

Abstract
Adenovirus (Ad) infection triggers a cell-specific antiviral response following exposure of viral DNA to the intracellular compartment. A variety of DNA sensors (DAI, AIM2, DDx41, RNA polymerase [Pol] III, and IFI16 [p204]) have been identified in recent years; however, the DNA sensor involved in detection of adenovirus has not been established. Cyclic GMP-AMP synthase (cGAS), a DNA sensor that produces a cyclic guanine-adenine dinucleotide (cGAMP) inducer of STING, has been examined to determine its role in generating an antiadenoviral response. Short hairpin RNA (shRNA) lentiviral vectors targeting TBK1, STING, and cGAS were established in murine MS1 endothelial and RAW 264.7 macrophage cell lines. Knockdown of TBK1, STING, and cGAS results in a dramatic reduction in the activation of the primary antiviral response marker phosphorylated interferon (IFN) response factor 3 (IRF3) following exposure to adenovirus. Furthermore, activation of secondary type I IFN signaling targets (ptyrSTAT1 and ptyrSTAT2 [ptyrSTAT1/2]) was also compromised. Consistent with compromised activation of primary and secondary response markers, transcriptional activation of IRF3-responsive genes (beta IFN [IFN-β], ISG15, ISG54) and secondary response transcripts were diminished in cells knocked down in cGAS, STING, or TBK1. These data establish cGAS as the dominant cytosolic DNA sensor responsible for detection of internalized adenovirus leading to induction of the type I interferon antiviral cascade.
INTRODUCTION
The human serotype 5 adenovirus (Ad5) is a nonenveloped linear double-stranded DNA virus associated with upper respiratory tract disease in humans. It has been extensively studied as a model for virus and host cell interactions. Replication-defective recombinant Ad5 vectors (rAdV) deleted in E1 and E3 coding domains have been characterized in gene therapy, vaccine, and oncolytic vector strategies in the murine model. Although nonpermissive for Ad5 replication, the murine model of rAdV infection provides a valuable resource for characterizing how the innate and adaptive immune response orchestrates an antiviral response to nonenveloped DNA viruses.
Virus uptake by immune sentinel cells such as macrophage and dendritic cells is vital to initiating the antiviral immune response. In addition to antigen-presenting cells (APCs), other cell types, including endothelial cells or tissue-specific cells such as hepatocytes, when exposed to virus, also contribute to the host antiviral response. In vitro studies of isolated bone marrow-derived APCs or representative cell lines have revealed a cell-specific antiviral innate response, where activation of the type I interferon (IFN) cascade is a dominant feature (1–4). A valuable marker for early events in the antiviral recognition response is activation of the transcription factor interferon response factor 3 (IRF3). Following infection, cytosolic IRF3 undergoes phosphorylation as a primary response to adenovirus uptake. Activation occurs in a MyD88/TRIF-independent manner; it requires integrin-dependent endosomal entry, escape, and presentation of viral DNA to the cytosolic compartment (3).
In rAdV-responsive murine cell lines, the STING/TBK1 cascade is required for IRF3 phosphorylation (5, 6). STING (7, 8) functions as an adaptor linking DNA recognition signaling to activation of the TBK1 kinase. TBK1 activation (9) leads to C-terminal IRF3 phosphorylation, dimerization, and translocation to the nucleus (10, 11). In the nucleus, IRF3, in collaboration with additional transcription factors (NF-κB and AP1), results in transcriptional activation of IRF3-responsive genes (including IFN-β) (12). This sequence of events contributes to the primary antiviral response to adenovirus infection. The translation of primary response transcripts such as IFN-β leads to autocrine/paracrine secondary signaling. The combination of primary and secondary response functions leads to expression of a complete antiviral response, which is distinct for different cell types.
Using various screening protocols, cell lines, and output assays, an extensive list of cytosolic DNA sensors, including DAI, RNA polymerase (Pol) III, IFI16, DDX41, and Aim 2, has been established (reviewed in reference 13). However, the DNA sensor involved in recognizing infection by adenovirus leading to early IRF3 activation has not been convincingly established. The recent identification of cyclic dinucleotide activation of STING (14–18) and the elegant discovery of cyclic-GMP-AMP synthase (cGAS) as a DNA sensor (19, 20) provide an important bridge between DNA detection and downstream signaling. cGAS in complex with duplex DNA (21–23) leads to enzyme activation and production of a novel cyclic guanine-adenine dinucleotide (cGAMP) (24–27) and STING activation.
In this report, we establish that knockdown of cGAS using short hairpin RNA (shRNA)-expressing lentiviral vectors results in loss of IRF3 activation following infection by first-generation recombinant adenovirus vector (rAdV). The magnitude of suppression is equivalent to that observed with knockdown of TBK1 or STING in the responsive cell lines tested. Secondary signaling as well as induction of antiviral gene expression is severely compromised as a consequence, disrupting the cGAS/STING/TBK1 pathway. These data lead us to conclude that cGAS is a primary cytosolic antiviral pattern recognition receptor (PRR) responding to adenovirus infection.
