

Abstract
Apoptosis is an important mechanism of West Nile virus (WNV) pathogenesis within the central nervous system (CNS). The signaling pathways that result in WNV-induced apoptotic neuronal death within the CNS have not been established. In this study, we identified death receptor (DR)-induced apoptosis as a pathway that may be important in WNV pathogenesis, based on the pattern of differential gene expression in WNV-infected, compared to uninfected, brains. Reverse transcription-PCR (RT-PCR) and Western blotting confirmed that genes involved in DR-induced apoptotic signaling are upregulated in the brain following WNV infection. Activity of the DR-associated initiator caspase, caspase 8, was also increased in the brains of WNV-infected mice and occurred in association with cleavage of Bid and activation of caspase 9. These results demonstrate that DR-induced apoptotic signaling is activated in the brain following WNV infection and suggest that the caspase 8-dependent cleavage of Bid promotes intrinsic apoptotic signaling within the brains of infected animals. Utilization of a novel ex vivo brain slice culture (BSC) model of WNV encephalitis revealed that inhibition of caspase 8 decreases virus-induced activation of caspase 3 and tissue injury. The BSC model allows us to examine WNV-induced pathogenesis in the absence of a peripheral immune response. Thus, our results indicate that WNV-induced neuronal injury in the brain is mediated by DR-induced apoptosis signaling and can occur in the absence of infiltrating immune cells. However, astrocytes and microglia were activated in WNV-infected BSC, suggesting that local immune responses influence WNV pathogenesis.
INTRODUCTION
Neuronal apoptosis is an important mechanism of virus-induced pathogenesis within the central nervous system (CNS) (1). During West Nile virus (WNV) encephalitis, the proapoptotic executioner caspase, caspase 3, is activated, and mice lacking caspase 3 have reduced neuronal death and tissue injury following West Nile virus infection (2). Despite the importance of apoptosis in WNV pathogenesis, the exact pathways involved in triggering apoptotic cell death in the CNS have not yet been defined.
Activation of initiator caspases 8 (3, 4) and 9 (3) occurs in cultured neuronal cells infected with WNV, and inhibition of these initiator caspases leads to reduced cleavage of the caspase 3 substrate poly(ADP-ribose) polymerase (PARP) (3, 4). These studies indicate that both extrinsic and intrinsic apoptotic signaling pathways are activated in vitro following WNV infection and are consistent with in vitro studies demonstrating that mitochondrial apoptotic signaling protein Bax is upregulated in neuronal cells following WNV infection (5) and that cytochrome c is released from the mitochondria (3). However, it remains to be seen whether the same apoptotic pathways are also activated in the intact brain during WNV encephalitis.
Innate and adaptive immune responses also influence WNV pathogenesis within the CNS. Toll-like receptors 3 and 7 (TLR3 and TLR7) and the cytoplasmic proteins encoded by retinoic acid-inducible gene I (RIG-I) and melanoma-differentiation-associated gene 5 (MDA5) are important for detection of WNV within the CNS, and defects in these, or additional, components of the interferon (IFN) response show enhanced viral burden and increased lethality (6). Although IFN restricts infection, pathogenic WNV strains attenuate IFN function at several steps of the induction and signaling cascade, allowing the virus to establish infection (6). Studies performed in mice (7–11) and humans (12–14) have also highlighted the role of CD8+ T cells and the associated importance of pathways involving Fas ligand (10) and tumor necrosis factor (TNF)-related apoptosis-inducing factor ligand (TRAIL) (15) effector mechanisms in containing WNV CNS infection.
In this report, we demonstrate that genes involved in death receptor (DR) apoptotic signaling are upregulated in the mouse brain following WNV infection. We also show for the first time that the activity of the DR-associated initiator caspase, caspase 8, is increased in the brain following WNV infection. WNV-induced activation of caspase 8 in the CNS is associated with cleavage of the proapoptotic Bcl-2 family protein Bid and with activation of caspase 9, suggesting that the caspase 8-dependent cleavage of Bid promotes intrinsic apoptotic signaling within the brains of infected animals. Utilization of WNV-infected ex vivo brain slice cultures (BSC), a novel model of WNV encephalitis, revealed that inhibition of caspase 8 decreased virus-induced activation of caspase 3 and reduced WNV-associated CNS tissue injury. These results provide the first demonstration of the role of DR apoptotic signaling in WNV-induced apoptosis and tissue injury within the brain. The use of BSC also shows that WNV-induced apoptosis occurs in infected brain tissue in the absence of infiltrating inflammatory cells or other components of the peripheral immune response. Our results suggest that local immune responses within the CNS, including the activation of microglia and astrocytes, contribute to WNV pathogenesis through the release of chemokines, cytokines, and neurotoxic factors. WNV infection induces astrocyte cell death, which may further contribute to neuronal injury through reduction of astrocyte-associated trophic support.
MATERIALS AND METHODS
Mouse infections.
West Nile virus stocks were obtained from clone-derived strain 382-99 (NY99) as previously described (16). Adult mice were infected with 100 PFU WNV by intracerebral (i.c.) inoculation.
Western blotting.
Brains were removed from WNV-infected and mock-infected mice and were transferred to 1 ml of PBS and stored at −80°C. Vials were thawed gently to room temperature, and the phosphate-buffered saline (PBS) was aspirated off. Brains were then homogenized in 300 μl of tissue protein extraction reagent supplemented with Halt protease inhibitor cocktail (Thermo Fisher Scientific, Inc., Rockford, IL), using a Dounce homogenizer. Lysates were then transferred to 1.5 ml Eppendorf tubes and were stored on ice until centrifugation (20,000 × g for 3 min). The supernatant was then transferred to a fresh tube containing 300 μl of 2× Laemmli buffer (125 mM Tris [pH 6.8], 4% SDS, 10% mM β-mercaptoethanol, 20% glycerol, and 0.004% bromophenol blue). Brain lysates were boiled for 5 min and stored at −70°C. Proteins were electrophoresed overnight by SDS-PAGE at a constant voltage of 70 V. Proteins were then electroblotted onto Hybond-C nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ). Immunoblots were blocked with 5% nonfat dry milk (NFDM)–Tris-buffered saline Tween 20 (TBST) for 2 h at room temperature before being probed with antibodies directed against death-associated protein 6 (Daxx), Bid, and cleaved caspase 3 (Cell Signaling Technology catalog no. 4533, 2003, and 9661). Antibodies were diluted in 3% NFDM–TBST according to the manufacturer's directions. All lysates were standardized for protein concentration with antibodies directed against actin (Oncogene, Cambridge, MA; catalog no. CP01) diluted 1:10,000 in 3% NFDM–TBST. Autoradiographs were quantitated by densitometric analysis using a Fluor-S MultiImager (Bio-Rad Laboratories, Hercules, CA). Statistical analysis was performed using Graphpad software (Instat).
