

Abstract
In primary effusion lymphoma (PEL) cells infected with latent Kaposi's sarcoma-associated herpesvirus (KSHV), the promoter of the viral lytic switch gene, Rta, is organized into bivalent chromatin, similar to cellular developmental switch genes. Histone deacetylase (HDAC) inhibitors (HDACis) reactivate latent KSHV and dramatically remodel the viral genome topology and chromatin architecture. However, reactivation is not uniform across a population of infected cells. We sought to identify an HDACi cocktail that would uniformly reactivate KSHV and reveal the regulatory HDACs. Using HDACis with various specificities, we found that class I HDACis were sufficient to reactivate the virus but differed in potency. Valproic acid (VPA) was the most effective HDACi, inducing lytic cycle gene expression in 75% of cells, while trichostatin A (TSA) induced less widespread lytic gene expression and inhibited VPA-stimulated reactivation. VPA was only slightly superior to TSA in inducing histone acetylation of Rta's promoter, but only VPA induced significant production of infectious virus, suggesting that HDAC regulation after Rta expression has a dramatic effect on reactivation progression. Ectopic HDACs 1, 3, and 6 inhibited TPA-stimulated KSHV reactivation. Surprisingly, ectopic HDACs 1 and 6 stimulated reactivation independently, suggesting that the stoichiometries of HDAC complexes are critical for the switch. Tubacin, a specific inhibitor of the ubiquitin-binding, proautophagic HDAC6, also inhibited VPA-stimulated reactivation. Immunofluorescence indicated that HDAC6 is expressed diffusely throughout latently infected cells, but its expression level and nuclear localization is increased during reactivation. Overall, our data suggest that inhibition of HDAC classes I and IIa and maintenance of HDAC6 (IIb) activity are required for optimal KSHV reactivation.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) is the etiologic agent of KS and other lymphoproliferative diseases. Epidemiologic studies suggest that reactivation of KSHV from latency is essential for progression to disease. Primary effusion lymphoma (PEL) cell lines have been the predominant model system for understanding the molecular basis of KSHV latency and reactivation (1–3). Treatment of PEL cells with histone deacetylase (HDAC) inhibitors (HDACis) activates productive viral reactivation, including viral gene expression, viral DNA replication, and release of mature virions (4–6).
HDACi compounds inhibit HDACs (also known as protein lysine deacetylates) (7). HDACs are a group of enzymes whose members largely share the ability to remove acetyl groups from ε-N-acetyl lysine amino acids in histones and other proteins (8). HDACs are critical regulators of both nuclear and cytoplasmic processes, including transcriptional initiation and elongation, protein stability, and multiprotein complex formation. The opposing actions of HDACs and histone acetyltransferases (HATs) converge positively and negatively with other histone modifications, such as methylation and phosphorylation, to establish, maintain, and modify the histone code (9–12). The histone code provides specificity for multiprotein complexes that read the code and bind to histones to modify the code and remodel chromatin, resulting in dynamic changes to chromatin structure.
Eighteen HDACs are classified into four groups on the basis of sequence phylogeny and function (7, 8, 13). The class I HDACs can be recruited to promoters as the enzymatic subunits of multiprotein transcriptional corepressor complexes called silencing mediator for retinoid and thyroid receptors/nuclear corepressor (SMRT/NCoR), corepressor of REST (CoREST), Mi-2/nucleosome remodeling deacetylase complex (Mi-2/NuRD), and SWI-independent 3 (Sin3). Class I HDACs regulate transcription by modulating acetylation of lysines in histone tails and, thus, chromatin compaction and remodeling. Class II HDACs are nuclear/cytoplasmic shuttling signal transducers that can regulate myogenic and lymphocytic differentiation, cytoskeletal dynamics, homologous recombination, and autophagy. Class III HDACs (sirtuins) generally participate in DNA metabolic responses to nutritional stimuli. The class IV HDAC11 regulates cellular origins of replication and transcription (14, 15).
Studies of the mechanism by which HDACis regulate KSHV reactivation have largely focused on the broad-spectrum inhibitor sodium butyrate (nBA). nBA induces expression of the KSHV lytic switch protein, Rta, leading to Rta-dependent production of mature viruses (5, 16–18). These data suggest that modulation of the chromatin in the Rta promoter is a critical mechanism for regulating the KSHV latent/lytic switch. During latency, Rta's 3.0-kb promoter is decorated with histones that bear modifications associated with both repression of transcription (histone H3 trimethylated at lysine 27 [H3K27-me3] and H3K9-me3) and activation of transcription (acetylation of histone H3 [H3K9/K14-ac] and H3K4-me3) (19–22). A similar pattern of bivalent histone modifications is seen on cellular genes that are transcriptionally repressed but are poised to respond to signals that switch on their transcription (23). Also during latency, Rta's promoter is physically linked to that of the major latency locus by the cellular cohesins and CTCF protein (19, 24).
nBA treatment of latently infected cells changes the histone modification patterns broadly on the KSHV genome, remodels nucleosomes, and alters the topology of the viral chromatin. On Rta's promoter, nBA treatment increases acetylation of histones H3 and H4 and decreases H3K27-me3 (5, 22, 25). nBA induction of Rta expression requires the cellular transcription factor Sp1 to bind DNA and recruit the CREB-binding protein (CBP) HAT and the chromatin remodeler Ini1/Snf5 (5, 25). Rta expression also corresponds to the repositioning of a nucleosome that occludes the Rta start site during latency (5). Furthermore, nBA treatment reduces CTCF and cohesin binding to Rta's promoter and disrupts the physical linkage of Rta to the major latency locus (19, 24). The cohesins repress Rta expression, as their depletion reactivates the virus, reduces histone modifications on its promoter, and results in increased binding of RNA polymerase II. Demethylation of H3K27-me3 or depletion of the histone methyltransferase EZH2 induces Rta expression to reactivate KSHV (22). The literature thus suggests that H3K27 must be continuously methylated to maintain KSHV latency and that the HDACi nBA reactivates the virus by increasing histone acetylation on the Rta promoter and reducing the ability of histone methyltransferases to modify H3K27.
Despite these seminal insights into chromatin dynamics associated with KSHV reactivation, the specific HDACs that regulate reactivation remain elusive. In this regard, nBA inhibits multiple HDAC classes (26), and HDACs 1, 5, and 7 remain bound to Rta's promoter regardless of nBA treatment (5). Further, it is unknown how non-HDACi chemicals that reactivate KSHV overcome HDAC-mediated repression. Finally, the postulated broad effects of HDACis on chromatin are not reflected in the response of the virus to nBA; single-cell assays suggest that KSHV does not respond equivalently to the chemical in all infected cells in a tissue culture population (16).
In this report, we demonstrate that valproic acid (VPA) is the most potent KSHV-reactivating HDACi among 7 HDACis with various specificities. The potencies of the HDACis in inducing production of infectious virus were not directly proportional to levels of acetylated histone H3 on Rta's promoter, suggesting that a major point of HDAC regulation of KSHV reactivation follows Rta expression. Nonetheless, reactivation was not achieved in 100% of cells. VPA effected different patterns of histone H3 modifications on different viral genes. Overexpression of HDACs revealed that the stoichiometries of HDACs 1, 3, and 6 were critical in regulating KSHV reactivation. Inhibition of class I HDAC was sufficient to reactivate KSHV, but optimal reactivation required inhibition of class IIa activity and maintenance of class IIb activity.
MATERIALS AND METHODS
Tissue culture, transfections, and HDACis.
Vero rKSHV.294, Vero rKSHV.219, 293 MSR tet-OFF, BCBL-1, and BC-3 cells were propagated as previously described (27–29). BCBL-1 cells were transfected by mixing 30 μg of total plasmid DNA with 3.0 × 106 cells, electroporating with a square-wave pulse of 250 V for 35 ms, and transferring the cells to T-25 flasks. Total DNA was normalized with empty pcDNA3.
Sources of HDAC inhibitors were as follows: VPA (catalog no. P4543), trichostatin A (TSA; catalog no. T-8552), nicotinamide (catalog no. N0636), sirtinol (catalog no. S7942), tubacin (catalog no. SML0065), and sodium butyrate (nBA; catalog no. B5887) were from Sigma; HC toxin (sc-200884) was from Santa Cruz Biotechnology; and MC 1568 was a gift from Lucia Altucci (30).
Plasmids and bacmid.
pcDNA3, pcDNA3-FLg50, and pcDNA-FLc50 were previously described (16, 31–33). HDAC1-FLAG, HDAC3-FLAG, HDAC6-FLAG, HDAC7-FLAG, and SIRT1-FLAG (34–36) (gifts of Eric Verdin) and H2b-green fluorescent protein (GFP) (37) (a gift of Geoff Wahl) were obtained from the AddGene repository (plasmids 13820, 13819, 13823, 13824, 13812, and 11680, respectively). All plasmids were verified by DNA sequencing and restriction digestion. Bacmid BAC36 was grown and purified as described previously (38).
Infectious virus quantitation.