MATERIALS AND METHODS
Viruses.
Ad5CiG (28) was previously described. Viruses weres grown at a large scale in 293 cells, followed by 2 rounds of CsCl banding and dialysis against 4% sucrose–50 mM Tris (pH 8.0)–2 mM MgCl2, and stored at −80°C. Viral particle numbers were quantified by spectrophotometric detection of intact virions according to the optical density at 260 nm (OD260) (1012 particles [p]/OD260 unit).
Cells.
RAW264.7 macrophage-like cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Cellgro) with 5% fetal bovine serum (FBS) (characterized; HyClone); MS1 murine endothelial cells were maintained in DMEM containing 10% FBS. All media were supplemented with 1% penicillin-streptomycin (Gibco).
Cell treatment.
For protein cell lysate assays, 106 cells per well were plated in 1 ml media in 12-well plates 24 h before treatment. For RNA cell harvest assays, 2 × 106 cells per well were plated in 2 ml media in 6-well plates 24 h before treatment. At time point 0, 2 × 104 Ad5CiG particles/cell diluted in 100 μl of Opti-MEM (Gibco) were added to each sample. Mock samples were treated with 100 μl Opti-MEM.
sh knockdown.
sh constructs for Scrambled (SCR), TBK1, and STING were previously described (5). Target sequences for murine cGAS (sh1 series [AGGATTGAGCTACAAGAATAT] and sh2 series [GCTGTAACACTTCTTATCAGG]) were identified with the small interfering RNA (siRNA) at the Whitehead website (29) and cloned into pLKO.1 (which includes a puromycin selection cassette) as previously described. The final sequenced constructs were used to generate lentiviral particles in 293T cells following cotransfection with vesicular stomatitis virus G glycoprotein (VSVG) and Δ8.9 packaging plasmids according to standard protocols. Cells targeted for knockdown were infected with sh-lentivirus and, 48 h postinfection (pi), selected with 4 μg/ml puromycin (Sigma) for approximately 7 days, at which point there was no survival of control cells lacking the puromycin cassette. The puromycin-selected sh cells were used in experiments as described below.
Western blots.
Whole-cell extracts were prepared by washing cells twice with ice-cold phosphate-buffered saline (PBS) and incubating them in lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40) with the addition of Phosphatase Inhibitor Cocktails 1 and 2 (Sigma catalog no. P2850 and P5726) and protease inhibitors (30 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM benzamidine) for 30 min at 4°C on a rocking platform before scraping and transferring to tubes were performed. The lysates were cleared by centrifugation at 13,000 × g for 20 min at 4°C, and protein quantification was performed with a detergent-compatible (DC) protein assay kit (Bio-Rad Laboratories).
For Western blot analysis, 20 μg total protein was separated using standard 10% SDS polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon P from Millipore). All blots were blocked in 5% skim milk–Tris-buffered saline (TBS)-Tween (0.1%) at room temperature for 1 h. Phospho-IRF3 (Ser396; catalog no. 4961), total STAT1 (catalog no. 9172), phospho-STAT1 (58D6) (Tyr701; catalog no. 9167), beta-actin (catalog no. 4967), total IRF3 (catalog no. 4302), STING (catalog no. 3337), TBK1 (catalog no. 3013), and horseradish peroxidase (HRP)-linked anti-rabbit IgG (catalog no. 7074) antibodies were from Cell Signaling. pSTAT2 (Tyr689; catalog no. 07-224) antibody was from Millipore. All primary antibodies were used at a dilution of 1:3,000 in 5% bovine serum albumin (BSA)–TBS. The HRP-linked secondary antibody was diluted 1:4,000 in 5% milk Tween–TBS. Signals were visualized by the use of Luminator Western HRP substrate (Millipore) and exposed to BioExcell autoradiography film. Exposed representative films were scanned and stored as TIFF files for figure production or quantitation using GelEval 1.35 software.
SYBR green I RT-qPCR.
Total cellular RNA was isolated using RNAzol reverse transcriptase (RT) (Molecular Research Center) as instructed by the manufacturer. For RT-quantitative PCR (RT-qPCR), a two-step protocol was employed. First, cDNA was synthesized from 2 μg total RNA in a volume of 20 μl using random hexamer primers with a Maxima First Strand cDNA Synthesis kit (Fermentas); second, amplifications were carried out in a total volume of 15 μl by using Maxima SYBR green/ROX qPCR Master Mix (Fermentas) in an Applied Biosystems Prism 7900H sequence detection system with SDS 2.1 software. Cycles consisted of an initial incubation step at 95°C for 10 min, 40 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, and a melting curve analysis cycle. Data acquisition was performed during the extension step. All determinations were performed in technical triplicate. Nontemplate controls and controls lacking RT (−RT) were run with every assay and had cycle threshold (CT) values which were significantly higher than those of experimental samples or were undetermined. The relative abundance of each mRNA was calculated by the ΔΔCT method (30, 31), normalizing to Tata-binding protein (TBP) expression with standardization to one reference sample as indicated. Sequences of primers are available on request.
