

Abstract
Programmed cell death (apoptosis) is an important host defense mechanism against intracellular pathogens, such as viruses. Accordingly, viruses have evolved multiple mechanisms to modulate apoptosis to enhance replication. Varicella-zoster virus (VZV) induces apoptosis in human fibroblasts and melanoma cells. We found that VZV triggered the phosphorylation of the proapoptotic proteins Bim and BAD but had little or no effect on other Bcl-2 family members. Since phosphorylation of Bim and BAD reduces their proapoptotic activity, this may prevent or delay apoptosis in VZV-infected cells. Phosphorylation of Bim but not BAD in VZV-infected cells was dependent on activation of the MEK/extracellular signal-regulated kinase (ERK) pathway. Cells knocked down for Bim showed delayed VZV plaque formation, resulting in longer survival of VZV-infected cells and increased replication of virus, compared with wild-type cells infected with virus. Conversely, overexpression of Bim resulted in earlier plaque formation, smaller plaques, reduced virus replication, and increased caspase 3 activity. Inhibition of caspase activity in VZV-infected cells overexpressing Bim restored levels of virus production similar to those seen with virus-infected wild-type cells. Previously we showed that VZV ORF12 activates ERK and inhibits apoptosis in virus-infected cells. Here we found that VZV ORF12 contributes to Bim and BAD phosphorylation. In summary, VZV triggers Bim phosphorylation; reduction of Bim levels results in longer survival of VZV-infected cells and increased VZV replication.
INTRODUCTION
Apoptosis is an important host defense mechanism against intracellular pathogens, such as viruses (1–3). Accordingly, viruses have evolved ways to manipulate this pathway to allow efficient virus replication and production of progeny (4). Apoptosis can be triggered by extracellular stimuli, such as tumor necrosis factor alpha (TNF-α), Fas ligand, or nutrient depletion, which is termed extrinsic apoptosis, or by intracellular stresses, such as endoplasmic reticulum (ER) stress, hypoxia, or DNA damage, which is termed intrinsic apoptosis. The intrinsic apoptosis or mitochondrial pathway is controlled by the interplay between several Bcl-2 family proteins: the prosurvival proteins Bcl-2 (B-cell lymphoma 2), Bcl-xL (Bcl-2 extra large), and Mcl-1 (myeloid cell leukemia 1), as well as the proapoptotic proteins Bax (Bcl-2-associated X protein), Bak (Bcl-2 homologous antagonist/killer), Bim (Bcl-2-interacting mediator of cell death), PUMA (p53-upregulated modulator of apoptosis), NOXA (NADPH oxidase activator 1), Bid (BH3-interacting domain death agonist), and BAD (Bcl-2-associated death promoter). Bax and Bak are effectors of apoptosis that form pores on mitochondrial membranes, resulting in release of cytochrome C and triggering apoptosis (5).
Apoptosis is a complex process, and many cellular components and signaling pathways are involved to ensure that it is properly controlled (6). Viruses regulate apoptosis using different mechanisms; most viruses encode proteins to suppress apoptosis, while some RNA viruses trigger apoptosis for virus spread (4). Alphaherpesviruses trigger different apoptosis responses depending on the cell types they infect (7, 8). Herpes simplex virus 1 (HSV-1) and HSV-2 encode a number of proteins that inhibit apoptosis (9, 10), including protein kinase US3 (11, 12), glycoprotein J (gJ) (13), and latency-associated transcript (LAT) (14). In addition, HSV mutants deleted for ICP4, ICP27, UL39, and gD undergo apoptosis in a cell-type-specific manner (15–19).
Varicella-zoster virus (VZV) is a ubiquitous human alphaherpesvirus that causes varicella (chickenpox) during primary infection and zoster (shingles) when the virus reactivates. VZV rapidly induces apoptosis (24 to 48 h after infection) in primary human foreskin fibroblasts (HFF) (20) and slowly induces apoptosis in melanoma cells (64 to 72 h after infection, (21) and in Vero cells (72 to 96 h after infection, (22)). VZV also induces apoptosis in B and T cells (23) but not in neurons (20, 24). The VZV ORF66 protein inhibits apoptosis in T cells (25), the ORF12 protein inhibits apoptosis in melanoma cells (26), and ORF63 was thought to be important to protect virus-infected neurons from apoptosis in one study (27) but not in another report (28). Here, we measured expression of Bcl-2 family member proteins during VZV infection and found that VZV induces phosphorylation of Bim which was dependent on activation of the MEK/ERK pathway, that overexpression of Bim induces caspase 3 cleavage and inhibits virus replication, and that VZV ORF12 contributes to phosphorylation of Bim.
MATERIALS AND METHODS
Cells, viruses, and chemical inhibitors.
Human melanoma (MeWo) and diploid fibroblast (MRC-5) cells were grown in minimal essential medium (MEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and HEK293T and human foreskin fibroblast (HFF) cells were grown in Dulbecco's modified eagle medium (DMEM) containing 10% FBS and 1% penicillin-streptomycin. VZV ROka (recombinant VZV derived from the Oka vaccine strain) and ROka12D (deleted for VZV ORF12) and MEK (U0126) and phosphatidylinositol 3-kinase (PI3K) (LY294002) inhibitors were described previously (26). The caspase inhibitor Q-VD-OPh was obtained from R&D systems.
Plasmids.