Vero-rKSHV.294 cells were seeded at a density of 2 × 105 cells/well in a 6-well plate and grown for 24 h. Cells were left untreated or treated with 12-O-tetradecanoyl phorbol-13-acetate (TPA; 20 ng/ml) or HDACis for 24 h. The medium was replaced with fresh supplemented Dulbecco modified Eagle medium and allowed to incubate for 48 h. Medium was collected and transferred to 293 MSR tet-OFF cells, which had been plated 24 h previously at a density of 2 × 105 cells/well in a 6-well plate. After incubation for 72 h, the medium was transferred to a 96-well plate to measure secreted alkaline phosphatase (SeAP) using a Great EscAPe SeAP fluorescence detection kit (Clontech).
Virion DNA preparation.
BCBL-1 cells (2 × 107) in T175 flasks or Vero-rKSHV.294 cells in 150-mm dishes were treated with VPA (1.0 mM), TSA (2 μM), or TPA (20 ng/ml). At 5 days posttreatment, cell supernatants were separated from cells and debris by centrifugation at 1,000 rpm for 5 min, followed by centrifugation at 3,000 rpm for 30 min and then passage through a 0.45-μm-pore-size syringe filter. To pellet viral particles, pure supernatants were centrifuged at 15,000 × g for 2 h in a swinging bucket rotor. Supernatants were carefully aspirated, and viral pellets were resuspended in 120 μl of solution I (40 mM Tris-HCl [pH 7], 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2). Free, unencapsidated DNA was degraded by addition of 1 unit of RQ1 DNase (Promega) for 30 min at 37°C. To inactivate the DNase, 20 mM EDTA was added and the mixture was incubated at 80°C for 5 min. Virions were lysed by adding 120 μl 2× virion lysis buffer (40 mM Tris-HCl [pH 7], 20 mM EDTA, 200 mM NaCl, 1% SDS) and 100 μg/ml proteinase K, and the mixture was incubated overnight at 37°C. Viral DNA was purified by extraction 2 times with phenol-chloroform and 1 time with chloroform and then precipitation with 0.1 volume 3 M sodium acetate, 2 volumes 100% ethanol, and 200 ng glycogen. Each precipitated viral DNA pellet was resuspended in 100 μl distilled water.
Virion DNA quantitation.
For PEL virions, equal volumes of virion-encapsidated DNA were electrophoresed on 0.7% agarose, stained by ethidium bromide, and visualized and quantitated using the ImageJ program (39). For Vero-rKSHV.294 virions, 50 μl viral DNA was denatured by adding an equal volume of 0.2 N NaOH, and the mixture was incubated for 10 min at room temperature (RT). Eight hundred microliters of 20× SSC (3 M sodium chloride, 300 mM sodium citrate) was added to each denatured viral DNA, and DNAs were suctioned onto a presoaked Hybond XL membrane (Amersham Biosciences) in a dot blotting apparatus. Each well was rinsed twice with 500 μl 20× SSC. The membrane was dried and placed on a UV chamber to cross-link the nucleic acid to the membrane. For prehybridization, the membrane was incubated with Church buffer (1% bovine serum albumin, 1 mM EDTA, 0.5 M phosphate buffer, 7% SDS) for 2 h at 68°C. The KSHV Rta probe was labeled by random priming to incorporate [32P]dCTP. The probe (106 cpm) was boiled and added to the prehybridization solution, and the mixture was then incubated at 68°C overnight. The membrane was washed twice with prewarmed Church wash buffer (0.04 M phosphate buffer, 1% SDS, 0.001 M EDTA, 0.014% phosphoric acid) for 1 h each time at 68°C. The membrane was dried at RT and then exposed to autoradiography film or a phosphorimager screen.
Reactivation quantitation by immunofluorescence in PEL cells.
BCBL-1 cells (3.0 × 106) were left untreated or treated in quadruplicate with VPA (1.0 mM) or in duplicate with TPA (20 ng/ml). In transfected BCBL-1 cells, TPA was added at 24 h postelectroporation. At 72 h posttreatment, 2.5 × 105 cells were adhered to polylysine-coated glass slides within 0.5-in.-diameter circles drawn with a Pap pen. Reactivation was scored by quantitating the percentage of cells expressing the K8.1 protein by immunofluorescence.
Immunofluorescence.
Cells were washed twice with 1× phosphate-buffered saline (PBS), and 2.5 × 105 cells were adhered for 30 min to polylysine-coated glass slides within 0.5-in.-diameter circles drawn with a Pap pen. Slides were fixed in 50% methanol–50% acetone for 5 min at −20°C and then air dried under a fume hood for 5 min. Cells were blocked in blocking buffer (3% bovine serum albumin [BSA], 1% glycine, PBS) for 30 min. Primary antibodies to K8.1 (diluted 1:1,000; Santa Cruz Biotechnology) and HDAC6 (diluted 1:50; Santa Cruz Biotechnology) were diluted in blocking buffer and incubated with the slides for 1 h at RT. The slides were washed twice for 30 min in PBS plus 0.4% Tween 20 and once for 30 min in 1× PBS. The secondary antibodies fluorescein isothiocyanate-conjugated goat antimouse antibody (diluted 1:500; ICN) and DyLight 550-conjugated goat antirabbit antibody (diluted 1:200; Thermo Scientific) were diluted in blocking buffer and incubated with the slides for 1 h at RT. The slides were washed as described above, and coverslips were mounted and sealed as described previously (33). Images were captured using a Zeiss Axiovert 200M with z frames start and a Nikon A1 confocal microscope point scanner with NIS Element AR (version 3.2) software.
Western blotting.
Cells were lysed in 100 μl radioimmunoprecipitation assay buffer (50 mM Tris, pH 8.0, 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma) with shaking at 4°C for 20 min. The debris was cleared by centrifugation, supernatants were transferred to new tubes, and total protein concentrations were determined by Bradford assays (Bio-Rad). Five hundred micrograms of each lysate was boiled in 1× Laemmli buffer and separated by SDS-PAGE. Proteins were electrophoretically transferred to a nitrocellulose membrane in transfer buffer (25 mM Tris base, 190 mM glycine, 20% methanol). The membrane was blocked for 1 h in PBS-T (PBS plus 0.1% Tween 20) containing 5% skim milk and washed with PBS-T. The membrane was incubated with polyclonal anti-FLAG antibody (diluted 1:320, Sigma) or anti-α-tubulin antibody (diluted 1:1,000; Sigma) at RT for 1 h. The membrane was washed twice with PBS-T for 30 min and one time with 1× PBS for 30 min. It was then incubated with secondary HRP-conjugated antimouse or antirabbit antibody (diluted 1:5,000; Bethyl) at RT for 1 h and washed as described above. The secondary antibody was detected by enhanced chemiluminescence (Thermo Scientific).
Chromatin immunoprecipitation (ChIP) and real-time PCR.
BC-3 cells (8.0 × 106) were stimulated with sodium butyrate (2.5 μM) and VPA (1.0 mM), alone and together, or left untreated for 24 h. Cells were treated with 1% formaldehyde at RT for 20 min to cross-link the DNA. Chromatin from lysed cells was sheared by a Bioruptor apparatus (Diagenode), using four 8-min cycles of 30 s on and 30 s off. After cell extracts were precleared with 60 μl of 50% (vol/vol) protein A-agarose at 4°C for 1 h, protein/DNA complexes were immunoprecipitated with 4 μg of anti-H3k9/k14-ac (catalog no. 06-599), anti-H3k9-me3 (catalog no. 17-625), anti-H3k4-me3 (catalog no. 04-745), and anti-H3k27-me3 (catalog no. 07-449) (all from Millipore), anti-GFP (Santa Cruz Biotechnology), or rabbit immunoglobulin G (IgG; Sigma) at 4°C overnight with nutation. Immunoprecipitated complexes were collected with 60 μl protein A-agarose for 1 h at 4°C. Protein/DNA complexes were washed one time with 1 ml low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl) at RT for 10 min, one time with 1 ml high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl) at RT for 10 min, one time with LiCl wash buffer (0.25 M LiCl, 1% Nonidet P-40, 1% Na-deoxycholate, 1 mM EDTA, 10 mM Tris-HCl) at RT for 10 min, and two times with 1× TE (Tris-EDTA) buffer at RT for 10 min. Chromatin was eluted from the beads in 210 μl elution buffer (50 mM Tris-HCl, 10 mM EDTA, 1% SDS) for 30 min at 37°C. Eight microliters of 5 M NaCl was added to the supernatant, and chromatin was de-cross-linked overnight at 65°C. Samples were diluted by adding 200 μl TE buffer. RNase A (1.6 μl; 50 mg/ml) was added to the samples, and the mixtures were incubated for 2 h at 37°C. Seven microliters of CaCl2 solution and 4 μl of proteinase K (20 mg/ml) were added, and the mixture was incubated for 1 h at 55°C. Supernatants were collected by centrifugation for 1 min at 6,000 rpm. DNA was purified by successive phenol-chloroform-iso-amyl alcohol (IAA) and chloroform-IAA extractions. DNA was precipitated with 1 ml 100% ethanol, 24 μl NaCl (5 M), and 1.5 μl glycogen (20 mg/ml) at −80°C for at least 30 min. After centrifugation for 15 min at 14,000 rpm, pellets were washed once with 1 ml 70% ethanol, centrifuged again, and dried in a hood. Immunoprecipitated chromatin was resuspended in 50 μl of double-distilled H2O and analyzed by real-time PCR, and amplicons were detected by SYBR green (Qiagen QuantiTec SYBR green PCR kit) or molecular beacons (Applied Biosystems AmpliTaq Gold polymerase). Primer sequences (5′ to 3′) were as follows: TCCGAGGTAATGTGCTCTATGAAG (forward) and ACAGACACCGGAGCAATACCC (reverse) (p50-800), AATGCACGACAACTCCCTCT (forward) and GACAACCGACTGGCAAAAAT (reverse) (ORF21), TGCATCAGCTGCCTAACCCAG (forward) and CGCCTAATAGCTGCTGCTACGG (reverse) (K6), and ATGTACGCTAGGGTCTCCCC (71413 forward) and CTGCTCCCACCTACACCATT (71700 reverse). The cycling parameters were 10 min at 95°C, followed by 40 cycles consisting of a 20-s denaturation at 95°C, a 30-s annealing at 60°C, and a 20-s extension at 72°C. Molecular beacon sequences were as follows (target sequences are underlined): GCGATCTCAAGCTAAGCTAAGCTCCCGCCGGATCGCG (p50-800) and GCGATCAGACCTCTCATACGGCACCCGATCGCG (ORF21). Fold (n-fold) enrichment was calculated by the ΔΔCT threshold cycle (CT) method (40), with specific antibody minus IgG used as the first ΔCT and VPA minus mock used as the second ΔCT. The positive-control template was BAC36, and the negative control lacked template. PCR products were visualized by the use of an agarose gel and ethidium bromide.
