

Abstract
Productive replication of human papillomavirus type 16 (HPV16) occurs only in differentiated keratinocyte cells. In addition to the viral E2 activator protein, HPV16 and related HPV types express transcripts coding for an E8^E2C fusion protein, which limits genome replication in undifferentiated keratinocytes. To address E8^E2C's role in productive replication of HPV16, stable keratinocyte cell lines containing wild-type (wt), E8^E2C knockout (E8−), or E8 KWK mutant (mt) genomes, in which conserved E8 residues were inactivated, were established. Copy numbers of E8− and E8 KWK mt genomes and amounts of early and late viral transcripts were greatly increased compared to those for the wt in undifferentiated keratinocytes, suggesting that HPV16 E8^E2C activities are highly dependent upon the E8 part. Upon differentiation in organotypic cultures, E8 mt genomes displayed higher early viral transcript levels, but no changes in cellular differentiation or virus-induced cellular DNA replication in suprabasal cells were observed. E8 mt genomes were amplified to higher copy numbers and showed increased L1 transcripts compared to wt genomes. Furthermore, the number of cells expressing the viral late protein E4 or L1 or amplifying viral genomes was greatly increased in E8 mt cell lines. In wild-type cells, E8^E2C transcript levels did not decrease by differentiation. Our data indicate that the E8^E2C repressor limits viral transcription and replication throughout the complete life cycle of HPV16.
INTRODUCTION
Persistent infections with high-risk (HR) human papillomaviruses (HPV) are a necessary risk factor for the development of cancer of the cervix uteri and are also responsible for a fraction of anal, vaginal, penile, and oropharyngeal cancers (1–3). Notably, HPV16 is an extremely powerful human carcinogen that is responsible for approximately 50% of cervical cancers and is also represented in other HR-HPV-related cancers at high frequencies (2).
HPV have a double-stranded DNA genome that is covalently closed and organized in nucleosomes. HPV possess eight to nine open reading frames (E1, E2, E4 to E8, L1, and L2) that are transcribed into polycistronic messages which are further processed by alternative splicing (4). It is currently assumed that HR-HPV encode at least 10 different viral proteins.
The HR-HPV replication cycle is tightly linked to the differentiation state of the infected epithelium. In undifferentiated basal-cell-like cells, HR-HPV genomes are maintained in the nucleus at 10 to 100 extrachromosomal copies per cell, and only early genes, mainly transcribed from the major early viral promoter located immediately upstream of the E6 gene (P97 for HPV16), but not the late viral L1 and L2 genes are expressed (5). Replication of the viral genome in undifferentiated cells requires the viral E1 and E2 proteins (6). E1 and E2 are sequence-specific DNA binding proteins which form a complex that recognizes the viral origin of replication (6). E1 assembles into a hexamer that acts as the replicative helicase and unwinds the viral DNA and also recruits cellular factors to enable replication of the viral genomes (6). E2's prime function for viral DNA replication is to enhance E1 binding to the origin, but it also recruits cellular proteins that contribute to viral DNA replication (6–9). Furthermore, E2 serves as a segregation factor for viral genomes during cell division (10).
Upon cellular differentiation, a series of events takes place that results in the synthesis of infectious virions in the uppermost layers of the infected epithelium. HR-HPV-infected keratinocytes reenter the S phase of the cell cycle and induce cellular DNA replication in the first suprabasal layers and proceed to the G2/M phase in middle layers (11, 12). In middle and upper epithelial layers, the viral late promoter (HPV16: P670) located in the E7 gene becomes active, and this coincides with the abundant expression of the viral E4 (E1^E4) protein and the amplification of the viral genomes (13–16). The viral late promoter is also responsible for the expression of L1 and L2 transcripts, and thus L1 and L2 protein expression and virus capsid synthesis can be observed in upper epithelial layers (12, 13, 15, 17). The contributions of viral gene products to the differentiation-dependent activation of the late promoter and amplification of viral genomes have been investigated by the analysis of viral mutant (mt) genomes, revealing that the E6, E7, E4, and E5 genes contribute to differentiation-dependent genome amplification and late gene expression. The knockout of E7 in HPV16 or HPV18 leads to a loss of genome amplification and L1 expression in differentiated cells, which is most likely due to the inability to induce DNA replication in suprabasal cells (18, 19). Similarly, the knockout of E6 in the context of the HPV18 genome resulted in a lack of genome amplification in differentiated cells, and this correlated with increased p53 protein levels and the absence of DNA replication in suprabasal cells (20). The knockout of E5 in HPV16 and -31 decreases but does not abolish late gene expression, and this correlates with reduced numbers of suprabasal cells in which DNA replication is induced (21, 22). HPV16 and -31 E4 knockout genomes also displayed decreased genome amplification and late gene expression, which also correlated with reduced DNA replication levels in suprabasal cells (23, 24).
In addition to full-length-form E2, several papillomaviruses (PV) express a spliced mRNA which links a splice donor site in E1 to the major splice acceptor site in the E2/E4 region. This transcript encodes an E8^E2C (or E8/E2) protein which consists of the E8 gene product fused to the C-terminal half of E2, which mediates sequence-specific DNA binding and dimerization of E2 proteins (16, 25–34). Genetic analyses have revealed that E8^E2C is a potent inhibitor of the genome replication of HR-HPV16, -18 and -31 in undifferentiated cells (29, 30, 34, 35). HR-HPV E8^E2C proteins are efficient repressors of the major viral early promoter (27, 30, 34, 36, 37). In addition, E8^E2C proteins inhibit the E1- and E2-dependent replication of the viral origin (30, 34, 35). Thus, the overreplication phenotype of E8^E2C mt genomes is most likely due to a derepression of both viral transcription and E1/E2-dependent replication.
Mutational analysis of the HPV31 E8^E2C protein has indicated that residues K5, W6, and K7 of the E8 domain, which are conserved among HR-HPV16, -18, -31, and -33 and many other alphapapillomaviruses, are required for both the repression of transcription and the inhibition of E1/E2-dependent replication of HPV31 (35, 37, 38). Recent studies have demonstrated that the E8 domain of HPV31 functionally interacts with cellular transcriptional repressor proteins, such as histone deacetylase 3 (HDAC3) and NCoR (38, 39). This suggests that E8^E2C proteins recruit cellular proteins to inhibit viral transcription and replication.
Currently, it is not known if E8^E2C proteins are also involved in the differentiation-dependent regulation of the late promoter and in viral genome amplification. Based on E8^E2C's inhibitory activities in undifferentiated cells, a differentiation-dependent downregulation of E8^E2C activity might contribute to the activation of the late promoter and/or genome amplification.
Our studies reveal that HPV16 E8 mt genomes not only display higher copy numbers and viral transcript levels than the wild type (wt) in undifferentiated cells but also in cells differentiated in organotypic cultures. This strongly suggests that the E8^E2C protein limits genome replication of HPV16 at all stages of the viral life cycle in an E8 domain-dependent manner.
MATERIALS AND METHODS
Recombinant plasmids.
