

Abstract
Vesicular stomatitis virus (VSV) vectors that express heterologous antigens have shown promise as vaccines in preclinical studies. The efficacy of VSV-based vaccines can be improved by engineering vectors that enhance innate immune responses. We previously generated a VSV vaccine vector that incorporates two enhancing strategies: an M protein mutation (M51R) that prevents the virus from suppressing host antiviral responses and a gene encoding bacterial flagellin (M51R-F vector). The rationale was that intracellular expression of flagellin would activate innate immune pathways in addition to those activated by virus alone. This was tested with dendritic cells (DCs) from mice containing deletions in key signaling molecules. Infection of DC with either M51R or M51R-F vector induced the production of interleukin-12 (IL-12) and IL-6 and increased surface expression of T cell costimulatory molecules. These responses were dramatically reduced in DCs from IPS-1−/− mice. Infection with M51R-F vector also induced the production of IL-1β. In addition, in approximately half of the DCs, M51R-F vector induced pyroptosis, a proinflammatory-type of cell death. These responses to flagellin were ablated in DCs from NLRC4−/− mice but not Toll-like receptor 5-deficient (TLR5−/−) mice, indicating that they resulted from inflammasome activation. These results demonstrate that flagellin induces additional innate immune mechanisms over those induced by VSV alone.
INTRODUCTION
Activation of innate immune cells is key to the activity of vaccine adjuvants as well as the success of live-virus vaccines. Thus, a strategy for developing improved vaccines is to enhance their ability to activate the innate immune system. There are currently multiple viruses being developed as live-virus vaccine vectors for the delivery of heterologous antigens that are effective activators of the innate immune system. Examples include the DNA viruses adenoviruses and poxviruses and the RNA viruses Newcastle disease virus, Sendai virus, and vesicular stomatitis virus (VSV) (1–4). In an effort to further stimulate the innate response and trigger a greater adaptive response, many viral vectors have also incorporated vaccine adjuvants. For example, influenza virus, simian virus 5 (SV5), poxvirus, rabies virus (5), and as we have reported, VSV, have been engineered to express bacterial flagellin (6–10). The purpose of the current study was to test the hypothesis that the expression of flagellin by VSV will lead to the activation of additional pathways not triggered by the virus alone.
VSV is currently being developed as a vaccine vector for the delivery of a wide array of heterologous antigens (1, 2, 11–15). Although laboratory strains of recombinant VSV are not usually pathogenic in humans, the potential for VSV to cause disease in humans has been addressed by attenuation of VSV vectors. Multiple methods of attenuation have been developed, including introducing mutations into the viral M protein (5, 16–18). Substitution of arginine for methionine at position 51 of the M protein prevents VSV from inhibiting host gene expression, thus allowing cells to mount an antiviral response. This leads to an increased immune response to VSV in the absence of neurovirulence (19). VSV's ability to elicit an immune response was further enhanced by insertion of the gene for flagellin from Salmonella enterica as a separate gene transcript. This allows the expression of full-length flagellin from the viral genome (6). We have previously reported that VSV encoding flagellin induces higher antibody titers in mice than does VSV vector without flagellin (6).
The hypothesis that flagellin activates additional pathways in innate immune cells compared to those triggered by the virus alone was tested by analyzing the ability of VSV encoding flagellin to activate dendritic cells (DCs) from mice with deletions in key signaling molecules. DCs provide the critical link between the innate and adaptive immune systems. Previous studies have shown the ability of VSV that expresses flagellin to elicit a higher level of cytokine production in human monocyte-derived DCs (6). VSV has been shown to signal in DCs primarily through the pattern recognition receptors (PRR) Toll-like receptor 7 (TLR7) and RIG-I (20, 21). Not all subsets of DCs express TLR7, and in these cells, the RIG-I pathway is likely the major inducer of antiviral responses (21). The RIG-I response to VSV is mediated through the mitochondrial adapter protein IPS-1 (22). In the case of flagellin, two different pathways have been shown to mediate the response in DCs (23–25). If the flagellin is extracellular, the response is mediated by the Toll-like receptor TLR5. If the flagellin is intracellular, the NOD-like receptor NLRC4 mediates the response. Depending on which molecule detects flagellin, different outcomes for the cell can result. TLR5 activation leads primarily to NF-κB activation and cytokine production (26). NLRC4 activation leads to formation of an inflammasome. The inflammasome is a multiprotein complex that activates innate immune response pathways. The composition of an inflammasome and the biological outcome is dependent on the sensor that is activated. In the case of NLRC4 activated by flagellin, the inflammasome is formed with pro-caspase-1 and adapter proteins, such as ASC. Formation of the NLRC4 inflammasome leads to processing and secretion of interleukin-1β (IL-1β) and IL-18 and can also lead to a form of cell death called pyroptosis (27–29). Pyroptosis is an inflammatory cell death that can promote enhanced immune responses (29, 30). This form of cell death differs from apoptosis in that it is more rapid and is manifested by lysis of the cell, which leads to release of cellular contents into the surrounding environment. Pyroptosis is detected by other innate immune cells as an inflammatory signal.