RNA purification, microarray, and RT-PCR.
In order to generate the most consistent data for our microarray studies, particular care was taken to standardize infections for RNA extractions. Twelve-week-old male Swiss Webster mice were infected with 100 PFU WNV by i.c. inoculation. At 5 to 6 days postinfection (dpi), when infected mice were showing neurologic symptoms, mice were sacrificed and brains from mock- and virus-infected mice were immediately placed in RNAlater and stored (short term) at −20°C. Approximately 160 mg of brain tissue (about half of the brain after the cerebellum was removed) was homogenized in 1 ml of Qiazol (Qiagen RNeasy Lipid Tisssue Minikit; catalog no. 74804) for at least 20 strokes until completely emulsified. The emulsion was transferred to a clean 1.5-ml tube and set aside for at least 5 min before addition of 200 μl chloroform. The mixture was shaken for 15 s and set aside for 5 min before centrifugation at 12,000 × g for 15 min at 4°C. The upper aqueous phase was then carefully removed and transferred to a new tube. One volume of 70% ethanol (prepared with diethyl pyrocarbonate [DEPC] water) was then added, and the solution was mixed before being transferred to an RNeasy column. RNA was purified following the manufacturer's specifications, and 0.5 to 1 μl of RNasin was added to the sample, which was then stored at −80°C. Affymetrix 1.0 ST mouse whole-genome chips (Santa Clara, CA) were used for microarray analysis of RNA extracted from WNV-infected and mock-infected brains. Individual Affymetrix chips were used for each WNV-infected and mock-infected sample according to the manufacturer's specifications. All microarray experiments and the initial analysis were performed at the microarray core facility of the University of Colorado Denver. Analysis was performed using R statistical computation software and packages from Bioconductor open source software for bioinformatics (17). Prior to statistical analysis, two preprocessing steps involving normalization and gene filtering were performed. Raw data from array scans were processed using the Robust Multichip Average (RMA) normalization method to subtract a background value that is based on modeling the perfect match (PM) signal intensities as a convolution of an exponential distribution of signal and a normal distribution of nonspecific signal while ignoring the mismatch (MM) signal (18). After normalization, data were filtered using two criteria: (i) Affymetrix mRNA detection calls were used to exclude all probe sets with an “absent” call in all samples, and (ii) transcripts that demonstrated little variation across all arrays were removed. This was performed by comparing the variance of the log intensities for each gene with the median of all variance for the entire array. Genes not significantly more variable than the median were filtered out. Microarray data have been uploaded onto GEO (www.ncbi.nlm.nih.gov/geo). Differentially regulated genes were analyzed using Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA). The Ingenuity Pathways Analysis knowledge base is a curated database constructed on the basis of scientific evidence from hundreds and thousands of journal articles, textbooks, and other data sources. IPA software was used to define which well-characterized cell signaling pathways are most relevant during WNV encephalitis. The significance (P values) of the association between the data set and the canonical pathway was measured by comparing the number of genes that are differentially regulated during reovirus encephalitis that participate in a pathway to the total number of genes in all pathway annotations stored in the Ingenuity Pathways knowledge base. Fisher's exact test was used to calculate a P value determining the probability that the association between the genes in the data and in the canonical pathway is explained by chance only.
For reverse transcription-PCR (RT-PCR), cDNA was prepared from purified RNA by reverse transcribing 1.0 μg of each RNA sample using a SABiosciences First Strand kit (C-03; SABiosciences, Frederick, MD). The 20-μl final volume of cDNA was diluted with 1,275 μl of water and added to 1,275 μl of 2× SABiosciences RT2 SYBR green master mix (PA-010). A 25-μl volume was dispensed into each well of a SABiosciences RT2 profile alpha/beta interferon (IFN-α/β) signaling (PAMM-14) or cytokine (PAMM-11) PCR array. RT-PCR was carried out on a Bio-Rad Opticon2 machine (Bio-Rad, Hercules, CA).
Fluorogenic caspase activity assays.
Caspase 3, 8, and 9 activity was determined by utilizing a fluorogenic assay (B&D Biosciences, San Jose, CA). The reaction products were measured on a Cytofluor Series 4000 spectrofluorometer (Applied Biosystems, Carlsbad, CA) at an excitation wavelength of 450 nm and an emission wavelength of 530 nm.
LDH assay.
Lactate dehydrogenase (LDH) efflux is commonly used as a measurement of injury in organotypic slices (19, 20). BSC media were collected at various times postinfection, and triplicate 10-μl samples were analyzed with LDH Cytotoxicity Assay kit II (Biovision, Mountain View, CA), in which water was used as a blanking control and purified LDH used as a positive control. LDH activity was quantified on a microplate reader (Emax; Molecular Devices, Sunnyvale, CA) as a function of the optical density (OD) measured at 450 nm with wavelength control at 650 nm.
Preparation of organotypic brain slice cultures and infections.
Brain slice cultures were prepared from 3-to-4-day-old NIH Swiss Webster mice (21, 22) in compliance with institutional guidelines. Mice were euthanized by decapitation and brains rapidly removed into ice-cold slicing media (minimal essential medium [MEM], 10 mM Tris, 28 mM d-glucose, pH 7.2, equilibrated with 95% O2–5% CO2). Under semisterile conditions, the frontal cortex and cerebellum were removed and 400-μm-thick coronal sections cut through cerebral regions containing the hippocampus and thalamus using a vibratome (Classic 1000; Leica Microsystems Inc., Bannockburn, IL). Four slices from a single animal were carefully positioned on top of a single semiporous membrane insert (catalog no. PICMORG50; Millipore, Billerica, MA) as previously described (21, 22). Membranes were placed in 35-mm-diameter tissue culture dishes containing 1.2 ml serum-containing media (Neurobasal medium supplemented with 10 mM HEPES, 1× B27, 10% fetal bovine serum [FBS], 400 μM l-glutamine, 600 μM GlutaMAX, 60 U/ml penicillin, 60 μg/ml streptomycin, 6 U/ml nystatin) such that the slices were at the medium-air interface. Media were refreshed with 5% serum-containing media (5% FBS) approximately 12 h after slicing. All subsequent medium changes were made with serum-free media every 2 days (i.e., at 3, 5, 7, and 9 dpi). Cultures were maintained in a humidified incubator at 5% CO2 and 36.5°C. At 1 day after plating, slices were infected with 105 PFU WNV (NY99) per slice. Virus was diluted into 20 μl PBS and applied dropwise to the top (air interface) of each slice. Mock infections were performed in a similar manner using PBS alone. The caspase 8 inhibitor Z-IETD-FMK (R and D Biosciences) was used at a concentration of 10 μM and was added to the media 2 h before infection. Inhibitor was reapplied with all subsequent medium changes.