The plasmid pBS 16 114b:1, harboring the HPV16 114/b wt genome, was kindly provided by M. Dürst (40), and plasmid hPGK1 pEF-IRES-P, encoding the cDNA for human phosphoglycerate kinase 1 (PGK1), was kindly provided by D. Zieker (41). The HPV16 E8− and E8 KWK mt genomes were constructed by overlap extension PCR using specific primers and verified by DNA sequencing. The E8− mutation replaces the codon for W6 with a stop codon in the E8 open reading frame (ORF) (TGG to TAG) and is silent in the overlapping E1 gene (30). In the E8 KWK mt, E8 residues K5/W6/K7 were changed to AAA, which also changes the E1 coding sequence from E138/V139/E140 to GSG. The pGL 16URR-luc and pC18-Sp1-luc luciferase reporter plasmids and the expression plasmids for HPV16 E2 and HPV16 E8^E2C and the respective hemagglutinin (HA)-tagged versions (HPV16 E2-HA and HPV16 E8^E2C-HA) have been previously described (27). Plasmid pSG 16 E8^E2C KWK mt and 16 E8^E2C-KWK mt –HA were constructed by PCR. The HPV16 E1 gene was amplified using plasmid pBS 16 114b:1, and the HPV16 E1 E138G/V139S/E140G gene was amplified using plasmid pHPV16 E8 KWK mt as a template and primers adding EcoRI and BglII restriction sites and then inserted into pSG5, giving rise to pSG 16 E1 and pSG 16 E1 E138G/V139S/E140G, respectively. Plasmid pSG 16 UTR E8^E2C was constructed by PCR and has a short 5′-untranslated region (HPV16 nucleotides [nt] 1257 to 1264) in addition to the E8^E2C coding region.
Cell culture.
The HPV-negative human keratinocyte cell line RTS3b and HeLa cells were maintained as previously described (42). HPV16-positive keratinocytes were established with minor modifications as described previously (34). Briefly, HPV16 genomes were released by BamHI digestion from the vector and gel purified. Linearized genomes were self-ligated at a concentration of 10 μg/ml using T4 DNA ligase and then concentrated by ethanol precipitation. Normal human foreskin keratinocytes (NHKs) from two different donors were cotransfected with HPV16 wt or mt genomes (1.5 μg) and pSV2 neo (1.5 μg) using the Fugene HD reagent (Promega). Cells were selected with G418 (150 μg/ml) for 14 days in the presence of mitomycin C-treated, neomycin-resistant 3T3 J2NHP cells (43) and E medium supplemented with 5% fetal bovine serum as previously described (15, 34). After drug selection, cells were cocultured with mitomycin C-treated NIH 3T3 J2 (34). To induce differentiation, NHKs or HPV16-positive keratinocytes were seeded onto collagen plugs in which NIH 3T3 J2 cells were embedded in cell culture inserts (140663; Thermo Fisher Scientific). After the keratinocytes reached confluence, the medium was completely removed from the inside of the inserts. Inserts were then placed in 100-mm culture dishes, and complete E medium without epidermal growth factor (EGF) was added to the culture dishes and changed every other day. Organotypic cultures were harvested 12 days after exposure to the air-liquid interface and cut into pieces to isolate RNA or DNA or to obtain cryosections for antibody staining.
Reporter gene assays.
HeLa or RTS3b cells (3 × 104) were seeded into 24-well dishes 1 day before transfection. Cells were transfected with reporter plasmids alone or together with pSG 16 E1, pSG 16 E2, pSG 16 E8^E2C, or 16 E8^E2C KWK mt expression constructs or the empty vector pSG5 using the Fugene HD reagent (Promega) and Opti-MEM (Life Technologies). Furthermore, 0.1 ng of pCMV-Gluc plasmid (New England BioLabs) was cotransfected as an internal control. Gaussia and firefly luciferase assays were carried out 48 h after transfection.
Immunoblot analysis.
To detect E8^E2C and E2 proteins by immunoblotting, 3 × 105 RTS3b cells were transfected with 0.4 μg of pSG expression plasmids and lysed 48 h after transfection in SDS-PAGE gel loading buffer. Equal aliquots were separated in a 12% SDS-PAGE gel and transferred onto a nitrocellulose membrane. Membranes were incubated with 1:1,000-diluted primary antibodies (anti-HA [MMS-101P; Covance] and anti-heat shock protein 90 [HSP90] [sc-69703; Santa Cruz Biotechnology]). Bound antibodies were detected with fluorescence-labeled antibodies (IRDye 680RD goat anti-mouse IgG and IRDye 800CW goat anti-rabbit IgG; Li-Cor) and an OdysseyFC infrared imaging system (Li-Cor Biosciences).
qPCR.
RNA was isolated from monolayer or organotypic cell cultures using the RNeasy minikit (Qiagen). RNA (1 μg) was reverse transcribed using the QuantiTect reverse transcription kit (Qiagen). Fifty nanograms of cDNA was analyzed in duplicate reaction mixtures by quantitative PCR (qPCR) using 0.3 μM gene-specific primers and 1× LightCycler 480 SYBR green I Master (Roche Applied Science) in a total volume of 20 μl. Reactions were carried out in a Light Cycler 480 instrument (Roche Applied Science) using a thermal profile of 10 min at 95°C followed by 45 cycles for 10 s at 95°C, 15 s at 55°C, 15 s at 72°C, melting curve for 10 s at 95°C, 30 s at 60°C, 90°C, and cooling for 30 s at 40°C and analyzed using the LightCycler 480 software program, version 1.5 (Roche Applied Science). Expression levels were determined using standard curves of plasmids pBS 16 114b:1, pSG 16 UTR E8^E2C, or hPGK1 pEF-IRES-P. The following primers were designed using the Primer 3 plus software program (44): PGK1 sense (5′-CTGTGGGGGTATTTGAATGG-3′) and antisense (5′-CTTCCAGGAGCTCCAAACTG-3′); HPV16 E6 (HPV16 E6i 237F, 5′-TTGCTTTTCGGGATTTATGC-3′; HPV16 E6 439R, 5′-CAGGACACAGTGGCTTTTGA-3′), HPV16 E7 (HPV16 E7 649F, 5′-GACAGCTCAGAGGAGGAGGA-3′; HPV16 E7 816R, 5′-GCCCATTAACAGGTCTTCCA-3′), HPV16 E1 (HPV16 E1C 1557F, 5′-AACGTGTTGCGATTGGTGTA-3′; HPV16 E1C 1804R, 5′-TACGCAATTTTGGAGGCTCT-3′), HPV16 E2 (16 E2N 3105F, 5′-TGGAAGTGCAGTTTGATGGA-3′; HPV16 E2N 3320R, 5′-CCGCATGAACTTCCCATACT-3′), HPV16 E8^E2C (16E8 1259F, 5′-GCGGGTATGGCAATACTGAA-3′; 16E2C 3475R, 5′-TCGCTGGATAGTCGTCTGTG-3′), and HPV16 L1 (HPV16 L1 6365F, 5′-GAACCATATGGCGACAGCTT-3′; HPV16 L1 6601R, 5′-ATTATTGTGGCCCTGTGCTC-3′).
