Brain Dis. 24:197–210. 10.1007/s11011-008-9130-5 [DOI] [PubMed] [Google Scholar]
- 6.Zignego AL, Giannini C, Monti M, Gragnani L. 2007. Hepatitis C virus lymphotropism: lessons from a decade of studies. Dig. Liver Dis. 39(Suppl 1):S38–S45. 10.1016/S1590-8658(07)80009-0 [DOI] [PubMed] [Google Scholar]
- 7.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeisel MB, Felmlee DJ, Baumert TF. 2013. Hepatitis C virus entry. Curr. Top. Microbiol. Immunol. 369:87–112. 10.1007/978-3-642-27340-7_4 [DOI] [PubMed] [Google Scholar]
- 9.Haid S, Grethe C, Dill MT, Heim M, Kaderali L, Pietschmann T. Isolate-dependent use of Claudins for cell entry by hepatitis C virus. Hepatology, in press. 10.1002/hep.26567 [DOI] [PubMed] [Google Scholar]
- 10.Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL, Dragic T. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 82:3555–3560. 10.1128/JVI.01977-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 81:12465–12471. 10.1128/JVI.01457-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, von Weizsacker F, Blum HE, Baumert TF. 2003. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278:41003–41012. 10.1074/jbc.M302267200 [DOI] [PubMed] [Google Scholar]
- 13.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80:5308–5320. 10.1128/JVI.02460-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. U. S. A. 110:10777–10782. 10.1073/pnas.1301764110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17:589–595. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sainz B, Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285. 10.1038/nm.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. U. S. A. 96:12766–12771. 10.1073/pnas.96.22.12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J. 2012. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55:998–1007. 10.1002/hep.25501 [DOI] [PubMed] [Google Scholar]
- 19.Owen DM, Huang H, Ye J, Gale M., Jr 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. 10.1016/j.virol.2009.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinmann E, Pietschmann T. 2013. Cell culture systems for hepatitis C virus. Curr. Top. Microbiol. Immunol. 369:17–48. 10.1007/978-3-642-27340-7_2 [DOI] [PubMed] [Google Scholar]
- 21.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. 10.1126/science.1113329 [DOI] [PubMed] [Google Scholar]
- 22.Chang J, Guo JT, Jiang D, Guo H, Taylor JM, Block TM. 2008. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J. Virol. 82:8215–8223. 10.1128/JVI.02575-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frentzen A, Kusuma A, Guerlevik E, Hueging K, Knocke S, Ginkel C, Brown RJ, Heim M, Dill MT, Kroger A, Kalinke U, Kaderali L, Kuehnel F, Pietschmann T. Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology, in press. 10.1002/hep.26626 [DOI] [PubMed] [Google Scholar]
- 24.Lin LT, Noyce RS, Pham TN, Wilson JA, Sisson GR, Michalak TI, Mossman KL, Richardson CD. 2010. Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J. Virol. 84:9170–9180. 10.1128/JVI.00559-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286:3018–3032. 10.1074/jbc.M110.175018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 80:2418–2428. 10.1128/JVI.80.5.2418-2428.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomssen R, Bonk S, Propfe C, Heermann KH, Kochel HG, Uy A. 1992. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. Berl. 181:293–300. 10.1007/BF00198849 [DOI] [PubMed] [Google Scholar]
- 28.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. U. S. A. 110:9505–9510. 10.1073/pnas.1307527110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kono Y, Hayashida K, Tanaka H, Ishibashi H, Harada M. 2003. High-density lipoprotein binding rate differs greatly between genotypes 1b and 2a/2b of hepatitis C virus. J. Med. Virol. 70:42–48. 10.1002/jmv.10372 [DOI] [PubMed] [Google Scholar]
- 30.Lindenbach BD. 2013. Virion assembly and release. Curr. Top. Microbiol. Immunol. 369:199–218. 10.1007/978-3-642-27340-7_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793. 10.1128/JVI.01091-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129. 10.1128/JVI.02053-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr., Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853. 10.1073/pnas.0700760104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meunier JC, Russell RS, Engle RE, Faulk KN, Purcell RH, Emerson SU. 2008. Apolipoprotein c1 association with hepatitis C virus. J. Virol. 82:9647–9656. 10.1128/JVI.00914-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. 2008. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 47:1437–1445. 10.1002/hep.22197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413. 10.1073/pnas.0504877103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. 10.1016/j.chom.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 39.Long G, Hiet MS, Windisch MP, Lee JY, Lohmann V, Bartenschlager R. 2011. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 141:1057–1066. 10.1053/j.gastro.2011.06.010 [DOI] [PubMed] [Google Scholar]
- 40.Narbus CM, Israelow B, Sourisseau M, Michta ML, Hopcraft SE, Zeiner GM, Evans MJ. 2011. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J. Virol. 85:12087–12092. 10.1128/JVI.05843-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034–7046. 10.1128/JVI.00118-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalajzic I, Stover ML, Liu P, Kalajzic Z, Rowe DW, Lichtler AC. 2001. Use of VSV-G pseudotyped retroviral vectors to target murine osteoprogenitor cells. Virology 284:37–45. 10.1006/viro.2001.0903 [DOI] [PubMed] [Google Scholar]