Plasmids pEN-TmiR and pSILK-Venus, used to construct lentiviral plasmids expressing short hairpin RNA (shRNA) to knock down Bim (shBim), were kindly provided by Iain Fraser at NIH (29). Oligonucleotides 5′-AGCGCAAGCAACCTTCTGATGTAAGTTAGTGAAGCCACAGATGTAACTTACATCAGAAGGTTGCTTT-3′ and its complementary oligonucleotide, 5′ GGCAAAAGCAACCTTCTGATGTAAGTTACATCTGTGGCTTCACTAACTTACATCAGAAGGTTGCTTG-3′, which contain Bim nucleotides 297 to 318 (underlined letters) with BfuAI linkers (bold letters), were synthesized and introduced into cloning entry vector pEN-TmiR between BfuA1 sites according to a method described previously (30). Briefly, the two oligonucleotides were annealed, phosphorylated with T4 polynucleotide kinase (New England BioLabs), and ligated to pEN-TmiR after the latter had been digested with BfuAI and treated with calf intestinal phosphatase. The resulting plasmid was termed pEN-TmiR-shBim. The cassette containing shBim driven by the TmiR promoter from pEN-TmiR-shBim was inserted into the lentiviral vector pSILK-Venus by recombination using Gateway LR Clonase II (Invitrogen), resulting in plasmid pSILK-TmiR-shBim, in which shBim is driven by a cytomegalovirus (CMV) promoter and doxycycline-inducible element. Other lentiviral vectors, pLentipuro/H1-TO/GW/BimshRNA, expressing Bim shRNA targeting a Bim site different than that of pSILK-TmiR-shBim, and pLenti6/Ubc-GW/BimEL WT, expressing wild-type (wt) Bim, were kindly provided by Andrew Aplin (Thomas Jefferson University, Philadelphia, PA) (31).
Lentivirus production.
HEK293T cells in 10-cm dishes were transfected with 7.5 μg of lentiviral vectors expressing shBim (pLentipuro/H1-TO/GW/BimshRNA or pSILK-TmiR-shBim) or wt Bim (pLenti6/Ubc-GW/BimEL WT) along with 5 μg VSV glycoprotein G-expressing plasmid pVSV-G and 7.5 μg of each of the two packaging plasmids, pMDLg/pRRE (32) and pRSVREV (32), diluted in Opti-MEM (Gibco Invitrogen) using the Lipofectamine 2000 reagent (Invitrogen). Sixteen hours after transfection, the medium was replaced with DMEM containing 10% FBS, and 48 h after transfection, supernatant containing virus was centrifuged, filtered, and stored at −80°C.
Cell line construction.
MeWo cells were transduced with lentivirus expressing wild-type Bim (obtained from plasmid pLenti6/Ubc-GW/BimEL WT) and selected with blasticidin to obtain a cell line stably expressing wt Bim, termed MeWo-wt-Bim. A cell line stably expressing shBim was constructed by transducing MeWo cells with lentivirus obtained from plasmid pLentipuro/H1-TO/GW/BimshRNA, selecting puromycin-resistant cells, and then transducing the puromycin-resistant cells with lentivirus obtained from plasmid pSILK-TmiR-shBim. The resulting cell line was termed MeWo-shBim. Induction of Bim shRNA in cells transduced with lentivirus from plasmid pSILK-TmiR-shBim was achieved by adding doxycycline to the medium 24 h before infections.
Immunoblotting.
Infected cell lysates were fractionated on polyacrylamide gels, transferred to nitrocellulose membranes, and incubated with rabbit anti-VZV IE62 antibody (a gift from Paul Kinchington, University of Pittsburgh), rabbit anti-p-ERK1/2, p-Akt, Bim, p-Bim(S69), BAD, p-BAD(S136), Bcl-2, Bcl-xL, Bax, Mcl-1, cleaved caspase 3 (Cell Signaling Technology), or mouse anti-actin antibody (Sigma).
Quantitation of Western blots and statistics.
ImageQuant TL image analysis software v7.0 (GE Healthcare) was used to quantify bands on Western blots. The two-tailed t test was used to calculate P values (see Fig. 3E and 5).
FIG 3.

Overexpression of Bim inhibits VZV replication. (A) Doxycycline (Dox) was added to MeWo, MeWo-shBim, or MeWo-wt-Bim cells, and 24 h later, the cells were infected with VZV ROka at 0.1 PFU/cell. The cells were incubated for 24 or 36 h at 37°C in MEM with 10% FBS, lysates were prepared, and Western blotting was performed to detect Bim, VZV IE62, and actin. The experiment was repeated with similar results. (B) MeWo, MeWo-shBim (shBim), or MeWo-wt-Bim (Bim wt) cells were infected with VZV ROka at 0.0004 PFU/cell and incubated at 34°C in MEM with 2% serum, doxycycline was added 24 h after infection, virus-infected cells were collected at different days after infection, and virus was titrated on MeWo cells by plaque assay. (C) VZV-infected cells expressing GFP were photographed on days 4 to 6 after infection. Magnification, ×100. (D) MeWo, MeWo-shBim, or MeWo-wt-Bim cells were infected with VZV ROka at 0.001 PFU/cell and incubated at 34°C with MEM containing 2% serum, doxycycline was added 24 h after infection, and the numbers of infectious foci (syncytia) with or without central plaques (holes in the monolayer) were counted on days 4 to 6 after infection. (E) Plaque size was measured for 10 plaques in each cell line at day 7. The bar shows the mean plaque size.
FIG 5.

Inhibition of caspase activity restores virus production in cells overexpressing Bim. Doxycycline was added to MeWo, MeWo-shBim, or MeWo-wt-Bim cells, and 24 h later, cells were infected with cell-associated VZV at 0.001 PFU/cell. Cells were incubated at 37°C with MEM containing 10% serum, Q-VD-OPh (50 μM) or dimethyl sulfoxide (DMSO) (the solvent for Q-VD-OPh) was added 24 h postinfection, and virus-infected cells were collected 4 days after infection for virus titration.