RESULTS
HDACis vary in their abilities to reactivate KSHV from latency.
HDACs are classified into four groups on the basis of their sequence phylogeny and function (Table 1). To determine which HDAC classes regulate KSHV latency and reactivation, we treated infected cells with a series of HDACis that have various specificities (Table 2) (7, 26, 30, 41). We measured KSHV reactivation in the Vero-rKSHV.219 cell line, which enables easy quantitation by scoring induction of an early (E) PAN promoter reporter cassette that drives expression of the red fluorescent protein (RFP) (29). We determined the optimal HDACi concentrations by scoring the percentage of RFP-positive cells in duplicate cultures of 3 × 105 cells that were treated with each HDACi for a 10-day period. Our criterion for determining the upper limits of HDACi concentrations was to exclude those doses that killed 100% of cells without inducing E-gene expression in greater than 10% of the cells within 6 days of addition. Cell death in these cases was the result of nonspecific HDACi toxicity rather than lysis by viral reactivation. Under these conditions, three of the seven HDACis were sufficient to reactivate KSHV (Table 2). Valproic acid (VPA), trichostatin A (TSA), and HC toxin reactivated virus in 73.5, 7.8, and 2.3% of cells, respectively. The dose-dependent response of the virus differed for VPA and TSA: reactivation generally increased over time for all 3 VPA doses (Fig. 1A) but for only 1 TSA dose (Fig. 1B). The positive control, TPA plus sodium butyrate (nBA), induced RFP expression in 12.3% of cells (not shown). These data suggest that inhibition of HDAC class I alone is sufficient to reactivate KSHV from latency but inhibition of classes I and IIa together is optimal.
TABLE 1.
Classification of HDACs
| Class | HDAC(s) |
|---|---|
| I | 1–3, 8 |
| IIa | 4, 5, 7, 9 |
| IIb | 6, 10 |
| III | Sirtuins 1–7 |
| IV | 11 |
TABLE 2.
HDACi-specific reactivation of KSHV from latency
| HDACi | Concn |
% maximum reactivation (SD)c | HDAC class specificity | |
|---|---|---|---|---|
| Range testeda | Optimumb | |||
| HC toxin | 2.0–10.0 nM | 10.0 nM | 2.3 (0.1) | I |
| VPA | 0.5–5.0 mM | 1.0 mM | 73.5 (1.4) | I, IIa, IIb (except HDAC 6) |
| TSA | 0.1–2.0 μM | 2.0 μM | 7.8 (0.8) | I, IIb |
| MC 1658 | 1.0–5.0 μM | 5.0 μM | 0.7 (0.1) | IIa |
| Nicotinamide | 100.0–900.0 μM | 300.0 μM | 0.3 (0.0) | III |
| Sirtinol | 5.0–500.0 μM | 50.0 μM | 0.5 (0.2) | III |
| Mock | 0.2 (0.0) | |||
HDACis were added to the medium of duplicate cultures of 3 × 105 Vero-rKSHV.219 cells in 35-mm dishes cells at the indicated concentrations. Reactivation was scored by quantitating the percentages of GFP-positive (total) cells that were also RFP positive (in which the E gene was induced) at 0 to 10 days following addition of the HDACis.
Optimal HDACi concentrations.
Maximum reactivation efficiencies.
FIG 1.
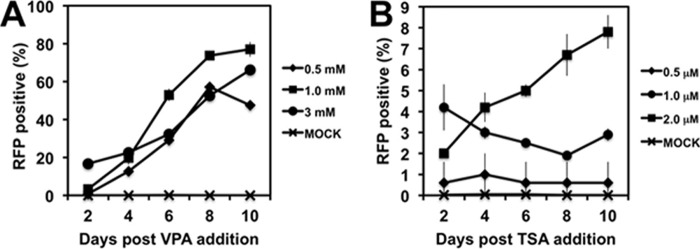
Reactivation of KSHV by treatment of infected cells with VPA (A) or TSA (B) at the indicated concentrations. See Table 2 for a description of the protocol and analysis.
To compare E-gene induction with the level of production of infectious KSHV, we tested the effects of the HDACis on a reporter KSHV. In this system, Vero cells were uniformly infected with a recombinant KSHV that contained the secreted alkaline phosphatase (SeAP) gene under the control of a tetracycline-responsive promoter (28). If the complete viral reactivation program is induced, the cells release mature virus to the medium. The medium was then transferred to uninfected 293 MSR tet-OFF reporter cells that constitutively express the tetracycline transactivator, and infectious virus was quantitated by measuring SeAP. These experiments demonstrated a dramatic difference between the effects of VPA and TSA on KSHV reactivation. VPA treatment resulted in a 230-fold increase in infectious virus in the cell medium after 3 days, while TSA treatment yielded only about a 5-fold increase (Fig. 2A). Differential viral production by VPA and TSA was also observed by Southern/dot blotting of encapsidated viral DNA purified from the Vero cell medium (Fig. 2B). While the dot blots agreed with the relative superiority of VPA induction, the amount of VPA-induced virus detected was less dramatic (ca. 7-fold) than the amount of SeAP detected.
FIG 2.
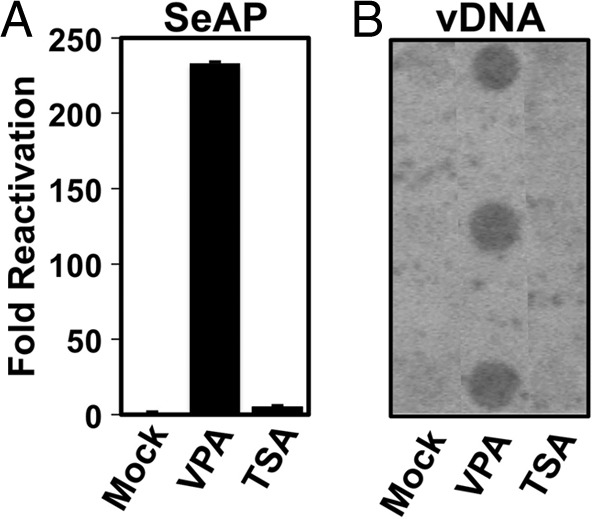
Production of infectious virus by VPA treatment of infected cells. Infected Vero rKSHV.294 cells were treated with 1 mM VPA or 2 μM TSA, and infectious virus in the cell medium was quantitated by (A), SeAP assay from serially infected 293 MSR tet-OFF cells, as described in Materials and Methods. SeAP activity was normalized to that for negative-control cells, which had been incubated with supernatants from Vero rKSHV.294 cells treated with solvent only (mock). Alternatively, (B) virion-associated DNA (vDNA) was purified from triplicate infected cultures, and equal volumes were quantitated by dot blotting using an Rta-specific probe.
TSA inhibits VPA-stimulated reactivation.
Despite the robust induction of virus production by VPA treatment, only about 70% of the infected cells expressed productive cycle gene products. To attempt to increase percent reactivation, we added a VPA boost in fresh medium 4 h after the initial treatment. The VPA boost had little effect on the percentage of cells expressing RFP (Fig. 3).
FIG 3.