Total genomic DNA from monolayer or organotypic cell cultures was isolated using the EZ1 instrument and EZ1 DNA tissue kit (Qiagen). HPV16 copy numbers were determined using a multiplex qPCR consisting of a mixture of primer/probe sets to detect HPV16 E6 (95F, 5′-AGAACTGCAATGTTTCAGGACC-3′; 174R, 5′-TGTATAGTTGTTTGCAGCTCTGTGC-3′; probe, 5′-FAM-CAGGAGCGACCCAGAAAGTTACCACAGTT-DQ-3′), HPV16 E2 (3362F, 5′-AACGAAGTATCCTCTCCTGAAATTATTAG-3′; 3443R, 5′-CCAAGGCGACGGCTTTG-3′; probe, 5′-Cy5-CACCCCGCCGCGACCCATA-DQ-3′), and cellular beta-actin (TaqMan gene expression assay Hs03023880_g1; Applied Biosystems). Standard curves of plasmid pBS 16 114b:1 or total genomic DNA from HPV-negative NHKs were run in parallel and used to calculate viral copy numbers (E2) per cell (assuming a DNA content of 6.6 pg for a diploid human keratinocyte cell and 2 copies of beta-actin per cell).
Southern blot hybridization.
Low-molecular-weight DNA from HPV16-positive cell lines was isolated as described previously (34). The equivalent of a half 100-mm tissue culture plate was digested with XhoI, a noncutter for HPV16, and run in a 0.7% agarose gel. Blotting and hybridization to a 32P-labeled HPV16 probe were carried out as described previously (34). After exposure of the membrane to phosphorimager screens, HPV16 genomes were visualized using the AIDA software package (Raytest).
Immunofluorescence analysis.
Organotypic cultures were harvested by detaching the epithelium from the collagen plug. The epithelium was embedded in OCT compound (Tissue-Tek), and frozen, and 6-μm-thick sections were obtained with a cryostat microtome (Reichert-Jung). Sections were fixed in 4% paraformaldehyde–phosphate-buffered saline (PBS) for 15 min at room temperature (RT). After washing steps, sections were incubated in blocking buffer (PBS–5% goat serum–0.3% Triton X-100) for 60 min. Primary antibodies (anti-PCNA [2586; Cell Signaling], anti-MCM2 [3619; Cell Signaling], anti-MCM7 [3735; Cell Signaling], anti-HPV16 E4 [TGV 402] [sc-53324; Santa Cruz Biotechnology], anti-HPV L1 [K1H8, DLN-14563; Dianova], and anti-keratin 10 [RKSE60; Research Diagnostics]) were diluted in PBS–bovine serum albumin (BSA)–0.3% (vol/vol) Triton X-100 and then incubated with the sections overnight at 4°C. After washing with PBS, diluted secondary goat Alexa-488-labeled anti-mouse or donkey Alexa-555-labeled anti-mouse antibodies were added and incubated for 60 min. Samples were then washed several times with PBS, counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted with FluoPrep medium (bioMérieux). Immunofluorescence signals were recorded with an Axiovert 200 M microscope (Zeiss), the appropriate fluorescence filter sets, and a 20× objective. Images were obtained using fixed exposure times for a given primary antibody and the Zeiss Axiovision 4.8 software program. All stainings were performed multiple times with independent organotypic cultures to ensure reproducibility.
Fluorescence in situ hybridization analysis.
Biotin-labeled HPV16 DNA was generated using the Biotin DecaLabel DNA labeling kit (Thermo Fisher Scientific) and then purified using a nucleotide removal kit (Qiagen). Rehydrated cryosections (6 μm) were fixed in 2% formaldehyde for 15 min at RT and then quenched with H2O2 in PBS. Biotinylated HPV16 DNA (5 mg/ml) was added to the sections in hybridization buffer (50% formamide, 4× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 1× Denhardt's, 5% [wt/vol] dextrane sulfate, and 200 μg/ml salmon sperm DNA) covered with a cover slide, heated to 80°C for 7 min and then incubated at 37°C overnight. Cover slides were removed by incubation with 0.5× SSC at RT, followed by washes with wash buffer (50% formamide, 2× SSC, and 0.05% Tween 20) at 42°C and then 2× SSC at 42°C. The hybridized HPV16 DNA was detected with streptavidin-horseradish peroxidase (HRP) and Alexa Fluor 488 tyramide substrate according to the manufacturer's instructions (TSA kit 22; Life Technologies). Images were obtained as described for the immunofluorescence analyses.
RESULTS
The KWK motif of the HPV16 E8^E2C protein is required for repression activities.
To address the role of the E8 domain in E8^E2C for the replication of HPV16, we first generated an HPV16 E8^E2C KWK mt expression construct and compared its activities in transient-transfection experiments to those of the HPV16 E8^E2C wt protein. wt HPV16 E8^E2C repressed the synthetic reporter construct pC18-Sp1-luc, which consists of 4 E2 binding sites and a minimal promoter, in both HPV-positive HeLa cells and HPV-negative RTS3b cells (Fig. 1A). In contrast, E8^E2C KWK mt failed to repress but rather slightly activated the reporter (Fig. 1A). Similarly, an HPV16 upstream regulatory region (URR) construct, in which the P97 major viral early promoter drives luciferase expression, was 5-fold inhibited in HeLa and RTS3b cells at 0.1 ng of HPV16 E8^E2C expression vector, whereas the E8^E2C KWK mt plasmid repressed less than 2-fold (Fig. 1B). Also, at higher concentrations, the wt repressed the URR construct much more efficiently than E8^E2C KWK mt (Fig. 1B). We recently developed a reporter gene expression-based assay for the E1 and E2-dependent replication of the HPV16 and -31 origin which is comparable to an assay described by Fradet-Turcotte and coworkers (45) (M. Dreer and F. Stubenrauch, unpublished observation). wt E8^E2C repressed E1/E2-dependent replication efficiently, whereas E8^E2C KWK mt displayed only weak replication repression activity in HeLa cells (Fig. 1C). To test whether the mutation of the KWK motif influences the abundance of the E8^E2C protein, expression plasmids for HA-tagged versions of E2, E8^E2C, and E8^E2C-KWK mt were transfected into RTS3b cells, and protein expression was analyzed by immunoblotting. HPV16 E2-HA and E8^E2C-HA were present at comparable levels, similar to findings obtained in HeLa cells (27), whereas much higher levels of the E8^E2C-KWK mt –HA protein could be detected (Fig. 1c). This has also been observed with HPV31 E8^E2C KWK mt (36, 37) and strongly suggests that the loss of repression activity of HPV16 E8^E2C KWK mt is not due to decreased protein levels. In summary, these data reveal that the E8 KWK mt of HPV16 E8^E2C behaves very similarly to the corresponding HPV31 mt (35, 37).
FIG 1.

Characterization of HPV16 E8^E2C wt and KWK mutant proteins. (A) HeLa or RTS3b cells were cotransfected with pC18-Sp1-luc (100 ng), 10 ng of pSG5 (vec), pSG 16 E8^E2C, or pSG 16 E8^E2C KWK mt and 0.1 ng of pCMV-Gluc. (B) HeLa or RTS3b cells were cotransfected with pGL 16URR-luc (100 ng), pCMV-Gluc (0.1 ng), and only pSG5 (0 ng) or the indicated amounts of pSG 16 E8^E2C or pSG 16 E8^E2C KWK mt (0.1, 1, and 10 ng). The total amount of DNA was kept constant by adding the pSG5 plasmid. (C) HeLa cells were cotransfected with 10 ng of pGL 16URR-luc (URR) and pCMV-Gluc (0.1 ng) and combinations of pSG 16 E1 or pSG E1 E138G/V139S/E140G (E1 mt) (100 ng), pSG 16 E2 (10 ng), pSG 16 E8^E2C (1 ng), or pSG 16 E8^E2C KWK mt (1 ng) as indicated. The total amount of DNA was kept constant by adding the pSG5 plasmid. Values are presented as the ratio of firefly luciferase (fluc) to Gaussia luciferase (gluc) activities. Data are derived from three (A and C) or four (B) independent experiments performed in duplicate. Error bars indicate the standard errors of the means. (D) RTS3b cells were transfected with 0.4 μg of the empty expression vector pSG5 (1), pSG 16 E2-HA (2), pSG 16 E8^E2C-HA (3), or pSG 16 E8^E2C KWK mt –HA (4) plasmid. Equal amounts of whole-cell lysates were analyzed by immunoblotting using an anti-HA antibody to detect E2 or E8^E2C proteins and anti-HSP90 to detect HSP90 as a reference protein. A molecular weight marker (in thousands) is indicated on the left.