RESULTS
VZV triggers phosphorylation of Bim and BAD.
VZV infection induces apoptosis of fibroblasts, Vero, MeWo, and T cells but does not induce apoptosis of neurons (33). Since the cellular targets responsible for VZV modulation of apoptosis are unknown, we measured levels of Bcl-2 family member proteins during infection. MeWo cells were infected with cell-associated VZV (strain ROka) at 0.1 PFU/cell, and virus-infected cells were collected at various times after infection. Levels of the prosurvival proteins Bcl-2, Bcl-xL, and Mcl-1 and proapoptotic proteins Bax, BAD, and Bim were measured. Levels of Bim increased in mock-infected MeWo cells over time, while cells infected with VZV had reduced levels of Bim compared with findings for uninfected cells (Fig. 1A). The ratio of phosphorylated Bim to total Bim was higher in VZV-infected cells than in mock-infected cells beginning at 12 h and peaking at 24 h after infection. Phosphorylated Bim (34, 35) and BAD (36) migrate at a lower rate than unphosphorylated proteins (Fig. 1A). Similarly, the ratio of phosphorylated BAD to total BAD was higher in VZV-infected cells than in mock-infected cells. The ratios of phosphorylated Bim to total Bim and phosphorylated BAD to total BAD were higher in VZV-infected MRC-5 cells at 24 h (Fig. 1B) and in VZV-infected HFF from 24 h to 48 h after infection (Fig. 1C) than in mock-infected cells. Since Bim and BAD are proapoptotic members of the BH3-only family of proteins and their proapoptotic activities are reduced by phosphorylation (37), modification of Bim and BAD in VZV-infected cells may prevent or delay apoptosis. The level of Bcl-2 increased slightly in MeWo cells after infection, at 12 and 24 h, while the level of Mcl-1 modestly increased at 24 and 36 h after infection. However, levels of Bcl-xL and Bax remained constant during infection.
FIG 1.

VZV infection increases Bim and BAD phosphorylation. MeWo (A), MRC-5 (B), or HFF (C) cells were infected with cell-associated VZV ROka at 0.1 PFU/cell or mock infected, and cells were collected at the indicated hours postinfection (HPI) for detection of Bcl-2 family proteins, VZV immediate early protein 62 (IE62), and actin by Western blotting. The ratios of phosphorylated Bim to total Bim and of phosphorylated BAD to total BAD were determined using ImageQuant and are shown below the Western blots.
The MEK/ERK pathway is required for increased levels of phosphorylated Bim in VZV-infected cells.
Phosphorylation of Bim and BAD is controlled by growth factors, cytokines, and cell signaling molecules, such as ERK and PI3K (37, 38). Since VZV activates the MEK/ERK pathway and PI3K (26, 39–41), we investigated whether VZV-induced phosphorylation of Bim and BAD is regulated by the MEK/ERK pathway and PI3K in virus-infected cells. MeWo cells were infected with cell-associated VZV at 0.1 PFU/cell, the cells were treated with PI3K or MEK inhibitor 24 h later, and lysates were prepared after an additional 6 h. Treatment with the MEK inhibitor U0126 or the PI3K inhibitor LY294002 abolished ERK or Akt phosphorylation, respectively. VZV-induced phosphorylation of Bim was inhibited by the MEK inhibitor but not the PI3K inhibitor, indicating that Bim phosphorylation is regulated by the MEK/ERK pathway but not PI3K (Fig. 2A). Similar results were observed in MRC-5 cells (Fig. 2B). In contrast, BAD phosphorylation was not reduced by either MEK or PI3K inhibitor.
FIG 2.

VZV triggering of Bim phosphorylation is dependent on MEK activation. MeWo (A) or MRC-5 (B) cells were infected with cell-associated VZV ROka at 0.1 PFU/cell or mock infected; 24 h later, the cells were treated with the PI3K inhibitor LY294002 or the MEK inhibitor U0216 for 6 h, lysates were prepared, and proteins were detected by Western blotting. p-ERK is downstream of MEK signaling; therefore, inhibition of MEK results in reduction of p-ERK. The ratios of phosphorylated Bim to total Bim and of phosphorylated BAD to total BAD were determined using ImageQuant and are shown below the Western blots.
Reduced expression of Bim increases VZV replication.