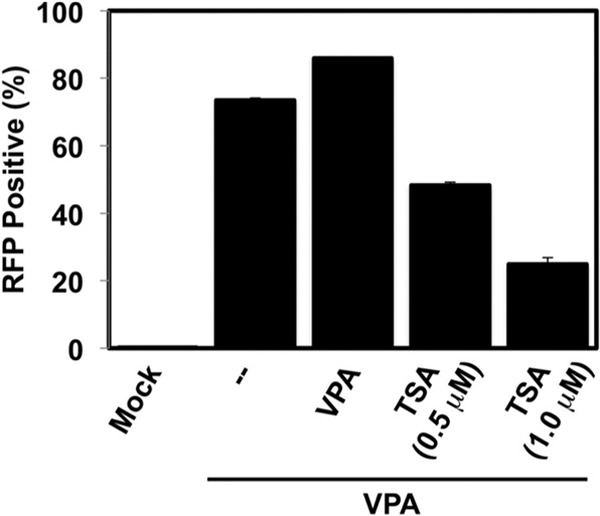
TSA inhibits VPA stimulation of KSHV reactivation. Vero rKSHV.219 cells were treated with 1 mM VPA alone or with solvent (mock) for 4 h. The medium was changed, and an additional 1 mM VPA was added alone or together with 0.5 μM or 1.0 μM TSA, as indicated. Reactivation was scored as described in Table 2.
Since TSA alone also induced E-gene expression (Table 2), we asked whether the combination of VPA and TSA would cooperate to reactivate KSHV. TSA caused a dose-dependent reduction in the amount of E-gene expression induced by VPA (Fig. 3). TSA at 0.5 and 1.0 μM reduced the proportion of RFP-expressing cells from 73% to 52% and 25%, respectively (Fig. 3). Further, the combination of 1 mM VPA and 2.0 μM TSA killed the entire cell population within 2 days of treatment (not shown). Together with the results presented in Fig. 2, we concluded from the results in Fig. 3 that, although TSA alone is sufficient to induce E-gene expression, it inefficiently induces complete reactivation and inhibits VPA-stimulated reactivation.
HDACi reactivation specificity is similar in Vero and PEL cells.
Since the major KSHV reservoir in vivo is B cells, we tested the response of the virus to HDACis in infected PEL cells by measuring the production of mature viruses. VPA induced a nearly 20-fold increase in the amount of encapsidated viral DNA released to the medium, while TSA had no effect (Fig. 4A and B). To confirm that the quantitated bands represented KSHV DNA (Fig. 4A), we compared the HindIII and NcoI restriction patterns of the virion-associated DNA with the pattern of BAC36, and the patterns matched each other (Fig. 4C and not shown). Interestingly, despite the dramatic difference between VPA and TSA in inducing virus production in Vero and BCBL-1 cells, the difference between VPA and TSA in inducing E-gene expression was not as dramatic.
FIG 4.
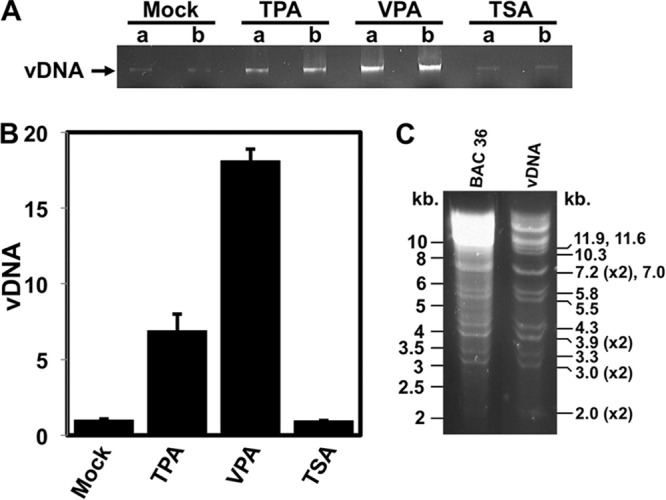
HDAC inhibitor-specific reactivation of KSHV. The indicated inducers were added to the medium of BCBL-1 cells. Reactivation was scored 5 days following treatment by quantitating equal volumes of virion-encapsidated DNA by agarose gel electrophoresis and ethidium bromide staining (A) and by ImageJ densitometry (B). Fold reactivation was calculated by comparison to the level for solvent (mock)-treated cells, which was normalized to 1-fold. In panel A, lanes a and b denote experimental replicates. (C) Purified virion-associated DNAs (from the experiment whose results are shown in panel A) and BAC36 (control) were digested with HindIII and compared by agarose gel electrophoresis and ethidium bromide staining. Numbers to the left indicate the sizes of the bands in DNA molecular size standards. Numbers to the right indicate the sizes of the HindIII restriction fragments of the KSHV genome.
Histone acetylation on the ORF50 promoter varies relative to the magnitude of KSHV reactivation.
nBA-stimulated KSHV reactivation is accompanied by an increase in histone H3 acetylation on ORF50's promoter (5, 22, 25). To determine whether VPA caused a similar change in histone H3, we performed chromatin immunoprecipitation and quantitated 2 previously identified promoter elements by quantitative PCR (ChIP/qPCR). At position −837/−720, histone H3K9/K14 acetylation was increased by a greater magnitude in response to VPA than in response to TSA (Fig. 5A). However, at position −170/+188, histone H3 acetylation was changed equivalently in response to both HDACis. Therefore, the relative transcriptional permissiveness of the Rta promoter varies in proportion to the relative magnitudes of reactivation stimulated by these HDACis in a site-specific manner. However, this ∼2.5-fold difference in induction of histone acetylation between VPA and TSA was much less dramatic than the difference in induction of E-gene expression (Fig. 1) and viral production (Fig. 2 and 4) between VPA and TSA. Similar, small differences between VPA- and TSA-induced H3 acetylation from BCBL-1 and Vero cells were observed in ChIP assays, demonstrating a general phenomenon in KSHV-infected cells (Fig. 5B).
FIG 5.
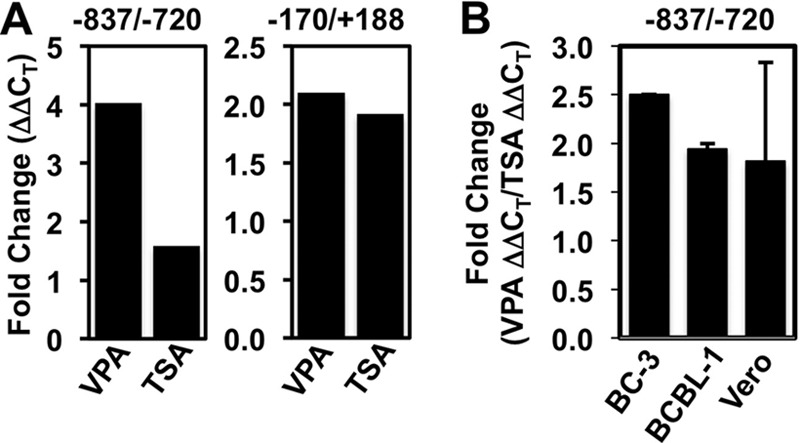
VPA is slightly better than TSA in inducing histone acetylation on the Rta promoter. (A) BC-3 cells were treated with the indicated HDACi or mock treated. ChIP assays were performed using anti-H3K9/K14-ac and total rabbit IgG (negative-control) antibodies. Chromatin immunoprecipitated DNA was quantitated by real-time PCR using primer/molecular beacon sets (−837/−720) or SYBR green (−170/+188) specific for the indicated regions of the Rta promoter. Fold changes were calculated using the ΔΔCT method and represent the increased ChIP of H3-acetylated chromatin relative to that of IgG in the presence or absence of the inducing chemical. (B) The indicated KSHV-infected cells were treated with VPA or TSA or left untreated. ChIP assays were performed as described for panel A, and the fold changes in H3 acetylation at the −837/−720 Rta promoter element were determined. The fold changes for VPA and TSA were calculated individually using the ΔΔCT method and represent the increased ChIP of H3-acetylated chromatin relative to the ChIP of IgG in the presence or absence of the inducing chemical. Then, the fold increase induced by VPA was divided by the fold increase induced by TPA for each cell line and plotted. Data represent the fold increase of acetylation induced by VPA divided by that induced by TSA; the results in the BC-3 bar were calculated from the data shown in panel A.
VPA increases euchromatin markers and decreases heterochromatin markers on the Rta promoter.
We extended the ChIP/qPCR analysis to additional histone modifications and included the bodies of two KSHV genes. At the 2 positions of the ORF50 promoter, VPA stimulation resulted in increases to the 2 transcriptionally permissive histone modifications (H3K9/K14-ac and H3K4-me3) and decreases in the 2 transcriptionally repressive histone modifications (H3K9-me3 and H3K27-me3; Fig. 6). While these data agree with the effects of nBA described in previous publications, the 15-fold increase in H3K4-me3 at −827/−720 is greater than the increase reported elsewhere (5, 22, 25).
FIG 6.
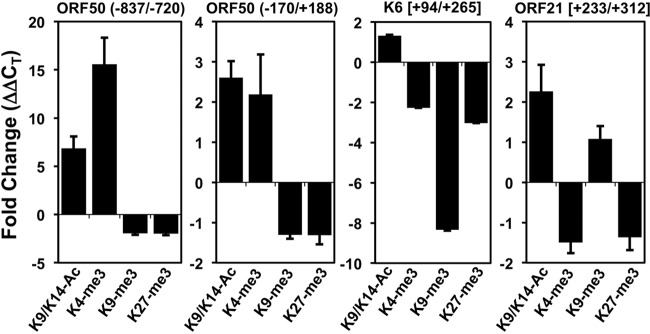
VPA increases euchromatin markers and decreases heterochromatin markers on the ORF50/Rta promoter. ChIP assays were performed with BC-3 cells using antibodies to various modifications of histone H3 (as shown) and rabbit IgG (negative control). Fold changes were calculated using the ΔΔCT method and represent the change in ChIP of each H3 modification relative to the ChIP of IgG in the presence or absence of the inducing chemical. Numbers in parentheses refer to the base pair positions relative to the ORF50 transcription start site. Numbers in brackets refer to the base pair positions relative to the translational start sites.
