

ABSTRACT
Human coronaviruses (HCoVs) are recognized respiratory pathogens with neuroinvasive and neurotropic properties in mice and humans. HCoV strain OC43 (HCoV-OC43) can infect and persist in human neural cells and activate neuroinflammatory and neurodegenerative mechanisms, suggesting that it could be involved in neurological disease of unknown etiology in humans. Moreover, we have shown that HCoV-OC43 is neurovirulent in susceptible mice, causing encephalitis, and that a viral mutant with a single point mutation in the viral surface spike (S) protein induces a paralytic disease that involves glutamate excitotoxicity in susceptible mice. Herein, we show that glutamate recycling via the glial transporter 1 protein transporter and glutamine synthetase are central to the dysregulation of glutamate homeostasis and development of motor dysfunctions and paralytic disease in HCoV-OC43-infected mice. Moreover, memantine, an N-methyl-d-aspartate receptor antagonist widely used in the treatment of neurological diseases in humans, improved clinical scores related to paralytic disease and motor disabilities by partially restoring the physiological neurofilament phosphorylation state in virus-infected mice. Interestingly, memantine attenuated mortality rates and body weight loss and reduced HCoV-OC43 replication in the central nervous system in a dose-dependent manner. This novel action of memantine on viral replication strongly suggests that it could be used as an antiviral agent to directly limit viral replication while improving neurological symptoms in various neurological diseases with a viral involvement.
IMPORTANCE
INTRODUCTION
Coronaviruses (CoVs) are enveloped RNA viruses recognized to induce respiratory, enteric, and neurological diseases in several animal species (1). Human coronaviruses (HCoVs) are pathogens responsible for upper and lower respiratory tract infections (2) and for the severe acute respiratory syndrome (SARS) epidemic (3). Over the years, HCoVs have also been associated with other serious human pathologies, such as development of pneumonia, myocarditis, and meningitis (4, 5), as well as, occasionally, acute disseminated encephalitis (6).
We have previously shown that HCoVs can infect and persist in human neural (neuronal and glial) cells and can activate glial cells to produce proinflammatory mediators (7–10). We have also shown that, like its murine counterpart, the mouse hepatitis virus (MHV), which causes a neurodegenerative and neuroinflammatory disease in susceptible mice and rats (1) and which is used to establish a viral animal model of multiple sclerosis (MS), HCoV strain OC43 (HCoV-OC43) has neuroinvasive properties in mice (11). Furthermore, as we have demonstrated that this strain of human coronavirus is neuroinvasive in humans (12), we hypothesized that HCoV-OC43 might be associated with neuroinflammatory and/or neurodegenerative human diseases.
Glutamate is a major excitatory neurotransmitter of the central nervous system (CNS) involved in several neurophysiological functions. A disruption of its homeostasis can lead to excitotoxicity, a pathological process by which neural cells may be damaged following an excessive stimulation of glutamate on its specific ionotropic receptors [the 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors] (13, 14). Glutamate is mainly synthesized from glutamine by two different isoforms of glutaminase, designated kidney-type glutaminase (KGA; also known as GLS1) and liver-type glutaminase (LGA; also known as GLS2) (15), and is stored in vesicles before being released into the synaptic cleft. In some pathologies, astrocytes and microglial cells can also synthesize and release glutamate by expressing glutaminase (16–18).
Overactivation of the AMPA and NMDA receptors may result in neural Ca2+ overload, which can mediate excitotoxicity by means of a cascade of events involving free radical production, mitochondrial dysfunction, and the activation of several enzymes, such as phospholipase A2, resulting in damage to cell membranes, the cytoskeleton, and DNA (19). Glutamate reuptake is necessary for the regulation of physiological extracellular glutamate concentrations and because it can potentially damage nerve cells. Glutamate homeostasis is mainly mediated by high-affinity sodium-dependent transporters, such as the glial transporter 1 (GLT-1) protein. In neurological diseases, disruption of GLT-1 expression was associated with an alteration in glutamate uptake (20). After reuptake, glutamate is converted to glutamine by the glial enzyme glutamine synthetase (GS) (21, 22), and glutamine is transported in nerve terminals and reconverted to glutamate (23).
NMDA receptors have been shown to play a significant physiological role by controlling synaptic plasticity and memory functions, and their overactivation by glutamate may lead to excitotoxicity because of their high permeability to calcium ions (24). Some high-affinity NMDA antagonists, such as dizocilpine (also designated (+)-MK-801 maleate), were shown to block excitotoxicity and cell death but also to induce severe side effects in humans, due to the blockade of normal neuronal function (25, 26). Memantine, a low-affinity voltage-dependent uncompetitive antagonist which blocks NMDA receptors only when they are overstimulated by glutamate (27), acts only under pathological conditions during prolonged activation of NMDA receptors without affecting normal neuronal NMDA receptor functions (28). Interestingly, glutamate excitotoxicity was reported to be involved in several human neurological diseases, such as Alzheimer's disease, MS, epilepsy, Huntington's disease, and Parkinson's disease (29, 30), and memantine was reported to have a beneficial effect in several of these human neuropathologies (31–34). Moreover, glutamate excitotoxicity could be involved after CNS infections by West Nile Virus (35), human immunodeficiency virus (HIV) (36), human herpesvirus 6 (HHV-6) (37), human T-lymphotropic virus type 1 (HTLV-1) (38), bornavirus (39), and Sindbis virus (40).
We have recently reported that mice infected with an HCoV-OC43 variant harboring one point mutation in its surface spike (S) glycoprotein (Y241H) (41, 42) suffered from glutamate excitotoxicity that induced a neuropathology characterized by an MS-like flaccid paralysis where motor dysfunctions and hind-limb paralysis were associated with dysregulation at the level of the AMPA receptors (42).
In the present study, we now demonstrate that NMDA receptors are also involved, further characterize the process that leads to a dysregulation of glutamate homeostasis during infection, and show that memantine improved clinical scores (CSs) related to paralytic disease and motor disabilities and reduced viral replication in the CNS of HCoV-OC43-infected mice in a dose-dependent manner.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were approved by the Institutional Animal Care and Use Ethics Committee (IACUC) of the Institut National de la Recherche Scientifique (INRS) and conformed to the Canadian Council on Animal Care (CCAC). Animal care and use protocol number 0902-01 was issued by the IACUC of INRS for the animal experiments described herein.
Viruses and cell lines.
The wild-type reference virus HCoV-OC43 (VR-759) was obtained in the 1980s from the American Type Culture Collection (ATCC). The recombinant virus HCoV-OC43 (rOC/ATCC) was generated using the full-length cDNA clone pBAC-OC43FL and displayed the same phenotypic properties as the wild-type virus, as previously described (43). This recombinant virus was used as the reference control virus for all experiments. Recombinant virus designated rOC/US241 results from the introduction of a single point mutation in the spike glycoprotein of HCoV-OC43, as previously described (42). The HRT-18 cell line (a gift from David Brian, University of Tennessee) was cultured in minimal essential medium alpha (MEM-alpha; Life Technologies) supplemented with 10% (vol/vol) fetal bovine serum (FBS; PAA GE Healthcare) and was used to produce viral stocks and in experiments with NMDA antagonists.
Primary cultures of mouse CNS cells.
Embryos at 11 to 13 days of gestation were removed from pregnant anesthetized BALB/c mice. The cortex and hippocampus of the embryonic pup brains were dissected and placed in Hanks balanced salt solution (HBSS) medium, without Ca2+ and Mg2+, supplemented with 1.0 mM sodium pyruvate and 10 mM HEPES buffer. The tissues were incubated in 5 ml of trypsin-EDTA (0.25%; Life Technologies) supplemented with 20 μg/ml of DNase I (0.1%; Sigma) for 20 min at 37°C. After digestion, the trypsin-DNase mix was removed from the tissue and enzyme was inactivated with HBSS medium supplemented with 10% (vol/vol) FBS for 2 min, and this medium was removed and replaced by fresh HBSS medium (without Ca2+ and Mg2+, supplemented with 1.0 mM sodium pyruvate and 10 mM HEPES buffer). Samples were then briefly centrifuged at 250 × g, and the supernatant was removed and replaced by 5 ml of HBSS. Tissues were gently pipetted up and down with a pipette with a 1-ml tip to dissociate the cells. Supernatants were then transferred into a 15-ml tube and centrifuged for 5 min at 1,000 × g. The pellet was resuspended in neurobasal medium (Invitrogen) supplemented with 0.5 mM GlutaMAX-I (Life Technologies) and B27 supplement (Life Technologies). Cells were then seeded at approximately 1 × 106/cm2 and grown on poly-d-lysine-treated glass coverslips in the same medium, which was replaced by fresh neurobasal medium with B27-GlutaMAX-I after 4 days in culture. The cultures were ready for infection after 10 days in culture.
Infection of human cell lines and primary murine CNS cell cultures.
HRT-18 cells and primary mouse CNS cell cultures were infected at a defined multiplicity of infection (MOI) or mock infected and then incubated at 33°C or 37°C, respectively, for 2 h (for virus adsorption), washed with phosphate-buffered saline (PBS), and incubated at 33°C with fresh MEM-alpha supplemented with 1% (vol/vol) FBS (for HRT-18 cells) or at 37°C with fresh neurobasal medium with B27-GlutaMAX-I (for primary murine CNS cell cultures) for different periods of time before processing at the indicated time points.
Endpoint reverse transcription-PCR (RT-PCR) for expression of NMDA-NR1 receptor subunit gene expression.