Since phosphorylation of Bim is dependent on VZV activation of MEK and inhibition of the MEK/ERK pathway suppresses viral replication (26, 40), we postulated that Bim may modulate VZV replication. We constructed MeWo cell lines that overexpress Bim (MeWo-wt-Bim cells) or are knocked down for Bim (MeWo-shBim cells) using lentivirus vectors that express wild-type Bim or Bim shRNA, respectively. ShBim was expressed under a doxycycline-inducible promoter in MeWo-shBim cells. MeWo, MeWo-shBim, or MeWo-wt-Bim cells were infected with VZV, and virus replication, plaque formation, syncytium formation, and plaque size were measured. Bim was not detected in doxycycline-induced Mewo-shBim cells, and Bim was expressed at increased levels in doxycycline-treated MeWo-wt-Bim cells compared to expression in MeWo cells (Fig. 3A). VZV replicated to higher titers in doxycycline-induced MeWo-shBim cells and to lower titers in doxycycline-treated MeWo-wt-Bim cells than in doxycycline-treated MeWo cells (Fig. 3B). In addition, infection of doxycycline-treated MeWo-shBim cells with green fluorescent protein (GFP)-expressing VZV (VZV-GFP) resulted in larger GFP-expressing syncytia on day 4 than with doxycycline-treated MeWo and MeWo-wt-Bim cells (Fig. 3C). Plaque formation was delayed in doxycycline-treated MeWo-shBim cells infected with VZV-ROka and accelerated in doxycycline-treated MeWo-wt-Bim cells compared with that in doxycycline-treated parental MeWo cells. To quantify these results, we counted the number of plaques formed in 100 infectious foci (based on syncytium formation) in MeWo-wt-Bim, MeWo-shBim, and parental MeWo cells from day 4 to day 6 after infection with VZV-ROka. At day 4 postinfection, 85% of infectious foci formed plaques (85 plaques per 100 infectious foci) on doxycycline-treated MeWo-wt-Bim cells, only 20% of infectious foci formed plaques on MeWo-shBim cells, and 45% of infectious foci formed plaques on parental MeWo cells. At day 5 postinfection, nearly all virus infectious foci became plaques on MeWo-wt-Bim cells, 85% of infectious foci became plaques on parental MeWo cells, and only 40% of infectious foci became plaques on MeWo-shBim cells. At day 6, nearly all infectious foci became plaques on all the cell lines (Fig. 3D). Significantly larger plaques were observed in doxycycline-treated MeWo-shBim cells (10.5 mm ± 0.17 mm [standard deviation {SD}]) and in doxycycline-treated MeWo cells (10.3 mm ± 0.53 mm [SD]) than in doxycycline-treated MeWo-wt-Bim cells (9.1 mm ± 1.17 mm [SD]) (Fig. 3E). Taken together, these results indicate that reduced Bim expression allows longer survival of VZV-infected cells, delayed plaque formation, and higher titers of cell-associated virus, while increased Bim expression results in shorter survival of virus-infected cells, smaller synctyia, earlier plaque formation, and lower titers of virus.
Overexpression of Bim induces caspase 3 cleavage in VZV-infected cells.
Bim enhances apoptosis by binding to antiapoptotic Bcl-2 proteins (35, 37). While VZV inhibits apoptosis early during infection, the mechanism by which VZV mediates this effect is unknown. Since VZV infection induces phosphorylation of Bim, which inhibits its proapoptotic activity, we postulated that regulation of Bim activity in virus-infected cells might affect apoptosis. MeWo, MeWo-shBim, and MeWo-wt-Bim cells were infected with cell-associated VZV at 0.05 PFU/cells or mock infected, and at 36 h, cells were assayed for cleaved caspase 3, Bim, and VZV IE62. Cleaved caspase 3 was present at much higher levels in VZV-infected, doxycycline-treated, MeWo-wt-Bim cells (which overexpress Bim) than in doxycycline-treated parental MeWo cells or doxycycline-treated MeWo-shBim cells (Fig. 4). Conversely, cleaved caspase 3 was lower in virus-infected, doxycycline-treated MeWo-shBim cells (knocked down for Bim) than in doxycycline-treated parental MeWo cells. VZV infection reduced Bim expression in doxycycline-treated MeWo and MeWo-shBim cells but not in doxycycline-treated MeWo-wt-Bim cells. VZV IE62 expression was reduced in doxycycline-treated MeWo-wt-Bim cells compared to that in the other cells (Fig. 4), likely due to lower virus yields in cells overexpressing Bim (Fig. 3B). In contrast, VZV IE62 expression was increased in doxycycline-treated MeWo-shBim cells compared to that in the other cells, presumably due to higher virus yields in cells knocked down for Bim. Similar results were also observed in HFF cells expressing shBim or wt Bim. Therefore, any inhibitory effect of VZV on apoptosis was superseded by the level of apoptosis induced by high-level expression of Bim. Taken together, these results indicate that overexpression of Bim activates caspase 3 and reduces VZV replication.
FIG 4.
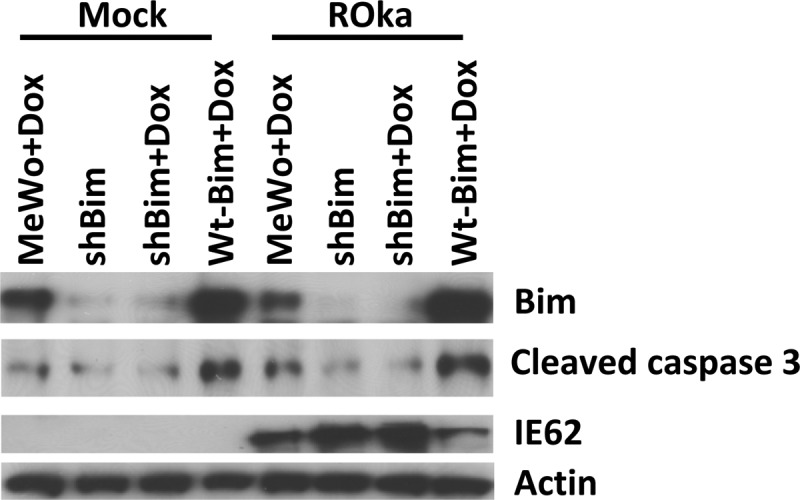
Overexpression of Bim induces caspase 3 activity. Doxycycline (Dox) was added to MeWo, MeWo-shBim, or MeWo-wt-Bim cells, and 24 h later, the cells were infected with VZV ROka at 0.05 PFU/cell. The cells were collected 36 h postinfection and cleaved caspase 3, Bim, VZV IE62, and actin were detected by Western blotting. The experiment was repeated with similar results.