In the current report, we found that the expression of flagellin from the viral genome led to production of IL-1β that was not elicited by virus alone. In addition, expression of flagellin led to pyroptotic cell death in approximately 50% of the DCs. The remaining DCs matured and produced cytokines IL-12 and IL-6 and showed increased surface expression of the T cell costimulatory molecules CD86, CD80, and CD40. Production of IL-1β and induction of pyroptosis were ablated in DCs from NLRC4−/− mice but not TLR5−/− mice, indicating that they resulted from inflammasome activation. In contrast, the production of IL-12 and IL-6 and expression of costimulatory molecules were dependent on IPS-1 and, thus, resulted primarily from antiviral signaling.
MATERIALS AND METHODS
Reagents.
Recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) generated from a baculovirus vector was provided by S. Mizel, Wake Forest School of Medicine. Fluorescently labeled antibodies against mouse cell surface antigens (CD11c, clone N418; CD86, clone GL-1; I-Ab, clone AF6-120.1) were purchased from BioLegend. Fluorescently labeled antibodies against CD40 (clone 3/23) and CD80 (clone 16-1OA1) were purchased from BD Pharmingen. Caspase-1 p10 (M-20) antibody was purchased from Santa Cruz. IL-1β antibody was purchased from R&D Systems. Flagellin antibody and purified flagellin were generously provided by S. Mizel. Lipopolysaccharide (LPS) from Salmonella enterica serovar Minnesota was purchased from Sigma (St. Louis, MO). Monoclonal anti-VSV N protein antibody 10G4 was produced as described previously (31, 32).
Mice.
C57BL/6 mice were purchased from Charles River (Wilmington, MA). With the permission of S. Akira, TLR5−/− mice were acquired from S. Mizel (Wake Forest School of Medicine), and IPS-1−/− mice were acquired from M. Schnell (Thomas Jefferson University). NLRC4−/− mice were obtained from V. Dixit (Genentech). All mice were maintained and bred in the animal facility at Wake Forest School of Medicine, and experiments were conducted under protocols approved by the Institutional Animal Care and Use Committee and were in compliance with all relevant federal guidelines related to animal care and use.
Dendritic cells.
GM-CSF-derived DCs were cultured as previously described (33). Briefly, bone marrow was obtained from the femurs and tibias of 8- to 24-week-old mice. Red blood cells were lysed and washed, and progenitor cells were plated at a density of 5 × 105/ml in RPMI medium containing 10% fetal calf serum supplemented with 10 ng/ml of GM-CSF and 10 μg/ml of gentamicin. Cells were cultured at 37°C for 6 days and fed with fresh medium and GM-CSF on days 2, 4, and 5. On day 6, the nonadherent and loosely adherent cells were harvested. These cells were 90% (±5%) CD11c positive and showed low levels of CD40, CD80, CD86, and major histocompatibility complex class II (MHC-II) antigens, typical of immature DCs (34).
Virus growth and preparation.
Recombinant M51R and M51R-F viruses were isolated from infectious VSV cDNA clones, and virus stocks were prepared and maintained on BHK cell monolayers as previously described (35). The M51R-F virus was purified by sucrose gradient centrifugation to remove extracellular flagellin.
Measurement of costimulatory molecule and cytokine expression following viral infection.
DCs were plated in a 48-well plate at a density of 5 × 105 cells/ml in antibiotic-free medium and were infected with virus at the multiplicity of infection (MOI) indicated in the figures or legends for 6, 12, or 24 h. In a separate set of experiments, cells were treated with purified flagellin or LPS (both at 100 ng/ml) as controls. Supernatants were collected and stored at −80°C, and cytokine levels (IL-1β, IL-6, and IL-12p40) were measured by enzyme-linked immunosorbent assay (ELISA; BD Biosciences). DCs were analyzed by staining for MHC-II and T cell costimulatory molecules CD86, CD80, and CD40 using fluorescently labeled antibodies. Data were acquired using a BD FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc.).
Immunoblotting.
DCs were infected at an MOI of 10, supernatants were collected at 8 h postinfection, and cells were lysed using radioimmunoprecipitation assay (RIPA) buffer without SDS. Protein concentrations of the lysates and supernatant were measured (Bio-Rad), and equivalent amounts of lysate or supernatant protein per sample were analyzed by SDS-PAGE followed by transfer to 0.45-μm polyvinylidene difluoride (PVDF) membranes. Membranes were blocked and probed with antibodies against flagellin, caspase-1, IL-1β, or actin followed by the appropriate secondary antibody conjugated to horseradish peroxidase. Chemiluminescent substrate was used for detection (Thermo Scientific, SuperSignal West Dura). Membranes were stripped and reprobed the following day until each protein had been analyzed. Pixel intensities were analyzed utilizing ImageQuant software.
Cell viability and death.
DCs were infected at an MOI of 10 and at the time points indicated in the figures or legends were analyzed for cell viability utilizing a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Promega) or were stained with 7-aminoactinomycin D (7AAD) according to the manufacturer's instructions (BD Pharmingen). Data were acquired using a BD FACSCalibur flow cytometer and analyzed using FlowJo software.
VSV N-protein detection.
DCs were infected at an MOI of 10. At 24 h postinfection, cells were harvested, fixed, permeabilized, and stained with anti-VSV-N antibody followed by a fluorescently labeled secondary antibody as described previously (36). Data were then acquired by flow cytometry as described above.
Statistical analysis.
Comparisons were tested for statistical significance by calculating the t statistic and the probability (P) of the null hypothesis using Microsoft Excel software. P values of <0.05 were considered statistically significant.