RESULTS
Microarray analysis of gene expression in the brain following infection with WNV.
Twelve-week-old male Swiss Webster mice (n = 3) were inoculated with WNV (NY99) by i.c. injection (100 PFU). Mice were sacrificed when infected animals showed symptoms of neurologic disease (around 5 dpi). Brains were removed and processed to obtain RNA. RT-PCR indicated that infected brains contained between 109 and 1010 PFU equivalents of WNV (data not shown). RNA was also obtained from 12-week-old mock-infected male mice (n = 3). RNAs were processed on mouse gene 1.0 ST arrays (Affymetrix). Infection of the brain with WNV led to the differential regulation of many genes within the CNS (Table 1). Consistent with the known activation of IFN signaling by WNV, the 23 genes with the highest levels of upregulation (50+-fold) (Table 2), including genes expressing GTPases, antiviral proteins, immune cell attractants, and proteins involved in the regulation of IFN signaling, were all associated with the IFN response.
TABLE 1.
WNV-induced gene regulation in the mouse braina
| Gene category | No. of genes with specified fold changes (P < 0.05) |
|||||
|---|---|---|---|---|---|---|
| Total | >50× | 10×–50× | 5×–10× | 2×–5× | 1.25×–2× | |
| Upregulated | 3,036 | 23 | 117 | 131 | 582 | 2,183 |
| Downregulated | 3,719 | 0 | 0 | 2 | 312 | 3,405 |
Microarray analysis was performed on RNA extracted from the brains of 12-week-old SW mice infected with WNV (NY99) by i.c. inoculation (n = 3) and of mock-infected control mice (n = 3).
TABLE 2.
Genes associated with IFN signaling are highly (50+-fold) upregulated in the brains of mice infected with WNV
| Function | Gene designation | Name | Fold upregulation | P |
|---|---|---|---|---|
| Chemoattractant for immune cells | Cxcl10 | C-X-C motif chemokine 10 | 270 | 2.90E-06 |
| Ccl5 | Chemokine (C-C motif) ligand 5 | 77 | 1.18E-07 | |
| IFN-inducible GTPase | Tgtp | T cell-specific GTPase 1 | 98 | 3.81E-07 |
| Iigp1 | IFN-inducible GTPase 1 | 50 | 4.08E-06 | |
| Irgm1 | Immunity-related GTPase family M member 1 | 53 | 1.60E-06 | |
| Irgm2 | Immunity-related GTPase family M member 2 | 83 | 3.32E-06 | |
| Gbp2 | Guanylate-binding protein 2 | 69 | 3.04E-06 | |
| Gbp6 | Guanylate-binding protein 6 | 59 | 2.85E-07 | |
| Gm12250 | Predicted gene 12250 | 65 | 3.79E-06 | |
| Regulation of IFN signaling | Usp18 | Ubiquitin-specific peptidase 18 | 85 | 6.35E-07 |
| Nlrc5 | NLR family, CARD domain containing 5. | 66 | 2.83E-06 | |
| Ifna2 | Alpha interferon 2 | 55 | 2.61E-05 | |
| Acute-phase response | Saa3 | Serum amyloid A 3 | 74 | 3.69E-07 |
| IFN-induced antiviral activity | Rsad2 | Radical S-adenosyl methionine domain-containing 2 | 74 | 3.35E-06 |
| Ifit1 | IFN-induced protein with tetratricopeptide repeats 1 | 67 | 5.35e-08 | |
| Ifit3 | IFN-induced protein with tetratricopeptide repeats 3 | 62 | 1.05E-06 | |
| Oasl2 | 2′-5′ Oligoadenylate synthetase-like 2 | 53 | 5.94E-07 | |
| Antigen processing and presentation | H2-gs10 | Major histocompatibility complex (MHC) class I-like protein GS10 | 69 | 1.37E-06 |
| IFN-induced, unknown function | Gm4951 | Predicted gene 4951 | 67 | 5.70E-08 |
| Slfn4 | Schlafen 4 | 71 | 1.70E-07 | |
| Map2l | Microtubulin-associated protein 2-like | 65 | 2.27E-07 | |
| Ifi44 | Interferon-induced protein 44 | 60 | 8.24E-06 | |
| IFN-induced apoptosis | Lcn2 | Lipocalin2 | 56 | 0.0004 |
Pathways associated with IFN and death receptor apoptotic signaling are upregulated in the brain following infection with WNV.
Ingenuity Pathway Analysis (IPA; Ingenuity Systems) was performed on genes that were differentially regulated in the brain following infection with WNV. IPA calculates (i) the probability that genes belonging to a particular pathway are differentially regulated compared to other cellular genes and (ii) the number of differentially regulated genes divided by the total number of genes within a specific pathway (ratio). Both these values can be used to identify cellular pathways that may be activated based on the pattern of differential gene expression. The nine pathways, derived from upregulated genes, that had the highest ratio of differentially to non-differentially regulated genes (≥0.34) and the highest significance (-log P ≥ 10) in WNV-infected compared to noninfected mice had the general functions of DR-induced apoptosis and innate and adaptive immune responses (Table 3). To investigate direct mechanisms of virus-induced cell death within the brain, we focused on apoptotic signaling pathways that were identified by IPA. The IPA-identified pathways “TNFR1 signaling,” “TNFR2 signaling,” and “induction of apoptosis by HIV-1” are all subsets of “DR signaling.” Figure 1a shows that 17 genes from DR signaling pathways were identified by IPA as being upregulated in the brains of WNV-infected animals compared to mock-infected controls with a fold change value of >1.25 (P < 0.05). Genes encoding the transcription factor nuclear factor kappa B (NF-κB), its inhibitor IκB, and upstream activator IκB kinase (IKK) were also upregulated but were not included in this count since NF-κB signaling is common to multiple signaling pathways and cannot be considered specific for DR signaling. Nine of the 17 identified genes were differentially upregulated with fold changes of ≥2, and these changes were highly significant (P < 0.0007). RT-PCR of RNA extracted from the brains of additional WNV- and mock-infected mice confirmed the differential expression of DR-associated genes following virus infection (Fig. 1b). Western blotting (Fig. 1c) was also used to confirm the upregulation of the representative gene, death-associated protein 6 (Daxx), at the protein level. These results establish that genes associated with DR-induced apoptotic signaling pathways are upregulated in the brain following infection with WNV.
TABLE 3.