Inhibition of caspases restores VZV replication in cells overexpressing Bim.
Since overexpression of Bim activates caspase 3 in VZV-infected cells, we postulated that the reduction in virus replication in MeWo-wt-Bim cells (which overexpress Bim) may be due to increased apoptosis in these cells. Doxycycline-treated MeWo, MeWo-wt-Bim, or MeWo-shBim cells were infected with VZV and treated with the pan-caspase inhibitor Q-VD-OPh 24 h after infection, and cells were collected 3 days later to titrate the virus. While inhibition of caspase activity with Q-VD-OPh slightly reduced viral titers in both MeWo cells (1.6-fold reduction) and MeWo-shBim cells (1.8-fold reduction), Q-VD-OPh increased VZV titers in MeWo-wt-Bim cells by 6.7-fold, restoring virus production to a level nearly the same as that in MeWo cells either treated or not treated with Q-VD-OPh (Fig. 5). Thus, increased caspase activity induced by overexpression of Bim inhibits VZV replication.
VZV ORF12 is required for efficient phosphorylation of Bim.
Since VZV triggers phosphorylation of Bim which is dependent on ERK, and we reported previously that the VZV ORF12 protein activates ERK and inhibits apoptosis (26), we postulated that VZV ORF12 might contribute to phosphorylation of Bim and BAD. MRC-5 cells were infected with VZV ROka or ROka12D (deleted for VZV ORF12) at 0.1 PFU/cell with cell-associated virus, and infected cells were collected at different times after infection. The ratio of phosphorylated Bim to total Bim was lower in MRC-5 cells infected with ROka12D than in those infected with ROka, indicating that ORF12 triggers Bim phosphorylation (Fig. 6A). In contrast, the ratio of phosphorylated BAD to total BAD was not lower in cells infected with ROka12D than in those infected with ROka. Levels of Bcl-2 and Bcl-xL showed little or no difference in cells infected with ROka12D compared to those infected with ROka. A lower ratio of phosphorylated Bim to total Bim was also observed in ROka12D-infected MeWo cells compared with ROka-infected MeWo cells beginning at 12 h after infection (Fig. 6B). These results indicate that VZV ORF12 triggers phosphorylation of Bim.
FIG 6.

ORF12 is required for efficient phosphorylation of Bim in VZV-infected cells. MRC-5 (A) or MeWo (B) cells were infected with cell-associated VZV ROka or ROka12D (VZV deleted for ORF12) at 0.1 PFU/cell or mock infected. Cell lysates were prepared at different hours postinfection (HPI), and proteins were detected by Western blotting. The ratios of phosphorylated Bim to total Bim and of phosphorylated BAD to total BAD were determined using ImageQuant and are shown below the Western blots.
Since VZV ORF12 triggers Bim phosphorylation, we tested whether overexpression of Bim has a more inhibitory effect on replication of VZV ROka12D than on that of ROka and if knock down of Bim enhances replication of VZV ROka12D more than that of ROka. MeWo-wt-Bim, MeWo-shBim, and parental MeWo cells were infected with VZV ROka or ROka12D in medium containing 2% FBS at 34°C, virus-infected cells were collected on days 1 to 6, and titers of virus were determined on MeWo cells. Replication of VZV ROka (Fig. 7A) and that of ROka12D (Fig. 7B) were similarly reduced on MeWo-wt-Bim cells, and while replication of both viruses was enhanced in MeWo-shBim cells compared to that in parental MeWo cells, the degree of enhancement was similar for ROka and ROka12D. Thus, while the ORF12 protein is important for efficient phosphorylation of Bim, VZV deleted for ORF12 is not more impaired for growth in cells overexpressing Bim or more enhanced for growth in cells knocked down for Bim than parental virus. These results indicate that the effect of ORF12 on phosphorylation of Bim does not have a major role in the effect of Bim on virus replication.
FIG 7.

Replication of ROka and ROka12D in cells knocked down or overexpressing Bim. MeWo, MeWo-shBim (shBim), or MeWo-wt-Bim (Bim wt) cells were infected with VZV ROka (A) or ROka12D (B) and incubated at 34°C in MEM with 2% serum. Doxycycline was added 24 h after infection, virus-infected cells were collected on days 1 to 6 after infection, and virus was titrated on MeWo cells by plaque assay.
DISCUSSION
Apoptosis is important for host defense against virus infection, and herpesviruses manipulate the host to inhibit apoptosis (1–3, 42). We found that VZV infection regulates Bcl-2 family proteins. Infection of cells with VZV triggered phosphorylation of Bim and BAD, while little or no effect was observed on other Bcl-2 family proteins, including Mcl-1, Bcl-2, Bcl-xL, or Bax. While we did not look specifically for phosphorylated forms of other Bcl-2 family members, we did not see changes in migration of these proteins, unlike the changes noted with Bim and BAD.
We found that inhibition of Bim expression prolonged survival of VZV-infected cells. Bim is a proapoptotic BH3-only protein that binds to the antiapoptotic Bcl-2 proteins Bcl-2, Bcl-xL, and Mcl-1, resulting in the release of Bax/Bak to promote apoptosis (35, 37). Phosphorylation of Bim reduces its proapoptotic activity by promoting its disassociation from Bcl-2, Bcl-xL, or Mcl-1 (35, 43). Other herpesviruses, in addition to VZV, regulate expression of Bim. Infection of cells with Epstein-Barr virus (EBV) results in degradation of Bim (44). EBV EBNA3A and EBNA3C cooperate to downregulate Bim (45), and EBV BHRF1 (a homolog of Bcl-2) associates with and sequesters Bim (46, 47). The EBV BART microRNAs reduce expression of Bim (48), and Bim undergoes epigenetic repression and CpG methylation in EBV-latently infected B cells (49). Kaposi's sarcoma-associated herpesvirus (KHSV)-encoding viral interferon regulatory factor 1 (vIRF-1) promotes nuclear translocation of Bim resulting in its inactivation (50).