RESULTS
Incorporation of a gene encoding bacterial flagellin has been shown to enhance the immune response and DC function for a variety of live-virus vectors, including VSV (6–10). The mechanism of enhancement is thought to be due to activation of innate immune pathways in antigen-presenting cells in addition to those activated by virus vectors that lack flagellin. This hypothesis was tested by analyzing the response to VSV vectors in murine myeloid DCs that lack key elements of the signaling pathways that respond to virus and flagellin. Construction of the M protein mutant VSV vector and the vector encoding Salmonella enterica flagellin, depicted in Fig. 1A, have been described previously (6). The M51R M protein mutant vector has a single amino acid change, methionine to arginine, at position 51 of the M protein. This mutation prevents the virus from inhibiting host gene expression and allows infected cells to mount an antiviral response. The gene for flagellin (the fliC gene) was inserted into the M51R backbone as a separate transcriptional unit between the M and G genes. These vectors induce potent innate and adaptive immune responses in the absence of viral pathogenicity in mice, whereas vectors with wild-type (WT) M protein retain pathogenicity in mouse models (6, 17, 19). The flagellin gene does not encode a eukaryotic signal sequence, so the flagellin would be expected to be primarily intracellular. The expression of flagellin was tested in myeloid DCs cultured from bone marrow precursors in the presence of GM-CSF. Cells were infected with the flagellin-expressing vector (M51R-F) or control vector (M51R), and flagellin expression in cell lysates or culture supernatants was determined by immunoblots (Fig. 1B). As expected, flagellin produced by cells infected with M51R-F vector was present in the cell lysate and also the culture supernatant. When the relative amounts of flagellin in the supernatant and lysate were measured in the immunoblots and corrected for the relative volumes of the two samples, it was estimated that approximately 85% of the flagellin was in the supernatant. This was likely released upon cell lysis.
FIG 1.
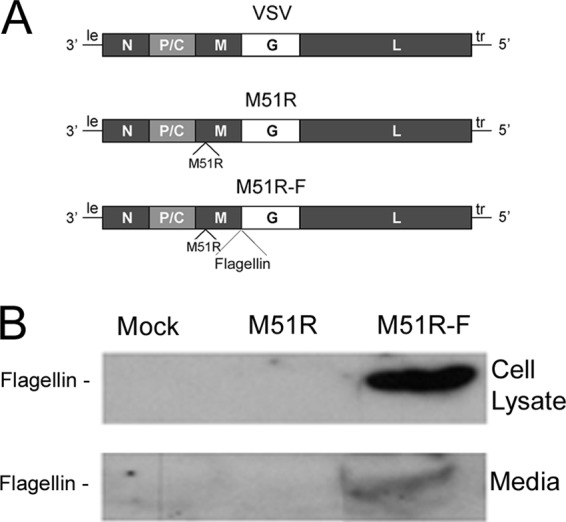
VSV vector that expresses flagellin. (A) Diagram of genomes of WT and M51R VSV and M51R-F VSV. Shown are the genes encoding the viral N, P, C, M, G, and L proteins and the leader (le) and trailer (tr) RNAs. The gene for flagellin (the fliC gene) was inserted into the M51R backbone as a separate transcriptional unit between the M and G genes. (B) Murine DCs were infected with M51R or M51R-F vector (MOI = 10) or mock infected. At 8 h postinfection, cell lysates (20 μg of protein) and media (70 μg of protein) were analyzed for flagellin expression by immunoblotting. The volumes analyzed in the figure shown are as follows: cell lysates, 15.7 μl from a total 120 μl; media, 4.3 μl from a total 2 ml.
The expression of flagellin by the M51R-F vector enhanced cytokine production by murine DCs over that induced by M51R vector, similar to published results with human DCs (Fig. 2) (6). DCs were infected with M51R and M51R-F at various MOIs for 24 h. Supernatants were analyzed for the cytokines IL-1β, IL-6, and IL-12 by ELISA. Infection of DCs with M51R vector did not result in detectable levels of IL-1β above the background of the ELISA. In contrast, the M51R-F vector elicited robust production of IL-1β. DCs infected with M51R vector showed a strong IL-6 and IL-12 response, which was increased in cells infected with M51R-F vector at MOIs of 0.1 and 1.0 (P values of 0.005 for IL-6 and 0.01 for IL-12, respectively). This shows that the presence of flagellin can enhance IL-6 and IL-12 production and is required to elicit production of IL-1β. Treatment of murine DCs with purified flagellin has been reported to elicit low levels of cytokine production compared to those seen with other TLR agonists (23, 26). This was confirmed in our experiments by comparing IL-6 and IL-12 production in response to flagellin with that in response to LPS (Fig. 2).
FIG 2.

Cytokine production by DCs following treatment with VSV vectors. DCs were treated at various MOIs for 24 h. Supernatants were collected, and cytokines were measured by ELISA. (A) IL-1β production, (B) IL-6 production, (C) IL-12 production. Three independent determinations are shown for each treatment. The line indicates the mean; asterisks indicate P < 0.05. Like symbols represent independent determinations for each treatment. In a separate set of experiments, DCs were treated with purified flagellin or lipopolysaccharide (LPS) as described previously (6) and analyzed for production of IL-6 and IL-12.