Ingenuity Pathway Analysis (IPA) of WNV-induced upregulated gene expression changes in the mouse braina
| Gene product general function(s) | Specific pathway | Ratiob (≥0.34) | -log Pc (≥10) |
|---|---|---|---|
| Death receptor apoptotic signaling | TNFR2 signaling | 0.50 | 12.0 |
| TNFR1 signaling | 0.38 | 10.1 | |
| Death receptor signaling | 0.38 | 11.5 | |
| Induction of apoptosis by HIV-1 | 0.35 | 10.5 | |
| Innate and adaptive immune responses | Interferon (IFN) signaling | 0.48 | 12.1 |
| Role of protein kinase R in IFN induction and antiviral response | 0.45 | 12.0 | |
| Role of pattern recognition receptors in recognition of bacteria and viruses | 0.35 | 14.0 | |
| Role of Rig-I (retinoic acid-inducible gene 1)-like vectors in innate viral immunity | 0.34 | 12.5 | |
| T helper cell differentiation | 0.36 | 12.4 |
IPA was performed on microarray data obtained from the analysis of RNA extracted from the brains of mice infected with WNV and mock-infected controls (see Table 1).
Data represent numbers of differentially regulated genes divided by the total number of genes within a specific pathway.
Data represent the probability that genes belonging to a particular pathway are differentially regulated compared to other cellular genes.
FIG 1.

Genes associated with DR-induced apoptotic signaling are upregulated in the brains of WNV-infected mice. Twelve-week-old mice were infected with WNV by i.c. inoculation. Microarray analysis was performed on RNA extracted from the brains of WNV-infected (n = 3) and mock-infected (n = 3) mice 5 to 6 dpi when infected animals were showing clinical signs of neurologic disease. (a) IPA was performed to identify cellular pathways which might play a role in WNV pathogenesis based on the pattern of differential gene expression levels seen in infected compared to uninfected brains. The diagrammatic representation of DR-associated gene expression shows genes which are upregulated (P < 0.05) by >2-fold (gray shading, bold outline) or by 1.25- to 2-fold (gray shading) or which are not upregulated (no shading). These genes include those encoding tumor necrosis factor alpha (TNF-α), TNF-related apoptosis-inducing factor ligand (TRAIL), Fas, TNF receptor 1 (TNFR1), TNFR2, death-associated protein 6 (Daxx), caspase 8, Bid, cellular caspase 8, Fas-associated protein with a death domain (FADD)-like apoptosis regulator (FLICE) inhibitory protein (FLIP), TNF-associated factor 2 (TRAF2), caspase 2, apoptosis activating factor 1 (Apaf1), caspase 9, caspase 3, cellular inhibitor of apoptosis protein 2 (cIAP2), TRAF family member-associated NF-κB activator (TANK), and TANK binding kinase (TBK). (b) RT-PCR confirmed the upregulation of DR-associated gene expression levels in the brains of an additional group of WNV-infected mice (n = 6) compared to additional mock-infected controls (n = 3). RNA from these mice was obtained 5 to 6 dpi. The graph shows the average increases in gene expression. Error bars represent standard errors of the mean. TRAIL (P < 0.0002), cIAP2 (P < 0.002), Daxx (P < 0.005), Fas (P < 0.01), cFLIP (P < 0.0002), TNFR1 (P < 0.0001), caspase 8 (P < 0.02), TNFR2 (P < 0.0007), and TNF (P < 0.001) were all significantly upregulated in the brains of WNV-infected mice. (c) Western blot analysis of lysates prepared from the brains of WNV-infected and mock-infected mice at 6 dpi demonstrated that the representative protein, Daxx, was upregulated at the protein level following WNV infection. The blot also shows that infected brains contained increased levels of cleaved (activated) caspase 3 (CC3). Actin was used as a loading control.
Caspase activity is upregulated in the brains of mice infected with WNV.
Having shown that genes associated with DR-induced apoptotic signaling were upregulated in the brains of mice infected with WNV compared to mock-infected controls, we next investigated the activity of initiator caspases within the brains of WNV-infected animals. Mice were infected by i.c. inoculation with 100 PFU WNV (NY99). Five days following infection, brains were harvested and caspase 8, 9, and 3 activity assays were performed. Figure 2a shows a significant increase in the activity of caspase 8, the initiator caspase associated with DR signaling, in the brains of WNV-infected animals compared to mock-infected controls. This suggests that DR apoptotic signaling is functional in the brain following infection with WNV. There was also a small, but significant, increase in the activity of caspase 9, the initiator caspase associated with mitochondrial apoptotic signaling, in the brain following WNV infection (Fig. 2a). The Bcl-2 family protein Bid, which is cleaved (activated) by caspase 8 and links extrinsic and intrinsic apoptotic signaling pathways, is also activated in the brain following WNV infection (Fig. 2c). The ratio of cleaved Bid to full-length Bid was significantly higher in infected brains than in mock-infected controls (Fig. 2d). This suggests that DR-associated activation of caspase 8 results in the cleavage of Bid and the subsequent activation of intrinsic apoptotic signaling following WNV infection. In agreement with previous studies (2), the activity of the effector caspase, caspase 3, was enhanced in the brain following WNV infection (Fig. 2b).
FIG 2.

Initiator caspases are activated in the brain following WNV infection. Adult mice were infected with WNV by i.c. inoculation. (a and b) Fluorogenic caspase activity assays were performed on lysates prepared from WNV-infected and mock-infected brains 5 dpi following infection. The graph shows the average increase in fluorescence (caspase activity) in WNV-infected compared to mock-infected brains. (c) Western blot analysis showed that Bid is cleaved in WNV-infected brains (6 dpi) compared to mock-infected controls. The blot also shows that infected brains contained increased levels of cleaved (activated) caspase 3 (CC3). Actin was used as a loading control. (d) Densitometric analysis demonstrated a significant (P < 0.05) increase in the average ratio of cleaved to full-length Bid in WNV-infected lysates compared to mock-infected controls. Error bars indicate standard errors of the mean.
Inhibition of caspase 8 blocks WNV-induced apoptosis in an ex vivo model of WNV pathogenesis.
