

Abstract
Arenaviruses merit significant interest as important human pathogens, since several of them cause severe hemorrhagic fever disease that is associated with high morbidity and significant mortality. Currently, there are no FDA-licensed arenavirus vaccines available, and current antiarenaviral therapy is limited to an off-labeled use of the nucleoside analog ribavirin, which has limited prophylactic efficacy. The pyrimidine biosynthesis inhibitor A3, which was identified in a high-throughput screen for compounds that blocked influenza virus replication, exhibits a broad-spectrum antiviral activity against negative- and positive-sense RNA viruses, retroviruses, and DNA viruses. In this study, we evaluated the antiviral activity of A3 against representative Old World (lymphocytic choriomeningitis virus) and New World (Junin virus) arenaviruses in rodent, monkey, and human cell lines. We show that A3 is significantly more efficient than ribavirin in controlling arenavirus multiplication and that the A3 inhibitory effect is in part due to its ability to interfere with viral RNA replication and transcription. We document an additive antiarenavirus effect of A3 and ribavirin, supporting the potential combination therapy of ribavirin and pyrimidine biosynthesis inhibitors for the treatment of arenavirus infections.
INTRODUCTION
Arenaviruses are enveloped viruses with a bisegmented negative-sense, single-stranded RNA genome (1). Each viral RNA segment uses an ambisense coding strategy to direct the synthesis of two viral proteins in opposite orientations, separated by a noncoding intergenic region (1). The small segment (S; 3.5 kb) encodes the viral glycoprotein precursor (GPC) and the viral nucleoprotein (NP), whereas the large segment (L; 7.2 kb) encodes the small RING finger protein (Z) and the RNA-dependent RNA polymerase (L) (1). GPC is processed by cellular site 1 protease (S1P) into the peripheral virion attachment protein GP1 and the fusion-active transmembrane protein GP2. Trimers of GP1/GP2 form the spikes that decorate the virus surface and mediate cell entry via receptor-mediated endocytosis (2). Z is the arenavirus counterpart of the matrix protein found in many negative-strand RNA viruses (3). NP, the most abundant viral protein in both infected cells and virions, together with the L segment and the viral genome RNA forms the viral ribonucleoprotein (vRNP) core, which is active in RNA replication and gene transcription (1, 4). The arenavirus NP has been also shown to counteract the type I interferon and inflammatory responses of the host against viral challenge (5–8).
There are, to date, over 35 recognized arenavirus species that are grouped, according to serologic, genomic, and geographic distribution, into Old World (OW) and New World (NW) arenaviruses. Arenaviruses are maintained as asymptomatic lifelong chronic infections in their rodent natural reservoirs (1). Chronically infected rodents move freely in their habitats and shed infectious virus. Infections of humans can occur by exposure of mucous membranes or abraded skin to aerosols or by direct contact with contaminated material (1). In addition, person-to-person transmission of arenaviruses can also occur via body secretions and excretions (1, 9). Several arenaviruses cause hemorrhagic fever (HF) disease in humans, which is associated with high morbidity and significant mortality (9–11). Thus, OW Lassa virus (LASV) is estimated to infect several hundred thousand individuals yearly in regions of West Africa where it is endemic, resulting in a high number of Lassa fever (LF) cases. Notably, increased travel to and from regions of endemicity has led to the importation of LF into metropolitan areas around the globe where the disease is not endemic (1, 12, 13). Likewise, NW Junín virus (JUNV) causes Argentine HF (AHF), a disease for which endemicity is mostly in the Pampas region of Argentina. AHF is associated with hemorrhagic and/or neurological manifestations and fatality rates of 15 to 30% (11). On the other hand, evidence indicates that the worldwide-distributed prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen of clinical significance in congenital infections (14–16). Moreover, LCMV poses a serious threat to immunocompromised individuals (17, 18). Besides a public health risk, arenaviruses are a biodefense threat, and six of them are recognized as category A agents by the Centers for Disease Control and Prevention (CDC) (19). Public health concerns posed by human pathogenic arenaviruses are further aggravated by the lack of Food and Drug Administration (FDA)-licensed arenavirus vaccines and current antiarenaviral therapy being limited to the off-label use of the nucleoside analog ribavirin, which is only partially effective and is often associated with hemolytic anemia and teratogenic side effects (11, 20–22). Moreover, ribavirin therapy requires early and intravenous administration for optimal efficacy (11, 23). The significance of arenaviruses in human health and biodefense readiness, together with the limited existing armamentarium to combat these infections, highlight the importance of development of novel and effective antiarenaviral drugs.
The antiviral activity of ribavirin is mediated by several, not mutually exclusive, mechanisms, including inhibition of the cellular inosine monophosphate dehydrogenase (IMPDH) and viral mutagenesis (24). Notably, the antiviral and mutagenic activities of ribavirin could not be accounted for solely by depletion of intracellular GTP levels, which are a consequence of IMPDH inhibition (24–28). Several other compounds, including cytidine analogs that target cytosine triphosphate synthetase (29), analogs of adenosine that target S-adenosylhomocysteine hydrolase and interfere with the 5′ cap of mRNA (30), and carbanucleoside analogs that target orotidylic acid decarboxylase (OMPD) and inhibit the conversion of OMP to UMP (31), have been shown to have antiarenaviral activities but did not exhibit advantages over ribavirin (25). Recently, a screen for inhibitors of influenza virus replication in cultured cells identified the de novo pyrimidine biosynthesis inhibitor compound A3 (32), which was found to have a broad inhibitory effect on multiplication of several other RNA viruses, including Newcastle disease virus, vesicular stomatitis virus, Sindbis virus, hepatitis C virus, West Nile virus, and dengue virus (32). The antiviral activity of A3 was counteracted by addition of exogenous orotic acid, suggesting that the activity of dihydroorotate dehydrogenase (DHODH) is the target of A3 (32). Here, we demonstrate a robust antiviral effect of A3 against OW (LCMV) and NW (JUNV) arenavirus infection in rodent (BHK-21), monkey (Vero), and human (A549) cell lines. The antiarenaviral activity of A3 was related to its ability to interfere with viral RNA replication and gene transcription, and it was reverted by the exogenous addition of orotic acid. Importantly, we present evidence that A3 and ribavirin are excellent candidates for combination antiarenaviral therapy to circumvent some of the limitations of ribavirin monotherapy.
MATERIALS AND METHODS
Cells and viruses.
A549 (ATCC CCL-185), BHK-21 (ATCC CCL-10), and Vero (ATCC CCL-81) cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Mediatech, Inc.) supplemented with 10% fetal bovine serum (FBS), l-glutamine (2 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 atmosphere. LCMV (Armstrong strain 53b), r3LCMV/CAT-GFP, r3LCMV/GFP-Gluc, live attenuated Candid#1 vaccine strain of JUNV, and r3Candid#1/CAT-GFP were produced by infecting either BHK-21 (LCMV) or Vero (Candid#1) cells at a multiplicity of infection (MOI) of 0.01 and harvesting tissue culture supernatants (TCS) at 72 h postinfection (h.p.i.) (33–36).
Compounds.