Statistical analysis.
Data were expressed as means ± standard errors of the means. Statistical analysis was performed with Student's t test. A P value of less than 0.05 was considered significant.
RESULTS
Characterization of shRNA knockdown of cGAS/STING/TBK1 in MS1 endothelial cells.
In prior studies investigating cell differences with respect to viral uptake (28) and induction of early antiviral responses (6), the murine MS1 endothelial cell line was found to be transduced by rAdV at modest levels but to undergo activation of IRF3 in a STING- and TBK1-dependent manner. Based on studies reported from the Chen laboratory (19, 20), STING activation has been found to occur through a cGAS-dependent recognition of DNA. To determine the role of cGAS as a pattern recognition receptor (PRR) for adenovirus, pLKO.1 shRNA lentiviral expression vectors targeting cGAS were constructed based on two cGAS sh target domains (1.5 and 2.1) as described in Materials and Methods. Lentiviral vectors targeting TBK1, STING, or cGAS were used to establish MS1 knockdown cell line pools. RNA from each pool was first characterized for target mRNA depletion. All transcripts were normalized to Tata-binding protein (TBP), and the relative level of targeted mRNA knockdown efficiency (in comparison to scramble shRNA [shSCR] control levels) was determined. TBK1 and STING knockdown efficiencies were 80% whereas mRNA knockdowns were 60% for both cGAS 1.5 and 2.1 shRNA cell pools (Fig. 1A). These cell line pools were used in an antiadenoviral response assay. Cells exposed to 2 × 104 viral particles/cell of the recombinant first-generation Ad5CiG vector (cytomegalovirus [CMV] promoter driving expression of chloramphenicol acetyltransferase [CAT] internal ribosome entry site [IRES]-green fluorescent protein [GFP] [CATiresGFP]) (28) were harvested at 6 and 24 h postinfection. Cell lysates were analyzed by Western blotting by first using a phosphoserine 388-specific anti-IRF3 antibody (murine p-ser 388 IRF3, corresponding to p-ser 396 in human IRF3). Phosphorylation at this site is a key indicator of IRF3 activation (10) and provides a robust, reliable marker for the early response to virus infection (2, 3, 5, 6). Wild-type (wt) MS1 cells and shSCR control cell pools revealed the presence of pIRF3 at both 6 and 24 h postinfection. In contrast, pIRF3 levels in virus-treated cell pools from TBK1, STING, and cGAS knockdowns were indistinguishable from those in mock-infected cells. Levels of secondary response targets such as pSTAT1/2 (STAT1 pTyr 701 and STAT2 pTyr 247) were too low at 6 h postinfection to provide an assessment of an altered response in the knockdown cell lines. At 24 h, when the pSTAT1 levels in wt and shSCR cells had increased, the TBK1, STING, and cGAS knockdown cell lines were indistinguishable from mock-infected cells. Western analysis of total TBK1 and STING confirmed the knockdown efficiency achieved within the knockdown pools (suitable anti-cGAS antibodies were not available).
FIG 1.
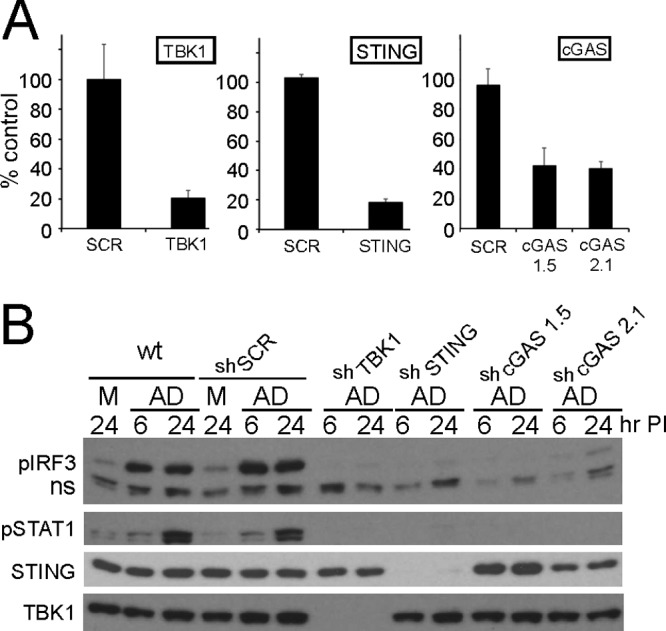
shRNA knockdown of cGAS, TBK1, and STING in MS1 endothelial cells. (A) RT-qPCR quantitation of mRNA levels in MS1 endothelial cells after knockdown and puromycin selection using shRNA lentiviral vectors targeting TBK1, STING, and cGAS. Samples were normalized to the cellular control TBP. One shSCR control sample was selected as representing a 100% standard. All qRT-PCR assays included biological triplicates as well as technical triplicates of each sample. (B) Western analysis of lysates harvested from wild-type (wt) or stable shRNA MS1 knockdown cell line pools at 6 or 24 h post-mock or -Ad5CiG infection using the indicated antibody. ns corresponds to a nonspecific band. M corresponds to mock treatment and AD to Ad5CiG infection. All experiments were carried out a minimum of three times; representative results are presented.