Knockout of E8^E2C or mutation of the E8 KWK motif increases HPV16 copy numbers and transcript levels in undifferentiated keratinocytes.
To address the role of E8^E2C for HPV16 replication, two different mt genomes were constructed in which E8^E2C expression was prevented by introducing a stop codon in the E8 gene as previously described (16 E8− [30]) or the E8 KWK motif was mutated (16 E8 KWK mt). HPV16 wt and mt genomes were transfected into normal human keratinocytes from two independent donors (1 and 2), and stable cell lines were established by drug selection. At passage 5 to 6, low-molecular-weight DNA was isolated and subjected to Southern blot analysis (Fig. 2A). This revealed that HPV16 wt and mt genomes were predominantly maintained extrachromosomally and suggested that both the E8 knockout and mutations in E8 resulted in increased copy numbers. To quantitate the differences more accurately, total cellular DNA was isolated and analyzed by a multiplex qPCR that simultaneously detects HPV16 E6, HPV16 E2, and cellular beta-actin, which allows correcting for cell numbers. This revealed that wt genomes were maintained at 55 and 56 copies/cell, respectively. In contrast, HPV16 E8− genomes were maintained at 360 and 347 copies per cell, respectively. This corresponded to 6.6- and 6.2-fold-increased copy numbers compared to those of the corresponding wt cell lines, which is very similar to the phenotype described by others (30). Interestingly, mutation of the E8 KWK residues resulted in HPV16 genomes that were maintained at 1,587 and 1,149 copies per cell, respectively, corresponding to 29- and 20.5-fold-higher copy numbers than those of the wt in both cell lines. This demonstrated that conserved E8 residues K5W6K7 are required to limit copy number in undifferentiated cells, as shown previously for HPV31 in short-term replication assays (35). To address if the differences in copy numbers between E8− and E8 KWK mt cell lines are due to the simultaneous mutations introduced into E1 (E138G/V139S/E140G), the E1 mt was cloned into an expression vector and functionally analyzed. This revealed that E1 E138G/V139S/E140G had replication activity similar to that of the wt (Fig. 1C). Furthermore, the activity of the E1 mt was also similar to that of the wt when combined with the E8^E2C KWK mt (Fig. 1C). This suggests that the increase in copy number in E8 KWK mt cells is not due to mutations in the overlapping E1 gene. HPV16 E8^E2C is a potent repressor of transcription (Fig. 1) (27, 30). We therefore evaluated the consequences of E8^E2C mutations for HPV16 transcription. Total RNA was isolated from the different cell lines and analyzed by qPCR using primers that detect full-length E6 (nt 237 to 439), E7 (nt 649 to 816), E1 (nt 1557 to 1804), full-length E2 (nt 3105 to 3320), or L1 (nt 6365 to 6601). This revealed that the levels of all early and late transcripts were increased by E8 mutations. Transcripts for E6, E7, E1, and E2 were similarly increased by the E8− mutation, ranging from 2.4- to 2.8-fold in set 1 and 2.1- to 3.2-fold in set 2. E8 KWK mt genome cell lines had similarly increased levels of transcripts, ranging from 3.4- to 6.5-fold for set 1 and from 2.8- to 5.7-fold for set 2 (Fig. 3). However, a difference was observed for L1 transcripts, which were 6.9- and 6.5-fold increased by the E8− mutation and 35.8- and 32.7-fold by the E8 KWK mutation. Taken together, these data confirm that E8^E2C is a negative regulator of viral genome replication but is not required for the episomal maintenance of HPV16 genomes in keratinocytes. Furthermore, the E8^E2C knockout phenotype can be mimicked by mutating conserved K5, W6, and K7 residues in the E8 gene, suggesting that the HPV16 E8 domain plays comparable roles in HPV16 and HPV31 (35). The mutation of E8^E2C not only raises the viral copy number but also increases transcription of viral early and late genes, suggesting that E8^E2C is a general inhibitor of HPV16 genome replication and transcription in undifferentiated cells.
FIG 2.
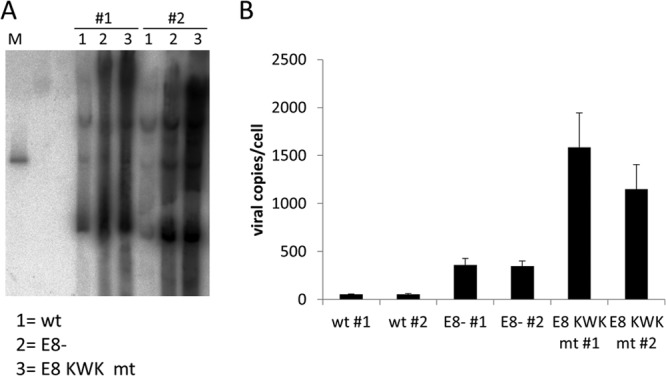
Mutations in E8 increase HPV16 episome copy numbers in normal human keratinocytes. (A) Southern blot analysis of low-molecular-weight DNA isolated from two different sets of low-passage-number HPV16 wt (wt) HPV16 E8− (E8−) or HPV16 E8 KWK mt (E8 KWK mt) cell lines. DNA was digested with XhoI, transferred to a membrane, and detected by hybridizing with a 32P-labeled HPV16 genomic probe. As a size marker, 100 pg of linearized HPV16 wt genome was used (M). (B) Quantitation of HPV16 copy numbers by qPCR. Equal amounts (50 ng) of total genomic DNA from the different cell lines were analyzed by multiplex qPCR. The values represent HPV16 E2 copies per 2 beta-actin copies. Data are derived from four independent DNA preparations for each cell line. Error bars indicate the standard errors of the means.
FIG 3.
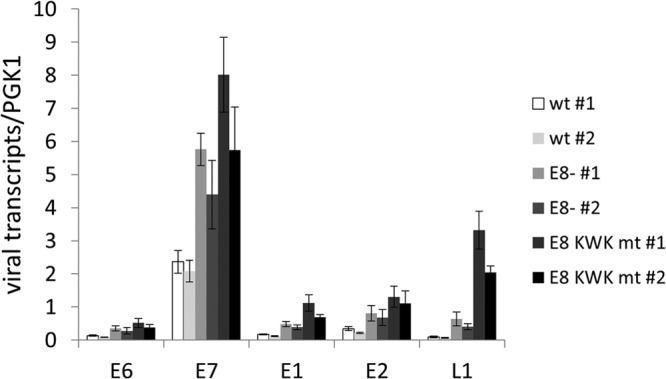
E8 mutations increase HPV16 early and late transcripts in undifferentiated cells. Total RNA from HPV16 wt (#1 and #2), HPV16 E8− (E8− #1 and #2) and HPV16 E8 KWK mt (E8 KWK mt #1 and #2) cells grown in monolayer culture was analyzed by qRT-PCR for the expression of E6, E7, E1, E2, and L1 transcripts using PGK1 as a reference gene. Copy numbers are derived from plasmid dilutions of pBS 16 114b:1 and hPGK1 pEF-IRES-P plasmids measured in parallel. Data are derived from four independent RNA preparations for each cell line. Error bars indicate the standard errors of the means.