Total RNA was extracted from HRT-18 cells and primary mouse CNS cell cultures using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. The cDNA was synthesized by use of a SuperScript III first-strand synthesis Supermix kit (Life Technologies) according to the manufacturer's instructions and was subsequently quantified using an ND1000 spectrophotometer (NanoDrop). The primers for the NMDA-NR1 subunit of the NMDA receptor were analyzed via the BLAST database to ensure the specificity of binding. The forward primer used was 5′-CAAGTATGCGGATGGGGTGA-3′, and the reverse primer used was 5′-CAGTCTGGTGGACATCTGGTA-3′ (NMDA-NR1; GenBank accession number NM_000832.6). Two micrograms of cDNA sample was added to a PCR mixture of Accuprime Pfx reaction mix, MgSO4, and deoxynucleoside triphosphates (Life Technologies). PCR was subsequently run on samples for 35 cycles under the following conditions: denaturation at 95°C for 35 s, annealing at 55°C for 45 s, and extension at 68°C for 40 s.
Mice, survival curves, body weight variations, and clinical scores.
Female BALB/c mice (Jackson Laboratories) with a postnatal age of 22 days were inoculated by the intracerebral route with 102.5 50% tissue culture infective doses of recombinant virus, as previously described (42). Groups of 10 mice infected with each recombinant virus were observed, and survival, body weight variations, and clinical scores related to motor dysfunctions were evaluated on a daily basis over a period of 21 days postinfection (dpi). Mice infected with rOC/US241 were evaluated and scored according to a scale based on experimental allergic encephalitis (EAE) clinical score (CS) evaluation (0 to 1, normal with no clinical signs; 1.5 to 2, partial hind-limb paralysis with a walk close to ground level; 2.5 to 3.5, complete hind-limb paralysis; 4 to 5, moribund state and death).
NMDA receptor antagonists.
The specific uncompetitive NMDA receptor antagonists memantine [3,5-dimethyl-tricyclo(3.3.1.13,7)decan-1-amine hydrochloride] and dizocilpine [chemical name, (+)-MK-801 maleate; (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate] were obtained from R&D Systems-Tocris.
For animal treatments, memantine and dizocilpine were dissolved in PBS. To investigate the effect of treatment with memantine or dizocilpine, groups of 10 BALB/c mice infected with rOC/ATCC or rOC/US241 were treated intraperitoneally with defined doses (μg/g of body weight) of memantine or dizocilpine at 12 h postinfection and then daily for 3 weeks, except when stated otherwise, or only with PBS (vehicle) to normalize experimental stress conditions. To verify the noncytotoxic effect of different doses of memantine and dizocilpine, sham-infected mice received the same doses of these NMDA receptor antagonists.
For treatment of cell cultures after virus adsorption, HRT-18 cells or primary murine CNS cell cultures were washed with sterile PBS and incubated with fresh MEM-alpha supplemented with 1% (vol/vol) FBS (for HRT-18 cells) or at 37°C with fresh neurobasal medium with B27-GlutaMAX-I (for primary mouse CNS cell cultures) containing defined concentrations (μM) of memantine or dizocilpine for different periods of time before processing at the time points indicated below.
Infectious virus assay using an IPA.
Mouse brain and spinal cord tissues or cell culture supernatants were processed for the presence and quantification of infectious virus by an indirect immunoperoxidase assay (IPA) on HRT-18 cells, as previously described (44). Briefly, cells were incubated with a mouse primary antibody used to detect the S protein of HCoV-OC43. After three PBS washes, cells were incubated with a secondary horseradish peroxidase-conjugated goat anti-mouse immunoglobulin antibody (Kirkegaard & Perry Laboratories). Finally, immune complexes were detected by incubation with 0.025% (wt/vol) 3,3′-diaminobenzidine tetrahydrochloride (Bio-Rad) and 0.01% (vol/vol) hydrogen peroxide in PBS, and infectious virus titers were calculated by the Karber method, as previously described (44). Statistical analyses were conducted by one-way analysis of variance (ANOVA), followed by Tukey's post hoc test.
Extracellular glutamate concentration assay.
Extracellular glutamate concentrations were measured using a colorimetric (optical density at 450 nm [OD450]) glutamate assay kit (ab83389; Abcam) according to the manufacturer's instructions. Statistical analyses were conducted by one-way ANOVA, followed by Tukey's post hoc test.
Cell viability and LDH release assay.
The viability of the cell cultures was assayed by use of a lactate dehydrogenase (LDH) cytotoxicity assay kit (Roche) according to the manufacturer's instructions. It consisted of a colorimetric assay (OD490) for quantification of cell death and cell lysis, based on the measurement of the lactate dehydrogenase activity released from the cytosol of damaged cells into the supernatant. Statistical analyses were conducted by one-way ANOVA, followed by Tukey's post hoc test.
Immunohistochemistry/immunofluorescence.
For immunohistochemistry, groups of three BALB/c mice either sham infected or infected with recombinant virus and treated with memantine, dizocilpine, or vehicle were randomly selected and perfused with a solution of 4% (wt/vol) paraformaldehyde at 10 dpi, which corresponds to the time of the peak of viral replication in the spinal cord and the outcome of clinical scores related to paralytic disease. Lumbar segments from spinal cords were cryoprotected in 30% (wt/vol) sucrose, frozen at −20°C, and processed with a cryostat (Microm HM 525) for sets with section sizes of 15 μm. Spinal cord sections were first blocked with horse serum in 1× PBS for 1 h at room temperature. Axonal damage was investigated by assessing the heavy neurofilament (NF-H) phosphorylation state. Tissue sections were incubated with a mouse antinonphosphorylated neurofilament monoclonal antibody (MAb; SMI 311; 1/1,000; Covance) for 2 h at room temperature. Activation of the glutaminase enzyme was investigated with a polyclonal goat anti-phosphate-activated glutaminase antibody (KGA; 1/500; Santa Cruz Biotechnology) or with a polyclonal rabbit anti-phosphate-activated glutaminase GLS2 (GLS2; 1/250; ab113509; Abcam) overnight at 4°C. Recycling of glutamate by the GS enzyme was investigated with a monoclonal mouse anti-GS antibody (clone GS-6; 1/500; MAB302; Millipore) overnight at 4°C. Tissue sections were then washed with 1× PBS and incubated with a biotinylated secondary antimouse, antigoat, or antirabbit antibody before revealing with stain from an ABC Vectastain kit (Vector Laboratories), as previously described (45).
For immunofluorescence staining, primary murine CNS cell cultures were washed with sterile PBS and then fixed with 4% (wt/vol) paraformaldehyde for 30 min at room temperature. After washing, cells were permeabilized with 100% methanol at −20°C for 5 min and washed with PBS. Before staining, cells were preincubated with a blocking solution containing horse serum in PBS for 1 h at room temperature and then incubated with primary antibodies: a polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) antibody (1/1,000; Dako), a polyclonal goat anti-phosphate-activated glutaminase antibody (isoform KGA; 1/500; Santa Cruz Biotechnology), a polyclonal rabbit anti-phosphate-activated glutaminase antibody (isoform GLS2; 1/250; ab113509; Abcam), or a monoclonal mouse anti-glutamine synthetase antibody (clone GS-6; 1/500; MAB302; Millipore) overnight at 4°C. After several washes with PBS, cells were incubated in the dark for 2 h at room temperature with the secondary fluorescent antibodies Alexa Fluor 568 antigoat, antimouse, or antirabbit (1/1,000; Life Technologies) or Alexa Fluor 488 antirabbit or antimouse (1/1,000; Life Technologies). After final PBS washes, tissue sections were incubated for 5 min at room temperature with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/ml; Life Technologies) and then mounted with Immuno-Mount mounting medium (Fisher Scientific). Immunohistochemical and fluorescent staining were observed under a Nikon Eclipse E800 microscope with a QImaging Retiga-EXi Fast 1394 digital camera using Procapture system software.
Protein extraction and Western blot analysis.
Spinal cords from groups of three mice selected randomly were homogenized in radioimmunoprecipitation assay buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 1% [vol/vol] NP-40, 0.25% [wt/vol] sodium deoxycholate, 1 mM EDTA) supplemented with a protease cocktail inhibitor (P8340; Sigma). Lysates were cleared by centrifugation for 5 min at 4°C and 17,000 × g, and supernatants were aliquoted and stored at −80°C until further analysis. A bicinchoninic acid protein assay kit (Novagen) was used to determine protein concentrations, according to the manufacturer's protocol. Proteins (20 μg per sample) were separated on a 4 to 12% gradient gel (Novex NuPage; Life Technologies) and transferred to a polyvinylidene difluoride membrane (Immobilon-P transfer membrane; Millipore) with a Bio-Rad Trans-Blot semidry transfer cell apparatus. Membranes were blocked with Tris-buffered saline (TBS) buffer containing 1% (vol/vol) Tween (TBS-T) and 5% (wt/vol) nonfat milk overnight at 4°C, and then the membranes were incubated with a polyclonal rabbit anti-GFAP antibody (1/1,000; Dako), a monoclonal guinea pig anti-glutamate transporter antibody (GLT-1; 1/1,000; AB1783; Millipore), a monoclonal rat anti-Mac-2 antibody (1/100; ATCC; Cedarlane), a polyclonal goat anti-phosphate-activated glutaminase antibody (KGA; 1/1,000; Santa Cruz Biotechnology), a polyclonal rabbit anti-phosphate-activated glutaminase antibody (GLS2; 1/1,000; ab113509; Abcam), a monoclonal mouse anti-GS antibody (clone GS-6; 1/1,000; MAB302; Millipore), or a polyclonal rabbit anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (1/1,000; Santa Cruz Biotechnology) for 1 h at room temperature.