A3 was purchased from Asinex. Uracil (catalog number U0750), orotic acid (catalog number O2750), and ribavirin (catalog number R9644) were purchased from Sigma-Aldrich. Uracil and orotic acid stocks were prepared at 50 mM in 0.1 M NaOH. Ribavirin was prepared at 4 mM in distilled water, and A3 was prepared at 100 mM in dimethyl sulfoxide (DMSO) (32, 37).
Virus growth kinetics and titrations.
Subconfluent A549, BHK-21, and Vero cells (2 × 105; 12-well plate format) were infected, in triplicate, with the indicated virus (MOI, 0.01) at 37°C in DMEM–2% FBS. After 90 min of adsorption, virus inoculum was removed and cells were washed twice with DMEM–2% FBS before adding fresh DMEM containing 10% FBS and the indicated compounds and concentrations, or the DMSO (0.1%) vehicle control. When specified, the medium was supplemented with uracil and orotic acid. At the indicated times postinfection, viral titers in TCS were determined. For LCMV and Candid#1, titers were determined based on immunofocus centers on Vero cells as described previously (37). For r3LCMV/CAT-GFP, r3LCMV/GFP-Gluc, and r3Candid#1/CAT-GFP, virus titers were determined on Vero cells based on green fluorescent protein (GFP) expression, which was directly visualized by fluorescence microscopy at 48 h.p.i (36). Expression levels of chloramphenicol acetyltransferase (CAT) and Gaussia luciferase (Gluc) were determined, at the same time points, as previously described (4, 36). Mean values and standard deviations were calculated using Microsoft Excel software. When indicated, nonlinear regression curves were determined, and the 50% inhibitory concentration (IC50) was calculated using GraphPad Prism software version 5.0c. Viral titers and representative images of at least three independent infections are shown for the experiments. The limit of detection, indicated by dotted lines in the figures, correspond to 100 focus-forming units (FFU)/ml. Graphed values below the limit of detection line correspond to averages when, for at least one of the triplicate experiments, there was viral detection.
MG assays.
For minigenome (MG) assays, BHK-21 and 293T cells were seeded (2.5 × 105/well) on 12-well plates 20 h prior to transfection. BHK-21 cells were transfected with Pol-I MG CAT (0.5 μg) together with either pCAGGS LCMV-NP (0.4 μg) and pCAGGS LCMV-L (0.6 μg) or pCAGGS JUNV-NP (0.4 μg) and pCAGGS JUNV-L (0.6 μg), by using Lipofectamine 2000 (Invitrogen). 293T cells were cotransfected with LCMV T7 MG CAT (0.5 μg) together with either pCAGGS-expressing plasmids for LCMV NP (0.4 μg) and L (0.6 μg) or with LASV T7 MG CAT (0.5 μG) together with pCAGGS-expressing plasmids for LASV NP (0.4 μg) and L (0.6 μg), plus pCAGGS T7 (0.5 μg). At 5 h posttransfection, medium was replaced with medium containing the indicated compounds and concentrations. At 72 h posttransfection, cell lysates were prepared to determine expression levels of CAT protein by using a CAT enzyme-linked immunosorbent assay (ELISA) kit (Roche) (38).
Cell viability.
The CellTiter 96 Aqueous One solution cell proliferation assay (Promega) was used to assess the effects of tested compounds on A549, BHK-21, and Vero cell viability. This method determines the number of viable cells based on levels of 3-(4,5-dimethylthiozol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium compound bioreduced to a colored formazan product, which is accomplished with NADPH or NADH in active cells (39). Viability of compound-treated cells was calculated as a percentage relative to values obtained with DMSO-treated cells (set as 100%). When indicated, nonlinear regression curves were determined and the 50% cytotoxic concentration (CC50) was calculated using GraphPad Prism software version 5.0c.
RNA analysis by Northern blotting hybridization.
Total cellular RNA was isolated by using TRI reagent (Molecular Research Center, Inc.) according to the manufacturer's instructions and analyzed by Northern blotting hybridization using a 32P-labeled DNA NP probe that hybridized to both the S genome RNA and NP mRNA. Methylene blue (MB) staining of the membrane was used to assess RNA quality and equal loading and transfer of RNA samples as determined by the amounts of 28 S rRNA (40).
Metabolomic assay.
A549 cells (3 × 106; 100-mm dish format) were serum starved for 24 h before A3 compound treatment (5 μM) or infection with LCMV (MOI, 1), or both. At 24 h.p.i., medium was replaced with fresh serum-free DMEM in the presence of 10 mM HEPES and the respective drug treatment. After 1 h at 37°C, medium was aspirated and dry ice cold methanol (80%) was added to extract metabolites. Samples were stored at −80°C for 10 min, and attached cells were collected. Samples were mixed by vortexing and centrifuged for 5 min at 2,500 rpm. TCS were collected, and pellets were processed twice as described above with 1 ml of dry ice cold methanol. Extracted metabolites were dried under nitrogen gas and resuspended in 200 μl of 50% methanol prior to centrifugation at 4°C for 5 min at full speed to pellet insoluble material. The resulting extracts were analyzed using reverse-phase chromatography with an ion-pairing reagent in a Shimadzu high-performance liquid chromatograph (HPLC) coupled to a Thermo Quantum triple-quadrupole mass spectrometer running in negative mode with selected reaction monitoring (SRM) specific scans. Specific chromatography conditions and mass spectrometry parameters were as described in reference 41. The resulting data were analyzed by using the publicly available mzrock machine learning tool kit, which automates SRM/HPLC feature detection, grouping, signal-to-noise classification, and comparison to known metabolite retention times (42).
Protein expression and purification of DHODH.
A soluble human DHODH, lacking the hydrophobic membrane-associated domain (43) and with a 6×His tag at its N terminus, was cloned into the pET-30a vector for bacterial expression and transformed into BL21(DE3) competent cells (Promega). Bacteria were grown in LB medium to an A600 of 0.6, induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (Fisher Scientific), and harvested by centrifugation at 6 h after induction. Cells were lysed, and proteins were purified using Ni-nitrilotriacetic acid resin according the manufacturer's instructions (Qiagen).
In vitro inhibition of DHODH.
The colorimetric in vitro DHODH activity assay was adapted from previous publications (44, 45). The assay monitors the reduction of 2,6-dichloroindophenol (DCIP) to DCIPH2. Serial dilutions of inhibitor in DMSO or DMSO only were added to the reaction solution containing 100 mM HEPES, 150 mM NaCl, 0.05% Triton X-100, 20 μM coenzyme Q10 (Sigma), 200 μM l-dihydroorotate (L-DHOA; Sigma), and 120 μM DCIP (Sigma). DHODH at 50 nM was added to start the reaction. Reactions were stopped 60 min later by addition of 1% sodium dodecyl sulfate. Absorbance was measured at 600 nm by using a microplate reader, and relative inhibition was calculated. Nonlinear regression curves were determined, and the IC50 was calculated using GraphPad Prism software version 5.0c.
RESULTS
Effect of A3 on arenavirus multiplication.