E8 mutations increase viral early transcript levels but do not influence DNA replication or differentiation markers in organotypic cultures.
To investigate the consequences of E8^E2C mutations in the productive life cycle of HPV16, HPV16-positive cell lines at low passages (4–6) or NHKs were grown in organotypic raft cultures and harvested 12 days after exposure of the keratinocytes to air. Total RNA was isolated from a part of the raft cultures, and early viral transcripts were quantitated by qPCR. Similar to undifferentiated cultures, the E6, E7, E1, and E2 transcripts were present in larger amounts in E8− and E8 KWK mt cell lines than in the wt (Fig. 4). E6 RNA levels were significantly increased, 6- and 8.3-fold, E7 RNA levels 5.7- and 8.1-fold, E1 RNA levels 5-fold, and E2 RNA levels 4- and 3.8-fold in E8− and E8 KWK mt cell lines, respectively (Fig. 4). However, differences between E8− and E8 KWK mt genomes were not significant. None of these transcripts was induced more than 2-fold in the wt or 3-fold E8 mt cell lines by differentiation (Fig. 4), consistent with the idea that P97 activity is not strongly regulated by cellular differentiation. Cryosections were stained for the differentiation marker keratin 10 and DNA replication proteins MCM2, MCM7, and PCNA (Fig. 5). Keratin 10 staining appeared in the first suprabasal cell layer and was present throughout the epithelium in NHK cells. A very similar pattern was observed in HPV16 wt, E8−, and E8 KWK mt cell lines, suggesting that the loss of E8^E2C does not dramatically influence differentiation. Furthermore, no consistent histological changes were observed between wt and mt cultures. Staining for the DNA replication proteins MCM2, MCM7, and PCNA, which are regarded as surrogate markers mainly for E7 activity (46–48), revealed that their expression is limited to the basal layer in NHK organotypic cultures. In HPV16 wt organotypic cultures MCM2, MCM7, and PCNA were also expressed throughout the epithelium, consistent with previous findings (46–48). Interestingly, no differences in the numbers of positive cells between wt and E8 mt cells were obvious despite significantly higher E7 transcript levels (Fig. 5). These results suggested that mutations of E8^E2C increase early transcript levels but do not change the histology, differentiation, or expression of cellular DNA replication proteins compared to those for wt HPV16.
FIG 4.
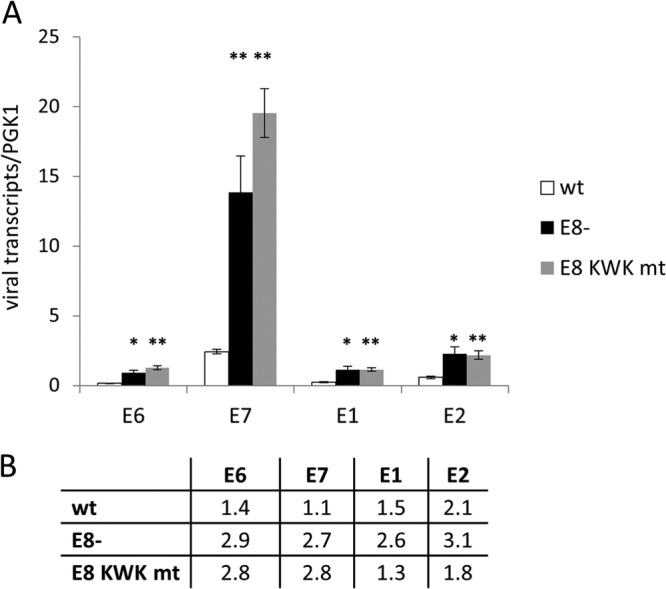
E8 mutations increase viral early transcription in differentiated organotypic cultures. (A) Total RNA from 12-day HPV16 wt (wt), HPV16 E8− (E8−), and HPV16 E8 KWK mt (E8 KWK mt) organotypic cultures was analyzed by qRT-PCR for the expression of E6, E7, E1, and E2 transcripts using PGK1 as a reference gene. Copy numbers are derived from plasmid dilutions of pBS 16 114b:1 and hPGK1 pEF-IRES-P plasmids measured in parallel. The data represent averages from six independent organotypic cultures, and error bars show the standard errors of the means. Statistical significance between results for the wt and mutants was determined by paired, two-sided Student's t test (∗, P < 0.05; ∗∗, P < 0.01). The differences between E8− and E8 KWK mt are not significant. (B) Relative changes of viral transcript levels (E6, E7, E1, and E2) induced by differentiation in HPV16 wt, E8−, or E8 KWK mt genome cell lines.
FIG 5.

E8 mutations do not influence differentiation or expression of cellular DNA replication proteins in organotypic cultures. Frozen sections of 12-day NHK, HPV16 wt (wt), HPV16 E8− (E8−), or HPV16 E8 KWK mt (E8 KWK mt) organotypic cultures were stained with antibodies against keratin 10 (K10), MCM2, MCM7, or PCNA and DAPI to detect cellular DNA.
E8^E2C limits viral genome amplification and expression of late transcripts and proteins.
Next, the extent of genome amplification and viral late protein expression was determined. Total genomic DNA was isolated from a part of the organotypic cultures, and viral copy numbers were quantitated by multiplex qPCR (Fig. 6). HPV16 wt genomes were present on average at 311 copies per cell, corresponding to a 5.6-fold differentiation-dependent increase (Fig. 2 and 6). E8− genomes were present on average at 1,693 copies per cell and E8 KWK mt genomes at 2,539 copies per cell upon differentiation (Fig. 6A). This revealed that E8− genomes were amplified 4.8-fold and E8 KWK mt genomes 1.9-fold upon differentiation, resulting in 5.4- and 8.2-fold-higher viral copy numbers than those of the wt. This surprisingly demonstrated that E8 mt genomes displayed significantly higher copy numbers than wt genomes upon differentiation. In contrast to early transcripts, late L1 transcripts were induced 71.7-fold upon differentiation in wt cells (Fig. 6B). However, L1 transcripts were induced to significantly higher levels in E8− cell lines (7.2-fold versus wt levels) and E8 KWK mt cells (14.5-fold versus wt levels) (Fig. 3 and 6). To further substantiate these findings, sections of organotypic cultures were stained for the late viral proteins E4 and L1 (Fig. 7). Some E4-positive cells appeared in upper layers of the epithelium of HPV16 wt cells. In contrast, nearly all cells in the middle to upper layers of E8− and E8 KWK mt organotypic cultures were strongly E4 positive. Similar results were obtained when staining for L1 (Fig. 7). In wt organotypic cultures, few cells were L1 positive, whereas more L1-positive cell could be observed in E8− and even more in E8 KWK mt cultures, mirroring the expression levels of L1 transcripts (Fig. 6 and 7). In addition, fluorescence in situ hybridization (FISH) analyses demonstrated that the number of cells amplifying viral genomes was increased in E8− and E8 KWK mt cell lines compared to that for the HPV16 wt (Fig. 7). Interestingly, the appearance of markers for productive HPV16 replication occurred in similar layers of wt and mt cells. This suggests that despite increased viral copy numbers and transcript levels, mt genomes are still highly dependent upon differentiation to induce productive replication. In summary, these data suggest that E8^E2C limits genome amplification and late protein expression of HPV16 in differentiating keratinocytes.