RNA isolated at 6 and 24 h post-adenovirus infection was used to evaluate the influence of shRNA treatments on the activation of established antiviral early-response genes (2, 3, 6). TBP was used for normalization of all RT-qPCR assays. For each mRNA transcript, a scrambled mock sample (shSCR) was given an arbitrary unit value of 100, allowing direct comparison to all other samples (Fig. 2). At 6 h postinfection, Ad5CiG induction of IFN-β and ISG15 transcripts was evident in wt and shSCR MS1 cell line pools but absent in cells knocked down in TBK1, STING, and cGAS. The IFN-β, ISG15, ISG54, and STAT1 transcripts by 24 h postinfection were significantly induced (P < 0.01) in wt and shSCR-treated cells, whereas TBK1, STING, or cGAS shRNA cell line pools were nonresponsive to rAdV infection with respect to IFN-β, ISG15, ISG54, and STAT1 induction. Additionally, in cell line pools treated with TBK1, STING, and cGAS shRNA, the basal level of the steady state of ISG15 was well below that found in shSCR-treated or wt cells. These data provide the first indication that cGAS is involved in adenoviral recognition and in subsequent induction of primary response transcription units in the infected cell.
FIG 2.

Comparative RT-qPCR of RNA isolated from shSCR, cGAS, and STING and TBK1 shRNA knockdown MS1 endothelial cell pools infected with adenovirus. Data represent the results of two-step RT-qPCR of RNA isolated from mock- or Ad5CiG-infected MS1 and shRNA knockdown cell pools harvested at 6 (A) and 24 (B) h pi. Results are shown for PCR primers corresponding to IFN-β-, ISG15-, ISG54-, and STAT1-inducible transcripts. All samples were normalized to TBP using the ΔΔCT method as described in Materials and Methods. The value for scramble sample 1 was set as an arbitrary unit of 100.
Adenovirus detection by cGAS in the RAW 264.7 macrophage-like cell line.
RAW 264.7 cells are a well-established murine macrophage-like cell line that undergoes an early response to rAdV infection (5). shRNA knockdown cell pools corresponding to the shSCR control, shTBK1, shSTING, and the two shcGAS 1.5 and 2.1 constructs were established in RAW 264.7 cells. Following puromycin selection, cell pools were analyzed for mRNA knockdown efficiency. As previously described, all transcripts were normalized to TBP, and the relative levels of targeted mRNA knockdown efficiency (in comparison to shSCR control levels) were 65% for TBK1, 80% for STING, 50% for cGAS 1.5, and 50% for cGAS 2.1 (Fig. 3A). These cell line pools were used in our standard rAd5CiG infection assay and harvested at 4 and 6 h postinfection to assess the impact of shRNA knockdowns. In the shSCR control cell pool, the RAW cells revealed detectable phosphorylation of IRF3 (Fig. 3B). In lysates harvested from rAdV-infected shTBK1, shSTING, and shcGAS cell pools, pIRF3 levels were diminished at 6 h postinfection (Fig. 3B and C). For quantitation of pIRF3 levels, 5 similar but separate experiments were used to determine the average decrease of pIRF3 at 6 h postinfection compared to the shSCR control level. In all shRNA knockdown assays, the levels of secondary response target phosphorylation (pSTAT1/2) were diminished in a manner consistent with the decrease in pIRF3.
FIG 3.