To determine the signaling pathways that contribute to cytokine production, DCs were cultured from bone marrow of WT, TLR5−/−, NLRC4−/−, or IPS-1−/− mice and were treated with M51R or M51R-F vector. Supernatants were analyzed for the production of IL-1β, IL-6, and IL-12 at 6 or 24 h postinfection. The data obtained at 24 h are shown in Fig. 3. As in Fig. 2, only the M51R-F vector stimulated production of IL-1β in WT DCs. IL-1 production was reduced slightly in TLR5−/− DCs but was completely abolished in NLRC4−/− DCs (P = 0.001) (Fig. 3A). Production of IL-1β by IPS-1−/− DCs was similar to that of WT DCs. The same results were obtained at 6 h, except that overall cytokine levels were lower (data not shown). These results indicate that NLRC4 is the major PRR for flagellin induction of IL-1β production in these DCs, whereas antiviral signaling through IPS-1 does not play a role.
FIG 3.

Cytokine production by DCs with deletions of key signaling molecules. Dendritic cells were obtained from bone marrow of WT, TLR5−/−, NLRC4−/−, or IPS-1−/− mice. DCs were infected for 24 h (MOI = 10), and cytokines were measured in supernatants by ELISA. (A) IL-1β, (B) IL-6, (C) IL-12. Three independent determinations are shown for each treatment. The line indicates the mean. Like symbols represent independent determinations for each treatment.
The effects of the gene deletions on production of IL-6 and IL-12 (Fig. 3B and C) were in contrast to those for IL-1β. Both TLR5−/− and NLRC4−/− DCs produced IL-6 and IL-12 at levels similar to those produced by WT DCs after infection with M51R and M51R-F. However, IPS-1−/− DCs treated with M51R vector produced no detectable IL-6 or IL-12 above the levels seen for mock-infected controls. IPS-1−/− cells treated with M51R-F virus did produce detectable amounts of IL-6 and IL-12, but they were well below the levels produced by cells of the other genotypes. Collectively, the data in Fig. 3 indicate that production of IL-1β in response to M51R-F vector primarily involves signaling through NLRC4, whereas production of IL-6 and IL-12 primarily involves signaling through IPS-1.
Production of IL-1β requires stimulation of prointerleukin-1β (proIL-1β) production, followed by caspase-1-mediated cleavage to generate the mature form of the cytokine. The latter process results from inflammasome activation. To determine the signaling pathways involved in expression of proIL-1β, DCs from WT, NLRC4−/−, or IPS-1−/− mice were treated with M51R or M51R-F vector for 8 h, and cell lysates and supernatants were analyzed by immunoblotting (Fig. 4). Only uncleaved proIL-1β was detected in cell lysates, and only the cleaved IL-1β was detected in the supernatants. There was a significant increase (P = 0.003 compared to results of mock treatment) in proIL-1β in the WT cells treated with M51R-F vector, whereas cells treated with M51R vector were similar to mock-treated cells, indicating that virus infection alone in the absence of flagellin does not trigger production of proIL-1β. Cells deficient in NLRC4 and treated with M51R-F had a significant reduction in proIL-1β expression (P = 0.007 compared to results for the WT), indicating that flagellin utilizes NLRC4 to trigger proIL-1β production. DCs deficient in IPS-1 also had a significant reduction in proIL-1β expression (P = 0.04), although secreted levels of IL-1β were similar between WT and IPS-1−/− DCs treated with M51R-F vector, indicating that expression of total IL-1β (proIL-1β + cleaved IL-1β) was higher than that for mock- or M51R vector-treated cells.
FIG 4.

Production of proIL-1β and mature IL-1β following VSV treatment. DCs were cultured from bone marrow of WT, NLRC4−/−, or IPS-1−/− mice and treated for 8 h with M51R or M51R-F vector (MOI = 10). (A) Immunoblot of cells and media from infected cells, probed with antibodies to detect pro-IL-1β, mature IL-1β, and actin as an internal reference. (B) Quantitation of pro-IL-1β in immunoblots. Integrated pixel densities were measured using ImageQuant software, and results are expressed as the maximal response (WT DCs treated with M51R-F vector). Data shown are means ± SD from 3 experiments.
An important indicator of DC maturation is the upregulation of costimulatory molecules and MHC-II molecules on the cell surface. To determine the effect of flagellin on expression of these surface markers, DCs were treated with M51R or M51R-F vector for 24 h and then harvested and stained for CD11c, CD80, CD86, and MHC-II molecules (Fig. 5). The cells were analyzed by flow cytometry with gating on CD11c-positive cells. Data are shown as a fold increase in mean fluorescence intensity (MFI) over that of mock-infected cells. WT cells treated with M51R or M51R-F vector upregulated CD86, CD80, and MHC-II molecules. There was no significant enhancement due to the presence of flagellin. DCs from TLR5- and NLRC4-deficient mice showed no difference in comparison to WT cells. However, DCs deficient in IPS-1 failed to respond to either M51R or M51R-F vector, with a statistically significant difference in the CD86 responses (P < 0.04 for WT, NLRC4−/−, and TLR5−/− mice versus IPS-1−/−, M51R-treated, and M51R-F-treated mice). Expression of CD80 and MHC-II molecules by IPS-1−/− DCs also trended toward lower values but did not reach statistical significance in these experiments. These results indicate that the upregulation of costimulatory molecules, particularly CD86, was dependent primarily on antiviral signaling through IPS-1.
FIG 5.