Here we showed that VZV phosphorylation of Bim is dependent on activation of the MEK/ERK pathway. ERK is critical for phosphorylation of Bim (37). Many herpesviruses activate ERK to regulate survival of infected cells. Human cytomegalovirus (HCMV) gB activates ERK to induce expression of Mcl-1 (51), the HSV-2 large subunit of ribonucleotide reductase (ICP10PK) triggers ERK activation to upregulate the X-linked inhibitor of apoptosis (XIAP) and the antiapoptosis protein Bag-1 (52, 53), EBV LMP2A activates ERK to degrade Bim (54), and KSHV LANA induces ERK expression to increase phosphorylation of c-Myc (55). VZV activates ERK, and suppression of ERK inhibits VZV replication (26, 40).
We found that VZV infection also triggered phosphorylation of BAD. BAD is a BH3-only proapoptotic protein that is tightly regulated by survival factors, and phosphorylation of BAD inhibits its apoptotic function (56, 57). BAD can be phosphorylated by Akt (58), by ERK (36), or by protein kinase A (59). In contrast to phosphorylation of Bim in VZV-infected cells, which is controlled by virus activation of ERK, phosphorylation of BAD in VZV-infected cells was not reduced with either PI3K or MEK inhibitors, indicating that VZV triggers phosphorylation of BAD through other mechanisms. Previously, Rahaus et al. showed that VZV infection induced phosphorylation of BAD at serine 112 (40) but inhibited phosphorylation at serine 136 (39). In contrast, we found that VZV induced phosphorylation of BAD at serine 136. While Rahaus et al. infected confluent monolayers of melanoma cells (39, 40), we found increased phosphorylation of BAD after infection of subconfluent melanoma cells, as well as infection of MRC-5 and HFF cells. BAD is known to be constitutively phosphorylated in melanoma cells through activation of the mitogen-activated protein kinase pathway (60). Thus, our results may differ from those of Rahaus et al. due to the cell lines used or the conditions of infection.
We found that knockdown of Bim enhanced survival of VZV-infected cells, delayed plaque formation, and resulted in higher levels of virus replication. In contrast, HSV did not grow to higher titers in cells knocked down for Bim (R. Godbout, K. Wang, X. Liu, and J. I. Cohen, unpublished data). A prior study showed that expression of a viral antiapoptotic Bcl-2 homolog in human cells enhanced the growth of murine cytomegalovirus (61). VZV grows to relatively low titers in cell culture, and production of cell-free virus is difficult. Thus, inhibition of Bim or other proapoptotic proteins (e.g., BAD and Bax) or expression of antiapoptotic proteins (e.g., Bcl-2 and Bcl-xL) might also increase levels of VZV replication and be used to enhance virus production.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
We thank Linda Kang (University of Philadelphia) for technical assistance, Iain Fraser (National Institutes of Health) for plasmids, Paul Kinchington for antibody to VZV IE62, and Rachel Godbout and Kening Wang for performing experiments with HSV-2.
Footnotes
Published ahead of print 13 November 2013
REFERENCES
- 1.Koyama AH, Irie H, Fukumori T, Hata S, Iida S, Akari H, Adachi A. 1998. Role of virus-induced apoptosis in a host defense mechanism against virus infection. J. Med. Invest. 45:37–45 [PubMed] [Google Scholar]
- 2.Everett H, McFadden G. 1999. Apoptosis: an innate immune response to virus infection. Trends Microbiol. 7:160–165. 10.1016/S0966-842X(99)01487-0 [DOI] [PubMed] [Google Scholar]
- 3.Perkins D. 2005. Virus signaling and apoptosis in the central nervous system infection. Front. Biosci. 10:2804–2819. 10.2741/1737 [DOI] [PubMed] [Google Scholar]
- 4.O'Brien V. 1998. Viruses and apoptosis. J. Gen. Virol. 79(Part 8):1833–1845 [DOI] [PubMed] [Google Scholar]
- 5.Basanez G, Soane L, Hardwick JM. 2012. A new view of the lethal apoptotic pore. PLoS Biol. 10:e1001399. 10.1371/journal.pbio.1001399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korsmeyer SJ, Gross A, Harada H, Zha J, Wang K, Yin XM, Wei M, Zinkel S. 1999. Death and survival signals determine active/inactive conformations of pro-apoptotic BAX, BAD, and BID molecules. Cold Spring Harbor Symp. Quant. Biol. 64:343–350. 10.1101/sqb.1999.64.343 [DOI] [PubMed] [Google Scholar]
- 7.Goodkin ML, Morton ER, Blaho JA. 2004. Herpes simplex virus infection and apoptosis. Int. Rev. Immunol. 23:141–172. 10.1080/08830180490265574 [DOI] [PubMed] [Google Scholar]
- 8.Esaki S, Goshima F, Katsumi S, Watanabe D, Ozaki N, Murakami S, Nishiyama Y. 2010. Apoptosis induction after herpes simplex virus infection differs according to cell type in vivo. Arch. Virol. 155:1235–1245. 10.1007/s00705-010-0712-2 [DOI] [PubMed] [Google Scholar]
- 9.Nguyen ML, Blaho JA. 2007. Apoptosis during herpes simplex virus infection. Adv. Virus Res. 69:67–97. 10.1016/S0065-3527(06)69002-7 [DOI] [PubMed] [Google Scholar]
- 10.Aubert M, Blaho JA. 2001. Modulation of apoptosis during herpes simplex virus infection in human cells. Microbes Infect. 3:859–866. 10.1016/S1286-4579(01)01444-7 [DOI] [PubMed] [Google Scholar]
- 11.Leopardi R, Van Sant C, Roizman B. 1997. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. U. S. A. 94:7891–7896. 10.1073/pnas.94.15.7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartier A, Komai T, Masucci MG. 2003. The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosphorylation of the Bcl-2 family member Bad. Exp. Cell Res. 291:242–250. 10.1016/S0014-4827(03)00375-6 [DOI] [PubMed] [Google Scholar]