Our results revealed that a 2-fold increase in caspase 8 activity was associated with a >10-fold increase in activated caspase 3 levels. To determine the role of caspase 8 in caspase 3 activation, we next wanted to determine the effect of inhibition of caspase 8 activity on WNV-induced activation of caspase 3. In vivo experiments using caspase inhibitors are problematic because of poor blood-brain-barrier (BBB) penetration after systemic administration. To facilitate our investigations, we therefore developed an ex vivo model of WNV pathogenesis using brain slice cultures (BSC) that was based on our recent work describing reovirus infection of BSC (22). Four 400-μm-thick tissue slices were prepared from individual brains extracted from 3-day-old SW mice and were infected with WNV (105 PFU per slice) after 24 h. Figure 3a shows increasing levels of viral antigen in infected BSC between 3 and 5 dpi. At 5 dpi, WNV antigen was visible in the majority (>90%) of neurons (Fig. 3a and b), mirroring in vivo observations (16). At 7 dpi, mitogen-activated protein 2 (MAP2) staining decreased, indicating neuronal death (Fig. 3c). Astrocytes become activated in infected BSC, as evidenced by larger nuclei and shorter, thicker processes (Fig. 3d). It is also clear that WNV was able to infect astrocytes in BSC (Fig. 3d) and that by 5 dpi there was significant astrocyte death within WNV-infected BSC. In contrast, although microglia became activated in WNV-infected BSC and contained WNV antigen, the pattern of staining is more consistent with phagocytosis of WNV-infected material than with actual infection of the microglia themselves (Fig. 3e). Infection of neurons and astrocytes with WNV allowed viral growth until about 5 to 6 dpi, when the growth curve flattened (Fig. 4a).
FIG 3.
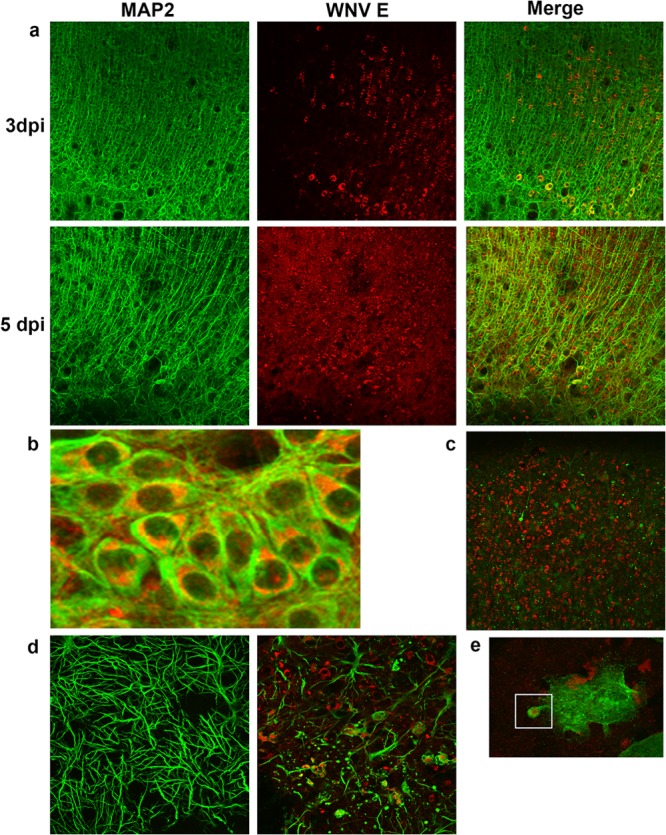
Characterization of an ex vivo model of WNV pathogenesis. Tissue slices (400 μm thick) were prepared from brains extracted from 3-day-old SW mice and were infected with WNV (105 PFU per slice) after 24 h. (a to c) Slices were prepared for immunohistochemical analysis (IHC) and were stained with antibodies directed against WNV E protein (red) and the neuronal marker MAP2 (green). The images show that WNV antigen was present in neurons. By 7 dpi, neuronal death was evidenced by the disappearance of MAP2-staining neurons. (d) Staining of slices with antibodies to WNV E protein (red) and the astrocyte marker GFAP (green) at 5 dpi demonstrates astrocyte activation (more-prominent cell bodies along with shorter, thicker processes) and astrocyte death in infected BSC. WNV also appears to infect astrocytes in BSC (arrows). (e) Staining of slices with antibodies to WNV E protein (red) and the microglial activation marker Iba1 (green) demonstrates microglial activation (no green staining in mock-infected cells—not shown). Microglia can be seen engulfing cellular debris containing WNV antigen.
FIG 4.

WNV induces injury and apoptosis in infected BSC. Four 400-μm-thick tissue slices were prepared from individual brains extracted from 3-day-old SW mice and were infected with WNV (105 PFU per slice) after 24 h. (a) PCR was used to determine levels of WNV RNA, which were compared to a standard curve to ascertain viral titer. (b) RT-PCR was used to investigate the expression of DR-associated genes in WNV-infected BSC compared to mock-infected controls at 7 dpi. The graph shows the average increase in gene expression in WNV-infected BSC (black bars) compared to mock-infected controls (gray bars). Error bars represent the standard errors of the means. (c) At 3, 5, 7, and 9 dpi, medium from the slices was analyzed for the presence of lactate dehydrogenase (LDH). This medium contained any LDH released since the previous collection. The graph shows the fold increase in absorbance (LDH levels) in WNV-infected BSC compared to mock-infected controls. Error bars represent the standard errors of the means. (d) At 3, 5, 7, and 9 dpi, caspase 3 activity assays were used to determine the amount of WNV-induced apoptosis in BSC. The graph shows the increase in fluorescence (caspase 3 activity) in WNV-infected BSC compared to mock-infected controls. Error bars represent the standard errors of the means.
WNV infection of BSC led to an increase in the expression of DR-associated genes, providing evidence that infection of BSC accurately represents in vivo findings (Fig. 4b). Virus-induced injury was also investigated by determining the levels of lactate dehydrogenase (LDH) in the media of WNV-infected cultures compared to mock-infected controls. Medium was collected at 3, 5, 7, and 9 days following infection and contained any LDH released since the previous collection. No increase in LDH was observed in the media collected from WNV-infected cultures, compared to mock-infected controls, at 3 dpi. However, increases in LDH were seen in the media collected from WNV-infected cultures, compared to mock-infected controls, at 5, 7, and 9 dpi (Fig. 4c). WNV infection of BSC also led to an increase in caspase 3 activity compared to mock-infected controls at 5, 7, and 9 dpi (Fig. 4d). These results demonstrate the presence of virus-induced injury and apoptosis in WNV-infected BSC and indicate that these ex vivo cultures can be used as a model for WNV pathogenesis within the CNS.
Pretreatment of BSC with a cell-permeative inhibitor of caspase 8 activity (Z-IETD-FMK) completely blocked WNV-induced activation of caspase 3, suggesting that caspase 8 activity is required for caspase 3 activation following WNV infection. Inhibition of caspase 8 activity also led to a decrease in WNV-induced LDH, demonstrating that caspase 8 also mediates WNV-induced tissue injury in an ex vivo model of WNV pathogenesis (Fig. 5). Caspase 8 inhibition had no effect on viral growth in the BSC (not shown).
FIG 5.