M51R and M51R-F vectors increase costimulatory molecule expression. WT, NLRC4−/−, TLR5−/−, and IPS-1−/− DCs were treated with M51R and M51R-F vectors (MOI = 10). At 24 h, the cells were harvested, stained for cell surface markers, and analyzed by flow cytometry. (A) Representative histograms depicting CD86 expression levels in arbitrary fluorescence units. Solid line, M51R treated; dotted line, M51R-F treated; shaded histogram, mock treated. (B) Mean fluorescence intensities (MFIs) from multiple experiments indicated as fold induction in MFI over that of mock-infected controls. Like symbols represent independent determinations for each treatment. The line represents the mean.
Activation of the NLRC4 inflammasome can lead to cell death by pyroptosis in some cell types. The observation that DCs treated with M51R-F vector elicit an IL-1β response is an indicator of activation of caspase-1, which is a requirement for pyroptosis. The major characteristics of pyroptosis that distinguish it from other forms of cell death are rapid permeabilization of the plasma membrane, inflammasome activation, and IL-1β release (29). The release of IL-1β is shown in Fig. 4. Evidence for rapid membrane permeabilization and inflammasome activation is shown in Fig. 6. WT and mutant DCs were treated for 6 h with M51R or M51R-F vector and then stained with 7AAD to measure membrane permeabilization. Cells were analyzed by flow cytometry for 7AAD fluorescence and forward light scattering (FSC-H). In the WT cells, approximately 20% of both mock-treated and M51R vector-treated DCs were 7AAD positive. In contrast, approximately 50% of DCs treated with M51R-F vector were 7AAD positive. This indicates that DCs treated with flagellin-expressing virus elicited a rapid permeabilization of the plasma membrane that suggests the induction of pyroptosis. When NLRC4−/− cells were treated with either vector, this early membrane permeabilization did not occur, and all treatments resembled levels seen with mock-infected cells. Consistent with these results, flagellin was not detected in the supernatant of NLRC4−/− DCs (data not shown). Therefore, the early cell death triggered by flagellin is dependent on NLRC4. In both TLR5−/− and IPS-1−/− cells, the effect of flagellin expression was similar to that in WT cells. This shows that signaling through NLRC4, but not IPS-1 or TLR5, plays a role in triggering cell death that is consistent with the induction of pyroptosis in response to M51R-F virus infection.
FIG 6.

Membrane permeabilization in DCs following viral vector treatment. DCs from WT, NLRC4−/−, IPS-1−/−, and TLR5−/− mice were treated with either M51R or M51R-F vector (MOI = 10) for 6 h. Cells were stained with 7AAD to measure membrane permeability and analyzed by flow cytometry. (A) Representative dot plots depicting forward scatter (FSC-H) versus 7AAD in arbitrary fluorescence units. Numbers indicate the percentage of positive cells in the gate. (B) Results of multiple experiments expressed as percentage of CD11c positive that were also 7AAD positive (means ± SD; n ≥ 3 experiments per group).
The observation that the M51R-F vector induces rapid membrane permeabilization raises the question of how many cells remain viable after treatment with these viruses. DCs were treated with M51R or M51R-F vector, and cell viability was determined at 6, 12, and 24 h after infection using an MTT assay (Fig. 7). There was lower cell viability after treatment with M51R-F than with M51R vector at each time point. However, there was no further decrease in cell viability between 12 and 24 h, with M51R vector-treated cells stabilized at approximately 65% viability and M51R-F vector-treated cells at approximately 48% viability.
FIG 7.

Viability of DCs following infection with M51R or M51R-F vector. WT DCs were infected with M51R or M51R-F vector for 6, 12, and 24 h. Cell viability was measured by MTT assay. Each point indicates the mean ± SD from 3 experiments.
The cell viability data raise the question of whether the surviving cells express viral antigens. Alternatively, they may be cells that are resistant to infection. To address these questions, DCs infected for 24 h were stained for surface CD11c and CD86 and intracellular VSV N protein. Cells were analyzed by flow cytometry with gating on live, CD11c-positive cells. Figure 8 shows representative histograms and data from multiple experiments expressed as the mean percentage of N protein-positive cells. At 24 h, roughly 70% of M51R vector-treated DCs and 55% of M51R-F vector-treated DCs were positive for viral N protein. This result was largely independent of DC genotype. In addition, of those N protein-positive cells, approximately 90% were also positive for CD86 (data not shown). This demonstrates that M51R-flagellin-infected DCs not only were activated to mature but also expressed viral antigen.
FIG 8.

Viral N protein expression in DCs following treatment with VSV. WT, NLRC4−/−, TLR5−/−, and IPS-1−/− DCs were treated with M51R and M51R-F vectors (MOI = 10) for 24 h. Cells were labeled for CD11c and were then fixed, permeabilized, and stained for VSV N protein. (A) Representative histograms depicting N protein expression in infected cells in arbitrary fluorescence units. (B) Results of multiple experiments expressed as means ± SD of the percentage of CD11c-positive cells that were also N protein positive (n = 3 experiments per group).