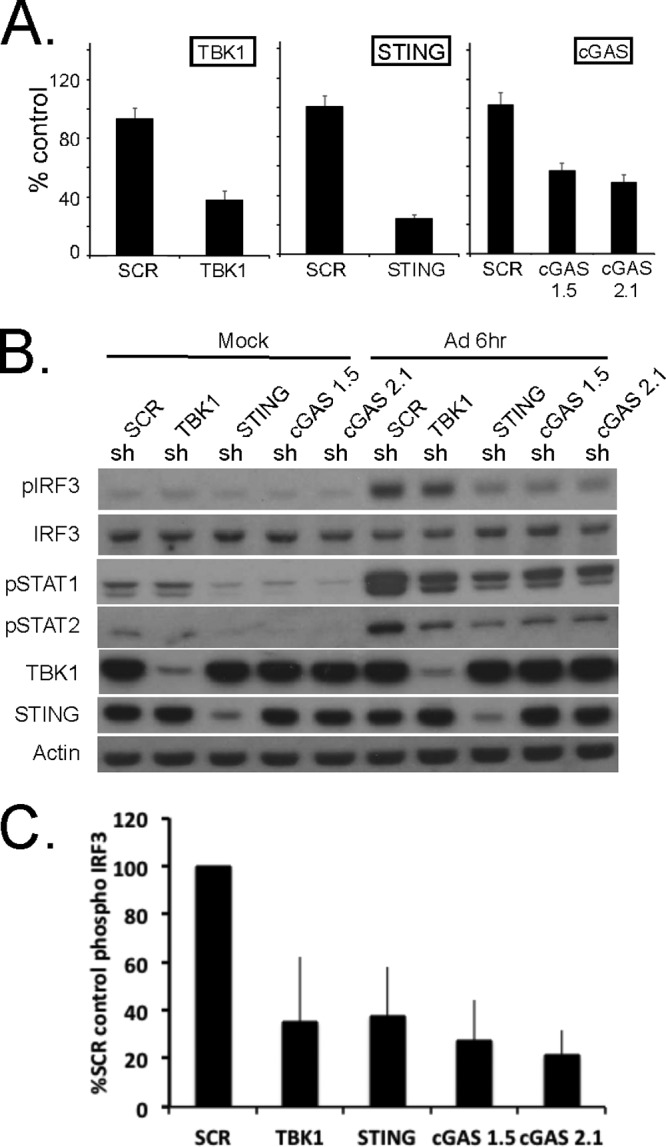
Stable shRNA knockdown of cGAS, TBK1, and STING in the RAW264.7 macrophage cell line. (A) RT-qPCR quantitation of mRNA levels in the RAW264.7 macrophage cell line after knockdown and puromycin selection using shRNA lentiviral vectors targeting TBK1, STING, and cGAS. shSCR corresponds to scramble control shRNA. Samples were normalized to the cellular control TBP. One shSCR control sample was selected as representing a 100% standard. (B) Western analysis of lysates harvested from wild-type or stable shRNA macrophage knockdown cell line pools 6 h post-Ad5CiG infection using the indicated antibody. (C) Quantitation of AdV-induced phospho-IRF3 for each sh knockdown. Determinations were averaged from the results of 5 or more experiments (as described for panel B), where the shSCR value was set as 100% for each experiment.
RNA was harvested 6 h postinfection from mock- or Ad5CiG-infected shRNA knockdown cell pools to assess the early antiviral response to virus infection. wt RAW264.7 cells were included as a second positive control. IRF3-dependent transcripts (IFN-β, ISG15, and ISG54) were strongly induced by Ad5CiG infection of shSCR and wt cell pools. In contrast, levels of these transcripts were significantly reduced in TBK1, STING, and cGAS cell pools exposed to Ad5CiG (Fig. 4A). Secondary response transcripts (TNF-α, IRF-7, and STAT1) were upregulated in Ad5CiG-infected wt and shSCR cell pools. Levels of these transcripts were comparatively lower in RNA isolated from STING and cGAS knockdowns. In this assay, the shTBK1 knockdown influence on TNF-α expression levels was not significantly different from that of the shSCR control. Furthermore, as noted in MS1 cells, basal steady-state levels of ISG15 in STING and cGAS RAW 264.7 cell pools were markedly lower than those found in shSCR pools. We also determined mRNA levels of TBK1, STING, and cGAS in each cell pool (Fig. 4B) and found cGAS to be inducible at 6 h postinfection. Finally, in analyzing the quantitative RT-PCR (qRT-PCR) data, rAdV-induced transcript levels were greatly reduced for each of the knockdown cell pools. However, we also detected a residual low-level induction response following Ad5CiG infection. We attribute a portion of the residual Ad5CiG induction response in these cell pools to incomplete knockdowns in the RAW 264.7 cells. This was most evident in the TBK1 and cGAS 1.5 knockdowns.
FIG 4.

Comparative RT-qPCR of RNA isolated from shSCR, cGAS, STING, and TBK1 shRNA knockdown RAW264.7 shRNA cell pools infected with adenovirus. Data represent the results of two-step RT-qPCR of RNA isolated from mock- or Ad5CiG-infected RAW264.7 cell lines and shRNA knockdown cell pools harvested at 6 h pi. Results are shown for PCR primers corresponding to primary response genes IFN-β, ISG15, and ISG54 and secondary response-inducible transcripts STAT1, TNF-α, and IRF7 (A) and to shRNA-targeted genes (B). All samples were normalized to TBP using the ΔΔCT method as described in Materials and Methods. The value for scramble sample 1 was set as an arbitrary unit of 100.