Membranes were washed three times with TBS-T and then incubated with antirabbit (1/1,000; GE Healthcare Lifesciences), antimouse (1/1,000; GE Healthcare Lifesciences), anti-guinea pig (1/1,000; Millipore), antirat (1/1,000; Kirkegaard & Perry Laboratories), or antigoat (1/1,000; R&D Systems) secondary antibodies coupled to horseradish peroxidase for 1 h at room temperature, and antibodies were detected by chemiluminescence using a Bio-Rad Immun-Star horseradish peroxidase substrate kit. Band detection and semiquantification were done using GeneSnap software from a Chemi Genius Syngene apparatus. ANOVA tests followed by Tukey's post hoc analysis were performed to determine the statistical significances in the differences in protein expression between different groups of mice using SPSS software (version 16.0).
RESULTS
Daily treatment of mice with NMDA receptor antagonists (memantine and dizocilpine) attenuates the motor dysfunctions and severe paralysis induced following infection rOC/US241.
We have recently reported that a viral variant with a single point mutation (Y241H) in the surface spike (S) glycoprotein of HCoV-OC43 (rOC/US241), acquired during viral persistence in human neural cells (41), led to the induction of motor dysfunctions and a paralytic disease in infected BALB/c mice (42). In order to characterize the involvement of NMDA receptors in rOC/US241-induced hind-limb paralysis, mice infected with rOC/US241 were treated with various doses of the NMDA receptor antagonist memantine or dizocilpine or with vehicle (PBS). The highest dose of dizocilpine that could be administered was 0.5 μg/g; above that dose, the drug was toxic to mice (catatonia, weight loss; data not shown). Both dizocilpine and memantine treatments attenuated clinical scores related to the motor dysfunctions and paralytic disease induced following rOC/US241 infection, with a more effective effect for memantine treatment than dizocilpine treatment (Fig. 1A and B). Indeed, fewer mice treated with memantine presented mild paralysis (CSs, 1.5 to 2), and they recovered more rapidly than dizocilpine-treated mice or control vehicle-treated mice. Moreover, daily memantine treatment attenuated (dose, up to 10 μg/g) and even abolished (dose, 30 μg/g) clinical scores related to the paralytic disease of mice infected with rOC/US241 in a dose-dependent manner (Fig. 1B).
FIG 1.

Daily memantine and dizocilpine treatment attenuates the motor dysfunctions and severe paralysis induced following infection of mice by S-mutant human coronavirus OC43. Groups of 10 BALB/c mice infected with rOC/US241 were treated daily by intraperitoneal injection of a defined dose of memantine or dizocilpine or of PBS (vehicle). Motor dysfunctions were evaluated and scored according to a scale based on experimental allergic encephalitis (EAE) clinical score (CS) evaluation (0 to 1, normal with no clinical signs; 1.5 to 2, partial hind-limb paralysis with a walk close to ground level; 2.5 to 3.5, complete hind-limb paralysis; 4 to 5, moribund state and death). (A) Daily treatment with 0.5 μg/g dizocilpine attenuated the clinical scores in mice infected with rOC/US241. Higher doses of dizocilpine had strong side effects on mice and increased the mortality rate, as illustrated in Fig. 2. (B) Daily memantine treatment attenuated (doses, up to 10 μg/g) and even abolished (dose, 30 μg/g) the clinical scores in a dose-dependent manner. Results are representative of three independent experiments.
Daily memantine treatment attenuates mortality rates and body weight loss of infected mice compared to those achieved by dizocilpine and vehicle (control) treatment.
To investigate the effect of treatment with memantine or dizocilpine on survival rates, groups of 10 BALB/c mice infected with reference wild-type OC43 coronavirus (rOC/ATCC) or S-mutant OC43 virus (rOC/US241) were treated intraperitoneally daily with defined doses of memantine or dizocilpine or with PBS (vehicle). Mice infected with rOC/ATCC or rOC/US241 and treated with PBS showed, respectively, 10% and 20% mortality. Daily memantine treatment attenuated (doses, up to 5 μg/g) and totally suppressed (doses, 10 μg/g and over) the mortality of mice infected with either virus in a dose-dependent manner (Fig. 2A) compared to that achieved with dizocilpine treatment, which did not attenuate mortality compared to that achieved with PBS (Fig. 2B). Moreover, a higher dose of dizocilpine (1 μg/g) by itself increased mortality in noninfected mice (Fig. 2B). We had verified beforehand that the treatment of sham-infected mice with various doses of memantine (3 to 30 μg/g) or dizocilpine (0.5 μg/g) was not toxic and did not affect the survival rate of mice (data not shown).
FIG 2.

Daily memantine treatment attenuates the mortality rates of infected mice compared to those achieved with dizocilpine and vehicle (control) treatment. Groups of 10 BALB/c mice infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) were treated daily with an intraperitoneal injection of a defined dose of memantine or dizocilpine or of PBS (vehicle). (A) Daily memantine treatment attenuated (doses, up to 5 μg/g) and totally suppressed (doses, 10 μg/g and above) the mortality rates for mice infected with either virus in a dose-dependent manner. (B) Daily dizocilpine treatment did not attenuate the mortality rates for mice infected with either virus. A higher dose of dizocilpine (1 μg/g) by itself increased the mortality rate. Results are representative of three independent experiments.
Mice were also investigated for weight variations because, as previously described (45), weight loss is a specific symptom of mice infected with HCoV-OC43. Mice infected with rOC/ATCC or rOC/US241 and treated with PBS showed a loss of body weight of about 10 to 15% at 10 days postinfection and recovered at between 12 and 22 days postinfection. Daily memantine treatment significantly attenuated (dose, 5 μg/g; P < 0.05 and P < 0.01) and even abolished (doses, 10 μg/g and greater) the weight loss symptoms of mice infected with either virus in a dose-dependent manner (Fig. 3A) compared to the effects of dizocilpine treatment, which did not attenuate the weight loss of mice infected with either virus (Fig. 3B). As mentioned above, we made sure that the treatment of sham-infected mice with various doses of memantine (3 to 30 μg/g) or dizocilpine (0.5 μg/g) had no effect and that it did not affect the relative weight of the mice (data not shown).
FIG 3.

Daily memantine treatment attenuates/abolishes the weight loss of infected mice compared to that achieved with dizocilpine and vehicle (control) treatments. Groups of 10 BALB/c mice infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) were intraperitoneally treated daily with a defined dose of memantine or dizocilpine or with PBS (vehicle). (A) Daily memantine treatment attenuated (doses, up to 5 μg/g) and abolished (dose, 10 μg/g and above) weight loss in mice infected with either virus in a dose-dependent manner. (B) Daily dizocilpine treatment did not attenuate weight loss in mice infected with either virus. *, P < 0.05 (Dukey's test) for comparison with infected mice treated with vehicle for a defined day postinfection; **, P < 0.01 (Dukey's test) for comparison with infected mice treated with vehicle for a defined day postinfection. Results are representative of three independent experiments.
Increase in extracellular glutamate levels and LDH release following infection of mouse primary CNS cell cultures.
In order to investigate whether HCoV-OC43 causes a dysregulation of glutamate homeostasis that may lead to neuronal loss, primary murine CNS cell cultures were infected with rOC/ATCC or rOC/US241 at various multiplicities of infection (MOIs) and treated with memantine (20 μM) or vehicle (medium). At 24 h postinfection, cell culture supernatants were assessed for extracellular glutamate and lactate dehydrogenase (LDH) release. Increasing MOIs of each virus led to a significant increase in extracellular glutamate levels following infection with rOC/ATCC (P < 0.05 for MOIs of 1 and 3) or rOC/US241 (P < 0.05 for MOIs of 0.1 and 1; P < 0.01 for an MOI of 3) compared to the level for mock-infected cells (Fig. 4A). Moreover, infection with rOC/US241 usually led to a more significant extracellular glutamate release (P < 0.05) than rOC/ATCC infection (Fig. 4A). Memantine treatment led to a significant decrease in the extracellular glutamate concentration following infection of primary cultures of mouse CNS cells with rOC/ATCC (P < 0.05 for an MOI of 1) and rOC/US241 (P < 0.05 for MOIs of 0.1 and 1) (Fig. 4A).
FIG 4.

Increase in extracellular glutamate levels and LDH release following infection of primary cultures of mouse CNS. Primary cultures of mouse CNS cells were infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) at different MOIs and treated with memantine (20 μM) or vehicle (medium). (A) Increasing MOIs of both viruses led to an increase in the extracellular glutamate level that was significantly decreased by memantine (MOIs, 0.1 and 1). (B) Increasing MOIs of both viruses also led to an increase in LDH release that was also significantly reduced by memantine (MOIs, 0.1 and 1). #, P < 0.05 (Dukey's test) for comparison to mock-infected cells; ##, P < 0.01 (Dukey's test) for comparison to mock-infected cells; &, P < 0.05 (Dukey's test) for comparison of rOC/ATCC- or rOC/US241-infected cells at a defined MOI; *, P < 0.05 (Dukey's test) for comparison of memantine- and vehicle-treated infected cells.
The viability of cell cultures was assayed via the LDH release assay. Increasing MOIs of each virus led to a significant increase in LDH release following infection with rOC/ATCC (P < 0.05 for an MOI of 1 and 3) or rOC/US241 (P < 0.05 for MOIs of 0.1, 1, and 3) compared to that for mock-infected cells (Fig. 4B). Infection of primary cultures of mouse CNS cells with rOC/US241 usually led to a more significant LDH release (P < 0.05) compared to that achieved with infection with rOC/ATCC (Fig. 4B). Memantine treatment led to a significant decrease in LDH release following infection of primary cultures of mouse CNS cells by both rOC/ATCC (P < 0.05 for an MOI of 1) and rOC/US241 (P < 0.05 for MOIs of 0.1 and 1) (Fig. 4B).