We first examined whether the broad-spectrum antiviral activity of compound A3 (32) extended to arenaviruses (Fig. 1). For this, we infected (MOI, 0.01) Vero cells with either LCMV (Fig. 2A) or the Candid#1 strain of JUNV (Fig. 2B) for 90 min and then treated them with the indicated concentrations of A3 (Fig. 2A and B, i graphs) or ribavirin (Fig. 2A and B, ii graphs). At the indicated times postinfection, we determined virus titers in TCS. Treatment with 1 μM A3 caused 4-log reductions in both LCMV and Candid#1 titers at 48 h.p.i., and with A3 concentrations of 5 μM or higher, treatment resulted in nondetectable titers of infectious progeny of both LCMV (Fig. 2A, graph i) and Candid#1 (Fig. 2B, graph i). In contrast, LCMV and Candid#1 titers at 48 h.p.i. were reduced by only 2 and 1 log, respectively, in cells treated with 40 μM ribavirin. Abrogation of virus multiplication in ribavirin-treated cells was only observed at a drug concentration of 100 μM (Fig. 2A and B, graphs ii).
FIG 1.

Chemical structure of A3 and its molecular mass.
FIG 2.

A3 inhibits arenavirus multiplication in Vero cells. Subconfluent monolayers of Vero cells were infected (MOI, 0.01) with LCMV (A) or Candid#1 (B). After 90 min of infection, viral inocula were removed and cells were treated with the indicated concentrations of A3 (i) or ribavirin (Rib) (ii). Aliquots of TCS were collected at the indicated times (hours postinfection), and virus titers (reported in FFU/ml) were determined via an immunofocus assay. Dotted lines indicate the limits of detection for the assay. Viral infections were performed in triplicates. Mean values and standard deviations are shown. Statistical analyses were conducted using a paired, two-tailed Student's t test within Microsoft Excel. *, P < 0.05.
To assess possible cell-type- and species-dependent differences in the antiarenaviral activity of A3, we examined its effects on LCMV multiplication in human A549 (Fig. 3A) and rodent BHK-21 (Fig. 3B) cells. The magnitude of the anti-LCMV activity of A3 in A549 cells was similar to that observed in Vero cells (Fig. 3A, graph i), whereas in BHK-21 cells higher concentrations of A3 were required to inhibit LCMV multiplication to the same degree observed in Vero and A549 cells (Fig. 3B, graph i). On the other hand, lower ribavirin concentrations were required to inhibit LCMV multiplication in A549 (Fig. 3A, graph ii) and BHK-21 (Fig. 3B, graph ii) cells, compared to Vero cells.
FIG 3.

A3 inhibits LCMV multiplication in A549 and BHK-21 cells. Subconfluent monolayers of A549 (A) and BHK-21 (B) cells were infected with LCMV (MOI, 0.01) and treated with the indicated concentrations of A3 (i) or ribavirin (Rib) (ii). Aliquots of TCS were obtained at the indicated times (hours postinfection) and titrated (resulting in data reported in FFU/ml) by using immunofluorescence. Dotted lines indicate the limits of detection for the assay. Viral infections were performed in triplicates. Mean values and standard deviations are shown. Statistical analyses were conducted using Student's t test within Microsoft Excel. *, P < 0.05.
For the concentrations and treatment periods tested, A3 exhibited minimal effects on the number of viable Vero (Fig. 4A), BHK-21 (Fig. 4B), and A549 (Fig. 4C) cells, indicating that the antiarenaviral activity of A3 was not due to drug-induced cytotoxicity. To make a better comparison of the antiviral activity of A3 (Fig. 5A) and ribavirin (Fig. 5B), we determined their IC50 (Fig. 5A and B, graphs i) and CC50 (Fig. 5A and B, graphs ii) in A549 cells. The results obtained against r3LCMV-GFP/Gluc showed that the IC50 for A3 was about 100-fold lower than for ribavirin (compare the i graphs in Fig. 5A and B). Notably, readouts based on Gluc expression levels (dependent on viral replication and transcription) and titers of infectious viral progeny gave similar IC50 values for each compound, validating the use of r3LCMV for the screening of antiviral compounds by using readouts based on reporter gene activity. A3 had a CC50 of 63.5 μM (Fig. 5A, graph ii) whereas, consistent with published data, ribavirin had a CC50 of >400 μM (Fig. 5B, graph ii) (46). A3 had an estimated selectivity index (SI; the CC50/IC50 ratio) of 774, based on the A3 IC50 derived from drug-mediated reduction on production of infectious progeny. A slightly lower SI of 616 for A3 was estimated when using the A3 IC50 that was based on drug-mediated inhibition of virus-driven Gluc gene reporter activity. The SI for ribavirin was >20. We could not determine a precise SI for ribavirin, as a precise CC50 was not obtained. Nevertheless, SI values for ribavirin have been reported for different virus/cell systems and are consistently found to be below 100 (46).
FIG 4.
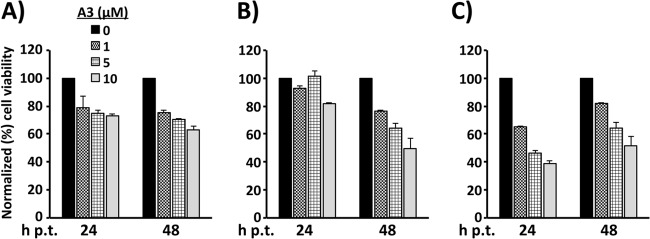
Effects of A3 on cell viability. Vero (A), A549 (B), and BHK-21 (C) cells were seeded in 96-well plates and cultured for 16 h prior to starting treatment with the indicated concentrations of A3 for 24 h or 48 h, followed by addition of 15 μl/well of the CellTiter 96 Aqueous One solution reagent. After incubation at 37°C for 20 min, the quantity of formazan product generated in each well, which is directly proportional to the numbers of viable cells, was determined in an ELISA. Viability of A3-treated cells was calculated as the percentage of values obtained with DMSO-treated cells (set at 100%). Mean values and standard deviations of three independent assays are shown.
FIG 5.

Evaluation of IC50 and CC50 levels for A3 in A549 cells. (i) To determine the IC50, subconfluent monolayers of A549 cells were infected with r3LCMV GFP/Gluc (MOI, 0.01) in the presence of increasing concentrations of A3 (A) or ribavirin (B). Viral titers (bars) and Gluc expression (lines) were determined at 48 h.p.i. Dotted lines indicate both the viral limits of detection and background levels of Gaussia luciferase expression for the assay. Viral infections were performed in triplicates. Mean values and standard deviations are shown. (ii) To determine CC50 values, A549 cells were treated as described in the legend for Fig. 4, with the indicated concentrations of A3 (A) or ribavirin (B) for 24 h, followed by cell viability analysis. Mean values of three independent assays are shown. Nonlinear regression curves were determined, and the respective IC50 and CC50 values were calculated.
Effects of A3 on viral RNA replication and gene transcription.