DISCUSSION
The activity of bacterial flagellin as a vaccine adjuvant is well established both as a purified protein and when expressed in viral vaccine vectors (reviewed in references 25 and 26). There are many cell types in the immune system whose activity is enhanced by flagellin. The experiments presented here address the mechanisms involved in the activity of DCs infected with VSV vectors that express flagellin. Previous experiments demonstrated the enhanced activation of human DCs compared to VSV vectors that do not express flagellin (6). The results presented here extend these results to murine DCs, with which we demonstrated that the expression of flagellin in the intracellular compartment leads to the activation of pathways distinct from those activated by virus alone and leads to an increased production of mediators of inflammatory responses. The enhanced release of inflammatory mediators included the production of IL-1β (Fig. 2 to 4) and the triggering of pyroptosis (Fig. 6 and 7). Additionally, it was shown that the main pathway that contributes to IL-1β production and pyroptosis is the NLRC4 inflammasome (Fig. 3 and 7).
The pattern of expression of cytokines and costimulatory molecules provided a clear distinction between pathways induced by virus and those induced by flagellin. The production of IL-6, IL-12, and costimulatory molecules was primarily through antiviral signaling, as shown by the effects of deletion of IPS-1. In contrast, production of IL-1β and induction of pyroptosis were primarily through inflammasome activation, as shown by deletion of NLRC4, which ablated both responses (Fig. 3 and 7). Deletion of TLR5, the other major PRR for flagellin, had only minor effects in this study. There was certainly the potential for activation of TLR5, since a substantial proportion of the flagellin produced by infected DCs was released into the extracellular environment. In contrast to human DCs, which respond robustly to extracellular flagellin, murine DCs generated in culture respond less well to extracellular flagellin than other TLR agonists, presumably because of their low levels of TLR5 expression (6, 23, 25, 26). Nonetheless, it was important for us to test the role of TLR5, because of the potential for virus infection to stimulate its expression. TLR5 may play a larger role in vivo due to the presence of subsets of DCs that do express TLR5 as well as other cell types that respond effectively to flagellin (23, 25, 26).
Previously published experiments have demonstrated VSV triggering the production of IL-1β through activation of the NLRP3 inflammasome in DCs similar to those studied here (37). In contrast, we detected little if any IL-1β production induced by VSV vectors that do not express flagellin. The discrepancy in our results could be due to minor differences in the culture technique for these DCs. This might lead to a difference in differentiation of DCs prior to infection, which could lead to a different result postinfection. Despite these differences in results, it appears safe to conclude that activation of the NLRC4 inflammasome by flagellin is a much more potent inducer of IL-1β production than activation of the NLRP3 inflammasome by VSV.
In addition to production of IL-1β, the other important effect of activation of the NLRC4 inflammasome in DCs by flagellin was the induction of cell death consistent with pyroptosis. This form of cell death depends on activation of caspase-1 (which also cleaves pro-IL-1β to generate mature IL-1β) or caspase-11 by NLRs containing Pyrin domains or caspase activation and recruitment domains (CARDs), such as NLRC4 (29, 30). Evidence for the induction of pyroptosis was the NLRC4-dependent permeabilization of the plasma membrane by 6 h postinfection and by the rapid induction of cell death as determined by MTT assay (Fig. 6 and 7). The principal mechanism of cell death induced by VSV in the absence of flagellin expression is apoptosis, which is effectively induced by M51R viruses through activation of both the death receptor and mitochondrial pathways (38–40). Induction of apoptosis in cell culture typically leads to membrane permeabilization and cell lysis in the late stages (38). However, apoptotic cells are usually taken up by phagocytic cells in vivo before their intracellular contents are released. In contrast to induction of cell death by apoptosis, induction of pyroptosis leads to rapid release of intracellular contents both in vivo and in cell culture (29, 30). Many of these intracellular contents represent “danger” signals that are potent activators of innate immune cells. These include metabolites such as ATP and uric acid, and proteins, such as heat shock proteins Hsp70 and Hsp90 and high-mobility group box 1 (HMGB1) protein (29, 30). It is also possible that factors released from infected cells induce cell death in adjacent cells. In addition to release of these activators of inflammatory responses, lysis of cells by pyroptosis leads to release of intracellular viral antigens, which can be cross-presented by other classes of DCs, such as CD8+ DCs (41, 42). For example, in the experiments in Fig. 8, the nonviable cells were positive for viral N protein. Cell death by pyroptosis has been described most often for myeloid cells infected with a variety of pathogens, but it is likely that other nonmyeloid cells are also capable of undergoing pyroptosis in response to vectors that express flagellin (29, 30).
The results in Fig. 7 show that approximately 50% of DCs infected with M51R-F virus remain viable at 24 h postinfection. This raises the question of why some cells survive the infection while others undergo cell death by pyroptosis. We ruled out the trivial explanation that the surviving cells were resistant to infection by demonstrating the expression of viral antigen in the surviving cells (Fig. 8). These were also the cells that express costimulatory molecules such as CD86, which were assayed at 24 h postinfection (Fig. 5). There is a similar dichotomy between cells that die as a result of apoptosis and those that survive the induction of apoptosis. This is also apparent in Fig. 7 as partial loss of cell viability following infection with M51R vector that does not express flagellin. In the case of the induction of apoptosis, individual cells vary in the extent to which they express both the inducers of apoptosis, such as caspases, and the inhibitors of apoptosis, such as XIAP. The cells that survive are likely those in which the inducers are expressed at a lower level, the inhibitors at a higher level, or both. Similarly, the cells that survive the induction of pyroptosis are likely to be those in which the balance of regulators favors survival. In addition, there is variation among cells in the level of flagellin expression, so that cells with relatively less flagellin would be expected to survive. One of the key questions about this mechanism of cell death is the identity of the regulators of pyroptosis, which could have a profound effect on the inflammatory response to signals such as flagellin.