FIG 6.
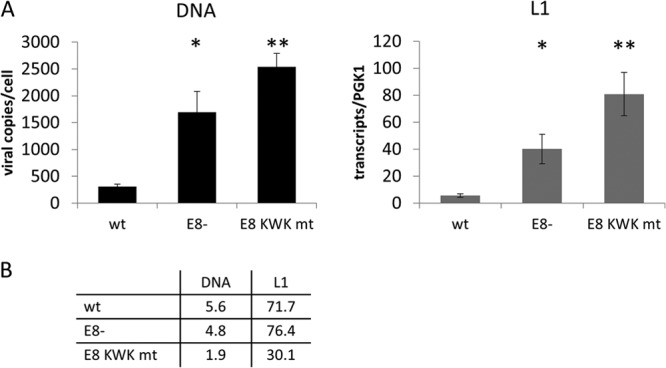
E8 mutant genomes amplify to higher copy numbers and express more L1 transcripts than the wt upon differentiation. (A) (Left panel) Total cellular DNA was isolated from 12-day organotypic cultures, and 50 ng was analyzed by multiplex qPCR as described in the legend for Fig. 2. (Right panel) Quantitation of L1 transcripts by qPCR using PGK1 as a reference gene. Data represent the averages for six independent organotypic cultures, and error bars indicate the standard errors of means. Statistical significance between the wt and mutants was determined by paired, two-sided Student's t test (∗, P < 0.05; ∗∗, P < 0.01). The differences between E8− and E8 KWK mt are not significant. (B) Relative changes in viral genome copies (DNA) and L1 transcript levels (L1) induced by differentiation in HPV16 wt, E8−, or E8 KWK mt genome cell lines.
FIG 7.

E8 mutations increase E4 and L1 protein expression. Frozen sections of 12-day NHK, HPV16 wt (wt), HPV16 E8− (E8−), or HPV16 E8 KWK mt (E8 KWK mt) organotypic cultures were stained with antibodies against HPV16 E4 (E4) or L1. HPV DNA was detected by FISH analyses. DAPI was used to counterstain cellular DNA.
E8^E2C transcripts are present in organotypic cultures of HPV16 wt cells.
To further confirm that E8^E2C limits productive replication of wt HPV16 in differentiating keratinocytes, the amounts of E8^E2C transcripts were determined by qPCR in undifferentiated cells and cells grown in organotypic cultures. In undifferentiated HPV16 wt cells, 0.011 E8^E2C/PGK1 transcripts were present (Fig. 8). In organotypic cultures, slightly higher levels of 0.024 E8^E2C/PGK1 transcripts could be observed. This strongly indicates that E8^E2C transcripts are also present in suprabasal, differentiated cells, since the basal cell layer of organotypic cultures, which is regarded to be similar to undifferentiated monolayer cells, makes up only ∼10% of the total cell number and thus cannot solely account for the amount of E8^E2C transcripts.
FIG 8.
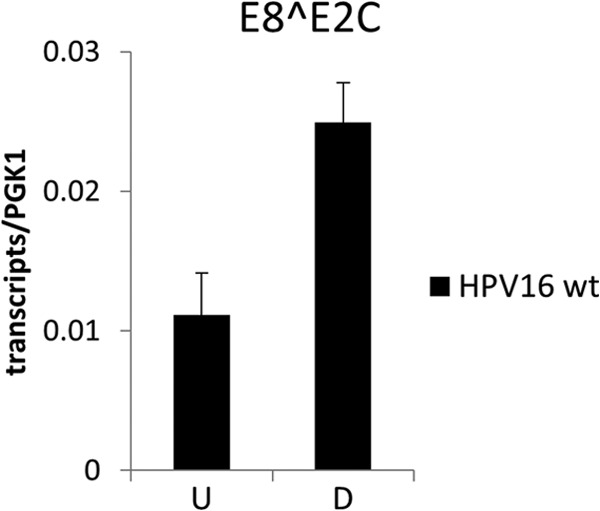
E8^E2C transcripts are present in organotypic cultures. Quantitation of HPV16 E8^E2C transcripts in total RNA from undifferentiated (U) or differentiated (D) HPV16 wt cell lines by qPCR using PGK1 as a reference gene. Copy numbers are derived from plasmid dilutions of pSG 16 UTR E8^E2C and hPGK1 pEF-IRES-P plasmids measured in parallel. Data represent the averages for four (monolayer) or three (organotypic cultures) independent RNA preparations for each cell line, and error bars indicate the standard errors of means.
DISCUSSION
The HPV replication cycle is strictly dependent upon the differentiation of the infected keratinocyte. In upper layers of the epithelium, viral genome amplification and expression of the capsid proteins L1 and L2 are first detected, which results in the production of infectious virions. It is now well established that the reentry of infected cells into the cell cycle in suprabasal layers is a prerequisite for HPV genome amplification and late protein expression. In the context of replicating HR-HPV genomes, the combined activities of E4, E5, E6, and E7 are required to achieve this, and mutations in one of these genes result in decreased or absent viral DNA amplification and late gene expression levels (18–24). Based on their essential replication functions in undifferentiated cells, E1 and E2 are very likely required for differentiation-dependent genome amplification. Thus, six viral ORFs located in the early region are required to allow productive replication.
In addition to positive replication regulators, HR-HPV16, -18, and -31 (and most likely all human alphapapillomaviruses) encode an alternative E2 protein, E8^E2C (E8/E2), which limits genome replication in undifferentiated cells (29, 30, 34). In line with a previous report (30), HPV16 E8^E2C knockout (E8−) genomes replicated at 6.4-fold-higher copy numbers as extrachromosomal elements in undifferentiated keratinocytes. Mutating the conserved E8 residues K5/W6/K7 in HPV16 E8^E2C led to a specific loss of transcription and replication repression (Fig. 1) comparable to that of HPV31 E8^E2C KWK mt (35, 37). HPV16 E8 KWK mt genomes replicated at even higher levels than E8− genomes, demonstrating that the E8 domain is largely responsible for the inhibition of HPV16 genome replication in undifferentiated cells. The increased copy number of E8 KWK mt genomes compared to that of E8− genomes is most likely not a consequence of the mutations in the overlapping E1 gene (Fig. 1C). Both HPV16 and HPV31 E8^E2C KWK mt not only lose the ability to repress transcription and E1/E2-dependent replication but slightly activate transcription from synthetic promoters but not the URR (Fig. 1B) (37). Thus, it is possible that the E8^E2C KWK mt moderately activates viral promoters other than P97 and that this contributes to the increase in copy numbers compared to those of the E8− genome. The overreplication of HPV16 E8 KWK mt genomes confirms and extends observations obtained for HPV31 and, since the HPV16 E8^E2C KWK mt was expressed at higher levels than the wt protein, challenges the view that alternative E2 proteins act mainly via their DNA-binding domain/dimerization domain by binding site competition or formation of inactive heterodimers with E2 (35, 37). It rather suggests that HPV16 E8^E2C, like HPV31 E8^E2C, functionally interacts with the cellular corepressors HDAC3 and NCoR to inhibit transcription and DNA replication (38, 39). Interestingly, E8 mutations not only increased viral copy numbers but also upregulated viral early and late transcription in undifferentiated cells (Fig. 3).