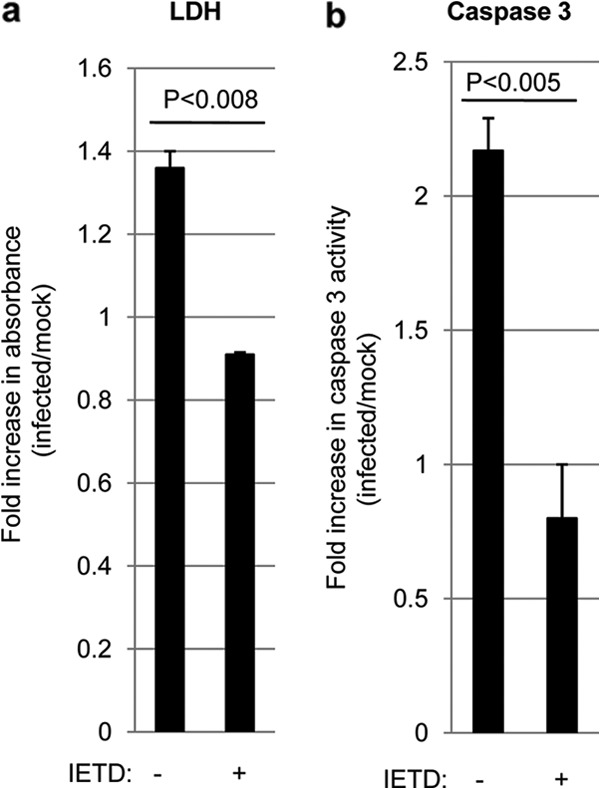
Inhibition of caspase 8 blocks WNV-induced apoptosis and tissue injury in an ex vivo model of WNV pathogenesis. Four 400-μm-thick tissue slices were prepared from individual brains extracted from 3-day-old SW mice. Slices were treated with 10 μM Z-IETD-FMK (IETD) for 24 h before being infected with WNV (105 PFU per slice). At 7 dpi, medium from the cultures was analyzed for the presence of lactate dehydrogenase (LDH). This medium contained any LDH released over the prior 2 days. The graphs show the fold increases in LDH levels (a) and caspase 3 activation (b) in WNV-infected BSC compared to mock-infected controls. Error bars represent the standard errors of the means.
DISCUSSION
Affymetrix 2.0 whole-genome chips were used to compare levels of gene expression in the brains of WNV-infected and mock-infected mice. A total of 3,037 genes were found to be upregulated and 3,719 genes were found to be downregulated in the brain following WNV infection (fold change > 1.25, P < 0.05). Ingenuity Pathway Analysis (Ingenuity Systems) identified pathways associated with DR apoptotic signaling and IFN signaling as having the highest predicted relevance for WNV infection based on the ratio of the number of upregulated genes within the pathway to the number of genes whose expression was not changed.
Since our current interest was in identifying pathways involved in WNV-induced apoptosis within the CNS system, we focused on the role of DR apoptotic signaling pathways in WNV pathogenesis. Pathway analysis of gene expression data identified 4 pathways associated with DR apoptotic signaling that were predicted to have relevance for WNV pathogenesis, including TNFR2 signaling (ratio = 0.5), TNFR1 signaling (ratio = 0.38), DR signaling (ratio = 0.38), and induction of apoptosis by HIV-1 (ratio = 0.35). These pathways also had a high predicted relevance for WNV pathogenesis based on P value (-log P = 10.0 to 12.0). TNFR1 signaling, TNFR2 signaling, and induction of apoptosis by HIV-1 are all subsets of DR apoptotic signaling. Disregarding IκB, IKK, and NF-κB, which are ubiquitous in many of the IPA pathways, DR apoptotic signaling involves 34 genes, 8 of which were upregulated between 1.25- and 2-fold following WNV infection and 9 of which were upregulated more than 2-fold. All genes that were upregulated more than 2-fold were confirmed by RT-PCR. Prior gene expression studies have also identified DR-associated genes that are differentially regulated following WNV infection. For example, (i) Daxx is upregulated in the spleens of mice infected with neuroinvasive strains of WNV compared to noninvasive strains (23); (ii) the death receptor-associated genes encoding cIAP2, TRAIL, TNF, and TRAF1 are upregulated in WNV-infected human retinal pigment epithelium (24), and (iii) multiple pathways involved in apoptosis, including induction of apoptosis by HIV-1, were identified in the brains of horses with exposure to WNV (25). In addition, we previously performed microarray analysis on RNA extracted from the brains of reovirus-infected mice (26). The expression of all DR-associated WNV-induced genes with an expression change of ≥2 was also significantly changed following infection of the brain with reovirus (not shown), including the gene encoding Daxx (27), suggesting that similar patterns of gene expression are seen in the brain following infection with a variety of viruses.
The upregulation of the DR-associated genes encoding Fas and TRAIL is consistent with previous reports highlighting the upregulation of Fas in cortical neurons following WNV infection (10) and the presence of TRAIL on CD8+ T cells from the brains of WNV-infected animals (15). Both these observations were linked with the ability of CD8+ T cells to mediate virus clearance from the CNS (10, 15). Increased TNF-α expression in WNV-infected neurons in vivo has also been previously demonstrated and may increase neuronal survival by downregulating expression of the CXCL10 receptor, CXCR3 (28).
We now show that the upregulation of DR-associated genes in the brain following WNV infection is associated with a significant increase in activity of the DR-associated initiator caspase, caspase 8, which in turn activates the effector caspase, caspase 3. Increased caspase 3 activation is consistent with prior studies highlighting the importance of caspase 3 in WNV pathogenesis (2). Increased caspase 8 activity in WNV-infected brains was also found to be associated with a small but significant increase in the activation of the initiator caspase, caspase 9. This suggests that caspase 8 also activates mitochondrial apoptotic signaling in WNV-infected brains, and we further show that, consistent with earlier reports from studies performed in vitro (3), Bid is cleaved in the brains of WNV-infected mice. Cleaved Bid is known to translocate to the mitochondria, where it interacts with other Bcl-2 family proteins to create pores in the mitochondrial membrane through which proapoptotic mitochondrial proteins, including cytochrome c, are released. Cytochrome c associates with Apaf 1 to activate caspase 9. Bid, Apaf1, and caspase 9 are all encoded by genes that were shown by microarray analysis to be upregulated in the brain following WNV infection (Fig. 1). Prior studies in vitro have also suggested that both DR and mitochondrial pathways of apoptosis are activated following WNV infection (3).