During latency, Rta's promoter, but not the body of ORF21, contains bivalent chromatin co-occupied by histones bearing transcriptionally activating and repressing modifications (22). We observed that the VPA-induced chromatin modification of Rta's promoter also differed from that of the ORF21 and K6 genes. The K6 gene showed decreases in only H3K4-me3, H3K9-me3, and H3K27-me3, while the ORF21gene showed an increase in H3K9/K14-ac and decreases in H3K4-me3 and H3K27-me3. Overall, the data demonstrate that VPA treatment leads to diverse changes to histone modifications at different loci in the KSHV genome during reactivation. VPA's effect on histone modification in ORF50 was qualitatively but not quantitatively similar to that described in previous reports of studies that used nBA (5, 22, 25).
Overexpression of HDACs 1, 3, and 6 inhibits KSHV reactivation.
To identify particular members of each HDAC class that participated in reactivation, we transfected expression vectors for individual HDACs into PEL cells and tested whether they could block TPA-induced reactivation. We employed TPA stimulation since TPA is not a direct HDAC inhibitor. We quantitated reactivation by scoring induction of the true late KSHV protein K8.1, whose expression depends upon Rta induction and prior DNA synthesis (31). We scored reactivation in each transfected cell population by dividing the percentage of K8.1-positive cells with TPA treatment by the percentage of K8.1-positive cells without TPA treatment for each plasmid. Overexpression of HDACs 1, 3, and 6 all potently inhibited TPA-induced reactivation, while HDAC7 and SIRT1 had no effect (Fig. 7A). Expression of all proteins was confirmed by Western blotting (Fig. 7B). For FLAG-HDAC7, we detected two protein bands that were not present in empty vector-transfected cells (Fig. 7B, right). The predicted molecular mass of HDAC7 is 100 kDa, which corresponds to the faster-migrating band; since we confirmed the DNA sequence of the HDAC7 clone, the identity of the slower protein band is unknown. Differential transfection efficiency did not account for the different reactivation phenotypes of the ectopic HDACs or the weaker expression of HDAC7 (not shown); we consistently achieved a 20 to 25% transfection efficiency with our PEL electroporation method (an example of the results is shown in Fig. 7C).
FIG 7.

Ectopic HDACs 1, 3, and 6 inhibit TPA-induced KSHV reactivation. (A) BCBL-1 cells were electroporated with 30 μg plasmids expressing the indicated HDACs or empty vector (Vec). Cells either were left untreated or were treated with TPA at 24 h postelectroporation. At 72 h posttreatment, reactivation was scored by measuring the percentage of cells expressing K8.1 by immunofluorescence; fold reactivation was scored by dividing the percentage of K8.1-positive cells transfected with each plasmid and treated with TPA by the percentage of identically transfected cells not treated with TPA. Cells were transfected with 30 μg of each plasmid. (B) Cellular protein extracts corresponding to the electroporations whose results are shown in panel A were tested by SDS-PAGE/Western blotting for expression of the indicated proteins. Asterisks in the right panel, ectopically expressed proteins. (C) BCBL-1 cells were electroporated with 30 μg histone H2b-GFP-expressing plasmid, fixed, and stained with DAPI (4′,6-diamidino-2-phenylindole) at 72 h postelectroporation. Using fluorescence microscopy, transfection efficiency was quantitated by dividing the number of GFP-positive cells by the total number of DAPI-positive cells. A representative field is shown in gray scale. (D) BCBL-1 cells electroporated with 10, 20, or 30 μg plasmids expressing the indicated HDACs were treated with TPA, and reactivation was scored as described for panel A.
HDAC1 repression was dose dependent and culminated in about a 50% reduction, while HDAC3 reached maximal repression at the lowest input plasmid amount (Fig. 7D, left and center). HDAC6 repression was more complex and varied nonproportionally to the input plasmid amount (Fig. 7D, right).
Overexpression of HDACs 1 and 6 is sufficient to reactivate KSHV.
When we expressed each of the HDACs ectopically in PEL cells, we noticed that HDACs 1 and 6 also increased the expression of K8.1 in the absence of any chemical treatment (Fig. 8A). Conversely, ectopic HDAC7 and SIRT1 did not affect K8.1 expression. To determine whether this effect was dose responsive, we repeated the experiment by titrating the HDAC1, 3, and 6 expression vectors in PEL electroporations. HDACs 1 and 6, but not HDAC3, demonstrated a dose-dependent increase in induction of K8.1 (Fig. 8B and not shown), suggesting that their expression was sufficient to reactivate the virus from latency. We stress, however, that the 3.5- to 5-fold increases in K8.1 expression induced by HDACs 1 and 6 were significantly less than those induced by similar amounts of the KSHV Rta plasmid, which were 12- to 15-fold in these experiments (not shown).
FIG 8.
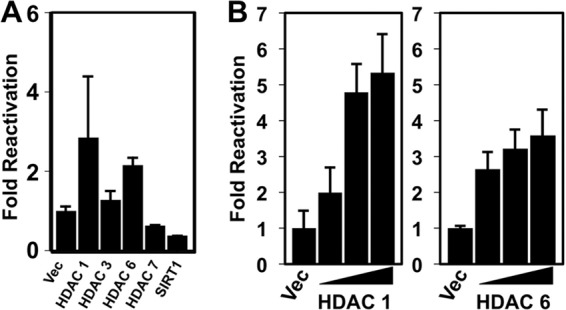
Ectopic HDACs 1 and 6 stimulate KSHV reactivation. BCBL-1 cells were electroporated with plasmids expressing the indicated HDACs or empty vector. At 72 h postelectroporation, reactivation was scored by measuring the percentage of cells expressing K8.1 by immunofluorescence; fold reactivation was scored by dividing the percentage of K8.1-positive cells transfected with each plasmid by the percentage of K8.1-positive cells transfected with empty expression vector (Vec). Cells were transfected with 30 μg of each plasmid (A) or 10, 20, and 30 μg of the indicated plasmids (B).
The HDAC6-specific inhibitor tubacin inhibits VPA-stimulated reactivation.
Our data suggested that HDAC6 might have both positive and negative effects on KSHV reactivation. Interestingly, VPA was more potent than TSA in inducing viral E-gene expression and complete viral reactivation, and TSA inhibited VPA-stimulated reactivation. The respective HDAC specificities of VPA and TSA overlap with the single exception of HDAC6: only TSA and not VPA inhibits HDAC6 (26). We employed the HDAC6-specific inhibitor tubacin (41) to determine whether it was sufficient to block VPA-stimulated reactivation in PEL cells. VPA alone induced a 60-fold increase in K8.1 expression relative to that in mock-treated cells, while tubacin alone had no effect (Fig. 9). However, adding VPA and tubacin simultaneously reduced K8.1 induction 15-fold over that from mock treatment. These data show that inhibiting HDAC6 reduces VPA-stimulated reactivation.
FIG 9.
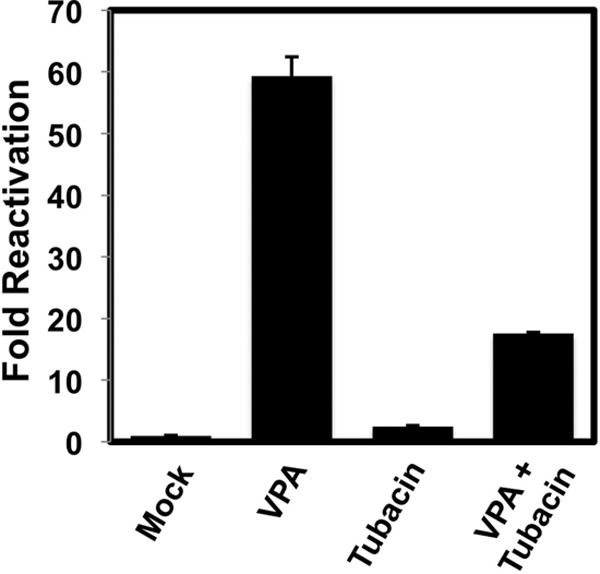
The HDAC6-specific inhibitor tubacin inhibits VPA-stimulated reactivation of KSHV. BCBL-1 cells were treated with the indicated HDACis. At 72 h posttreatment, reactivation was scored by measuring the percentage of cells expressing K8.1 by immunofluorescence; fold reactivation was scored by dividing the percentage of K8.1 positive cells treated with HDACi by the percentage of mock-treated (Mock) K8.1-expressing cells.
The subcellular localization of HDAC6 is altered by signals that reactivate KSHV from latency.