The increased extracellular glutamate level is not a result of an increase in expression of phosphate-activated glutaminase.
Given that an increase in the extracellular glutamate concentration could result from an increase in glutamate synthesis, groups of three BALB/c mice infected with rOC/ATCC or rOC/US241 and treated with memantine or vehicle were sacrificed and evaluated for phosphate-activated kidney-type glutaminase (KGA) or liver-type glutaminase (GLS2) in lumbar spinal cord grey matter (GM) segments at 10 dpi (the time of the peak of viral replication in the spinal cord and the outcome of clinical scores related to paralytic disease). Infection of mice with rOC/ATCC or rOC/US241 did not lead to changes in KGA or GLS2 expression in lumbar spinal cord GM (Fig. 5A and B). Western blot analysis of spinal cord proteins confirmed the histological findings. Infection of mice with rOC/ATCC or rOC/US241 did not lead to changes in expression of phosphate-activated KGA or GLS2, and memantine treatments did not affect glutaminase expression (Fig. 5C). To confirm these in vivo results, primary murine CNS cell cultures were infected with rOC/ATCC or rOC/US241 and treated with memantine or vehicle. Expression of KGA and GLS2 by immunofluorescence and immunohistochemistry revealed that it was restricted to neurons, with no changes in the level of expression of either enzyme at 24 h postinfection for both viruses (Fig. 5A and B). We had made sure beforehand that the treatment of sham-infected mice with 3 μg/g of memantine or primary murine CNS cell cultures with 20 μM memantine did not affect the basal level of expression of KGA and GLS2 compared to the level obtained after vehicle (control) treatment (data not shown).
FIG 5.

The increase in extracellular glutamate levels is not a result of an increase in expression of phosphate-activated glutaminase. Phosphate-activated kidney-type glutaminase (KGA) or liver-type glutaminase (GLS2) was detected in lumbar spinal cord GM segments in infected mice treated with memantine or vehicle at 10 dpi (black arrows). Primary cultures of murine CNS cells were infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) and treated with memantine (20 μM) or vehicle (medium). Immunofluorescence revealed no changes in KGA (A) or GLS2 (B) activation at 24 h postinfection in lumbar spinal cord GM in primary murine CNS cell cultures following infection by either virus. MAP2, microtubule-associated protein 2. Magnifications, ×400. (C) Semiquantitative Western blot analysis of spinal cord proteins confirmed that activation of KGA and GLS2 was not modulated and that the levels of astrogliosis (a significant increase in GFAP expression) and microglial cell activation (an increase in Mac-2 expression) compared to those in sham-infected mice occurred. Memantine treatment significantly reduced Mac-2 expression in mice infected with rOC/US241 compared to that in rOC/ATCC-treated mice. Data are represented as the mean ± SEM (n = 3). ###, P < 0.001 (Dukey's test) for comparison to sham-infected mice; ##, P < 0.01 (Dukey's test) for comparison to sham-infected mice; #, P < 0.05 (Dukey's test) for comparison to sham-infected mice; *, P < 0.05 (Dukey's test) for comparison of groups of memantine treated- or nontreated infected mice. 1, sham infection plus vehicle treatment; 2, rOC/ATCC infection plus vehicle treatment; 3, rOC/ATCC infection plus treatment with 3 μg/g memantine; 4, rOC/US241 infection plus vehicle treatment; 5, rOC/US241 infection plus treatment with 3 μg/g memantine. Results are representative of those from three independent experiments.
Astrocytes and microglial cells may contribute to an excess of glutamate release by expressing glutaminase. Mice infected with either virus showed significant GFAP expression (P < 0.001) compared to that for sham-infected mice, as previously shown (42), and memantine treatment did not attenuate this astrocytic activation (Fig. 5C). However, mice infected with either virus showed significant (P < 0.01 and P < 0.05) microglial cell activation (as monitored by Mac-2 expression) compared to that for sham-infected mice, which was increased significantly (P < 0.05) more following infection with rOC/US241 than following infection with rOC/ATCC, as previously shown (42). Memantine significantly (P < 0.05) attenuated this microglial cell activation in mice infected with rOC/US241 (Fig. 5C).
Expression of GS and GLT-1 is downregulated in mice infected with rOC/US241 and partially restored following treatment with memantine.
Groups of three BALB/c mice infected with rOC/ATCC or rOC/US241 treated with memantine or vehicle were sacrificed and evaluated by immunohistochemistry for GS and glutamate transporter 1 (GLT-1) of lumbar spinal cord GM segments at 10 dpi. GS staining was downregulated in the GM of mice infected with rOC/US241 (Fig. 6A). To confirm these in vivo results, primary cultures of murine CNS cells were infected with rOC/ATCC or rOC/US241 (MOI = 1), treated with memantine (20 μM) or vehicle (medium), and assessed for GS expression by immunofluorescence at 24 h postinfection. GS was exclusively expressed in astrocytes (GFAP marker) and was dramatically downregulated in GM and in infected murine CNS cell cultures infected with rOC/US241, and memantine treatment led to an increase in GS expression (Fig. 6A). Western blot analysis of spinal cord proteins confirmed the histological findings. Infection of mice with rOC/US241 led to a significant (P < 0.01 and P < 0.01) downregulation of GS expression compared to that in sham-infected mice or rOC/ATCC-infected mice treated with vehicle. Memantine treatment led to a significant (P < 0.01) increase in GS expression (Fig. 6B). Moreover, infection of mice with rOC/US241 led to a significant (P < 0.01 and P < 0.01) downregulation of GLT-1 expression compared to that in sham-infected mice or rOC/ATCC-infected mice treated with vehicle, as previously shown (42). Memantine treatment led to a significant (P < 0.01) increase in GLT-1 expression (Fig. 6B). We had made sure beforehand that the treatment of sham-infected mice with 3 μg/g of memantine or primary murine CNS cell cultures with 20 μM memantine did not affect the basal level of expression of GS and GLT-1 compared to the level obtained after the control treatment (data not shown).
FIG 6.

Expression of GS and GLT-1 is downregulated in mice infected with S-mutant human coronavirus OC43 and partially restored following treatment with memantine. GS was evaluated by immunohistochemistry in lumbar spinal cord GM segments of mice infected with rOC/ATCC or rOC/US241 and treated with memantine or vehicle at 10 dpi (black arrows) and by immunofluorescence at 24 h postinfection in primary cultures of murine CNS cells infected and treated with memantine (20 μM) or vehicle (medium). (A) GS expression was dramatically downregulated in GM and in primary cultures of murine CNS cells infected with the S-mutant virus (rOC/US241), and memantine treatment partially restored GS expression. GFAP, glial fibrillary acidic protein. Magnifications, ×400. (B) Semiquantitative Western blot analysis of spinal cord proteins confirmed the histological findings. Infection of mice with rOC/US241 led to a significant downregulation of GS and GLT-1 expression that was significantly restored by memantine. Data are represented as the mean ± SEM (n = 3). ##, P < 0.01 (Dukey's test) for comparison to sham-infected mice; **, P < 0.01 (Dukey's test) for comparison of groups of memantine-treated or nontreated infected mice. 1, sham infection plus vehicle treatment; 2, rOC/ATCC infection plus vehicle treatment; 3, rOC/ATCC infection plus treatment with 3 μg/g memantine; 4, rOC/US241 infection plus vehicle treatment; 5, rOC/US241 infection plus treatment with 3 μg/g memantine. Results are representative of three independent experiments.
Daily treatment with memantine reduces neuronal dysfunction in mice infected with rOC/ATCC or rOC/US241.
Spinal cords from mice infected with rOC/US241 or rOC/ATCC and treated with memantine or vehicle were harvested at 10 dpi (the time of peak of viral replication in the spinal cord and the outcome of motor dysfunctions and severe paralysis) to evaluate whether a neuronal alteration associated with excitotoxicity was under way in infected mice. Neuronal dysregulation was investigated by evaluating the axonal neurofilament phosphorylation state. Infection of mice with rOC/ATCC or rOC/US241 resulted in an abnormal loss of soma nonphosphorylated NF-H immunoreactivity in the spinal cord GM compared to that in sham-infected mice. This modification in the phosphorylation state was more pronounced following infection with rOC/US2410 than following infection with rOC/ATCC (Fig. 7; SMI 311 GM, white arrows). On the other hand, the spinal cord white matter (WM) of mice infected with rOC/ATCC and rOC/US241 showed an abnormal presence of nonphosphorylated NF-H immunoreactivity, and axonal swelling in WM was more evident following rOC/US241 infection than following rOC/ATCC infection (Fig. 7; SMI 311 WM, black arrows).
FIG 7.