Although apoptosis is induced in primary neurons following WNV infection, the role of Fas and TRAIL in CD8+ T cell-mediated clearance of WNV infection suggests that infiltrating immune cells may be required for DR apoptotic signaling in the brain. We have recently developed an ex vivo brain slice culture system to investigate reovirus-induced pathogenesis in isolation from peripheral immunity and associated immune cells (22). We now show that this culture system can be used for investigations of WNV pathogenesis. West Nile virus-infected BSC released more LDH than mock-infected controls, indicating that WNV causes tissue injury in this ex vivo model (Fig. 4). In addition, we showed that WNV induces the activation of caspase 3 in infected slices (Fig. 4). Levels of LDH release and caspase 3 activation peak at 7 dpi, suggesting that caspase 3 activation is linked to virus-induced tissue injury, as has been shown in vivo (2). West Nile virus-infected BSC also show increased expression of DR-associated genes, indicating that WNV infection of ex vivo brain slices appropriately mirrors WNV infection in vivo. Using this culture system, we were able to show that WNV-induced activation of caspase 3 was inhibited in the presence of a cell-permeative inhibitor of caspase 8, indicating that caspase 8 contributes to the activation of caspase 3 and the onset of WNV-induced apoptosis. The ex vivo isolation of brain tissue allows us to infer that these phenotypes are not dependent upon infiltration of immune cells.
This report focuses on apoptotic signaling pathways that were identified by IPA as having relevance for WNV infection based on differential gene regulation. However, IPA also identified pathways involved in IFN signaling. The genes with the highest levels of upregulation (50+ fold) were all associated with the IFN response (Table 2), including genes expressing GTPases, antiviral proteins, immune cell attractants, and proteins involved in the regulation of IFN signaling. Mammalian cells have been shown to detect WNV and induce type 1 IFNs (IFN-α/β) by processes involving RIG-I (29), MDA5 (29), and IPS-1 (30, 31), with lesser contributions from protein kinase R (PKR) (32), TLR3 (33, 34), TLR7 (35), and MyD88 (36). IFN-α and -β are produced during the earliest stages of WNV infection after host cell recognition of viral RNA, and mice with genetic defects in individual RNA sensors, the receptor for IFN-α and -β, or constituents of its signaling cascade (e.g., STAT1) show markedly enhanced viral burden in tissues, leading to rapid lethality (37). Although IFN restricts infection, WNV has evolved countermeasures to limit its efficacy and attenuates IFN function at multiple steps of the induction and signaling cascade (38–44). Indeed, WNV is resistant to the antiviral effects of IFN in cell culture once infection is established, which may explain the relatively narrow therapeutic window for IFN administration that has been observed in animal models or humans infected with WNV (45).
The precise role of microglia and astrocytes following WNV infection is unclear. Although WNV does not appear to replicate in microglia (46), increased WNV replication is associated with decreased microglial activation in SARM (sterile alpha and HEAT/Armadillo motif) −/− mice (47), suggesting that microglia are important in limiting WNV growth in the CNS. Infection of astrocytes is also thought to influence WNV pathogenesis in the CNS. Humans and hamsters with WNV-induced spinal cord disease had greater numbers of glial fibrillary acidic protein (GFAP)-positive astrocytes than controls (48). In addition, pathogenic, but not nonpathogenic, WNV strains are able to replicate in cultured astrocytes (49), which may lead to the induction of multiple matrix metalloproteinases (MMPs) and blood-brain-barrier (BBB) disruption (50). Our report demonstrates that microglia and astrocytes are activated in BSC following WNV infection. We also show that infection of astrocytes leads to astrocyte death. Local immune responses thus probably influence WNV pathogenesis. The release of chemokines and cytokines from activated microglia and astrocytes likely contributes to inflammation following WNV infection. In addition, the death of infected astrocytes may increase neuronal cell death.
ACKNOWLEDGMENTS
This publication was supported by NIH grants R01 NS076512 (K.L.T.), R21 AL101064 (K.L.T.), F30 NS071630 (K.R.D.), T32 GM008497 (K.R.D.), and U54 AI065357/RMDP009 (D.B.) and by a VA merit grant (K.L.T.). K.L.T. is supported by the Reuler-Lewin Family Professorship.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Clarke P, Tyler KL. 2009. Apoptosis in animal models of virus-induced disease. Nat. Rev. Microbiol. 7:144–155. 10.1038/nrmicro2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samuel MA, Morrey JD, Diamond MS. 2007. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J. Virol. 81:2614–2623. 10.1128/JVI.02311-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleinschmidt MC, Michaelis M, Ogbomo H, Doerr HW, Cinatl J., Jr 2007. Inhibition of apoptosis prevents West Nile virus induced cell death. BMC Microbiol. 7:49. 10.1186/1471-2180-7-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramanathan MP, Chambers JA, Pankhong P, Chattergoon M, Attatippaholkun W, Dang K, Shah N, Weiner DB. 2006. Host cell killing by the West Nile virus NS2B-NS3 proteolytic complex: NS3 alone is sufficient to recruit caspase-8-based apoptotic pathway. Virology 345:56–72. 10.1016/j.virol.2005.08.043 [DOI] [PubMed] [Google Scholar]
- 5.Parquet MC, Kumatori A, Hasebe F, Morita K, Igarashi A. 2001. West Nile virus-induced bax-dependent apoptosis. FEBS Lett. 500:17–24. 10.1016/S0014-5793(01)02573-X [DOI] [PubMed] [Google Scholar]
- 6.Diamond MS. 2009. Virus and host determinants of West Nile virus pathogenesis. PLoS Pathog. 5:e1000452. 10.1371/journal.ppat.1000452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shrestha B, Diamond MS. 2004. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 78:8312–8321. 10.1128/JVI.78.15.8312-8321.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrestha B, Samuel MA, Diamond MS. 2006. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 80:119–129. 10.1128/JVI.80.1.119-129.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shrestha B, Wang T, Samuel MA, Whitby K, Craft J, Fikrig E, Diamond MS. 2006. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J. Virol. 80:5338–5348. 10.1128/JVI.00274-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrestha B, Diamond MS. 2007. Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J. Virol. 81:11749–11757. 10.1128/JVI.01136-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brien JD, Uhrlaub JL, Nikolich-Zugich J. 2007. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur. J. Immunol. 37:1855–1863. 10.1002/eji.200737196 [DOI] [PubMed] [Google Scholar]
- 12.Kleinschmidt-DeMasters BK, Marder BA, Levi ME, Laird SP, McNutt JT, Escott EJ, Everson GT, Tyler KL. 2004. Naturally acquired West Nile virus encephalomyelitis in transplant recipients: clinical, laboratory, diagnostic, and neuropathological features. Arch. Neurol. 61:1210–1220. 10.1001/archneur.61.8.1210 [DOI] [PubMed] [Google Scholar]