- 44.Pham HM, Arganaraz ER, Groschel B, Trono D, Lama J. 2004. Lentiviral vectors interfering with virus-induced CD4 down-modulation potently block human immunodeficiency virus type 1 replication in primary lymphocytes. J. Virol. 78:13072–13081. 10.1128/JVI.78.23.13072-13081.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haid S, Pietschmann T, Pecheur EI. 2009. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition and density of virus particles. J. Biol. Chem. 284:17657–17667. 10.1074/jbc.M109.014647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van den Hoff MJ, Labruyere WT, Moorman AF, Lamers WH. 1990. The osmolarity of the electroporation medium affects the transient expression of genes. Nucleic Acids Res. 18:6464. 10.1093/nar/18.21.6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vieyres G, Pietschmann T. 2013. Entry and replication of recombinant hepatitis C viruses in cell culture. Methods 59:233–248. 10.1016/j.ymeth.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 48.Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Archiv. Exp. Pathol. Pharmakol. 162:480–487. 10.1007/BF01863914 [DOI] [Google Scholar]
- 49.Spearman C. 1908. The method of “right and wrong cases” (“constant stimuli”) without Gauss's formulae. Br. J. Psychol. 2:227–242 [Google Scholar]
- 50.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 109:6205–6210. 10.1073/pnas.1114927109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 9:e1003355. 10.1371/journal.ppat.1003355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menzel N, Fischl W, Hueging K, Bankwitz D, Frentzen A, Haid S, Gentzsch J, Kaderali L, Bartenschlager R, Pietschmann T. 2012. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 8:e1002829. 10.1371/journal.ppat.1002829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3:e103. 10.1371/journal.ppat.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 55.Moradpour D, Englert C, Wakita T, Wands JR. 1996. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology 222:51–63. 10.1006/viro.1996.0397 [DOI] [PubMed] [Google Scholar]
- 56.Hope RG, McLauchlan J. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81:1913–1925 [DOI] [PubMed] [Google Scholar]
- 57.Ciesek S, Westhaus S, Wicht M, Wappler I, Henschen S, Sarrazin C, Hamdi N, Abdelaziz AI, Strassburg CP, Wedemeyer H, Manns MP, Pietschmann T, von Hahn T. 2011. Impact of intra- and interspecies variation of occludin on its function as coreceptor for authentic hepatitis C virus particles. J. Virol. 85:7613–7621. 10.1128/JVI.00212-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM. 2010. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 28:167–171. 10.1038/nbt.1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F. 2011. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 54:48–55. 10.1016/j.jhep.2010.06.024 [DOI] [PubMed] [Google Scholar]
- 60.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097. 10.1038/ncb1631 [DOI] [PubMed] [Google Scholar]
- 61.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. 10.1002/hep.21959 [DOI] [PubMed] [Google Scholar]
- 62.Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 90:48–58. 10.1099/vir.0.006700-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 64.Da Costa D, Turek M, Felmlee DJ, Girardi E, Pfeffer S, Long G, Bartenschlager R, Zeisel MB, Baumert TF. 2012. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J. Virol. 86:11919–11925. 10.1128/JVI.01066-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86:7256–7267. 10.1128/JVI.07222-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jammart B, Michelet M, Pecheur EI, Parent R, Bartosch B, Zoulim F, Durantel D. 2013. Very-low-density lipoprotein (VLDL)-producing and hepatitis C virus-replicating HepG2 cells secrete no more lipoviroparticles than VLDL-deficient Huh7.5 cells. J. Virol. 87:5065–5080. 10.1128/JVI.01405-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomssen R, Bonk S, Thiele A. 1993. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. Berl. 182:329–334 [DOI] [PubMed] [Google Scholar]
- 68.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U. S. A. 103:3805–3809. 10.1073/pnas.0511218103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Podevin P, Carpentier A, Pene V, Aoudjehane L, Carriere M, Zaidi S, Hernandez C, Calle V, Meritet JF, Scatton O, Dreux M, Cosset FL, Wakita T, Bartenschlager R, Demignot S, Conti F, Rosenberg AR, Calmus Y. 2010. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 139:1355–1364. 10.1053/j.gastro.2010.06.058 [DOI] [PubMed] [Google Scholar]
- 70.Roelandt P, Obeid S, Paeshuyse J, Vanhove J, Van Lommel A, Nahmias Y, Nevens F, Neyts J, Verfaillie CM. 2012. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J. Hepatol. 57:246–251. 10.1016/j.jhep.2012.03.030 [DOI] [PubMed] [Google Scholar]
- 71.Schwartz RE, Trehan K, Andrus L, Sheahan TP, Ploss A, Duncan SA, Rice CM, Bhatia SN. 2012. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A. 109:2544–2548. 10.1073/pnas.1121400109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, Tang H. 2012. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 8:e1002617. 10.1371/journal.ppat.1002617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8:e1002466. 10.1371/journal.ppat.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7:e1002302. 10.1371/journal.ppat.1002302 [DOI] [PMC free article] [PubMed] [Google Scholar]