Currently, both VSV and flagellin are in clinical trials in humans as vaccine vectors and adjuvants for heterologous antigens (43) (http://clinicaltrials.gov/ct2/show/NCT01859325?spons=profectus&rank=1, http://clinicaltrials.gov/ct2/show/NCT01381744, http://clinicaltrials.gov/show/NCT01628640; sites accessed 18 September 2013). VSV has several qualities that make it an attractive candidate vaccine vector, such as a low seroprevalence in humans and the ability to replicate in the cytoplasm without genetic shift or recombination, and thus far the safety profile has been favorable. Flagellin also has many desirable qualities, such as retention of activity in the presence of preexisting antibody, when used as an adjuvant in the form of intact flagellin or as fusion proteins with heterologous antigens (25). Thus far, the safety profile for flagellin has also been favorable, without excessive side effects related to the induction of inflammatory responses. However, there is no vaccine vector that incorporates both VSV and flagellin in current clinical trials, although this is an attractive approach for the next generation of VSV vectors. There are other viruses that are also being developed as vectors expressing flagellin (SV5, influenza virus, rabies virus) that may also be subject to future clinical trials.
ACKNOWLEDGMENTS
We thank Matthias Schnell, Thomas Jefferson University School of Medicine, and Vishva Dixit, Genentech Corporation, for providing IPS-1−/− and NLRC4−/− mice, respectively. We thank Steven Mizel, Wake Forest School of Medicine, for providing TLR5−/− mice, purified flagellin, antibody against flagellin, and GM-CSF.
This study was supported by NIH grants R01 AI032983 and P01 AI082325.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Geisbert TW, Feldmann H. 2011. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg virus infections. J. Infect. Dis. 204(Suppl 3):S1075–S1081. 10.1093/infdis/jir349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz JA, Buonocore L, Suguitan AL, Jr, Silaghi A, Kobasa D, Kobinger G, Feldmann H, Subbarao K, Rose JK. 2010. Potent vesicular stomatitis virus-based avian influenza vaccines provide long-term sterilizing immunity against heterologous challenge. J. Virol. 84:4611–4618. 10.1128/JVI.02637-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. 2011. Viral vectors as vaccine platforms: deployment in sight. Curr. Opin. Immunol. 23:377–382. 10.1016/j.coi.2011.03.006 [DOI] [PubMed] [Google Scholar]
- 4.Kurihara K, Takahara Y, Nomura T, Ishii H, Iwamoto N, Takahashi N, Inoue M, Iida A, Hara H, Shu T, Hasegawa M, Moriya C, Matano T. 2012. Immunogenicity of repeated Sendai viral vector vaccination in macaques. Microbes Infect. 14:1169–1176. 10.1016/j.micinf.2012.07.016 [DOI] [PubMed] [Google Scholar]
- 5.Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J. Virol. 81:2056–2064. 10.1128/JVI.01911-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed M, Puckett S, Arimilli S, Braxton CL, Mizel SB, Lyles DS. 2010. Stimulation of human dendritic cells by wild-type and M protein mutant vesicular stomatitis viruses engineered to express bacterial flagellin. J. Virol. 84:12093–12098. 10.1128/JVI.00406-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arimilli S, Johnson JB, Clark KM, Graff AH, Alexander-Miller MA, Mizel SB, Parks GD. 2008. Engineered expression of the TLR5 ligand flagellin enhances paramyxovirus activation of human dendritic cell function. J. Virol. 82:10975–10985. 10.1128/JVI.01288-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney KN, Phipps JP, Johnson JB, Mizel SB. 2010. A recombinant flagellin-poxvirus fusion protein vaccine elicits complement-dependent protection against respiratory challenge with vaccinia virus in mice. Viral Immunol. 23:201–210. 10.1089/vim.2009.0107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leng J, Stout-Delgado HW, Kavita U, Jacobs A, Tang J, Du W, Tussey L, Goldstein DR. 2011. Efficacy of a vaccine that links viral epitopes to flagellin in protecting aged mice from influenza viral infection. Vaccine 29:8147–8155. 10.1016/j.vaccine.2011.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou M, Zhang G, Ren G, Gnanadurai CW, Li Z, Chai Q, Yang Y, Leyson CM, Wu W, Cui M, Fu ZF. 2013. Recombinant rabies viruses expressing GM-CSF or flagellin are effective vaccines for both intramuscular and oral immunizations. PLoS One 8:e63384. 10.1371/journal.pone.0063384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tani H, Komoda Y, Matsuo E, Suzuki K, Hamamoto I, Yamashita T, Moriishi K, Fujiyama K, Kanto T, Hayashi N, Owsianka A, Patel AH, Whitt MA, Matsuura Y. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J. Virol. 81:8601–8612. 10.1128/JVI.00608-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobleigh MA, Buonocore L, Uprichard SL, Rose JK, Robek MD. 2010. A vesicular stomatitis virus-based hepatitis B virus vaccine vector provides protection against challenge in a single dose. J. Virol. 84:7513–7522. 10.1128/JVI.00200-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schell JB, Rose NF, Bahl K, Diller K, Buonocore L, Hunter M, Marx PA, Gambhira R, Tang H, Montefiori DC, Johnson WE, Rose JK. 2011. Significant protection against high-dose simian immunodeficiency virus challenge conferred by a new prime-boost vaccine regimen. J. Virol. 85:5764–5772. 10.1128/JVI.00342-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapadia SU, Rose JK, Lamirande E, Vogel L, Subbarao K, Roberts A. 2005. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology 340:174–182. 