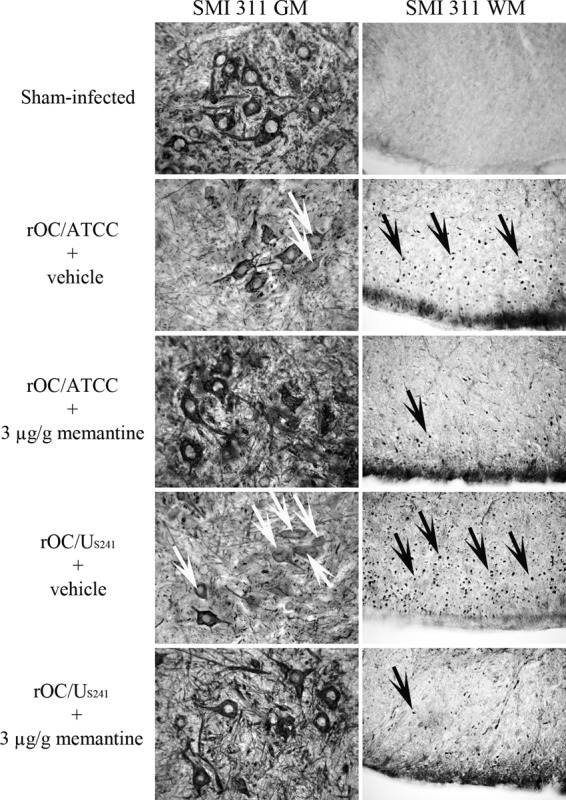
Daily treatment with a low dose of memantine reduces neuronal dysfunctions in infected mice. Groups of three BALB/c mice infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) and treated with vehicle or memantine (3 μg/g) were sacrificed and evaluated by immunohistochemistry for the levels of neurofilament phosphorylation in the lumbar spinal cord GM and WM segments at 10 dpi compared to those in control mice. Following infection, GM showed a loss of normal nonphosphorylated NF immunoreactivity compared to the GM of sham-infected mice. This loss was more pronounced following rOC/US241 infection than following rOC/ATCC infection (white arrows). The spinal cord WM of infected mice showed a higher level of abnormal axonal nonphosphorylated NF-H with abnormal axonal swelling, which was more pronounced following infection with rOC/US241 than following sham infection (black arrows) and infection with rOC/ATCC. Results are representative of two independent experiments with three mice per group. Magnification, ×400.
Higher doses of memantine treatment attenuate viral replication in the CNS of infected mice.
Because memantine treatment had a dose-response effect on various parameters, including mortality rates and body weight loss, and because memantine is a derivative of adamantane (an antiviral drug), we wished to evaluate whether the treatment may at some point affect viral replication in the CNS. Daily treatment over a period of 21 days with higher doses of memantine (10 μg/g and above) significantly reduced, in a dose-dependent manner, the replication of rOC/ATCC (P < 0.05 and P < 0.01) and rOC/US241 (P < 0.05, P < 0.01, and P < 0.001) in the brains and spinal cords of infected mice (Fig. 8A), indicative of an antiviral effect. However, dizocilpine treatment did not affect the replication of either virus (Fig. 8B).
FIG 8.

Daily treatment with high doses of memantine attenuates viral replication in the CNS of infected mice. Groups of three BALB/c mice infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) were sacrificed and evaluated for viral replication in brains and spinal cords every 2 days. (A) Memantine treatment reduced viral replication in the brains and spinal cords of infected mice in a dose-dependent manner (between 10 μg/g and 30 μg/g). (B) Dizocilpine treatment did not affect viral replication in the brains and spinal cords of infected mice. Data are represented as medians with ranges (n = 3). *, P < 0.05 (Dukey's test) for comparison to control mice (infected mice treated only with vehicle) for a defined day postinfection; **, P < 0.01 (Dukey's test) for comparison to control mice (infected mice treated only with vehicle) for a defined day postinfection; ***, P < 0.001 (Dukey's test) for comparison to control mice (infected mice treated only with vehicle) for a defined day postinfection. Each symbol on the graph represents a single mouse. L.O.D., limit of detection; TCID50, 50% tissue culture infective dose. Results are representative of those from two independent experiments.
Prophylactic transient treatment with memantine attenuates viral replication in the CNS of infected mice.
Having shown that the long-term use of high doses of memantine (daily treatment for 21 days) altered viral replication, we wished to evaluate whether a preventive treatment for a short period of time would also have the same effect. Groups of mice were treated with memantine (5 μg/g) 1 day prior to viral infection and daily for only 4 days postinfection; viral replication in brains and spinal cords was evaluated every 2 days. This preventive use of memantine for a shorter period of time significantly (P < 0.05) reduced viral replication in the brain and spinal cord of mice infected with rOC/ATCC or rOC/US241 (Fig. 9).
FIG 9.

Prophylactic transient treatment with memantine attenuates viral replication in the CNS of infected mice. Groups of three BALB/c mice were treated with memantine (5 μg/g) 1 day before viral infection and daily for 4 days postinfection. Mice infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) were sacrificed and evaluated for viral replication in brains and spinal cords every 2 days. The preventive memantine treatment reduced viral replication in the brains and spinal cords of infected mice. Data are represented as medians with ranges (n = 3). *, P < 0.05 (Dukey's test) for comparison to control mice (infected mice treated only with vehicle) for a defined day postinfection. Each symbol on the graph represents a single mouse. L.O.D., limit of detection; TCID50, 50% tissue culture infective dose. Results are representative of those from two independent experiments.
Memantine reduces viral replication in an NMDA blockade-independent manner.
In order to investigate the effect of memantine on HCoV-OC43 replication, primary cultures of mouse CNS cells (which express NMDA receptors, as verified by RT-PCR for the expression of the NMDA-NR1 receptor subunit; Fig. 10D) or cells of the HRT-18 cell line (which do not express NMDA receptors; Fig. 10D) were infected with rOC/ATCC or rOC/US241 and treated with increasing noncytotoxic doses (data not shown) of memantine or dizocilpine. Memantine treatment of cells significantly reduced the production of intra- and extracellular infectious virus in primary cultures of mouse CNS cells (P < 0.05 for 30 μM and P < 0.01 for 70 μM; Fig. 10A) and HRT-18 cells (P < 0.05 for 30 μM and P < 0.01 for 70 μM; Fig. 10B) compared to the levels of production by dizocilpine- or PBS-treated cells. To investigate whether memantine affected virus entry into cells, it was added to the cell medium either before, during, or after infection. Memantine treatment (70 μM) significantly reduced the intra- and extracellular production of infectious rOC/ATCC and rOC/US241, when administered after infection (P < 0.05), compared to that in the control (PBS-treated infected cells) or when memantine treatment was added before or during infection (Fig. 10C).
FIG 10.

High doses of memantine reduce viral replication in a NMDA-blockade-independent manner. Primary cultures of mouse CNS cells or the HRT-18 cell line were infected with reference wild-type virus (rOC/ATCC) or S-mutant virus (rOC/US241) and treated with increasing doses of memantine or dizocilpine. Memantine treatment significantly reduced intra- and extracellular infectious virus titers in primary cultures of mouse CNS cells (A) and in cells of the human epithelial HRT-18 cell line (B) compared to those achieved with dizocilpine and control (medium alone) treatment. h.p.i., hours postinfection. *, P < 0.05 (Dukey's test) for comparison of memantine- or dizocilpine-treated infected cells with PBS-treated infected cells; **, P < 0.01 (Dukey's test) for comparison of memantine- or dizocilpine-treated infected cells with PBS-treated infected cells. (C) Memantine treatment (70 μM) significantly reduced intra- and extracellular infectious virus titers when added after virus adsorption. B, before infection; D, during infection; A, after infection. *, P < 0.05 (Dukey's test) for comparison of memantine-treated infected cells and control (PBS-treated infected) cells. Results are representative of three independent experiments. (D) Expression of the NR1 subunit of the NMDA receptor was confirmed by RT-PCR in primary cultures of mouse CNS cells (lane 1; the 211-bp fragment size is indicated). No expression was detected in the HRT-18 cell line (lane 2). Lane MM, molecular size marker; lane −, negative control for PCR.
DISCUSSION
We have recently reported that a single point mutation in the surface spike (S) glycoprotein of human coronavirus OC43 (Y241H) modulates virus-induced neuropathology in a mouse model from an encephalitis to an MS-like paralytic disease related to glutamate excitotoxicity, with involvement of AMPA receptors (42). In the current study, we made use of two uncompetitive antagonists of NMDA receptors and demonstrate that these glutamate receptors are also involved in the neuropathology induced following infection of mice with HCoV-OC43. Moreover, we show that memantine protected mice from infection by having a dose-dependent effect on both neurological symptoms and viral replication.
Our results show that HCoV-OC43 infection causes a dysregulation of glutamate homeostasis that may lead to neuronal loss (neurodegeneration). The significant increase in the extracellular glutamate level and LDH release shown in Fig. 4 underlines the importance of glutamate dysregulation after infection, as shown in other virus infections (46–48). The fact that this increase is more significant following infection with rOC/US241 than following infection with rOC/ATCC strongly suggests that the viral S glycoprotein is an important factor in this dysregulation. The observation that memantine treatment led to a significant decrease in the extracellular release of glutamate and LDH at 24 h postinfection shows that glutamate homeostasis is altered during infection and that it appears to involve NMDA receptors, leading to excitotoxicity.
Glutamate homeostasis relies on two different mechanisms in order to prevent excitotoxicity. In order to decipher the possible mechanism leading to an increase in extracellular glutamate following infection by HCoV-OC43, we first investigated the synthesis of glutamate (Fig. 5). HCoV-OC43 infects neurons as primary targets of infection in the murine CNS (11) and in human neuron/astrocyte cocultures (49) and induces neuronal degeneration and eventual cell death. This could be associated with a stress that can disturb the phosphate-activated glutaminase enzyme expression responsible for glutamate synthesis, as was shown in HIV-1-infected microglia (18). Our results (Fig. 5) show that expression of the two glutaminase isoforms found in the CNS (50) is not modified in the spinal cord of mice or in primary cultures of mouse CNS cells infected with HCoV-OC43. Moreover, we show that expression of GFAP (an astrocyte marker) and Mac-2 (a marker of microglial cell activation) is upregulated following infection with HCoV-OC43 and that microglial activation is significantly increased following infection by the S mutant (rOC/US241) compared to that in the reference wild-type virus (rOC/ATCC). Some studies have shown that, in some circumstances, astrocytes and microglial cells may express glutaminase and release glutamate (16–18, 51, 52). However, our results clearly show that neither astrocytes nor microglial cells expressed any detectable phosphate-activated glutaminase (Fig. 5). This strongly suggests that the increase in extracellular glutamate release following infection by HCoV-OC43 is not a result of an increased expression of phosphate-activated glutaminase enzyme.