DISCUSSION
We have used the shRNA lentiviral knockdown strategy in RAW264.7 macrophage and MS1 endothelial cells to investigate the role of the newly discovered cGAS DNA sensor (19) as a PRR/DNA sensor for rAdV infection. Using IRF3 phosphorylation as our primary recognition response marker, cGAS shRNA knockdown pools from each of these murine cell lines were compromised in response to rAdV infection. Furthermore, knocking down cGAS resulted in diminished secondary signaling indicated by phosphorylation of STAT1 and STAT2. We have also included assays of cell pools knocked down in STING and TBK1. Knockdown of cGAS had an impact on the rAdV recognition response equal to or greater than that of STING and TBK1 (modeled in Fig. 5). In further support of the idea of a critical role played by each of these proteins in the early recognition/activation response to rAdV, qRT-PCR analysis of mRNA isolated from each shRNA knockdown following infection revealed a similarly muted induction of antiviral response transcripts. The shRNA knockdowns are remarkably effective in suppressing the antiadenoviral induction profile in the MS1 endothelial cell line and highly significant in RAW264.7 cells for IFN-β, ISG15, and ISG54.
FIG 5.

Summary model for rAdV DNA sensing. Following endosomal entry of rAdV into the cell (lower left), viral DNA can trigger the TLR9 pathway, which through the MyD88 adaptor leads to activation of IRF7 and IRF5 but not IRF3. Alternatively, adenovirus escapes the endosome, revealing virus and viral DNA complexes to the cytosolic compartment. An array of DNA sensors are available to bind viral DNA; one DNA sensor, cyclic GMP-AMP synthase (cGAS), has been shown to influence the antiviral response to adenovirus infection in murine endothelial and macrophage cell lines. DNA engagement by cGAS results in production of a unique cGAMP containing 2′5′ and 3′5 linkages that bind to the adaptor STING and leads to TBK1 activation. Active TBK1 mediates C-terminal phosphorylation of IRF3. IRF3 dimerizes, translocates to the nucleus (indicated by the double-line structure), and associates with transcription units (IFN-β or ISG15, -54, and -56). In collaboration with additional transcription factors (NF-κB, ATF/cJUN), gene expression is induced. AIM2 and p204 are Hin-200 proteins that complex with ASC through pyrin domains to form an inflammasome complex, leading to secretion of IL-1 and IL-18. DNA template present either in the cytosol or in the nucleus can be transcribed. Small viral RNAs have been implicated in stimulating the antiviral response through activation of RNA sensors (MDA-5 and RIG-I) which in turn complex with IPS (MAVS) and activate TBK1.
APCs are key early responders to rAdV infection, contributing to both innate and adaptive immune responses in the murine model; however, they are not the only cell type that participates in the antiviral innate immune response. In an earlier study, several murine cell lines were screened to characterize cell type-specific antiviral responses (6). Nonresponsive FL83 hepatocytes lacked the STING adaptor, whereas the MS1 endothelial cell line and RAW264.7 macrophage cell lines revealed rAdV DNA-dependent activation of IRF3. shRNA knockdown of STING and TBK1 compromised IRF3 activation in both of these cell lines (5, 6). However, knockdown of putative DNA sensors, including DAI, p204, DDX41, and AIM2, had not revealed a consistent or sustainable antiadenovirus phenotype (5, 6), implying either redundancy of function within the population of established DNA sensors or the existence of at least one additional sensor which functions as a dominant adenovirus PRR. Based on the data derived from cGAS knockdown in these two distinct cell lines, we conclude that cGAS functions as the dominant cytosolic DNA PRR for adenovirus in the murine cell line model.
Although members of the population of putative DNA sensors are not essential to the primary recognition response to rAdV, as a group they are inducible by rAdV infection as part of the secondary antiviral response (6). Several of these DNA sensors participate in inflammatory cell death cascades (32–34). Unlike the direct DNA-mediated induction of the cGAS/STING/TBK1 pathway, activation of AIM2 or NLRP3 inflammasome complexes (35–37) is greatly enhanced by pretreatment or by priming the cell with lipopolysaccharide (LPS). Stimulation of cells by such agents leads to induction of ISG-responsive transcripts, including components of the inflammasome complex. Absent this priming, adenovirus-induced inflammasome complexes are inefficiently formed (38). DAI is also an interferon-inducible transcript in the cell lines we are using (6). DAI was identified as the first DNA sensor contributing to IFN induction (39) but has also been shown to contribute to virally induced cell death pathways through DAI/RIP1/3 complexes (40–42). Therefore, stimulation of the cGAS/STING/TBK1/IRF3 pathway by rAdV induces several genes critical to inflammatory death-related pathways. Treatments that result in elevation of the basal state of cGAS/STING/TBK1/IRF3 pathway may result in a higher level of inflammatory priming and decrease the activation thresholds required to stimulate cell death cascades.