To determine if A3 affected RNA synthesis, both replication and transcription, mediated by the arenavirus polymerase, we performed LCMV (Fig. 6A, graph i) and Candid#1 (Fig. 6B, graph i) MG assays, in which expression of the CAT reporter gene served as the surrogate for the viral polymerase activity. For this, BHK-21 cells were cotransfected with plasmids expressing the minimal trans-active factors required for viral replication and transcription (L and NP) together with a polymerase I (Pol I)-based MG plasmid, which allowed for CAT expression mediated by the intracellularly reconstituted virus polymerase (47). After overnight incubation, the medium was changed and cells were treated with the indicated A3 concentrations; CAT expression levels were determined 36 h later. We used ribavirin as a validated inhibitor of LCMV (Fig. 6A, graph ii) and Candid#1 (Fig. 6B, graph ii) RNA synthesis. A3 exhibited a dose-dependent inhibitory effect on both LCMV (Fig. 6A, graph i) and Candid#1 (Fig. 6B, graph i) MG activities, which were higher in magnitude than that exerted by ribavirin (Fig. 6A and B, graphs ii).
FIG 6.

A3 exhibits a dose-dependent inhibitory effect on the activities of both the LCMV and Candid#1 minigenomes. BHK-21 cells in 12-well plates were transfected with LCMV (A) or Candid#1 (B) MG systems. At 5 h posttransfection, transfection medium was replaced with fresh medium containing the indicated concentrations of A3 (A and B, i graphs) or ribavirin (A and B, ii graphs). At 72 h posttransfection, cell lysates were prepared to determine CAT expression levels in an ELISA. Values correspond to mean values and standard deviations of two independent experiments. For each experiment, each sample was examined in duplicate.
To further confirm the effects of A3 on viral RNA replication and gene transcription, we analyzed RNA expression in BHK-21 cells infected with LCMV and treated with A3 (Fig. 7). At the indicated time p.i., total cellular RNA was isolated and analyzed by Northern blotting by using an NP-specific probe that hybridized to the NP mRNA and S genome RNA. Both viral RNA replication and gene transcription were inhibited by A3 in a dose-dependent manner.
FIG 7.

A3 exhibits a dose-dependent inhibitory effect on viral RNA replication and transcription in LCMV-infected BHK-21 cells. BHK-21 cells were infected (MOI, 0.1) with LCMV. After 90 min of adsorption, the virus inoculum was removed, cells were washed, and fresh medium was added that contained the indicated concentration of A3. At the indicated time postinfection, total cellular RNA was isolated and analyzed by Northern blotting hybridization using a 32P-labeled DNA NP probe. Methylene blue (MB) staining of the membrane was used to assess RNA quality and equal loading and transfer of RNA samples, determined by the amounts of 28 S rRNA.
To further examine the broad antiarenaviral activity of A3, we determined whether A3 inhibited multiplication of the New World arenavirus JUNV. For this, we examined the effect of A3 on cells infected with recombinant trisegmented (r3) LCMV (Fig. 8A) or Candid#1 (Fig. 8B) (36, 38, 48). At 48 h.p.i., A3 exhibited a dose-dependent inhibitory effect on expression of both GFP (Fig. 8A and B, panels i) and CAT (Fig. 8A and B, graphs ii), which correlated with A3-mediated reductions in titers of infectious viral progeny (Fig. 8A and B, graphs iii). In addition, we also investigated whether A3 displayed antiviral activity against the HF-causing LASV. For this, we compared the inhibitory effect of A3 on the activities of LCMV and LASV minigenomes (Fig. 9). A3 exhibited a dose-dependent inhibitory effect on both LCMV and LASV MG activities, demonstrating the antiviral activity of A3 on HF-causing arenavirus.
FIG 8.

Dose-dependent inhibitory activity of A3 on recombinant trisegmented arenavirus. Vero cells were infected (MOI, 0.1) with r3LCMV/CAT-GFP (A) or r3Candid#1/CAT-GFP (B). After 90 min of infection, the virus inoculum was removed, cells were washed, and fresh medium was added that contained the indicated concentration of A3. At 48 h.p.i. GFP expression was directly visualized by fluorescence microscopy (A and B, images i). After imaging, cell lysates were prepare to determine CAT expression levels (A and B, graphs ii). Virus titers in TCS (A and B, graphs iii) were determined on Vero cells, based on GFP expression, which was directly visualized by fluorescence microscopy at 48 h.p.i. Mean values and standard deviations were calculated using Microsoft Excel software. Viral titers and representative images of at least three independent infections are shown.
FIG 9.
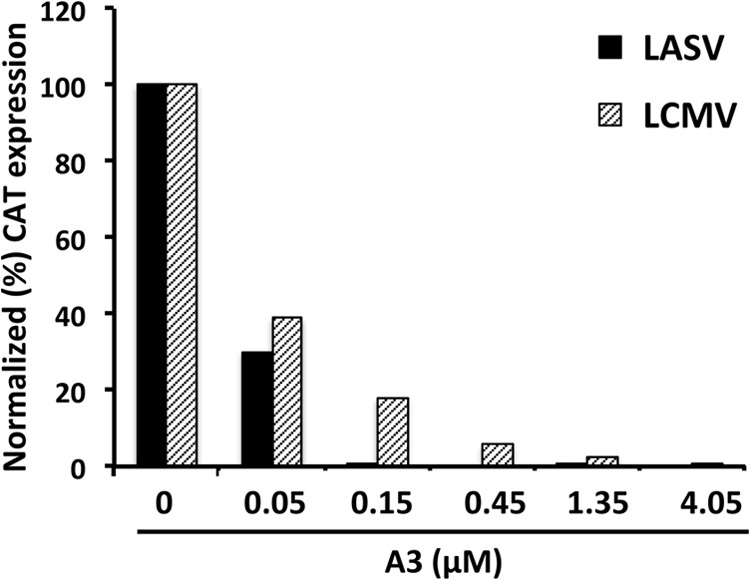
A3 exhibits a dose-dependent inhibitory effect on the activity of the LASV minigenome. 293T cells in 12-well plates were transfected with the components of the LCMV or LASV T7 MG systems. At 5 h posttransfection, transfection medium was replaced with fresh medium containing the indicated concentrations of A3. At 60 h posttransfection, cell lysates were prepared to determine CAT expression levels in an ELISA.
Effect of A3 on the de novo pyrimidine biosynthesis pathway.