10.1016/j.virol.2005.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts A, Reuter JD, Wilson JH, Baldwin S, Rose JK. 2004. Complete protection from papillomavirus challenge after a single vaccination with a vesicular stomatitis virus vector expressing high levels of L1 protein. J. Virol. 78:3196–3199. 10.1128/JVI.78.6.3196-3199.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flanagan EB, Zamparo JM, Ball LA, Rodriguez LL, Wertz GW. 2001. Rearrangement of the genes of vesicular stomatitis virus eliminates clinical disease in the natural host: new strategy for vaccine development. J. Virol. 75:6107–6114. 10.1128/JVI.75.13.6107-6114.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 77:4646–4657. 10.1128/JVI.77.8.4646-4657.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braxton CL, Puckett SH, Mizel SB, Lyles DS. 2010. Protection against lethal vaccinia virus challenge by using an attenuated matrix protein mutant vesicular stomatitis virus vaccine vector expressing poxvirus antigens. J. Virol. 84:3552–3561. 10.1128/JVI.01572-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. 2008. Immune response in the absence of neurovirulence in mice infected with M protein mutant vesicular stomatitis virus. J. Virol. 82:9273–9277. 10.1128/JVI.00915-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 101:5598–5603. 10.1073/pnas.0400937101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28. 10.1016/j.immuni.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988. 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 23.Bates JT, Uematsu S, Akira S, Mizel SB. 2009. Direct stimulation of tlr5+/+ CD11c+ cells is necessary for the adjuvant activity of flagellin. J. Immunol. 182:7539–7547. 10.4049/jimmunol.0804225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. 10.1038/nature10510 [DOI] [PubMed] [Google Scholar]
- 25.Mizel SB, Bates JT. 2010. Flagellin as an adjuvant: cellular mechanisms and potential. J. Immunol. 185:5677–5682. 10.4049/jimmunol.1002156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honko AN, Mizel SB. 2005. Effects of flagellin on innate and adaptive immunity. Immunol. Res. 33:83–101. 10.1385/IR:33:1:083 [DOI] [PubMed] [Google Scholar]
- 27.Koizumi Y, Toma C, Higa N, Nohara T, Nakasone N, Suzuki T. 2012. Inflammasome activation via intracellular NLRs triggered by bacterial infection. Cell Microbiol. 14:149–154. 10.1111/j.1462-5822.2011.01707.x [DOI] [PubMed] [Google Scholar]
- 28.Lamkanfi M, Dixit VM. 2011. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 187:597–602. 10.4049/jimmunol.1100229 [DOI] [PubMed] [Google Scholar]
- 29.Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. 2013. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 16:319–326. 10.1016/j.mib.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamkanfi M, Dixit VM. 2012. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28:137–161. 10.1146/annurev-cellbio-101011-155745 [DOI] [PubMed] [Google Scholar]
- 31.Lefrancois L, Lyles DS. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121:157–167 [PubMed] [Google Scholar]
- 32.McCreedy BJ, Jr, Lyles DS. 1989. Distribution of M protein and nucleocapsid protein of vesicular stomatitis virus in infected cell plasma membranes. Virus Res. 14:189–205. 10.1016/0168-1702(89)90001-4 [DOI] [PubMed] [Google Scholar]
- 33.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693–1702. 10.1084/jem.176.6.1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merad M, Sathe P, Helft J, Miller J, Mortha A. 2013. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 31:563–604. 10.1146/annurev-immunol-020711-074950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed M, Brzoza KL, Hiltbold EM. 2006. Matrix protein mutant of vesicular stomatitis virus stimulates maturation of myeloid dendritic cells. J. Virol. 80:2194–2205. 10.1128/JVI.80.5.2194-2205.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westcott MM, Ahmed M, Smedberg JR, Rajani KR, Hiltbold EM, Lyles DS. 2013. Preservation of dendritic cell function during vesicular stomatitis virus infection reflects both intrinsic and acquired mechanisms of resistance to suppression of host gene expression by viral M protein. J. Virol. 87:11730–11740. 10.1128/JVI.00680-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajan JV, Rodriguez D, Miao EA, Aderem A. 2011. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 85:4167–4172. 10.1128/JVI.01687-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopecky SA, Willingham MC, Lyles DS. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169–12181. 10.1128/JVI.75.24.12169-12181.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaddy DF, Lyles DS. 2007. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J. Virol. 81:2792–2804. 10.1128/JVI.01760-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaddy DF, Lyles DS. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170–4179. 10.1128/JVI.79.7.4170-4179.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shortman K, Heath WR. 2010. The CD8+ dendritic cell subset. Immunol. Rev. 234:18–31. 10.1111/j.0105-2896.2009.00870.x [DOI] [PubMed] [Google Scholar]
- 42.Joffre OP, Segura E, Savina A, Amigorena S. 2012. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12:557–569. 10.1038/nri3254 [DOI] [PubMed] [Google Scholar]
- 43.Turley CB, Rupp RE, Johnson C, Taylor DN, Wolfson J, Tussey L, Kavita U, Stanberry L, Shaw A. 2011. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine 29:5145–5152. 10.1016/j.vaccine.2011.05.041 [DOI] [PubMed] [Google Scholar]