In addition to transcript-specific mRNA degradation, expression of siRNAs and microRNAs (miRNAs) can suppress gene activity through blocking gene-specific mRNA processing and or translation (reviewed in references 43 and 44). In the case of shcGAS 1.5 and 2.1, these modes of RNA interference may contribute to inhibition of cGAS expression and subsequent IRF3 activation by rAdV. Although the level of shRNA depletion of cGAS mRNA was only 50% to 60%, the impact on IRF3 phosphorylation and on the level of inducible transcript expression indicated that the level of functional interference was greater than that of the mRNA degradation. The potency of shRNA interference may vary in a cell-dependent manner; we observed that the antiviral response in MS1 cells was more vulnerable to complete suppression by the shRNA knockdown strategy.
Separate from its role in the rAdV recognition response, targeting the cGAS/STING/TBK1 pathway was found to influence the homeostatic balance of ISG mRNAs. Following knockdown of cGAS, STING, or TBK1 in both cell lines, basal levels of several ISG transcripts were reduced (Fig. 2 and 4). ISG15 offers the most pronounced example of this influence (P < E-6). ISG is a ubiquitin-like protein that targets cellular and viral proteins as part of a posttranslational antiviral mechanism (45). In RAW264.7 cells, where the knockdowns were less pronounced, basal levels of rAdV-inducible genes were diminished but the genes were still inducible following rAdV infection, albeit to a lower level. We propose that the cGAS/STING/TBK1 cascade is an important contributor to basal maintenance of a primed state for ISGs. Interferon sensitization has been hypothesized to contribute to maintenance of a basal state priming for responsive cell lines that facilitates rapid induction of target genes (46–48). Changes in basal priming of ISGs may contribute to alterations in the antiviral response phenotype and provide an avenue for modifying the antiviral response in a cell-specific manner.
rAdV are deficient in E1 and E3 genes; they are nonreplicating and lack many of the viral gene products that suppress host cell antiviral strategies. The model that we have employed focuses on early host response functions, absent most known adenoviral immune evasion mechanisms. Insights gained from these studies are relevant to viral vectors for gene therapy, vaccine strategies, and strategies involving oncolytic vectors. However, the model does not allow us to address questions of Ad mechanisms that may function to downregulate the cGAS/STING/TBK1/IRF3 cascade. Such studies need to be carried out in human cell lines with replication-competent adenovirus. Studies have established Toll-like receptor 9 (TLR9)-mediated sensing of CD46-targeted Ad vectors in HeLa and human plasmacytoid dendritic cells (49); however, the role of the STING/TBK1/cGAS pathway in human cell lines needs to be fully characterized. Additionally, in both the murine and human models, if TLR9 and cGAS signaling cascades are equally available, the determinants guiding use of one pathway rather than the other are not known, and we do not know if the uses of these pathways are mutually exclusive. Having established the sensing pathways leading to rAdV induction of type I IFN, we are in position to address these issues. Finally, the identification of cGAS and cGAMP in activating STING following virus internalization provides new targets that may be used to manipulate the antiviral response to adenovirus vectors.
ACKNOWLEDGMENTS
E.F.-P. is a recipient of a Hearst Foundation Award. This work was supported by Public Health Service grants RO1 AI094050 to E.F.-P.