Evidence indicates that A3 interferes with the de novo pyrimidine biosynthesis pathway, likely via targeting the dihydroorotate dehydrogenase (DHODH) enzyme (32) through a mechanism yet to be determined. Uracil is converted by the pyrimidine salvage pathway into UMP (49). On the other hand, orotic acid is produced by DHODH as an intermediate in the de novo pyrimidine pathway. Both pathways converge at the generation of UMP; therefore, unlimited access to uracil or orotic acid might overcome the effects of inhibition of DHODH and restore normal pyrimidine pools (32). To assess whether the antiarenaviral activity of A3 was related to its effect on pyrimidine synthesis, we examined whether treatment with uracil or orotic acid restored normal production of infectious progeny in LCMV-infected Vero (Fig. 10A) and A549 (Fig. 10B) cells that were also treated with A3 (5 μM). Uracil (Ura), at the highest concentration tested, partially restored viral titers in Vero cells but not in A549 cells (Fig. 10A and B, graphs i, respectively), a finding similar to that reported for influenza A virus (32). In contrast, orotic acid (Oro) restored viral titers in both Vero cells (Fig. 10A, graph ii) and A549 (Fig. 10B, graph ii) cells in a dose-dependent manner. These results might reflect that the de novo pyrimidine biosynthesis, rather than the salvage, pathway is the primary target of A3 (Fig. 11). To further investigate the involvement of the de novo pyrimidine biosynthesis pathway in the antiarenaviral activity of A3, we used mass spectrometry to analyze changes in key metabolite concentrations (Fig. 11A) in A549 cells under the following conditions: (i) treatment with A3, (ii) LCMV infection, and (iii) treatment with A3 and infection with LCMV (Fig. 11B). Consistent with the predicted effects of A3 on the de novo pyrimidine biosynthesis pathway, A3-treated cells showed accumulation of metabolites N-carbamoyl aspartate and dihydroorotate, as well as a slight increase of orotate. Likewise, downstream metabolites, including UDP, UTP, and CTP, were significantly decreased by A3 treatment. LCMV infection did not affect levels of any of the metabolites analyzed, suggesting that LCMV infection itself does not substantially alter the pool sizes of the pyrimidine metabolites analyzed. To further confirm the contribution of the de novo pyrimidine biosynthesis pathway in arenavirus infections, we treated A549 and BHK-21 LCMV-infected cells (MOI, 0.01) with different concentrations of N-(phosphonoacetyl)-l-aspartate (PALA), an inhibitor of aspartate transcarbamyolase (ATC), the first enzyme of the de novo pyrimidine biosynthesis pathway. PALA exhibited a dose-dependent inhibitory effect on LCMV multiplication in both A549 cells (Fig. 12A) and BHK-21 cells (Fig. 12B), with a stronger inhibitory effect in BHK-21 cells. At the concentrations used, PALA did not have any significant effect on the numbers of viable A549 cells (Fig. 12C) or BHK-21 cells (Fig. 12D), indicating that the effect of PALA on LCMV multiplication was not related to drug-induced toxicity. To conclusively determine that A3 targets DHODH, we examined the effect of A3 on the in vitro activity of a purified soluble form of the human DHODH enzyme (43). The colorimetric assay analyzes the ability of DHODH to reduce 2,6-dichloroindophenol (DCIP) to DCIPH2 (Fig. 13A). DHODH activity was tested in the presence of a wide range of concentrations of A3 and several previously characterized DHODH inhibitors, leflunomide, teriflunomide, and brequinar (Fig. 13B) (50). Leflunomide is a prodrug, and teriflunomide it its active form and also, as expected, leflunomide showed limited activities in vitro, whereas teriflunomide inhibited DHODH activity with an IC50 of 1.33 μM. A3 showed a very similar inhibitory potency (IC50 of 1.13 μM) to that of teriflunomide but lower than that of brequinar (IC50 of 0.04 μM). These results confirmed that A3 affects the de novo pyrimidine biosynthesis pathway by targeting DHODH enzyme activity.
FIG 10.

Uracil and orotate addition reverses the antiviral effect of A3. Subconfluent monolayers of Vero (A) and A549 (B) cells were infected with LCMV (MOI, 0.01). After viral infection, cells were washed and treated with A3 (5 μM) and with the indicated increasing concentrations of uracil (Ura [A and B, graphs i]) and orotic acid (Oro [A and B, graphs ii]). Vehicle treatment (DMSO) was used as an internal control. Virus in TCS at the indicated hours postinfection were determined by immunofluorescence (results are reported as FFU/ml). Dotted lines indicate the limits of detection for the assay. Viral infections were performed in triplicates. Mean values and standard deviations are shown. Statistical analysis were conducted using Student's t test from Microsoft Excel. *, P < 0.05.
FIG 11.

A3 reduces metabolites of de novo pyrimidine biosynthesis during LCMV infection. (A) Schematic representation of de novo pyrimidine biosynthesis pathway: stars indicate compounds analyzed by mass spectrometry. PALA and A3 inhibitors work to block their enzymatic target. Solid squares represent enzymatic complexes. In gray, enzymatic reactions (arrows) and metabolites (italic) are shown from the salvage pathway that allow the incorporation of uracil into the de novo pathway. Modified from reference 49. (B) Metabolite changes upon A3, LCMV, or A3 plus LCMV treatment. A549 cells were treated with A3 compound (5 μM) and/or infected with LCMV (MOI, 1). At 24 h.p.i., metabolite changes were analyzed by LC/MS-MS. Experiments were performed in quadruplicates. Mean values and standard deviations are displayed as log2-converted values.
FIG 12.

Inhibition of LCMV infection by PALA. Subconfluent monolayers of A549 (A) and BHK-21 (B) cells were infected with LCMV (MOI, 0.01) and treated with the indicated concentrations of PALA. Aliquots of TCS were obtained at the indicated times postinfection and titrated (results in FFU/ml) by immunofluorescence. Viral infections were performed in triplicates. Dotted lines indicate the limits of detection for the assay. Mean values and standard deviations are shown. Statistical analyses were conducted using Student's t test within Microsoft Excel. *, P < 0.05. PALA cytotoxicity in A549 (C) and BHK-21 (D) cells was evaluated as described for Fig. 3. Cell viability was normalized to DMSO-treated cells (set as 100%). Mean values and standard deviations of three independent assays are shown.
FIG 13.
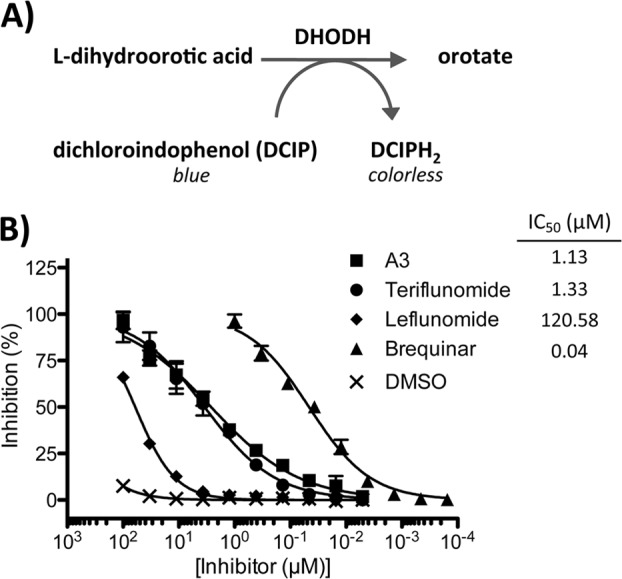
A3 inhibits human DHODH in vitro. (A) DHODH catalyzes the conversion of l-dihydroorotic acid to the oxidized orotate, thereby reducing dichloroindophenol (DCIP, blue) into DCIPH2 (colorless). (B) In vitro DHODH inhibition was measured by adding serial dilutions of A3 and the known DHODH inhibitors brequinar, teriflunomide, and leflunomide. DMSO was included as a negative control. IC50s were calculated based on four independent experiments.