The best-characterized role for HDAC6 is as a cytoplasmic regulator of autophagy (42, 43). In that role, HDAC6 deacetylates tubulin to promote transport of misfolded proteins to the autophagosome. A few examples have emerged, however, in which shuttling of HDAC6 to the nucleus or perinuclear area regulates nuclear processes (44–46). To begin to understand HDAC6's role in KSHV reactivation, we observed its subcellular localization by indirect immunofluorescence in VPA-treated PEL cells. HDAC6 was expressed in all PEL cells, but its localization differed in individual cells. In most cells, HDAC6 stained with a diffuse, cytoplasmic pattern (Fig. 10A, unfilled arrow). However, in cells in which virus was reactivating, indicated by K8.1 staining and solid arrows, HDAC6 stained more intensely and was usually concentrated in nuclear puncta or sometimes in perinuclear puncta (Fig. 10A, solid arrows). In some cells in which HDAC6 was primarily nuclear, a small fraction could be detected in the cytoplasm (Fig. 10B; see Video S1 and Video S2 in the supplemental material). These data demonstrate that HDAC6 relocalizes into or against the nuclei when KSHV reactivates.
FIG 10.
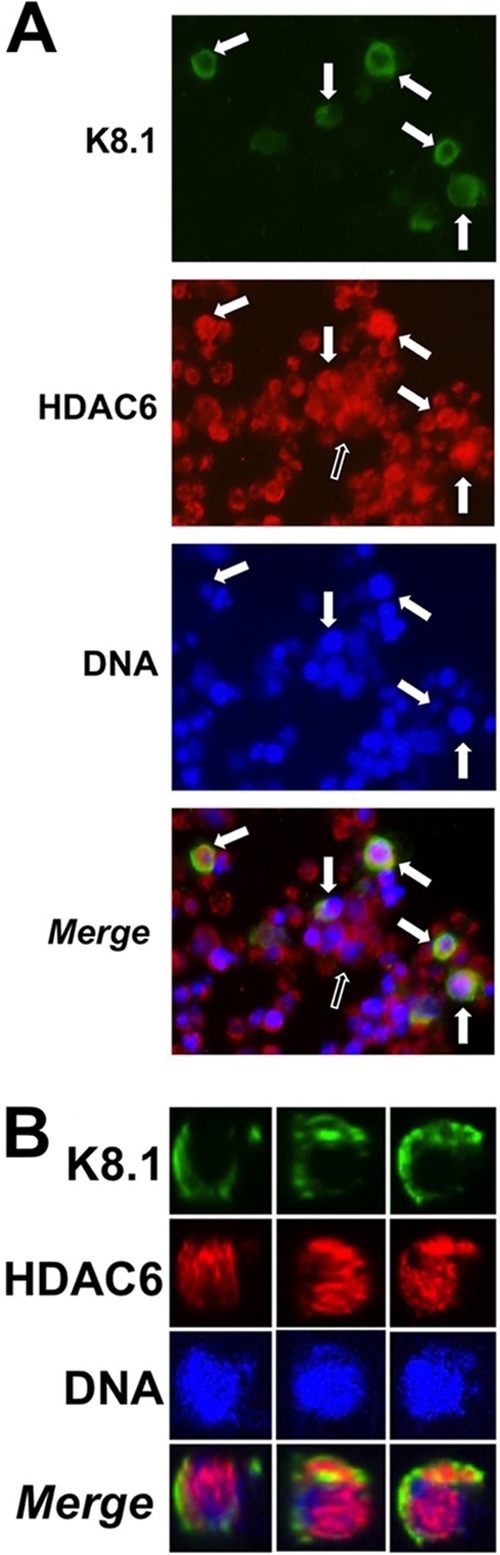
HDAC6 relocalizes during KSHV reactivation. BCBL-1 cells were treated with VPA, and HDAC6 and viral K8.1 were detected by indirect immunofluorescence. Nuclear DNA was stained with DAPI. (A) Solid arrows, K8.1-expressing cells in which HDAC6 relocalized to the perinuclear area or nucleus; unfilled arrow, appearance of HDAC6 in most cells. (B) A single K8.1-positive cell viewed from 3 perspectives.
DISCUSSION
HDACis have been recognized to be inducers of KSHV reactivation since the publication of the first tissue culture systems for KSHV infection. However, the identities of the particular HDACs that regulate KSHV reactivation have not been established. In our study, we employed HDACis with various specificities and overexpression of individual HDACs to identify HDACs 1, 3, and 6 as critical regulators of KSHV reactivation. Class I HDAC inhibitors were sufficient to reactivate the virus but differed in potency (Table 2). VPA was the most effective HDACi in inducing E-gene expression and was more potent than TSA in activating chromatin on the Rta promoter and producing mature virus (Fig. 1, 2, and 5). However, the difference between VPA and TSA in mature virus production was much more dramatic than the differences between VPA and TSA in inducing transcriptionally permissive chromatin on Rta's promoter and activating E genes. This observation suggests that the major mechanistic difference between VPA and TSA functions after Rta expression. Ectopic HDACs 1, 3, and 6 inhibited TPA-stimulated KSHV reactivation, and HDACs 1 and 6 stimulated reactivation independently (Fig. 8). HDAC6 was required for optimal VPA-stimulated reactivation (Fig. 9) and relocalized against and within the nucleus in reactivating cells (Fig. 10).
This study and others suggest the mechanism by which HDACis reactivate KSHV. During latency, Rta's promoter is organized as bivalent chromatin containing histones modified with both activating and repressing marks (19–22). Demethylation of H3K27-me3, expression of the histone demethylase JMJD3 or UTX, or inhibition of the H3K27-me3 methyltransferase EZH2 reactivates KSHV from latency (20, 22). In other systems, EZH2 and HDACs interact physically, and EZH2-mediated repression requires HDAC activity (47, 48). HDACs have been found to be constitutively associated with Rta's promoter (5). On a single histone, deacetylation of H3K9 is a prerequisite for H3K9 methylation, so HDACis prevent de novo H3K9 methylation. Therefore, we propose that VPA and TSA initiate KSHV reactivation by inhibiting continued repression of Rta expression by histone deacetylation and methylation.
Our data strongly suggest that the critical HDACi target that distinguishes the potency of VPA from that of TSA is HDAC6. Both TSA and VPA inhibit type I HDACs and expression and function of the H3K27 methyltransferase EZH2, but only TSA inhibits HDAC6 (41, 48–53). Furthermore, both TSA and tubacin, an HDAC6-specific HDACi, reduced VPA-stimulated reactivation (Fig. 3 and 9). While our data do not exclude alternative explanations for the differences in reactivation potency between VPA and TSA, they raise the possibility that HDAC6 activity might underlie previously described phenotypic differences between VPA and TSA. For example, TSA has a more potent negative effect on cell proliferation than VPA (41, 54). While both VPA and TSA inhibit HDAC2, only VPA causes its degradation (55). VPA also induces DNA demethylation (56), a modification with a potential, yet controversial, role in regulating KSHV latency (20, 57). Our data also do not exclude the possibility that differences in nonhistone targets of the drugs contribute to their different potencies (58–60).
VPA's potency relative to that of TSA was much greater in producing mature virions (Fig. 2) than in inducing the PAN promoter-driven RFP reporter (Fig. 1). It was recently shown that the HDACi nBA can stimulate transcriptional elongation of RNA polymerase II, which is prepoised on the PAN promoter during latency (61). This effect was independent of Rta expression and the production of mature virus. Since nBA and TSA have similar HDAC specificities, we think it is likely that TSA induces the PAN promoter reporter similarly, leading to the similar lack of a direct correspondence between E-gene expression and virus production. TSA's induction of E-gene markers without concomitant viral production in BCBL-1 cells occurs by an unknown mechanism but could reflect a requirement for HDAC6 after E-gene reactivation.
Our HDACi data also suggest that repression of both class I and IIa HDACs is required for optimal reactivation. HC toxin inhibited only class I HDACs and had the least effect on reactivation compared to the effects of VPA and TSA (Table 1). Class IIa HDACs have little activity against acetylated histones and restricted expression patterns (62). However, class IIa HDACs can recruit class I HDACs to DNA, which might compensate for HDAC class I inhibition in KSHV-infected cells.
The ability of HDACs 1, 3, and 6 to inhibit TPA-mediated reactivation suggests that they function downstream of TPA (Fig. 7). Furthermore, these data suggest that TPA has to overcome the combinatorial negative effects of HDACs 1, 3, and 6 to reactivate the virus. Indeed, overexpression of HDACs 1 and 6 independently reactivated KSHV (Fig. 8), but not as potently as overexpression of Rta, suggesting that multiple HDACs function combinatorially to regulate KSHV reactivation. Similarly, TPA treatment of HIV-infected cells induces acetylation of histones H3 and H4 on the long terminal repeat to induce viral reactivation (63).
Our data also do not distinguish between the HDAC mechanisms that function upstream or downstream of Rta expression. Reactivation of KSHV is associated with dramatic, genome-wide changes to histone modifications (22). While it is well-established that Rta's expression is induced by HDACis, Rta transactivates downstream promoters by recruiting HATs and chromatin remodeling proteins to DNA (64, 65); therefore, it is likely that part of the viral response to HDACis is mediated cooperatively with Rta. Indeed, VPA induces relatively modest alterations to chromatin in Rta's promoter (Fig. 5 and 6), while it induces a more dramatic increase in mature virus production (Fig. 2 and 4). Furthermore, depletion of the H3K9-me3 demethylase did not independently reactivate KSHV but reduced Rta-mediated reactivation (22, 66). Those data suggest that ongoing histone modifications are also required for successful progression down the lytic cascade. In fact, ectopic overexpression of Rta results in histone modifications on Rta's promoter that are similar to those induced by HDACis (22), and the Rta transcriptional target PAN recruits the demethylases JMJD3 and UTX1 to Rta's promoter to sustain Rta expression (67).