- 13.Zhou G, Galvan V, Campadelli-Fiume G, Roizman B. 2000. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 74:11782–11791. 10.1128/JVI.74.24.11782-11791.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C. 2004. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J. Neurovirol. 10:260–265. 10.1080/13550280490468690 [DOI] [PubMed] [Google Scholar]
- 15.Aiamkitsumrit B, Zhang X, Block TM, Norton P, Fraser NW, Su YH. 2007. Herpes simplex virus type 1 ICP4 deletion mutant virus d120 infection failed to induce apoptosis in nerve growth factor-differentiated PC12 cells. J. Neurovirol. 13:305–314. 10.1080/13550280701361490 [DOI] [PubMed] [Google Scholar]
- 16.Aubert M, Blaho JA. 1999. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 73:2803–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. 2011. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 16:256–271. 10.1007/s10495-010-0560-2 [DOI] [PubMed] [Google Scholar]
- 18.Zhou G, Roizman B. 2001. The domains of glycoprotein D required to block apoptosis depend on whether glycoprotein D is present in the virions carrying herpes simplex virus 1 genome lacking the gene encoding the glycoprotein. J. Virol. 75:6166–6172. 10.1128/JVI.75.13.6166-6172.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medici MA, Sciortino MT, Perri D, Amici C, Avitabile E, Ciotti M, Balestrieri E, De Smaele E, Franzoso G, Mastino A. 2003. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J. Biol. Chem. 278:36059–36067. 10.1074/jbc.M306198200 [DOI] [PubMed] [Google Scholar]
- 20.Hood C, Cunningham AL, Slobedman B, Boadle RA, Abendroth A. 2003. Varicella-zoster virus-infected human sensory neurons are resistant to apoptosis, yet human foreskin fibroblasts are susceptible: evidence for a cell-type-specific apoptotic response. J. Virol. 77:12852–12864. 10.1128/JVI.77.23.12852-12864.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brazeau E, Mahalingam R, Gilden D, Wellish M, Kaufer BB, Osterrieder N, Pugazhenthi S. 2010. Varicella-zoster virus-induced apoptosis in MeWo cells is accompanied by down-regulation of Bcl-2 expression. J. Neurovirol. 16:133–140. 10.3109/13550281003682547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadzot-Delvaux C, Thonard P, Schoonbroodt S, Piette J, Rentier B. 1995. Varicella-zoster virus induces apoptosis in cell culture. J. Gen. Virol. 76(Part 11):2875–2879. 10.1099/0022-1317-76-11-2875 [DOI] [PubMed] [Google Scholar]
- 23.Konig A, Homme C, Hauroder B, Dietrich A, Wolff MH. 2003. The varicella-zoster virus induces apoptosis in vitro in subpopulations of primary human peripheral blood mononuclear cells. Microbes Infect. 5:879–889. 10.1016/S1286-4579(03)00177-1 [DOI] [PubMed] [Google Scholar]
- 24.Pugazhenthi S, Nair S, Velmurugan K, Liang Q, Mahalingam R, Cohrs RJ, Nagel MA, Gilden D. 2011. Varicella-zoster virus infection of differentiated human neural stem cells. J. Virol. 85:6678–6686. 10.1128/JVI.00445-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaap A, Fortin JF, Sommer M, Zerboni L, Stamatis S, Ku CC, Nolan GP, Arvin AM. 2005. T-cell tropism and the role of ORF66 protein in pathogenesis of varicella-zoster virus infection. J. Virol. 79:12921–12933. 10.1128/JVI.79.20.12921-12933.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X, Li Q, Dowdell K, Fischer ER, Cohen JI. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J. Virol. 86:3143–3151. 10.1128/JVI.06923-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hood C, Cunningham AL, Slobedman B, Arvin AM, Sommer MH, Kinchington PR, Abendroth A. 2006. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 80:1025–1031. 10.1128/JVI.80.2.1025-1031.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen JI, Cox E, Pesnicak L, Srinivas S, Krogmann T. 2004. The varicella-zoster virus open reading frame 63 latency-associated protein is critical for establishment of latency. J. Virol. 78:11833–11840. 10.1128/JVI.78.21.11833-11840.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, Rebres R, Roach T, Seaman W, Simon MI, Fraser ID. 2006. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. U. S. A. 103:13759–13764. 10.1073/pnas.0606179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X, Santat LA, Chang MS, Liu J, Zavzavadjian JR, Wall EA, Kivork C, Simon MI, Fraser ID. 2007. A versatile approach to multiple gene RNA interference using microRNA-based short hairpin RNAs. BMC Mol. Biol. 8:98. 10.1186/1471-2199-8-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boisvert-Adamo K, Aplin AE. 2008. Mutant B-RAF mediates resistance to anoikis via Bad and Bim. Oncogene 27:3301–3312. 10.1038/sj.onc.1211003 [DOI] [PubMed] [Google Scholar]
- 32.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James SF, Mahalingam R, Gilden D. 2012. Does apoptosis play a role in varicella zoster virus latency and reactivation? Viruses 4:1509–1514. 10.3390/v4091509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129:1337–1349. 10.1016/j.cell.2007.04.027 [DOI] [PubMed] [Google Scholar]