Effect of ribavirin and A3 combination therapy on LCMV multiplication.
Ribavirin is known to affect the cellular GTP pool (51) and also to promote lethal mutagenesis (28), whereas A3 targets the pyrimidine biosynthesis pathway and thereby directly affects the UTP pools. Because different pathways appear to contribute to the antiarenaviral activities of A3 and ribavirin, we reasoned that ribavirin and A3 combination therapy might offer benefits over each single monotherapy. To evaluate this, we treated LCMV-infected A 549 cells (MOI, 0.01) (Fig. 14A) and BHK-21 cells (Fig. 14B) with increasing concentrations of ribavirin in the presence or absence of 0.5 μM A3 after LCMV infection (Fig. 14A and B, graphs i). Similarly, we treated LCMV-infected (MOI, 0.01) cells with increasing concentrations of A3 in the presence or absence of 20 μM ribavirin (Fig. 14A and B, graphs ii). We observed that combination therapy of A3 and ribavirin had a more potent antiarenavirus activity that either single-drug treatment in both human (A549) and rodent (BHK-21) cells.
FIG 14.

Additive antiviral effects of A3 and ribavirin. Subconfluent A549 (A) and BHK-21 (B) cells were infected with LCMV (MOI, 0.01) and treated with the indicated concentrations of ribavirin (Rib) (i) in the presence or absence of 0.5 μM A3. Similarly, LCMV-infected cells were treated with the indicated concentrations of A3 (ii) in the presence or absence of ribavirin (Rib; 20 μM in A549 cells [A, graph ii] and 1 μM in BHK-21 cells [B, graph ii]). Viral titers in TCS at the indicated times were determined based on immunofluorescence (reported as FFU/ml). Viral infections were performed in triplicates. Dotted lines indicate the limits of detection for the assay. Mean values and standard deviations are shown. Statistical analysis were conducted using Student's t test within Microsoft Excel. *, P < 0.05.
DISCUSSION
In this work, we have shown that A3 (Fig. 1), a compound shown to have broad-spectrum antiviral activity (32), also has a potent antiviral activity against representative members of the OW (LCMV) and NW (JUNV) arenavirus family. The antiarenaviral activity of A3 was observed in different cell types and species, including human A549 cells (Fig. 2 and 3), and at drug concentrations associated with minimal effects on cell viability (Fig. 4 and 5). The A3 inhibitory effect on arenavirus multiplication was, at least partly, related to its ability to inhibit virus RNA replication and transcription (Fig. 6, 7, 8, and 9). However, in LCMV-infected BHK-21 cells, the effect of A3 at intermediate (5 μM) concentrations on virus titers at 48 h p.i. appeared to be of a magnitude higher than the corresponding A3 effect on viral RNA synthesis levels (compare Fig. 3B, graph i, and 7). This observation suggests that in addition to its inhibitory effect on viral RNA synthesis, A3 could also exert a mutagenic effect on the viral genome RNA that would result in a decreased virus-specific infectivity, which would further contribute to the antiviral activity of A3. It should be noted that a precedent for this has been reported for ribavirin, a drug that, depending on the experimental conditions used, can both inhibit viral RNA synthesis and have a mutagenic effect on the arenavirus genome (28, 52). However, based on its structure (Fig. 1), it seems highly unlikely that A3 would be recognized as a substrate by the arenavirus polymerase. As recently reported for another inhibitor of the pyrimidine biosynthesis pathway, A3-mediated activation of components of the cell innate immune response could also contribute to its antiviral activity (53). The antiarenaviral activity of A3 was prevented by providing an excess of orotic acid (Fig. 10), strongly suggesting the involvement of the de novo pyrimidine biosynthesis pathway as the primary target of A3, which was further supported by the results of metabolite analysis in A3-treated cells (Fig. 11), the effect of the ACT inhibitor PALA on LCMV multiplication (Fig. 12), and the in vitro analysis of DHODH activity (Fig. 13). Moreover, in combination therapy, ribavirin and A3 exhibited more potent inhibitory activities on arenavirus multiplication than either single-drug treatment (Fig. 14). These findings suggest the potential of developing A3 or A3-related drugs as novel antiviral drugs against human pathogenic arenaviruses alone or in combination with current ribavirin monotherapy.
Previous studies have documented that inhibitors of cytosine triphosphate synthetase (29) and OMPD (31) exhibit antiarenaviral activity, but their potency is lower than that of ribavirin (25). We observed that the magnitude of the antiarenaviral activity of A3 was significantly higher in human A549 cells than in hamster BHK-21 cells, a finding consistent with reported cell-species-dependent differences on the antiviral activity of A3 against influenza virus (32). This could reflect documented differences among cell lines regarding their relative use of the de novo pyrimidine biosynthetic and salvage pathways (54) or differences in the ability of A3 to inhibit its human versus rodent target. We also observed that the inhibitory effect of A3 was less strong at late times after infection. However, it remains to be determined whether this observation was related to the A3 half-life. Notably, in human cells, A3 exhibited a significantly stronger antiviral activity than ribavirin.
As with influenza virus (32), A3 affected arenavirus replication and transcription. These effects are most likely related to A3 treatment limiting pyrimidine pools and thereby affecting the viral polymerase activity. We used mass spectrometry to confirm that under our experimental conditions, A3 treatment resulted in decreased pyrimidine pools. In addition, we observed the buildup of metabolites upstream of the DHODH enzymatic reaction, which is thought to be the target of A3 (32). We also observed a slight increase in orotate upon treatment with A3, which may seem contradictory based on the enzymatic target of A3. This finding could be due to compensatory effects in response to inhibition of the de novo pyrimidine biosynthesis pathway. In addition, orotate is the only metabolite generated in the inner membrane of the mitochondria and, therefore, cytoplasmic levels of orotate might not totally reflect the global effect of the A3 compound (49). Additional studies are required to dissect the details of the mechanisms by which A3 exerts its antiarenaviral activity.
The involvement of the pyrimidine biosynthesis pathway in A3-mediated antiarenaviral activity was further supported by the observation that the ACT inhibitor PALA also inhibited LCMV multiplication. Consistent with these findings, the FDA-approved weak inhibitor of DHODH, leflunomide (50), exhibited a lower antiarenaviral activity than A3 or PALA (data not shown). Likewise, the suppression of the A3 antiarenaviral activity in cells cultured in the presence of increasing concentrations of orotate provided additional support that A3 exerts its antiarenaviral activity by interfering with the pyrimidine biosynthesis pathway.
Combination therapy, using drugs that target different components of the virus life cycle, has proven to offer advantages over drug monotherapy against several viral infections, including HIV (55), hepatitis C virus (56), and influenza A virus (57). Ribavirin, currently the only antiviral drug used against arenaviruses, and A3 target different metabolic pathways within the cell. We therefore explored the value of a combination therapy with ribavirin and A3. Our results have raised the exciting possibility that combination treatment with ribavirin and A3, or other inhibitors of pyrimidine biosynthesis, can offer an alternative therapeutic approach to the current ribavirin-based monotherapy used to combat HF arenavirus infections. The generation of new drugs, as well as repurposing of already existing drugs, that target either purine and pyrimidine biosynthesis pathways, as represented by the recently FDA-approved drug teriflunomide (58), the active metabolite of leflunomide, expand the possibilities for combinatorial treatments targeting both biosynthetic pathways to increase the likelihood of controlling severe HF arenavirus infections.