The ability of HDAC1 to reactivate KSHV when it is overexpressed alone appears to contradict its inhibition of TPA-stimulated reactivation. In fact, TSA also reactivates KSHV and can activate expression of endogenous HDAC1 (68). We think that a likely function of HDAC overexpression is that the excess HDACs disrupt the functional stoichiometry of the multiprotein complexes that repress Rta transcription. This conclusion is most intuitive for HDAC1, which is part of the catalytic cores of the multiprotein transcriptional corepressor complexes CoREST, NuRD, and Sin3 (8). Each of these complexes contains at least 6 to 12 protein subunits, some of which have multiple isoforms (8, 69–71). HDAC1 has no activity alone but requires other members of the corepressor complexes (72), an ideal scenario for stoichiometric regulation. Therefore, we postulate that excess HDAC1 titrates away critical components of HDAC/HMT and/or HDAC/viral protein complexes that are essential for continued repression of Rta's promoter and maintenance of latency. In particular, H3K27 methyltransferase (EZH2) activity, depletion of which reactivates KSHV (20, 22), has been shown to require HDACs (48). Also, the viral protein LANA inhibits reactivation and forms multiprotein complexes with HDAC1 (73, 74).
Furthermore, both HDAC1 and Sin3 have positive and negative roles in transcription (75–77), and HDAC1 is found on the promoters of both positively and negatively regulated genes (78). It is also possible that the ectopic HDACs titrate away the posttranslational modifying enzymes of the endogenous HDACs, since posttranslational modification of HDACs modulates their functions, subcellular localizations, protein-protein interactions, and stabilities (79). Overall, we propose that the stoichiometry of complexes containing HDACs 1 and 6 is critical for regulation of the KSHV reactivation switch.
While it was not surprising that HDACs 1 and 3 regulate KSHV reactivation, the role of HDAC6 stoichiometry in reactivation is less clear. To this end, HDAC6 participates in four broad cellular processes with precedents for regulating KSHV reactivation: signal transduction, transcription, the DNA damage response (DDR), and autophagy. HDAC6 shuttles from the cytoplasm to the perinucleus and nucleus to regulate NF-κB and CBP (44, 45). HDAC6 can be recruited by Sp1 and Runx2 to gene promoters to repress transcription (80, 81). In the nucleus, HDAC6 is recruited to active RNA polymerase II on the bodies of transcribed genes (82). HDAC6 also deacetylates hsp90 to promote stabilization of its client proteins, including c-Raf and ErbB2 (83, 84).
HDAC6's best-described function is to respond to cell stress by stimulating autophagy (43). In this process, HDAC6 promotes cell survival by deacetylating tubulin and bringing misfolded, ubiquitylated proteins to aggresomes. However, since HDAC6 is also required for induction and maintenance of tumorigenesis, HDAC6 inhibitors like tubacin are inhibitors of cell growth in many models (85). Tubacin also induces the DDR (86), and DDR proteins are acetylated to promote cytoplasmic transport and degradation via autophagy (87).
Our data thus suggest that HDAC6 might promote KSHV reactivation by inhibiting the DDR, stimulating autophagy, and promoting cell survival. DDR induction is inconsistent with the lytic replication of many herpesviruses (88), and tubacin kills Epstein-Barr virus (EBV)-infected cells (89). DDR proteins like the SMCs directly repress Rta expression (19, 24), and VPA, which does not inhibit HDAC6, downregulates SMC expression (90). Interestingly, the DDR is induced during KSHV latency, and autophagy and reactivation are inhibited by vFLIP (91–94). Conversely, autophagy and reactivation are stimulated by Rta (95). Our data suggest that HDAC6 is a central cellular player in this regulation and raise the question whether inhibition of DDR and stimulation of autophagy are sufficient to reactivate KSHV. As VPA and TSA showed similar effects on histone H3 acetylation on Rta's promoter and only TSA inhibited HDAC6, HDAC6 is not likely a significant regulator of Rta transcription.
The dynamic subcellular localization of HDAC6 in our experiments is not conclusive in revealing HDAC6's role in reactivation. While HDAC6 expression was more intensely nuclear and perinuclear in K8.1-expressing cells, its diffuse cytoplasmic staining was still observed in those cells (Fig. 10). In fact, HDAC6 was also perinuclear in some K8.1-negative cells. We note that HDAC6 has been detected in nuclear aggresomes with viral and cellular proteins in EBV-infected cells (96). Any of these subcellular locales has the potential to influence KSHV reactivation, and we posit that HDAC6 may have multiple roles in regulating KSHV's latent-to-lytic switch.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Eric Verdin, Geoff Wahl, Jeff Vieira, and Lucia Altucci for the gifts of plasmids, cells, and reagents and Rachna Shah for technical support. We thank the members of the D. M. Lukac lab for critical reading of the manuscript.
This study was supported by NIH grant AI078138 to D.M.L. and New Jersey Commission on Cancer Research grant DFHS13PPC010 to J.D.
Footnotes
Published ahead of print 13 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02665-13.
REFERENCES
- 1.Boshoff C, Gao S-J, Healy L, Matthews T, Thomas A, Coignet L, Warnke R, Strauchen J, Matutes E, Kamel O, Moore P, Weiss R, Chang Y. 1998. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood 91:1671–1679 [PubMed] [Google Scholar]
- 2.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346. 10.1038/nm0396-342 [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714 [PubMed] [Google Scholar]
- 4.Shaw R, Arbiser J, Offermann MK. 2000. Valproic acid induced human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 14:899–902. 10.1097/00002030-200005050-00021 [DOI] [PubMed] [Google Scholar]
- 5.Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. 2003. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 77:11425–11435. 10.1128/JVI.77.21.11425-11435.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller G, Heston L, Grogan E, Gradoville L, Rigsby M, Sun R, Shedd D, Kushnaryov VM, Grossberg S, Chang Y. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan O, La Thangue NB. 2012. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 90:85–94. 10.1038/icb.2011.100 [DOI] [PubMed] [Google Scholar]
- 8.Yang XJ, Seto E. 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9:206–218. 10.1038/nrm2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080. 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 11.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. 2007. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14:1025–1040. 10.1038/nsmb1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young NL, Dimaggio PA, Garcia BA. 2010. The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67:3983–4000. 10.1007/s00018-010-0475-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imai S, Guarente L. 2010. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol. Sci. 31:212–220. 10.1016/j.tips.2010.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong PG, Glozak MA, Cao TV, Vaziri C, Seto E, Alexandrow M. 2010. Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle 9:4351–4363. 10.4161/cc.9.21.13596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. 2009. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 10:92–100. 10.1038/ni.1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212. 10.1128/JVI.74.13.6207-6212.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen HS, Wikramasinghe P, Showe L, Lieberman PM. 2012. Cohesins repress Kaposi's sarcoma-associated herpesvirus immediate early gene transcription during latency. J. Virol. 86:9454–9464. 10.1128/JVI.00787-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6:e1000935. 10.1371/journal.ppat.1000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566–12575. 10.1128/JVI.78.22.12566-12575.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013. 10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- 24.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 7:e1002140. 10.1371/journal.ppat.1002140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye J, Shedd D, Miller G. 2005. An Sp1 response element in the Kaposi's sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 79:1397–1408. 10.1128/JVI.79.3.1397-1408.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolden JE, Peart MJ, Johnstone RW. 2006. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5:769–784. 10.1038/nrd2133 [DOI] [PubMed] [Google Scholar]
- 27.Bu W, Carroll KD, Palmeri D, Lukac DM. 2007. The Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788–5806. 10.1128/JVI.00140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gantt S, Carlsson J, Ikoma M, Gachelet E, Gray M, Geballe AP, Corey L, Casper C, Lagunoff M, Vieira J. 2011. The HIV protease inhibitor nelfinavir inhibits Kaposi's sarcoma-associated herpesvirus replication in vitro. Antimicrob. Agents Chemother. 55:2696–2703. 10.1128/AAC.01295-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. 10.1016/j.virol.2004.03.049 [DOI] [PubMed] [Google Scholar]
- 30.Scognamiglio A, Nebbioso A, Manzo F, Valente S, Mai A, Altucci L. 2008. HDAC-class II specific inhibition involves HDAC proteasome-dependent degradation mediated by RANBP2. Biochim. Biophys. Acta 1783:2030–2038. 10.1016/j.bbamcr.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 31.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 32.Palmeri D, Carroll KD, Gonzalez-Lopez O, Lukac DM. 2011. Kaposi's sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jk/CSL. J. Virol. 85:11901–11915. 10.1128/JVI.05479-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmeri D, Spadavecchia S, Carroll K, Lukac DM. 2007. Promoter and cell-specific transcriptional activation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 81:13299–13314. 10.1128/JVI.00732-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emiliani S, Fischle W, Van Lint C, Al-Abed Y, Verdin E. 1998. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. U. S. A. 95:2795–2800. 10.1073/pnas.95.6.2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E. 1999. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J. Biol. Chem. 274:11713–11720. 10.1074/jbc.274.17.11713 [DOI] [PubMed] [Google Scholar]
- 36.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. 2003. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11:437–444. 10.1016/S1097-2765(03)00038-8 [DOI] [PubMed] [Google Scholar]