As previously reported, recycling of glutamate is also an important key factor for the physiological regulation of glutamate homeostasis to prevent excitotoxicity (53). Under physiological conditions, glutamate homeostasis is in large part regulated by glial glutamate transporter 1 (GLT-1), which is mainly expressed on astrocytes and which is responsible for up to 90% of the total glutamate clearance in adult CNS (54). As seen in Fig. 5, astrocytes are strongly activated following infection with either recombinant virus. However, expression of GLT-1 did not correlate with this activation associated with an increased number of astrocytes expressing high levels of GFAP, as previously shown (42). Indeed, the results presented in Fig. 6 show that GLT-1 expression was significantly downregulated following rOC/US241 infection compared to that in mice infected with rOC/ATCC or sham-infected mice. Interestingly, decreased expression of this transporter was reported in several neurological diseases, as well as in other viral models (35, 37, 55, 56). Of note, memantine treatment led to a major restoration of GLT-1 expression.
Glutamate homeostasis is also regulated by glutamine synthetase. Indeed, after reuptake, glutamate is converted to glutamine by the astroglial enzyme GS (21, 22). Glutamine is then transported into nerve terminals and reconverted to glutamate for an eventual new glutamate-glutamine cycle (23). As shown in Fig. 6, GS expression was significantly downregulated following rOC/US241 infection compared to that in mice infected with rOC/ATCC or sham-infected mice, and memantine treatment led to a major restoration of GS expression.
We have previously reported that infection of mice with HCoV-OC43 led to the release of proinflammatory cytokines (tumor necrosis factor alpha [TNF-α], interleukin-1 [IL-1], and IL-6) in the spinal cord of infected mice, with a significant increase in IL-6 in mice infected with an S mutant virus compared to that in mice infected with the reference wild-type virus (45). Proinflammatory cytokines such as IL-6 are known to downregulate glutamate transporter GLT-1 expression (56–58) and GS expression (59, 60). In the present study, we show that infection with the S-mutant virus rOC/US241 led to significant microglia/macrophage activation compared to what was observed with infection with the wild-type reference virus (rOC/ATCC) and that treatment with memantine significantly reduced microglial cell activation, as previously reported (61, 62).
We show that the increased microglial cell/macrophage activation correlated with the downregulation of expression of GLT-1 and GS (Fig. 5C and 6B). Our results suggest that memantine may prevent downregulation of GLT-1 and GS expression by downregulating microglial cell activation, which is responsible for the major release of some proinflammatory molecules, like IL-1, IL-6, and TNF-α, that could act on disruption of glutamate homeostasis by downregulating glutamate recycling (53, 63, 64).
Glutamate excitotoxicity may damage the cytoskeleton of axons and affect the axonal transport rate by disrupting the neurofilament phosphorylation state (65, 66). Signs of neuronal injury are monitored by evaluation for a change in the NF phosphorylation state, which is characterized by a loss of nonphosphorylated NF in soma and an increase in nonphosphorylated NF in axons (67). Mice infected with rOC/US241 or rOC/ATCC also showed an abnormal loss of nonphosphorylated NF-H in soma in the grey matter (GM), and this was more evident following rOC/US241 infection. Moreover, the spinal cord white matter (WM) of mice infected with rOC/US241 or rOC/ATCC presented abnormal axonal nonphosphorylated NF-H, which was also more evident following rOC/US241 infection, even leading to axonal beading and swelling (Fig. 7). It has already been reported that modifications in the phosphorylation state of neurofilaments contribute to motor neuron disease, as observed in amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS), as well as following infection with Theiler's murine encephalomyelitis virus (TMEV) in an animal model of MS (68, 69, 87). This abnormal disruption of the NF-H phosphorylation state in axons and cell bodies observed following infection could lead to neuronal dysfunction and disruption of axonal transport with perturbations in neuronal transmission, which was demonstrated to account for motor disabilities (70). Daily treatment with memantine (3 μg/g) significantly reduced the imbalance in NF-H phosphorylation and also improved motor dysfunctions, probably through both a direct effect on protection of neurons from glutamate overactivation on NMDA receptors (71) and an indirect effect by downregulating microglial cell activation, that result in the release of proinflammatory mediators or toxic molecules that may damage neurons (61, 62).
Memantine is an adamantane derivative. Some of these derivatives are used as drugs in human medicine and have antiviral properties. These include amantadine and rimantadine (with activity against influenza A virus [72]), tromantadine (with activity against herpes simplex virus [73]), and bananin (with activity against the SARS coronavirus [74]).
In order to explore the mode of action of memantine as an antiviral molecule, we made use of mouse primary CNS cell cultures (which express NMDA receptors) and of a human epithelial cell line (commonly used in the laboratory to amplify HCoV-OC43, which does not express NMDA receptors). The observation that memantine affects viral replication in both cell types strongly suggests that memantine acts as an antiviral drug without the involvement of NMDA receptors (Fig. 10A and B). Moreover, our observation that memantine shows a highly significant antiviral activity after primary infection of susceptible cells (Fig. 10C) indicates that it inhibits viral replication after virus attachment to the cell receptor, as was also shown for other adamantane derivatives. Indeed, amantadine and rimantadine inhibit the early steps of uncoating of influenza virus in endosomes (75, 76), tromantadine inhibits early steps in herpes simplex virus replication before macromolecular synthesis and a late event, such as assembly or release of virus (77), and bananin inhibits the helicase activities of SARS coronavirus (74). Memantine could act by inhibition of the ion channel activity of the HCoV-OC43 E protein, as amantadine and rimantadine do by binding the influenza virus M2 protein proton channel (75, 76), or it could inhibit the ATPase activity of the HCoV-OC43 helicase, as bananin does against the SARS coronavirus (74).
To our knowledge, our study shows the first evidence that memantine, widely used in humans to treat neurological diseases, shows antiviral properties against a human neuroinvasive and neurotropic virus. This novel action of memantine strongly suggests that it could be used as an antiviral agent in various neurological diseases with a viral involvement, such as herpes encephalitis or meningitis, or neurological diseases for which the etiology could involve viruses, such as multiple sclerosis (78), Alzheimer's disease (79), and Parkinson's disease (80).
Moreover, as shown in Fig. 9, transient treatment with memantine (5 μg/g) significantly (P < 0.05) attenuated viral replication in the CNS of infected mice, which strongly suggests that memantine could be used as a prophylactic agent for a short treatment in an attempt to control some viral outbreaks, as previously shown for other antiviral agents effective against influenza virus (81).
In summary, we show that memantine improved clinical scores related to paralytic disease and motor disabilities by partially restoring the physiological NF phosphorylation state in S-mutant-virus-infected mice. Moreover, memantine attenuated, in a dose-dependent manner, mortality rates and body weight loss and reduced viral replication in the central nervous system of HCoV-OC43-infected mice. Furthermore, glutamate recycling via the GLT-1 transporter and the GS enzyme, rather than glutamate synthesis, appears to be the key factor in glutamate homeostasis dysregulation that may lead to the motor dysfunctions and paralytic disease induced in mice following infection with HCoV-OC43. Even though we are aware of the potential limitations of studying a human virus in a mouse model and we are interpreting our results accordingly, we believe that this approach is relevant and important for predicting how a virus could interact with humans, as shown for several other human viruses used in mouse models (82–86). Our results also strongly suggest that memantine could be used as both a prophylactic and a therapeutic antiviral agent against neuroinvasive and neurotropic human viruses that cause viral encephalitis or meningitis or neurological diseases in which the etiology could involve viruses. Further studies are under way to investigate both the role of inflammation on the downregulation of glutamate recycling and the precise mechanism that triggers NMDA receptor-associated excitotoxicity, to identify the viral and cellular factors involved in the process following infection of mice by the human coronavirus OC43, and to understand how memantine plays such an important role in controlling both neurodegeneration and viral infection itself.
ACKNOWLEDGMENTS
This work was supported by grant no. MT-9203 from the Institute of Infection and Immunity (III) of the Canadian Institutes of Health Research (CIHR) to Pierre J. Talbot, who is the holder of the Tier-1 (Senior) Canada Research Chair in Neuroimmunovirology award. Elodie Brison gratefully acknowledges a doctoral studentship from the Multiple Sclerosis Society of Canada.