We thank Daniela Anghelina for critical reading of the manuscript.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Fejer G, Drechsel L, Liese J, Schleicher U, Ruzsics Z, Imelli N, Greber UF, Keck S, Hildenbrand B, Krug A, Bogdan C, Freudenberg MA. 2008. Key role of splenic myeloid DCs in the IFN-alphabeta response to adenoviruses in vivo. PLoS Pathog. 4:e1000208. 10.1371/journal.ppat.1000208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nociari M, Ocheretina O, Murphy M, Falck-Pedersen E. 11 February 2009. Adenovirus induction of IRF3 occurs through a binary trigger targeting JNK and TBK-1 kinase cascades and type I IFN autocrine signaling. J. Virol. 10.1128/JVI.02591-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nociari M, Ocheretina O, Schoggins JW, Falck-Pedersen E. 2007. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J. Virol. 81:4145–4157. 10.1128/JVI.02685-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu J, Huang X, Yang Y. 2007. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 81:3170–3180. 10.1128/JVI.02192-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein SC, Falck-Pedersen E. 2012. Sensing adenovirus infection: activation of interferon regulatory factor 3 in RAW 264.7 cells. J. Virol. 86:4527–4537. 10.1128/JVI.07071-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein SC, Lam E, Falck-Pedersen E. 2012. Cell-specific regulation of nucleic acid sensor cascades: a controlling interest in the antiviral response. J. Virol. 86:13303–13312. 10.1128/JVI.02296-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. 2004. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 101:233–238. 10.1073/pnas.2237236100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Servant MJ, Grandvaux N, ten Oever BR, Duguay D, Lin R, Hiscott J. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 278:9441–9447. 10.1074/jbc.M209851200 [DOI] [PubMed] [Google Scholar]
- 11.Clément JF, Bibeau-Poirier A, Gravel SP, Grandvaux N, Bonneil E, Thibault P, Meloche S, Servant MJ. 2008. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J. Virol. 82:3984–3996. 10.1128/JVI.02526-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-beta enhanceosome. Cell 129:1111–1123. 10.1016/j.cell.2007.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm CK, Paludan SR, Fitzgerald KA. 2013. DNA recognition in immunity and disease. Curr. Opin. Immunol. 25:13–18. 10.1016/j.coi.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. 10.1038/nature10429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79:688–694. 10.1128/IAI.00999-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang YH, Liu XY, Du XX, Jiang ZF, Su XD. 2012. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol. 19:728–730. 10.1038/nsmb.2333 [DOI] [PubMed] [Google Scholar]
- 17.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 2012. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J. Biol. Chem. 287:39776–39788. 10.1074/jbc.M112.382986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shu C, Yi G, Watts T, Kao CC, Li P. 2012. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19:722–724. 10.1038/nsmb.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. 10.1126/science.1232458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. 10.1126/science.1229963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP. 2013. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498:332–337. 10.1038/nature12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, Hartmann G, Barchet W, Tuschl T, Patel DJ. 2013. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154:748–762. 10.1016/j.cell.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kranzusch PJ, Lee AS, Berger JM, Doudna JA. 2013. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3:1362–1368. 10.1016/j.celrep.2013.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. 2013. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. 10.1038/nature12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ. 2013. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153:1094–1107. 10.1016/j.cell.2013.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. 2013. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51:226–235. 10.1016/j.molcel.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3:1355–1361. 10.1016/j.celrep.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schoggins JW, Falck-Pedersen E. 2006. Fiber and penton base capsid modifications yield diminished Ad5 transduction and proinflammatory gene expression with retention of antigen specific humoral immunity. J. Virol. 80:10634–10644. 10.1128/JVI.01359-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. 2004. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 32:W130–W134. 10.1093/nar/gkh366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 32.Choubey D, Duan X, Dickerson E, Ponomareva L, Panchanathan R, Shen H, Srivastava R. June 2010. Interferon-inducible p200-family proteins as novel sensors of cytoplasmic DNA: role in inflammation and autoimmunity. J. Interferon Cytokine Res. 10.1089/jir.2009.0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. 2010. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11:395–402. 10.1038/ni.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schattgen SA, Fitzgerald KA. 2011. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 243:109–118. 10.1111/j.1600-065X.2011.01053.x [DOI] [PubMed] [Google Scholar]
- 35.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. 2009. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183:787–791. 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. 2010. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361. 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe H, Gehrke S, Contassot E, Roques S, Tschopp J, Friedmann PS, French LE, Gaide O. 2008. Danger signaling through the inflammasome acts as a master switch between tolerance and sensitization. J. Immunol. 180:5826–5832 [DOI] [PubMed] [Google Scholar]
- 38.Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. 2008. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107. 10.1038/nature06664 [DOI] [PubMed] [Google Scholar]
- 39.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505. 10.1038/nature06013 [DOI] [PubMed] [Google Scholar]
- 40.Kaiser WJ, Upton JW, Mocarski ES. 2008. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 181:6427–6434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. 2009. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 10:916–922. 10.1038/embor.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–297. 10.1016/j.chom.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kole R, Krainer AR, Altman S. 2012. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 11:125–140. 10.1038/nrd3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou R, Rana TM. 2013. RNA-based mechanisms regulating host-virus interactions. Immunol. Rev. 253:97–111. 10.1111/imr.12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skaug B, Chen ZJ. 2010. Emerging role of ISG15 in antiviral immunity. Cell 143:187–190. 10.1016/j.cell.2010.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freaney JE, Kim R, Mandhana R, Horvath CM. 2013. Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription. Cell Rep. 4:959–973. 10.1016/j.celrep.2013.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, Woods JM, Haines GK, Ivashkiv LB. 2002. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat. Immunol. 3:859–866. 10.1038/ni828 [DOI] [PubMed] [Google Scholar]
- 48.Hu X, Ivashkiv LB. 2009. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31:539–550. 10.1016/j.immuni.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iacobelli-Martinez M, Nemerow GR. 2007. Preferential activation of Toll-like receptor nine by CD46-utilizing adenoviruses. J. Virol. 81:1305–1312. 10.1128/JVI.01926-06 [DOI] [PMC free article] [PubMed] [Google Scholar]