ACKNOWLEDGMENTS
We thank Snezhana Dimitrova for excellent technical support. The JUNV NP SA02-BG12 monoclonal antibody was obtained through the NIAID Biodefense and Emerging Infectious Research Resources Repository (BEI Resources).
E.O.-R. is a Rochester Vaccine Fellowship recipient (2012), and his current support is provided by a postdoctoral Fellowship for Diversity and Academic Excellence from the Office for Faculty Development and Diversity at the University of Rochester. S.D. was supported by the Cellular, Biochemical and Molecular Sciences grant T32-GM-68411-8. Current S.D. support is provided by the HIV T32-AI-049815 grant. Research in the L.M.-S. laboratory is funded by the NIH grants RO1 AI077719, R21NS075611-01, and R03AI099681-01A1, the NIAID Centers of Excellence for Influenza Research and Surveillance (HHSN266200700008C), and The University of Rochester Center for Biodefense Immune Modeling (HHSN272201000055C). Research in the J.C.T. laboratory was supported by grants RO1 AI047140, RO1 AI077719, and RO1 AI079665 from NIH. J.M. is supported by an NIH grant, AI081773, and is a Damon Runyon-Rachleff Innovator supported by the Damon Runyon Cancer Research Foundation (DRR-09-10).
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Buchmeier MJ, Peters CJ, de la Torre JC. 2007. Arenaviridae: the viruses and their replication, p 1792–1827 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Rojek JM, Kunz S. 2008. Cell entry by human pathogenic arenaviruses. Cell. Microbiol. 10:828–835. 10.1111/j.1462-5822.2007.01113.x [DOI] [PubMed] [Google Scholar]
- 3.Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. U. S. A. 100:12978–12983. 10.1073/pnas.2133782100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. 2000. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 74:3470–3477. 10.1128/JVI.74.8.3470-3477.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 80:9192–9199. 10.1128/JVI.00555-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Sobrido L, Giannakas P, Cubitt B, Garcia-Sastre A, de la Torre JC. 2007. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 81:12696–12703. 10.1128/JVI.00882-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodrigo WW, Ortiz-Riano E, Pythoud C, Kunz S, de la Torre JC, Martinez-Sobrido L. 2012. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 86:8185–8197. 10.1128/JVI.07240-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carnec X, Baize S, Reynard S, Diancourt L, Caro V, Tordo N, Bouloy M. 2011. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 85:12093–12097. 10.1128/JVI.00429-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormick JB, Fisher-Hoch SP. 2002. Lassa fever, p 75–110 In Oldstone MB. (ed), Arenaviruses I, vol 262 Springer-Verlag, Berlin, Germany [Google Scholar]
- 10.Gunther S, Lenz O. 2004. Lassa virus. Crit. Rev. Clin. Lab. Sci. 41:339–390. 10.1080/10408360490497456 [DOI] [PubMed] [Google Scholar]
- 11.Enria DA, Briggiler AM, Sanchez Z. 2008. Treatment of Argentine hemorrhagic fever. Antiviral Res. 78:132–139. 10.1016/j.antiviral.2007.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isaacson M. 2001. Viral hemorrhagic fever hazards for travelers in Africa. Clin. Infect. Dis. 33:1707–1712. 10.1086/322620 [DOI] [PubMed] [Google Scholar]
- 13.Holmes GP, McCormick JB, Trock SC, Chase RA, Lewis SM, Mason CA, Hall PA, Brammer LS, Perez-Oronoz GI, McDonnell MK, Paulissen JP, Schonberger LB, Fisher-Hoch SP. 1990. Lassa fever in the United States. Investigation of a case and new guidelines for management. N. Engl. J. Med. 323:1120–1123. 10.1056/NEJM199010183231607 [DOI] [PubMed] [Google Scholar]
- 14.Mets MB, Barton LL, Khan AS, Ksiazek TG. 2000. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am. J. Ophthalmol. 130:209–215. 10.1016/S0002-9394(00)00570-5 [DOI] [PubMed] [Google Scholar]
- 15.Barton LL. 1996. Lymphocytic choriomeningitis virus: a neglected central nervous system pathogen. Clin. Infect. Dis. 22:197. [DOI] [PubMed] [Google Scholar]
- 16.Jahrling PB, Peters CJ. 1992. Lymphocytic choriomeningitis virus: a neglected pathogen of man. Arch. Pathol. Lab. Med. 116:486–488 [PubMed] [Google Scholar]
- 17.Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. 2008. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 358:991–998. 10.1056/NEJMoa073785 [DOI] [PubMed] [Google Scholar]
- 18.Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, Shieh WJ, Erickson BR, Bandy U, DeMaria A, Jr, Davis JP, Delmonico FL, Pavlin B, Likos A, Vincent MJ, Sealy TK, Goldsmith CS, Jernigan DB, Rollin PE, Packard MM, Patel M, Rowland C, Helfand RF, Nichol ST, Fishman JA, Ksiazek T, Zaki SR. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354:2235–2249. 10.1056/NEJMoa053240 [DOI] [PubMed] [Google Scholar]
- 19.Federal Register 2012. Possession, use, and transfer of select agents and toxins; biennial review. Final rule. Fed. Regist. 77:61083–61115 [PubMed] [Google Scholar]
- 20.Rodriguez M, McCormick JB, Weissenbacher MC. 1986. Antiviral effect of ribavirin on Junin virus replication in vitro. Rev. Argent. Microbiol. 18:69–74 [PubMed] [Google Scholar]
- 21.Snell N. 1988. Ribavirin therapy for lassa fever. Practitioner 232:432. [PubMed] [Google Scholar]
- 22.Kowdley KV. 2005. Hematologic side effects of interferon and ribavirin therapy. J. Clin. Gastroenterol. 39:S3–S8. 10.1097/01.mcg.0000145494.76305.11 [DOI] [PubMed] [Google Scholar]
- 23.McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont-Williams R. 1986. Lassa fever: effective therapy with ribavirin. N. Engl. J. Med. 314:20–26 [DOI] [PubMed] [Google Scholar]
- 24.Graci JD, Cameron CE. 2006. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 16:37–48. 10.1002/rmv.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linero FN, Sepulveda CS, Giovannoni F, Castilla V, Garcia CC, Scolaro LA, Damonte EB. 2012. Host cell factors as antiviral targets in arenavirus infection. Viruses 4:1569–1591. 10.3390/v4091569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sepulveda CS, Garcia CC, Fascio ML, D'Accorso NB, Docampo Palacios ML, Pellon RF, Damonte EB. 2012. Inhibition of Junin virus RNA synthesis by an antiviral acridone derivative. Antiviral Res. 93:16–22. 10.1016/j.antiviral.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 27.Olschlager S, Neyts J, Gunther S. 2011. Depletion of GTP pool is not the predominant mechanism by which ribavirin exerts its antiviral effect on Lassa virus. Antiviral Res. 91:89–93. 10.1016/j.antiviral.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 28.Moreno H, Gallego I, Sevilla N, de la Torre JC, Domingo E, Martin V. 2011. Ribavirin can be mutagenic for arenaviruses. J. Virol. 85:7246–7255. 10.1128/JVI.00614-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrei G, De Clercq E. 1993. Molecular approaches for the treatment of hemorrhagic fever virus infections. Antiviral Res. 22:45–75. 10.1016/0166-3542(93)90085-W [DOI] [PubMed] [Google Scholar]