Footnotes
Published ahead of print 13 November 2013
REFERENCES
- 1.Buchmeier MJ, Lane TE. 1999. Viral-induced neurodegenerative disease. Curr. Opin. Microbiol. 2:398–402. 10.1016/S1369-5274(99)80070-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talbot PJ, Jacomy H, Desforges M. 2008. Pathogenesis of human coronaviruses other than severe acute respiratory syndrome coronavirus, p 313–324 In Perlman S, Gallagher T, Snilder EJ. (ed), The nidoviruses. ASM Press, Washington, DC [Google Scholar]
- 3.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. 10.1126/science.1085952 [DOI] [PubMed] [Google Scholar]
- 4.Riski H, Hovi T. 1980. Coronavirus infections of man associated with diseases other than the common cold. J. Med. Virol. 6:259–265. 10.1002/jmv.1890060309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forgie S, Marrie TJ. 2009. Healthcare-associated atypical pneumonia. Semin. Resp. Crit. Care 30:67–85. 10.1055/s-0028-1119811 [DOI] [PubMed] [Google Scholar]
- 6.Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H. 2004. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 113:e73–e76 [DOI] [PubMed] [Google Scholar]
- 7.Bonavia A, Arbour N, Yong VW, Talbot PJ. 1997. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J. Virol. 71:800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arbour N, Côté G, Lachance C, Tardieu M, Cashman NR, Talbot PJ. 1999. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J. Virol. 73:3338–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arbour N, Ekandé S, Côté G, Lachance C, Chagnon F, Tardieu M, Cashman NR, Talbot PJ. 1999. Persistent infection of human oligodendrocytic and neuroglial cell lines by human coronavirus 229E. J. Virol. 73:3326–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards JA, Denis F, Talbot PJ. 2000. Activation of glial cells by human coronavirus OC43 infection. J. Neuroimmunol. 108:73–81. 10.1016/S0165-5728(00)00266-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacomy H, Talbot PJ. 2003. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 315:20–33. 10.1016/S0042-6822(03)00323-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbour N, Day R, Newcombe J, Talbot PJ. 2000. Neuroinvasion by human respiratory coronaviruses. J. Virol. 74:8913–8921. 10.1128/JVI.74.19.8913-8921.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olney J. 1978. Neurotoxicity of excitatory amino acids, p 95–121 In McGeer EG, Olney JW, McGeer PL. (ed), Kainic acid as a tool in neurobiology. Raven Press, New York, NY [Google Scholar]
- 14.Mark LP, Prost RW, Ulmer JL, Smith MM, Daniels DL, Strottmann JM, Brown WD, Hacein-Bey L. 2001. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. AJNR Am. J. Neuroradiol. 22:1813–1824 [PMC free article] [PubMed] [Google Scholar]
- 15.Aledo JC, Gomez-Fabre PM, Olalla L, Marquez J. 2000. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm. Genome 11:1107–1110. 10.1007/s003350010190 [DOI] [PubMed] [Google Scholar]
- 16.Kvamme E, Nissen-Meyer LS, Roberg BA, Torgner IA. 2008. Novel form of phosphate activated glutaminase in cultured astrocytes and human neuroblastoma cells, PAG in brain pathology and localization in the mitochondria. Neurochem. Res. 33:1341–1345. 10.1007/s11064-008-9589-9 [DOI] [PubMed] [Google Scholar]
- 17.Shijie J, Takeuchi H, Yawata I, Harada Y, Sonobe Y, Doi Y, Liang J, Hua L, Yasuoka S, Zhou Y, Noda M, Kawanokuchi J, Mizuno T, Suzumura A. 2009. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J. Exp. Med. 217:87–92. 10.1620/tjem.217.87 [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Zhao L, Jia B, Wu L, Li Y, Curthoys N, Zheng JC. 2011. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J. Neurosci. 31:15195–15204. 10.1523/JNEUROSCI.2051-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sattler R, Tymianski M. 2000. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. (Berl.) 78:3–13. 10.1007/s001090000077 [DOI] [PubMed] [Google Scholar]
- 20.Tilleux S, Hermans E. 2007. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 85:2059–2070. 10.1002/jnr.21325 [DOI] [PubMed] [Google Scholar]
- 21.Norenberg MD. 1979. Distribution of glutamine synthetase in the rat central nervous system. J. Histochem. Cytochem. 27:756–762. 10.1177/27.3.39099 [DOI] [PubMed] [Google Scholar]
- 22.Norenberg MD, Martinez-Hernandez A. 1979. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 161:303–310. 10.1016/0006-8993(79)90071-4 [DOI] [PubMed] [Google Scholar]
- 23.Albrecht J, Sidoryk-Wegrzynowicz M, Zielinska M, Aschner M. 2010. Roles of glutamine in neurotransmission. Neuron Glia Biol. 6:263–276. 10.1017/S1740925X11000093 [DOI] [PubMed] [Google Scholar]
- 24.Arundine M, Tymianski M. 2003. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34:325–337. 10.1016/S0143-4160(03)00141-6 [DOI] [PubMed] [Google Scholar]
- 25.Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, Orgogozo JM, Whitehead J. 2000. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. GAIN International Investigators. Lancet 355:1949–1954. 10.1016/S0140-6736(00)02326-6 [DOI] [PubMed] [Google Scholar]
- 26.Kemp JA, McKernan RM. 2002. NMDA receptor pathways as drug targets. Nat. Neurosci. 5(Suppl):1039–1042. 10.1038/nn936 [DOI] [PubMed] [Google Scholar]
- 27.Parsons CG, Stoffler A, Danysz W. 2007. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—too little activation is bad, too much is even worse. Neuropharmacology 53:699–723. 10.1016/j.neuropharm.2007.07.013 [DOI] [PubMed] [Google Scholar]
- 28.Lipton SA. 2004. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 1:101–110. 10.1602/neurorx.1.1.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong XX, Wang Y, Qin ZH. 2009. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30:379–387. 10.1038/aps.2009.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. 2013. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 698:6–18. 10.1016/j.ejphar.2012.10.032 [DOI] [PubMed] [Google Scholar]
- 31.Wilkinson D. 2012. A review of the effects of memantine on clinical progression in Alzheimer's disease. Int. J. Geriatr. Psychiatry 27:769–776. 10.1002/gps.2788 [DOI] [PubMed] [Google Scholar]
- 32.Starck M, Albrecht H, Pollmann W, Dieterich M, Straube A. 2010. Acquired pendular nystagmus in multiple sclerosis: an examiner-blind cross-over treatment study of memantine and gabapentin. J. Neurol. 257:322–327. 10.1007/s00415-009-5309-x [DOI] [PubMed] [Google Scholar]
- 33.Emre M, Tsolaki M, Bonuccelli U, Destee A, Tolosa E, Kutzelnigg A, Ceballos-Baumann A, Zdravkovic S, Bladstrom A, Jones R. 2010. Memantine for patients with Parkinson's disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9:969–977. 10.1016/S1474-4422(10)70194-0 [DOI] [PubMed] [Google Scholar]
- 34.Anitha M, Nandhu MS, Anju TR, Jes P, Paulose CS. 2011. Targeting glutamate mediated excitotoxicity in Huntington's disease: neural progenitors and partial glutamate antagonist—memantine. Med. Hypotheses 76:138–140. 10.1016/j.mehy.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 35.Blakely PK, Kleinschmidt-DeMasters BK, Tyler KL, Irani DN. 2009. Disrupted glutamate transporter expression in the spinal cord with acute flaccid paralysis caused by West Nile virus infection. J. Neuropathol. Exp. Neurol. 68:1061–1072. 10.1097/NEN.0b013e3181b8ba14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cisneros IE, Ghorpade A. 2012. HIV-1, methamphetamine and astrocyte glutamate regulation: combined excitotoxic implications for neuro-AIDS. Curr. HIV Res. 10:392–406. 10.2174/157016212802138832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fotheringham J, Williams EL, Akhyani N, Jacobson S. 2008. Human herpesvirus 6 (HHV-6) induces dysregulation of glutamate uptake and transporter expression in astrocytes. J. Neuroimmune Pharmacol. 3:105–116. 10.1007/s11481-007-9084-0 [DOI] [PubMed] [Google Scholar]
- 38.Szymocha R, Akaoka H, Dutuit M, Malcus C, Didier-Bazes M, Belin MF, Giraudon P. 2000. Human T-cell lymphotropic virus type 1-infected T lymphocytes impair catabolism and uptake of glutamate by astrocytes via Tax-1 and tumor necrosis factor alpha. J. Virol. 74:6433–6441. 10.1128/JVI.74.14.6433-6441.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Billaud JN, Ly C, Phillips TR, de la Torre JC. 2000. Borna disease virus persistence causes inhibition of glutamate uptake by feline primary cortical astrocytes. J. Virol. 74:10438–10446. 10.1128/JVI.74.22.10438-10446.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darman J, Backovic S, Dike S, Maragakis NJ, Krishnan C, Rothstein JD, Irani DN, Kerr DA. 2004. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J. Neurosci. 24:7566–7575. 10.1523/JNEUROSCI.2002-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.St-Jean JR, Desforges M, Talbot PJ. 2006. Genetic evolution of human coronavirus OC43 in neural cell culture. Adv. Exp. Med. Biol. 581:499–502. 10.1007/978-0-387-33012-9_88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brison E, Jacomy H, Desforges M, Talbot PJ. 2011. Glutamate excitotoxicity is involved in the induction of paralysis in mice after infection by a human coronavirus with a single point mutation in its spike protein. J. Virol. 85:12464–12473. 10.1128/JVI.05576-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St-Jean JR, Desforges M, Almazan F, Jacomy H, Enjuanes L, Talbot PJ. 2006. Recovery of a neurovirulent human coronavirus OC43 from an infectious cDNA clone. J. Virol. 80:3670–3674. 10.1128/JVI.80.7.3670-3674.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert F, Jacomy H, Marceau G, Talbot PJ. 2008. Titration of human coronaviruses using an immunoperoxidase assay. Methods Mol. Biol. 454:93–102. 10.1007/978-1-59745-181-9_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacomy H, St-Jean JR, Brison E, Marceau G, Desforges M, Talbot PJ. 2010. Mutations in the spike glycoprotein of human coronavirus OC43 modulate disease in BALB/c mice from encephalitis to flaccid paralysis and demyelination. J. Neurovirol. 16:279–293. 10.3109/13550284.2010.497806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Power C, Moench T, Peeling J, Kong PA, Langelier T. 1997. Feline immunodeficiency virus causes increased glutamate levels and neuronal loss in brain. Neuroscience 77:1175–1185. 10.1016/S0306-4522(96)00531-3 [DOI] [PubMed] [Google Scholar]