Based on E8^E2C's functions in undifferentiated cells, we reasoned that a differentiation-dependent loss of E8^E2C repression activity might contribute to late promoter activation and/or genome amplification. If E8^E2C activity were completely abolished in suprabasal cells, the levels of viral genome replication and late viral transcripts should be similar in cell lines with wt or E8^E2C knockout HPV16 genomes upon differentiation.
After differentiation of HPV16 wt and E8 mt cell lines, we examined differentiation markers, cellular DNA replication proteins, viral genome amplification, viral early and late transcription, and late viral protein expression. Interestingly, no striking changes in morphology or differentiation were evident between wt and E8 mt organotypic cultures. Similar to undifferentiated cultures, both E8 mt cell lines showed greatly increased E6 and E7 transcript levels, but no obvious changes in the number of cells positive for or expression levels of the cellular DNA replication proteins MCM2, MCM7, and PCNA were evident. This may indicate that E6 and E7 levels produced from wt HPV16 genomes are sufficient to completely reactivate DNA replication in suprabasal cells and this cannot be further enhanced. Previous investigations also did not observe differences in the growth rates of HPV16 wt and HPV16 E8− cell lines in monolayer cultures, which further supports the idea that increased E6 and/or E7 levels do not necessarily affect cell growth (30).
Not only the wt but also E8− and E8 KWK mt genomes were further amplified by differentiation, reaching significantly higher levels than the wt (Fig. 6A). E8 mt cell lines also displayed an increased number of cells positive for the late viral proteins E4 and L1, which correlated with higher L1 transcript levels than those of wt cells (Fig. 6B and 7). This could be due to two different scenarios: (i) E8^E2C acts mainly in undifferentiated cells, and the higher copy numbers and viral transcript levels of the E8 mutants in differentiated cells are a consequence of the already increased copy numbers in undifferentiated cells, or (ii) E8^E2C acts in both undifferentiated and differentiated cells to limit viral transcription and genome replication. Our data favor the second model, since E8^E2C transcripts are also present in cells grown in organotypic cultures (Fig. 8). Unfortunately, no specific antibodies against HPV16 E8^E2C are currently available that would allow analysis of the distribution of E8^E2C during the productive replication cycle. Furthermore, a loss of E8^E2C repression activity upon differentiation is not likely because the wt never reached the copy numbers of the mt genomes and the relative genome amplifications upon differentiation are similar between wt (5.6-fold) and E8− (4.8-fold) genomes (Fig. 6B). The lower relative amplification of E8 KWK mt genomes (∼2-fold) upon differentiation (Fig. 6B) might be due to already higher copy numbers in undifferentiated cells and the possibility that cellular proteins required for HPV amplification are limiting in suprabasal cells.
Since the viral late promoter is mainly responsible for L1 and E4 expression upon differentiation, our data suggest that E8^E2C inhibits not only the major early promoter P97 but also the late promoter in differentiating cells. This could occur via conserved E2 binding sites in the URR, since E8^E2C, in contrast to E2, has a long-distance repression activity that is dependent upon the E8 domain (27, 37). However, inhibition of the late promoter could also be an indirect consequence of E8^E2C's ability to repress genome replication, since the HPV31 late promoter is influenced by viral copy number (49). Taken together, these data indicate that E8^E2C limits productive replication of HPV16 by inhibiting genome amplification and capsid protein synthesis.
The tissue culture experiments described here and by Lace et al. (30) have not revealed obvious disadvantages for the host cell when E8^E2C cannot be expressed by the HPV16 genome. This raises the intriguing question of why HPV16 maintains the expression of E8^E2C. One possibility is that in vivo, E8^E2C is required to prevent high-level expression of early proteins and spurious viral late protein expression in undifferentiated cells to minimize detection by the host immune system. As a trade-off, E8^E2C may also partially limit genome amplification and late protein expression in cells where virus production takes place. Alternatively, E8^E2C may be required for the regulation of cellular genes, and this may be relevant only for virus production in vivo. Interestingly, HPV16 E8− genomes can be stably maintained in NHK as episomes, whereas HPV31 E8− genomes integrate into the host cell genome (30, 34). This could also indicate that the inhibition of productive replication by E8^E2C is a peculiar feature of HPV16 and might be related to carcinogenicity in vivo. Future studies using E8^E2C knockout mutants of different PV types are required to solve these questions.
ACKNOWLEDGMENTS
We thank M. Dürst for providing the cloned HPV16 114b genome.
This work was supported by a grant by the Deutsche Forschungsgemeinschaft (Stu 218/4-1) to F.S.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F. 2005. Carcinogenicity of human papillomaviruses. Lancet Oncol. 6:204. 10.1016/S1470-2045(05)70086-3 [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F. 2006. Chapter 2. The burden of HPV-related cancers. Vaccine 24(Suppl 3):S3/11–S3/25. 10.1016/j.vaccine.2006.05.111 [DOI] [PubMed] [Google Scholar]
- 3.Schiffman M, Clifford G, Buonaguro FM. 2009. Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline. Infect. Agents Cancer 4:8. 10.1186/1750-9378-4-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz S. 2013. Papillomavirus transcripts and posttranscriptional regulation. Virology 445:187–196. 10.1016/j.virol.2013.04.034 [DOI] [PubMed] [Google Scholar]
- 5.Stubenrauch F, Laimins LA. 1999. Human papillomavirus life cycle: active and latent phases. Semin. Cancer Biol. 9:379–386. 10.1006/scbi.1999.0141 [DOI] [PubMed] [Google Scholar]
- 6.Stenlund A. 2003. Initiation of DNA replication: lessons from viral initiator proteins. Nat. Rev. Mol. Cell Biol. 4:777–785. 10.1038/nrm1226 [DOI] [PubMed] [Google Scholar]
- 7.Breiding DE, Sverdrup F, Grossel MJ, Moscufo N, Boonchai W, Androphy EJ. 1997. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol. Cell. Biol. 17:7208–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donaldson MM, Mackintosh LJ, Bodily JM, Dornan ES, Laimins LA, Morgan IM. 2012. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 86:12806–12815. 10.1128/JVI.01002-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 87:3871–3884. 10.1128/JVI.03068-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McBride AA, Sakakibara N, Stepp WH, Jang MK. 2012. Hitchhiking on host chromatin: how papillomaviruses persist. Biochim. Biophys. Acta 1819:820–825. 10.1016/j.bbagrm.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10:550–560. 10.1038/nrc2886 [DOI] [PubMed] [Google Scholar]
- 12.Wang HK, Duffy AA, Broker TR, Chow LT. 2009. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 23:181–194. 10.1101/gad.1735109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohm S, Wilczynski SP, Pfister H, Iftner T. 1993. The predominant mRNA class in HPV16-infected genital neoplasias does not encode the E6 or the E7 protein. Int. J. Cancer 55:791–798. 10.1002/ijc.2910550517 [DOI] [PubMed] [Google Scholar]