- 30.Gowen BB, Wong MH, Larson D, Ye W, Jung KH, Sefing EJ, Skirpstunas R, Smee DF, Morrey JD, Schneller SW. 2010. Development of a new tacaribe arenavirus infection model and its use to explore antiviral activity of a novel aristeromycin analog. PLoS One 5(9):e12760. 10.1371/journal.pone.0012760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guillerm G, Guillerm D, Vandenplas-Vitkowski C, Glapski C, De Clercq E. 2003. Inactivation of S-adenosyl-L-homocysteine hydrolase with novel 5′-thioadenosine derivatives: antiviral effects. Bioorg. Med. Chem. Lett. 13:1649–1652. 10.1016/S0960-894X(03)00279-8 [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. 2011. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 108:5777–5782. 10.1073/pnas.1101143108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortiz-Riano E, Cheng BY, de la Torre JC, Martinez-Sobrido L. 2011. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J. Virol. 85:13038–13048. 10.1128/JVI.05834-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emonet SF, Seregin AV, Yun NE, Poussard AL, Walker AG, de la Torre JC, Paessler S. 2011. Rescue from cloned cDNAs and in vivo characterization of recombinant pathogenic Romero and live-attenuated Candid #1 strains of Junin virus, the causative agent of Argentine hemorrhagic fever disease. J. Virol. 85:1473–1483. 10.1128/JVI.02102-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flatz L, Bergthaler A, de la Torre JC, Pinschewer DD. 2006. Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proc. Natl. Acad. Sci. U. S. A. 103:4663–4668. 10.1073/pnas.0600652103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortiz-Riano E, Cheng BY, Carlos de la Torre J, Martinez-Sobrido L. 2013. Arenavirus reverse genetics for vaccine development. J. Gen. Virol. 94:1175–1188. 10.1099/vir.0.051102-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodrigo WW, de la Torre JC, Martinez-Sobrido L. 2011. Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. J. Virol. 85:1684–1695. 10.1128/JVI.02229-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emonet SE, Urata S, de la Torre JC. 2011. Arenavirus reverse genetics: new approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 411:416–425. 10.1016/j.virol.2011.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berridge MV, Tan AS. 1993. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 303:474–482 [DOI] [PubMed] [Google Scholar]
- 40.Urata S, Ngo N, de la Torre JC. 2012. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 86:4578–4585. 10.1128/JVI.06604-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179–1186. 10.1038/nbt.1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melamud E, Vastag L, Rabinowitz JD. 2010. Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 82:9818–9826. 10.1021/ac10211166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA. 2002. Malarial dihydroorotate dehydrogenase. Substrate and inhibitor specificity. J. Biol. Chem. 277:41827–41834. 10.1074/jbc.M206854200 [DOI] [PubMed] [Google Scholar]
- 44.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. 2005. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 280:21847–21853. 10.1074/jbc.M501100200 [DOI] [PubMed] [Google Scholar]
- 45.Kulkarni OP, Sayyed SG, Kantner C, Ryu M, Schnurr M, Sardy M, Leban J, Jankowsky R, Ammendola A, Doblhofer R, Anders HJ. 2010. 4SC-101, a novel small molecule dihydroorotate dehydrogenase inhibitor, suppresses systemic lupus erythematosus in MRL-(Fas)lpr mice. Am. J. Pathol. 176:2840–2847. 10.2353/ajpath.2010.091227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markland W, McQuaid TJ, Jain J, Kwong AD. 2000. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: a comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 44:859–866. 10.1128/AAC.44.4.859-866.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinschewer DD, Perez M, de la Torre JC. 2003. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 77:3882–3887. 10.1128/JVI.77.6.3882-3887.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emonet SF, Garidou L, McGavern DB, de la Torre JC. 2009. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc. Natl. Acad. Sci. U. S. A. 106:3473–3478. 10.1073/pnas.0900088106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans DR, Guy HI. 2004. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 279:33035–33038. 10.1074/jbc.R400007200 [DOI] [PubMed] [Google Scholar]
- 50.Knecht W, Loffler M. 1998. Species-related inhibition of human and rat dihydroorotate dehydrogenase by immunosuppressive isoxazol and cinchoninic acid derivatives. Biochem. Pharmacol. 56:1259–1264 [DOI] [PubMed] [Google Scholar]
- 51.Leyssen P, Balzarini J, De Clercq E, Neyts J. 2005. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J. Virol. 79:1943–1947. 10.1128/JVI.79.3.1943-1947.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno H, Grande-Perez A, Domingo E, Martin V. 2012. Arenaviruses and lethal mutagenesis: prospects for new ribavirin-based interventions. Viruses 4:2786–2805. 10.3390/v4112786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucas-Hourani M, Dauzonne D, Jorda P, Cousin G, Lupan A, Helynck O, Caignard G, Janvier G, Andre-Leroux G, Khiar S, Escriou N, Despres P, Jacob Y, Munier-Lehmann H, Tangy F, Vidalain PO. 2013. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog. 9(10):e1003678. 10.1371/journal.ppat.1003678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Traut TW, Jones ME. 1996. Uracil metabolism. UMP synthesis from orotic acid or uridine and conversion of uracil to beta-alanine: enzymes and cDNAs. Prog. Nucleic Acid Res. Mol. Biol. 53:1–78 [DOI] [PubMed] [Google Scholar]
- 55.Volberding PA, Deeks SG. 1998. Antiretroviral therapy for HIV infection: promises and problems. JAMA 279:1343–1344. 10.1001/jama.279.17.1343 [DOI] [PubMed] [Google Scholar]
- 56.Hotho DM, de Bruijne J, Spaan M, Treitel MA, Boonstra A, de Knegt RJ, Janssen HL, Reesink HW. 2013. Sustained virologic response after therapy with the HCV protease inhibitor narlaprevir in combination with peginterferon and ribavirin is durable through long-term follow-up. J. Viral Hepat. 20:e78–e81. 10.1111/jvh.12012 [DOI] [PubMed] [Google Scholar]
- 57.Haasbach E, Hartmayer C, Planz O. 2013. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro. Antiviral Res. 98:319–324. 10.1016/j.antiviral.2013.03.006 [DOI] [PubMed] [Google Scholar]
- 58.Oh J, O'Connor PW. 2013. An update of teriflunomide for treatment of multiple sclerosis. Ther. Clin. Risk Manag. 9:177–190. 10.2147/TCRM.S30947 [DOI] [PMC free article] [PubMed] [Google Scholar]