- 47.Gupta S, Knight AG, Knapp PE, Hauser KF, Keller JN, Bruce-Keller AJ. 2010. HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci. Lett. 485:233–236. 10.1016/j.neulet.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen CJ, Ou YC, Chang CY, Pan HC, Liao SL, Chen SY, Raung SL, Lai CY. 2012. Glutamate released by Japanese encephalitis virus-infected microglia involves TNF-alpha signaling and contributes to neuronal death. Glia 60:487–501. 10.1002/glia.22282 [DOI] [PubMed] [Google Scholar]
- 49.Desforges M, Favreau DJ, Brison E, Desjardins J, Messen-Pinard M, Jacomy H, Talbot PJ. 2013. Human coronavirus: respiratory pathogens revisited as infectious neuroinvasive, neurotropic, and neurovirulent agents, p 93–121 In Singh SK, Ruzek D. (ed), Neuroviral infections: RNA viruses and retroviruses. CRC Press LLC, New York, NY [Google Scholar]
- 50.Kvamme E, Roberg B, Torgner IA. 2000. Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem. Res. 25:1407–1419. 10.1023/A:1007668801570 [DOI] [PubMed] [Google Scholar]
- 51.Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A. 1988. Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem. Res. 13:383–388 [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. 2006. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 281:21362–21368. 10.1074/jbc.M600504200 [DOI] [PubMed] [Google Scholar]
- 53.Danbolt NC. 2001. Glutamate uptake. Prog. Neurobiol. 65:1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- 54.Anderson CM, Swanson RA. 2000. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ. 2003. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312:60–73. 10.1016/S0042-6822(03)00181-8 [DOI] [PubMed] [Google Scholar]
- 56.Carmen J, Rothstein JD, Kerr DA. 2009. Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res. 1263:143–154. 10.1016/j.brainres.2009.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okada K, Yamashita U, Tsuji S. 2005. Modulation of Na(+)-dependent glutamate transporter of murine astrocytes by inflammatory mediators. J. UOEH 27:161–170 [DOI] [PubMed] [Google Scholar]
- 58.Prow NA, Irani DN. 2008. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J. Neurochem. 105:1276–1286. 10.1111/j.1471-4159.2008.05230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang TL, O'Banion MK. 1998. Interleukin-1 beta and tumor necrosis factor-alpha suppress dexamethasone induction of glutamine synthetase in primary mouse astrocytes. J. Neurochem. 71:1436–1442 [DOI] [PubMed] [Google Scholar]
- 60.Zou J, Wang YX, Dou FF, Lu HZ, Ma ZW, Lu PH, Xu XM. 2010. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem. Int. 56:577–584. 10.1016/j.neuint.2009.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeda K, Muramatsu M, Chikuma T, Kato T. 2009. Effect of memantine on the levels of neuropeptides and microglial cells in the brain regions of rats with neuropathic pain. J. Mol. Neurosci. 39:380–390. 10.1007/s12031-009-9224-5 [DOI] [PubMed] [Google Scholar]
- 62.Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, Rawls SM, Flood P, Hong JS, Lu RB. 2009. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34:2344–2357. 10.1038/npp.2009.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. 2000. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7:153–159. 10.1159/000026433 [DOI] [PubMed] [Google Scholar]
- 64.Liao SL, Chen CJ. 2001. Differential effects of cytokines and redox potential on glutamate uptake in rat cortical glial cultures. Neurosci. Lett. 299:113–116. 10.1016/S0304-3940(01)01499-9 [DOI] [PubMed] [Google Scholar]
- 65.Ackerley S, Grierson AJ, Brownlees J, Thornhill P, Anderton BH, Leigh PN, Shaw CE, Miller CCJ. 2000. Glutamate slows axonal transport of neurofilaments in transfected neurons. J. Cell Biol. 150:165–175. 10.1083/jcb.150.1.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller CCJ, Ackerley S, Brownlees J, Grierson AJ, Jacobsen NJO, Thornhill P. 2002. Axonal transport of neurofilaments in normal and disease states. Cell. Mol. Life Sci. 59:323–330. 10.1007/s00018-002-8425-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shea TB, Chan WK, Kushkuley J, Lee S. 2009. Organizational dynamics, functions, and pathobiological dysfunctions of neurofilaments. Results Probl. Cell Differ. 48:29–45. 10.1007/400_2009_8 [DOI] [PubMed] [Google Scholar]
- 68.Schirmer L, Antel JP, Bruck W, Stadelmann C. 2011. Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol. 21:428–440. 10.1111/j.1750-3639.2010.00466.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS. 2003. Axonal injury heralds virus-induced demyelination. Am. J. Pathol. 162:1259–1269. 10.1016/S0002-9440(10)63922-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perrot R, Berges R, Bocquet A, Eyer J. 2008. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 38:27–65. 10.1007/s12035-008-8033-0 [DOI] [PubMed] [Google Scholar]
- 71.Lipton SA. 2007. Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr. Drug Targets 8:621–632. 10.2174/138945007780618472 [DOI] [PubMed] [Google Scholar]
- 72.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. 2011. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J. Am. Chem. Soc. 133:4274–4284. 10.1021/ja102581n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ickes DE, Venetta TM, Phonphok Y, Rosenthal KS. 1990. Tromantadine inhibits a late step in herpes simplex virus type 1 replication and syncytium formation. Antiviral Res. 14:75–85. 10.1016/0166-3542(90)90045-9 [DOI] [PubMed] [Google Scholar]
- 74.Tanner JA, Zheng BJ, Zhou J, Watt RM, Jiang JQ, Wong KL, Lin YP, Lu LY, He ML, Kung HF, Kesel AJ, Huang JD. 2005. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 12:303–311. 10.1016/j.chembiol.2005.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bukrinskaya AG, Vorkunova NK, Narmanbetova RA. 1980. Rimantadine hydrochloride blocks the second step of influenza virus uncoating. Arch. Virol. 66:275–282. 10.1007/BF01314742 [DOI] [PubMed] [Google Scholar]
- 76.Takeda M, Pekosz A, Shuck K, Pinto LH, Lamb RA. 2002. Influenza A virus M2 ion channel activity is essential for efficient replication in tissue culture. J. Virol. 76:1391–1399. 10.1128/JVI.76.3.1391-1399.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosenthal KS, Sokol MS, Ingram RL, Subramanian R, Fort RC. 1982. Tromantadine: inhibitor of early and late events in herpes simplex virus replication. Antimicrob. Agents Chemother. 22:1031–1036. 10.1128/AAC.22.6.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tselis A. 2011. Evidence for viral etiology of multiple sclerosis. Semin. Neurol. 31:307–316. 10.1055/s-0031-1287656 [DOI] [PubMed] [Google Scholar]
- 79.Grant WB, Campbell A, Itzhaki RF, Savory J. 2002. The significance of environmental factors in the etiology of Alzheimer's disease. J. Alzheimer's Dis. 4:179–189 [DOI] [PubMed] [Google Scholar]
- 80.Tisch S, Brew BJ. 2010. HIV, HAART, and Parkinson's disease: co-incidence or pathogenetic link? Mov. Disord 25:2257–2258. 10.1002/mds.23277 [DOI] [PubMed] [Google Scholar]
- 81.Ison MG. 2013. Clinical use of approved influenza antivirals: therapy and prophylaxis. Influenza Other Respir. Viruses 7(Suppl 1):7–13. 10.1111/irv.12046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fett C, DeDiego ML, Regla-Nava JA, Enjuanes L, Perlman S. 2013. Complete protection against severe acute respiratory syndrome coronavirus-mediated lethal respiratory disease in aged mice by immunization with a mouse-adapted virus lacking E protein. J. Virol. 87:6551–6559. 10.1128/JVI.00087-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Page C, Goicochea L, Matthews K, Zhang Y, Klover P, Holtzman MJ, Hennighausen L, Frieman M. 2012. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J. Virol. 86:13334–13349. 10.1128/JVI.01689-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noçon AL, Ip JP, Terry R, Lim SL, Getts DR, Muller M, Hofer MJ, King NJ, Campbell IL. 30 October 2013. The bacteriostatic protein lipocalin 2 is induced in the central nervous system with West Nile virus encephalitis. J. Virol. [Epub ahead of print.]. 10.1128/JVI.02094-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li XF, Deng YQ, Yang HQ, Zhao H, Jiang T, Yu XD, Li SH, Ye Q, Zhu SY, Wang HJ, Zhang Y, Ma J, Yu YX, Liu ZY, Li YH, Qin ED, Shi PY, Qin CF. 2013. A chimeric dengue virus vaccine using Japanese encephalitis vaccine strain SA14-14-2 as backbone is immunogenic and protective against either parental virus in mice or nonhuman primates. J. Virol. 87:13694–13705. 10.1128/JVI.00931-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mott KR, Zandian M, Allen SJ, Ghiasi H. 2013. Role of interleukin-2 and herpes simplex virus 1 in central nervous system demyelination in mice. J. Virol. 87:12102–12109. 10.1128/JVI.02241-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boylan K, Yang C, Crook J, Overstreet K, Heckman M, Wang Y, Borchelt D, Shaw G. 2009. Immunoreactivity of the phosphorylated axonal neurofilament H subunit (pNF-H) in blood of ALS model rodents and ALS patients: evaluation of blood pNF-H as a potential ALS biomarker. J. Neurochem. 111:1182–1191. 10.1111/j.1471-4159.2009.06386.x [DOI] [PubMed] [Google Scholar]