- 35.Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. 2007. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 26:2856–2867. 10.1038/sj.emboj.7601723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheid MP, Schubert KM, Duronio V. 1999. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J. Biol. Chem. 274:31108–31113. 10.1074/jbc.274.43.31108 [DOI] [PubMed] [Google Scholar]
- 37.Ewings KE, Wiggins CM, Cook SJ. 2007. Bim and the pro-survival Bcl-2 proteins: opposites attract, ERK repels. Cell Cycle 6:2236–2240. 10.4161/cc.6.18.4728 [DOI] [PubMed] [Google Scholar]
- 38.Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. 2004. Extracellular signal-regulated kinases 1/2 are serum-stimulated “Bim(EL) kinases” that bind to the BH3-only protein Bim(EL) causing its phosphorylation and turnover. J. Biol. Chem. 279:8837–8847. 10.1074/jbc.M311578200 [DOI] [PubMed] [Google Scholar]
- 39.Rahaus M, Desloges N, Wolff MH. 2007. Varicella-zoster virus requires a functional PI3K/Akt/GSK-3alpha/beta signaling cascade for efficient replication. Cell Signal. 19:312–320. 10.1016/j.cellsig.2006.07.003 [DOI] [PubMed] [Google Scholar]
- 40.Rahaus M, Desloges N, Wolff MH. 2006. Varicella-zoster virus influences the activities of components and targets of the ERK signalling pathway. J. Gen. Virol. 87:749–758. 10.1099/vir.0.81571-0 [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Cohen JI. 2013. Varicella-zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J. Virol. 87:1842–1848. 10.1128/JVI.02395-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardwick JM. 1997. Virus-induced apoptosis. Adv. Pharmacol. 41:295–336. 10.1016/S1054-3589(08)61063-7 [DOI] [PubMed] [Google Scholar]
- 43.Kutuk O, Letai A. 2008. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med. 8:102–118. 10.2174/156652408783769599 [DOI] [PubMed] [Google Scholar]
- 44.Clybouw C, McHichi B, Mouhamad S, Auffredou MT, Bourgeade MF, Sharma S, Leca G, Vazquez A. 2005. EBV infection of human B lymphocytes leads to down-regulation of Bim expression: relationship to resistance to apoptosis. J. Immunol. 175:2968–2973 [DOI] [PubMed] [Google Scholar]
- 45.Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. 2008. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt's lymphoma. Oncogene 27:421–433. 10.1038/sj.onc.1210668 [DOI] [PubMed] [Google Scholar]
- 46.Desbien AL, Kappler JW, Marrack P. 2009. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. U. S. A. 106:5663–5668. 10.1073/pnas.0901036106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, Huang DC, Colman PM. 2010. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 6:e1001236. 10.1371/journal.ppat.1001236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marquitz AR, Mathur A, Nam CS, Raab-Traub N. 2011. The Epstein-Barr virus BART microRNAs target the pro-apoptotic protein Bim. Virology 412:392–400. 10.1016/j.virol.2011.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paschos K, Smith P, Anderton E, Middeldorp JM, White RE, Allday MJ. 2009. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 5:e1000492. 10.1371/journal.ppat.1000492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi YB, Nicholas J. 2010. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 6:e1001031. 10.1371/journal.ppat.1001031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reeves MB, Breidenstein A, Compton T. 2012. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 109:588–593. 10.1073/pnas.1114966108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith W, Tomasec P, Aicheler R, Loewendorf A, Nemcovicova I, Wang EC, Stanton RJ, Macauley M, Norris P, Willen L, Ruckova E, Nomoto A, Schneider P, Hahn G, Zajonc DM, Ware CF, Wilkinson GW, Benedict CA. 2013. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 13:324–335. 10.1016/j.chom.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perkins D, Pereira EF, Aurelian L. 2003. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J. Virol. 77:1292–1305. 10.1128/JVI.77.2.1292-1305.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwakiri D, Minamitani T, Samanta M. 2013. Epstein-Barr virus latent membrane protein 2A contributes to anoikis resistance through ERK activation. J. Virol. 87:8227–8234. 10.1128/JVI.01089-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 81:10451–10459. 10.1128/JVI.00804-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelekar A, Chang BS, Harlan JE, Fesik SW, Thompson CB. 1997. Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-XL. Mol. Cell. Biol. 17:7040–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruvolo PP, Deng X, May WS. 2001. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 15:515–522. 10.1038/sj/leu/2402090 [DOI] [PubMed] [Google Scholar]
- 58.Datta SR, Brunet A, Greenberg ME. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905–2927. 10.1101/gad.13.22.2905 [DOI] [PubMed] [Google Scholar]
- 59.Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. 1999. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 3:413–422. 10.1016/S1097-2765(00)80469-4 [DOI] [PubMed] [Google Scholar]
- 60.Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. 2003. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Res. 63:8330–8337 [PubMed] [Google Scholar]
- 61.Jurak I, Brune W. 2006. Induction of apoptosis limits cytomegalovirus cross-species infection. EMBO J. 25:2634–2642. 10.1038/sj.emboj.7601133 [DOI] [PMC free article] [PubMed] [Google Scholar]