- 13.Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. 2005. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202:1087–1098. 10.1084/jem.20042530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanteri MC, Heitman JW, Owen RE, Busch T, Gefter N, Kiely N, Kamel HT, Tobler LH, Busch MP, Norris PJ. 2008. Comprehensive analysis of West Nile virus-specific T cell responses in humans. J. Infect. Dis. 197:1296–1306. 10.1086/586898 [DOI] [PubMed] [Google Scholar]
- 15.Shrestha B, Pinto AK, Green S, Bosch I, Diamond MS. 2012. CD8+ T cells use TRAIL to restrict West Nile virus pathogenesis by controlling infection in neurons. J. Virol. 86:8937–8948. 10.1128/JVI.00673-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brault AC, Huang CY, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 39:1162–1166. 10.1038/ng2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. 10.1093/biostatistics/4.2.249 [DOI] [PubMed] [Google Scholar]
- 19.Bruce AJ, Sakhi S, Schreiber SS, Baudry M. 1995. Development of kainic acid and N-methyl-D-aspartic acid toxicity in organotypic hippocampal cultures. Exp. Neurol. 132:209–219. 10.1016/0014-4886(95)90026-8 [DOI] [PubMed] [Google Scholar]
- 20.Noraberg J, Kristensen BW, Zimmer J. 1999. Markers for neuronal degeneration in organotypic slice cultures. Brain Res. Brain Res. Protoc. 3:278–290. 10.1016/S1385-299X(98)00050-6 [DOI] [PubMed] [Google Scholar]
- 21.Stoppini L, Buchs PA, Muller D. 1991. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37:173–182. 10.1016/0165-0270(91)90128-M [DOI] [PubMed] [Google Scholar]
- 22.Dionne KR, Leser JS, Lorenzen KA, Beckham JD, Tyler KL. 2011. A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp. Neurol. 228:222–231. 10.1016/j.expneurol.2011.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venter M, Myers TG, Wilson MA, Kindt TJ, Paweska JT, Burt FJ, Leman PA, Swanepoel R. 2005. Gene expression in mice infected with West Nile virus strains of different neurovirulence. Virology 342:119–140. 10.1016/j.virol.2005.07.013 [DOI] [PubMed] [Google Scholar]
- 24.Munoz-Erazo L, Natoli R, Provis JM, Madigan MC, King NJ. 2012. Microarray analysis of gene expression in West Nile virus-infected human retinal pigment epithelium. Mol. Vis. 18:730–743 [PMC free article] [PubMed] [Google Scholar]
- 25.Bourgeois MA, Denslow ND, Seino KS, Barber DS, Long MT. 2011. Gene expression analysis in the thalamus and cerebrum of horses experimentally infected with West Nile virus. PLoS One 6:e24371. 10.1371/journal.pone.0024371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyler KL, Leser JS, Phang TL, Clarke P. 2010. Gene expression in the brain during reovirus encephalitis. J. Neurovirol. 16:56–71. 10.3109/13550280903586394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dionne KR, Zhuang Y, Leser JS, Tyler KL, Clarke P. 9 January 2013. Daxx upregulation within the cytoplasm of reovirus-infected cells is mediated by interferon and contributes to apoptosis. J. Virol. 10.1128/JVI.02324-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang B, Patel J, Croyle M, Diamond MS, Klein RS. 2010. TNF-alpha-dependent regulation of CXCR3 expression modulates neuronal survival during West Nile virus encephalitis. J. Neuroimmunol. 224:28–38. 10.1016/j.jneuroim.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr 2008. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 82:609–616. 10.1128/JVI.01305-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daffis S, Suthar MS, Szretter KJ, Gale M, Jr, Diamond MS. 2009. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 5:e1000607. 10.1371/journal.ppat.1000607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M., Jr 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 6:e1000757. 10.1371/journal.ppat.1000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilfoy FD, Mason PW. 2007. West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J. Virol. 81:11148–11158. 10.1128/JVI.00446-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daffis S, Samuel MA, Suthar MS, Gale M, Jr, Diamond MS. 2008. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 82:10349–10358. 10.1128/JVI.00935-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373. 10.1038/nm1140 [DOI] [PubMed] [Google Scholar]
- 35.Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. 2009. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 30:242–253. 10.1016/j.immuni.2008.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Jr, Diamond MS. 2010. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 84:12125–12138. 10.1128/JVI.01026-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daffis S, Suthar MS, Gale M, Jr, Diamond MS. 2009. Measure and countermeasure: type I IFN (IFN-alpha/beta) antiviral response against West Nile virus. J. Innate Immun. 1:435–445. 10.1159/000226248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu WJ, Wang XJ, Clark DC, Lobigs M, Hall RA, Khromykh AA. 2006. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 80:2396–2404. 10.1128/JVI.80.5.2396-2404.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scholle F, Mason PW. 2005. West Nile virus replication interferes with both poly(I:C)-induced interferon gene transcription and response to interferon treatment. Virology 342:77–87. 10.1016/j.virol.2005.07.021 [DOI] [PubMed] [Google Scholar]
- 40.Wilson JR, de Sessions PF, Leon MA, Scholle F. 2008. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 82:8262–8271. 10.1128/JVI.00226-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fredericksen BL, Gale M., Jr 2006West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 80:2913–2923. 10.1128/JVI.80.6.2913-2923.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo JT, Hayashi J, Seeger C. 2005. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 79:1343–1350. 10.1128/JVI.79.3.1343-1350.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, García-Sastre A. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013. 10.1128/JVI.79.13.8004-8013.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mackenzie JM, Khromykh AA, Parton RG. 2007. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2:229–239. 10.1016/j.chom.2007.09.003 [DOI] [PubMed] [Google Scholar]
- 45.Chan-Tack KM, Forrest G. 2005. Failure of interferon alpha-2b in a patient with West Nile virus meningoencephalitis and acute flaccid paralysis. Scand. J. Infect. Dis. 37:944–946. 10.1080/00365540500262690 [DOI] [PubMed] [Google Scholar]
- 46.Cheeran MC, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. 2005. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 11:512–524. 10.1080/13550280500384982 [DOI] [PubMed] [Google Scholar]
- 47.Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J. Virol. 83:9329–9338. 10.1128/JVI.00836-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blakely PK, Kleinschmidt-DeMasters BK, Tyler KL, Irani DN. 2009. Disrupted glutamate transporter expression in the spinal cord with acute flaccid paralysis caused by West Nile virus infection. J. Neuropathol. Exp. Neurol. 68:1061–1072. 10.1097/NEN.0b013e3181b8ba14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hussmann KL, Samuel MA, Kim KS, Diamond MS, Fredericksen BL. 2013. Differential replication of pathogenic and nonpathogenic strains of West Nile virus within astrocytes. J. Virol. 87:2814–2822. 10.1128/JVI.02577-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. 2010. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397:130–138. 10.1016/j.virol.2009.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]