- 37.Kanda T, Sullivan KF, Wahl GM. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. 10.1016/S0960-9822(98)70156-3 [DOI] [PubMed] [Google Scholar]
- 38.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185–6196. 10.1128/JVI.76.12.6185-6196.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 41.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. 2008. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409:581–589. 10.1042/BJ20070779 [DOI] [PubMed] [Google Scholar]
- 42.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. 2003. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U. S. A. 100:4389–4394. 10.1073/pnas.0430973100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. 2008. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 68:2557–2560. 10.1158/0008-5472.CAN-07-5989 [DOI] [PubMed] [Google Scholar]
- 44.Ma H, Nguyen C, Lee KS, Kahn M. 2005. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 24:3619–3631. 10.1038/sj.onc.1208433 [DOI] [PubMed] [Google Scholar]
- 45.Riolo MT, Cooper ZA, Holloway MP, Cheng Y, Bianchi C, Yakirevich E, Ma L, Chin YE, Altura RA. 2012. Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. J. Biol. Chem. 287:10885–10893. 10.1074/jbc.M111.308791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wickstrom SA, Masoumi KC, Khochbin S, Fassler R, Massoumi R. 2010. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 29:131–144. 10.1038/emboj.2009.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Vlag J, Otte AP. 1999. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23:474–478. 10.1038/70602 [DOI] [PubMed] [Google Scholar]
- 48.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419:624–629. 10.1038/nature01075 [DOI] [PubMed] [Google Scholar]
- 49.Bommi PV, Dimri M, Sahasrabuddhe AA, Khandekar J, Dimri GP. 2010. The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle 9:2663–2673. 10.4161/cc.9.13.12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA. 2008. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treatment Rev. 34:206–222. 10.1016/j.ctrv.2007.11.003 [DOI] [PubMed] [Google Scholar]
- 51.Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. 2004. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 64:1079–1086. 10.1158/0008-5472.CAN-03-0799 [DOI] [PubMed] [Google Scholar]
- 52.Jung JW, Lee S, Seo MS, Park SB, Kurtz A, Kang SK, Kang KS. 2010. Histone deacetylase controls adult stem cell aging by balancing the expression of polycomb genes and jumonji domain containing 3. Cell. Mol. Life Sci. 67:1165–1176. 10.1007/s00018-009-0242-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 100:11606–11611. 10.1073/pnas.1933744100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dannenberg LO, Edenberg HJ. 2006. Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics 7:181. 10.1186/1471-2164-7-181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, Gottlicher M. 2003. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 22:3411–3420. 10.1093/emboj/cdg315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Detich N, Bovenzi V, Szyf M. 2003. Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 278:27586–27592. 10.1074/jbc.M303740200 [DOI] [PubMed] [Google Scholar]
- 57.Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. 2001. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. U. S. A. 98:4119–4124. 10.1073/pnas.051004198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 59.Monti B, Polazzi E, Contestabile A. 2009. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2:95–109. 10.2174/1874467210902010095 [DOI] [PubMed] [Google Scholar]
- 60.Buchwald M, Kramer OH, Heinzel T. 2009. HDACi—targets beyond chromatin. Cancer Lett. 280:160–167. 10.1016/j.canlet.2009.02.028 [DOI] [PubMed] [Google Scholar]
- 61.Toth Z, Brulois KF, Wong LY, Lee HR, Chung B, Jung JU. 2012. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi's sarcoma-associated herpesvirus lytic gene expression. J. Virol. 86:9696–9707. 10.1128/JVI.01012-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10:32–42. 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lusic M, Marcello A, Cereseto A, Giacca M. 2003. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 22:6550–6561. 10.1093/emboj/cdg631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. 2009. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology 387:76–88. 10.1016/j.virol.2009.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gwack Y, Baek HJ, Nakamura H, Lee SH, Meisterernst M, Roeder RG, Jung JU. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055–2067. 10.1128/MCB.23.6.2055-2067.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang PC, Fitzgerald LD, Hsia DA, Izumiya Y, Wu CY, Hsieh WP, Lin SF, Campbell M, Lam KS, Luciw PA, Tepper CG, Kung HJ. 2011. Histone demethylase JMJD2A regulates Kaposi's sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 85:3283–3293. 10.1128/JVI.02485-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 8:e1002680. 10.1371/journal.ppat.1002680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hauser C, Schuettengruber B, Bartl S, Lagger G, Seiser C. 2002. Activation of the mouse histone deacetylase 1 gene by cooperative histone phosphorylation and acetylation. Mol. Cell. Biol. 22:7820–7830. 10.1128/MCB.22.22.7820-7830.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayakawa T, Nakayama J. 2011. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011:129383. 10.1155/2011/129383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. 2011. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29:255–265. 10.1038/nbt.1759 [DOI] [PubMed] [Google Scholar]
- 71.You A, Tong JK, Grozinger CM, Schreiber SL. 2001. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. U. S. A. 98:1454–1458. 10.1073/pnas.98.4.1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sengupta N, Seto E. 2004. Regulation of histone deacetylase activities. J. Cell. Biochem. 93:57–67. 10.1002/jcb.20179 [DOI] [PubMed] [Google Scholar]
- 73.Krithivas A, Young DB, Liao G, Greene D, Hayward SD. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637–9645. 10.1128/JVI.74.20.9637-9645.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. 2010. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 6:e1000818. 10.1371/journal.ppat.1000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, Vercauteren K, Saelens X, Meuleman P, Leroux-Roels G, De Bosscher K, Boutros M, Tavernier J. 2012. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. U. S. A. 109:12058–12063. 10.1073/pnas.1206458109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silverstein RA, Ekwall K. 2005. Sin3: a flexible regulator of global gene expression and genome stability. Curr. Genet. 47:1–17. 10.1007/s00294-004-0541-5 [DOI] [PubMed] [Google Scholar]
- 77.Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, Seiser C. 2006. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell. Biol. 26:7913–7928. 10.1128/MCB.01220-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kidder BL, Palmer S. 2012. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 40:2925–2939. 10.1093/nar/gkr1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Marcotullio L, Canettieri G, Infante P, Greco A, Gulino A. 2011. Protected from the inside: endogenous histone deacetylase inhibitors and the road to cancer. Biochim. Biophys. Acta 1815:241–252. 10.1016/j.bbcan.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 80.Westendorf JJ, Zaidi SK, Cascino JE, Kahler R, van Wijnen AJ, Lian JB, Yoshida M, Stein GS, Li X. 2002. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22:7982–7992. 10.1128/MCB.22.22.7982-7992.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chou CW, Chen CC. 2008. HDAC inhibition upregulates the expression of angiostatic ADAMTS1. FEBS Lett. 582:4059–4065. 10.1016/j.febslet.2008.10.048 [DOI] [PubMed] [Google Scholar]
- 82.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138:1019–1031. 10.1016/j.cell.2009.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280:26729–26734. 10.1074/jbc.C500186200 [DOI] [PubMed] [Google Scholar]
- 84.Chen CS, Weng SC, Tseng PH, Lin HP, Chen CS. 2005. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280:38879–38887. 10.1074/jbc.M505733200 [DOI] [PubMed] [Google Scholar]
- 85.Aldana-Masangkay GI, Sakamoto KM. 2011. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011:875824. 10.1155/2011/875824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Namdar M, Perez G, Ngo L, Marks PA. 2010. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. U. S. A. 107:20003–20008. 10.1073/pnas.1013754107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M. 2012. Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res. 72:1332–1335. 10.1158/0008-5472.CAN-11-3172 [DOI] [PubMed] [Google Scholar]
- 88.Nikitin PA, Luftig MA. 2011. At a crossroads: human DNA tumor viruses and the host DNA damage response. Future Virol. 6:813–830. 10.2217/fvl.11.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawada J, Zou P, Mazitschek R, Bradner JE, Cohen JI. 2009. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. J. Biol. Chem. 284:17102–17109. 10.1074/jbc.M809090200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. 2005. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 65:3815–3822. 10.1158/0008-5472.CAN-04-2478 [DOI] [PubMed] [Google Scholar]
- 91.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. 2009. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11:1355–1362. 10.1038/ncb1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. 2012. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 11:167–180. 10.1016/j.chom.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 93.Ye FC, Zhou FC, Xie JP, Kang T, Greene W, Kuhne K, Lei XF, Li QH, Gao SJ. 2008. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 82:4235–4249. 10.1128/JVI.02370-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199:993–1003. 10.1084/jem.20031467 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 95.Wen HJ, Yang Z, Zhou Y, Wood C. 2010. Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 84:7448–7458. 10.1128/JVI.00024-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park R, Wang'ondu R, Heston L, Shedd D, Miller G. 2011. Efficient induction of nuclear aggresomes by specific single missense mutations in the DNA-binding domain of a viral AP-1 homolog. J. Biol. Chem. 286:9748–9762. 10.1074/jbc.M110.198325 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.