- 14.Grassmann K, Rapp B, Maschek H, Petry KU, Iftner T. 1996. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 70:2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 66:6070–6080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Meyers C, Wang HK, Chow LT, Zheng ZM. 2011. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 85:8080–8092. 10.1128/JVI.00670-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doorbar J, Foo C, Coleman N, Medcalf L, Hartley O, Prospero T, Napthine S, Sterling J, Winter G, Griffin H. 1997. Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic Fabs to E4. Virology 238:40–52. 10.1006/viro.1997.8768 [DOI] [PubMed] [Google Scholar]
- 18.Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. 2000. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 74:6622–6631. 10.1128/JVI.74.14.6622-6631.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLaughlin-Drubin ME, Bromberg-White JL, Meyers C. 2005. The role of the human papillomavirus type 18 E7 oncoprotein during the complete viral life cycle. Virology 338:61–68. 10.1016/j.virol.2005.04.036 [DOI] [PubMed] [Google Scholar]
- 20.Kho EY, Wang HK, Banerjee NS, Broker TR, Chow LT. 2013. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc. Natl. Acad. Sci. U. S. A. 110:7542–7549. 10.1073/pnas.1304855110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fehrmann F, Klumpp DJ, Laimins LA. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 77:2819–2831. 10.1128/JVI.77.5.2819-2831.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genther SM, Sterling S, Duensing S, Munger K, Sattler C, Lambert PF. 2003. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 77:2832–2842. 10.1128/JVI.77.5.2832-2842.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. 2005. Human papillomavirus type 16 E1^E4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 79:13150–13165. 10.1128/JVI.79.20.13150-13165.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson R, Fehrmann F, Laimins LA. 2005. Role of the E1-E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 79:6732–6740. 10.1128/JVI.79.11.6732-6740.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choe J, Vaillancourt P, Stenlund A, Botchan M. 1989. Bovine papillomavirus type 1 encodes two forms of a transcriptional repressor: structural and functional analysis of new viral cDNAs. J. Virol. 63:1743–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. 1990. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178:254–262. 10.1016/0042-6822(90)90401-C [DOI] [PubMed] [Google Scholar]
- 27.Fertey J, Hurst J, Straub E, Schenker A, Iftner T, Stubenrauch F. 2011. Growth inhibition of HeLa cells is a conserved feature of high-risk human papillomavirus E8^E2C proteins and can also be achieved by an artificial repressor protein. J. Virol. 85:2918–2926. 10.1128/JVI.01647-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeckel S, Loetzsch E, Huber E, Stubenrauch F, Iftner T. 2003. Identification of the E9/E2C cDNA and functional characterization of the gene product reveal a new repressor of transcription and replication in cottontail rabbit papillomavirus. J. Virol. 77:8736–8744. 10.1128/JVI.77.16.8736-8744.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurg R, Uusen P, Vosa L, Ustav M. 2010. Human papillomavirus E2 protein with single activation domain initiates HPV18 genome replication, but is not sufficient for long-term maintenance of virus genome. Virology 408:159–166. 10.1016/j.virol.2010.09.010 [DOI] [PubMed] [Google Scholar]
- 30.Lace MJ, Anson JR, Thomas GS, Turek LP, Haugen TH. 2008. The E8-E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J. Virol. 82:10841–10853. 10.1128/JVI.01481-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palermo-Dilts DA, Broker TR, Chow LT. 1990. Human papillomavirus type 1 produces redundant as well as polycistronic mRNAs in plantar warts. J. Virol. 64:3144–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rotenberg MO, Chow LT, Broker TR. 1989. Characterization of rare human papillomavirus type 11 mRNAs coding for regulatory and structural proteins, using the polymerase chain reaction. Virology 172:489–497. 10.1016/0042-6822(89)90191-8 [DOI] [PubMed] [Google Scholar]
- 33.Snijders PJ, van den Brule AJ, Schrijnemakers HF, Raaphorst PM, Meijer CJ, Walboomers JM. 1992. Human papillomavirus type 33 in a tonsillar carcinoma generates its putative E7 mRNA via two E6* transcript species which are terminated at different early region poly(A) sites. J. Virol. 66:3172–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 74:1178–1186. 10.1128/JVI.74.3.1178-1186.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zobel T, Iftner T, Stubenrauch F. 2003. The papillomavirus E8-E2C protein represses DNA replication from extrachromosomal origins. Mol. Cell. Biol. 23:8352–8362. 10.1128/MCB.23.22.8352-8362.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stubenrauch F, Straub E, Fertey J, Iftner T. 2007. The E8 repression domain can replace the E2 transactivation domain for growth inhibition of HeLa cells by papillomavirus E2 proteins. Int. J. Cancer 121:2284–2292. 10.1002/ijc.22907 [DOI] [PubMed] [Google Scholar]
- 37.Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8E2C protein of high-risk human papillomavirus type 31. J. Virol. 75:4139–4149. 10.1128/JVI.75.9.4139-4149.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J. Virol. 84:4451–4460. 10.1128/JVI.02390-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ammermann I, Bruckner M, Matthes F, Iftner T, Stubenrauch F. 2008. Inhibition of transcription and DNA replication by the papillomavirus E8-E2C protein is mediated by interaction with corepressor molecules. J. Virol. 82:5127–5136. 10.1128/JVI.02647-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirnbauer R, Taub J, Greenstone H, Roden R, Durst M, Gissmann L, Lowy DR, Schiller JT. 1993. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J. Virol. 67:6929–6936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmad SS, Glatzle J, Bajaeifer K, Buhler S, Lehmann T, Konigsrainer I, Vollmer JP, Sipos B, Ahmad SS, Northoff H, Konigsrainer A, Zieker D. 2013. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 43:586–590. 10.3892/ijo.2013.1971 [DOI] [PubMed] [Google Scholar]
- 42.Schenker A, Straub E, Iftner T, Stubenrauch F. 2013. Cell-type-dependent activities of regulatory regions and E2 proteins derived from carcinogenic and non-carcinogenic human alphapapillomaviruses. J. Gen. Virol. 94:1343–1350. 10.1099/vir.0.049072-0 [DOI] [PubMed] [Google Scholar]
- 43.Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20:1436–1447. 10.1128/MCB.20.4.1436-1447.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rozen SS, Helen J. 2000. Primer3 on the WWW for general users and for biologist programmers, p 365–386 In Misener S, Krawetz SA. (ed), Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 45.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. 10.1016/j.virol.2009.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. 1995. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 9:2335–2349. 10.1101/gad.9.19.2335 [DOI] [PubMed] [Google Scholar]
- 47.Middleton K, Peh W, Southern S, Griffin H, Sotlar K, Nakahara T, El-Sherif A, Morris L, Seth R, Hibma M, Jenkins D, Lambert P, Coleman N, Doorbar J. 2003. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 77:10186–10201. 10.1128/JVI.77.19.10186-10201.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peh WL, Middleton K, Christensen N, Nicholls P, Egawa K, Sotlar K, Brandsma J, Percival A, Lewis J, Liu WJ, Doorbar J. 2002. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 76:10401–10416. 10.1128/JVI.76.20.10401-10416.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spink KM, Laimins LA. 2005. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J. Virol. 79:4918–4926. 10.1128/JVI.79.8.4918-4926.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]