

Abstract
The adenovirus death protein (ADP) is expressed at late times during a lytic infection of species C adenoviruses. ADP promotes the release of progeny virus by accelerating the lysis and death of the host cell. Since some human lymphocytes survive while maintaining a persistent infection with species C adenovirus, we compared ADP expression in these cells with ADP expression in lymphocytes that proceed with a lytic infection. Levels of ADP were low in KE37 and BJAB cells, which support a persistent infection. In contrast, levels of ADP mRNA and protein were higher in Jurkat cells, which proceed with a lytic infection. Epithelial cells infected with an ADP-overexpressing virus died more quickly than epithelial cells infected with an ADP-deleted virus. However, KE37, and BJAB cells remained viable after infection with the ADP-overexpressing virus. Although the levels of ADP mRNA increased in KE37 and BJAB cells infected with the ADP-overexpressing virus, the fraction of cells with detectable ADP was unchanged, suggesting that the control of ADP expression differs between epithelial and lymphocytic cells. When infected with an ADP-deleted adenovirus, Jurkat cells survived and maintained viral DNA for greater than 1 month. These findings are consistent with the notion that the level of ADP expression determines whether lymphocytic cells proceed with a lytic or a persistent adenovirus infection.
INTRODUCTION
Species C human adenoviruses (HAdV) (types 1, 2, 5, and 6) commonly infect the upper respiratory and gastrointestinal tracts. Species C adenoviruses usually cause mild disease in humans but are responsible for more-severe disease in immunocompromised individuals (1). Typically, infection of epithelial cells results in a lytic infection with release of newly synthesized virions and the death of infected cells after a few days (2, 3). Following a primary infection in children, species C adenoviruses enter a persistent stage during which infectious virus can be intermittently detected in the stool after virus is no longer detected in the nasopharyngeal cavity (4). Species C adenoviruses can also establish persistent and latent infections in human lymphocytes of the tonsil and adenoids (5–7). Based on these and other reports, infection with species C adenoviruses appears to be a two-step process characterized by acute replication in epithelial cells, followed by persistent infection of lymphocytes. Most studies characterizing the virus life cycle in vitro have been performed with epithelial cells.
In contrast to the lytic infection seen in epithelial cells, an adenovirus infection of human lymphocytes can follow a very different course. Species C genomes can be detected in lymphocytes of the tonsil and adenoids in 85% of children between 1 and 15 years of age (6). Species C adenoviruses can establish persistent and latent infections in human lymphocytes, as well as in some lymphocyte cell lines (8). Virus was found to persistently infect lymphocytes while the AdV genome was maintained as an episome in the absence of detectable infectious virus (4, 6). Infectious virus and viral transcripts could be detected when patient-derived lymphocytes were stimulated in vitro (7). Zhang et al. (8) modeled adenovirus latency in lymphocyte cell lines. Those authors identified several human lymphocyte cell lines in which the viral genome could be detected up to a year after infection whereas detectable expression of viral late proteins was lost shortly after initial infection. The mechanism of long-term viral genome maintenance is unknown. The kinetics of viral gene expression also appear to occur much slower in lymphocytes than in epithelial cells (9). Moreover, infection of epithelial cells proceeds rapidly and is lytic, whereas infection of lymphocytes is often not lytic to the cells (9–13).
The function of adenoviral proteins and the pattern of viral gene expression during a lytic infection have been well characterized in epithelial cells (14, 15). Broadly speaking, early viral genes are expressed before the viral DNA genome replicates and are designated by an “E.” Late genes are expressed after viral DNA replication and encode virion structural proteins. It should be noted that the temporal pattern of expression of a given gene is not strictly determined. The 11.6-kDa adenovirus death protein (ADP) is encoded by early region 3 (E3) of the genome but is expressed most abundantly during the late stages of infection from the major late promoter. ADP promotes lysis of the infected cell after progeny virus production, presumably to facilitate virion release and dissemination concomitant with the death of the cell (2).
Human lymphocytic cell lines have been identified in which an adenovirus infection proceeds with markedly different outcomes. Jurkat cells enter a productive, lytic infection resembling that observed in epithelial cells (8). In contrast, BJAB and KE37 cells establish a long-term persistent infection (8). Epithelial cells, which are lytically infected, express high levels of ADP, and it seems likely that cells in which the virus persists would need to prevent ADP expression and the accompanying cell lysis to maintain persistence. However, nothing is known about ADP expression in infected lymphocytes. In this study, we hypothesized that ADP expression plays a role in determining whether infected lymphocytes enter the lytic or persistent pathway. Results described here indicate that low-level expression of ADP may indeed permit establishment of a persistent infection. Furthermore, results of this study demonstrate that the dynamics of ADP expression differ greatly between infected epithelial cells and infected lymphocytes.
MATERIALS AND METHODS
Cell lines.
Cell culture media, supplements, and fetal bovine sera were obtained from Mediatech, Inc. (Manassas, VA). A549 lung carcinoma epithelial cells (ATCC CCL-185; American Type Culture Collection, Manassas, VA) were maintained as confluent monolayer cultures in Dulbecco-modified Eagle's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum and 10 mM glutamine and incubated at 37°C in a humidified atmosphere containing 8% CO2. The Burkitts lymphoma B-cell line (BJAB) and Jurkat T-cell lines were also obtained from the ATCC and maintained as suspension cultures in RPMI 1640 medium (RPMI) supplemented with 10% fetal bovine serum and 10 mM glutamine. The KE37 cell line (an immature T-cell acute lymphoblastic leukemia [ALL]) was purchased from the German Collection of Microorganisms and Cell Cultures. These cells were maintained by incubating at 37°C in a humidified atmosphere containing 5% CO2.
Viruses.
The wild-type (WT) virus used in this study is wt300, which is a type 5 adenovirus kindly provided by Tom Shenk (Princeton University) that served as the parent for the mutant viruses studied here. Some studies used the phenotypically wild-type virus dl309, which lacks a portion of E3 region (10.4K, 14.5K, and 14.7K) that has been shown to be dispensable for growth in tissue culture (16). wt300 and dl309 direct comparable levels of ADP expression and are designated ADP-WT in this report. The ADP-mutant virus pm534 (ADP-) was constructed by converting the methionine codons at position 34 and 42 to nonsense codons. Any truncated protein derived by initiation at subsequent methionine codons would be unable to incorporate into membranes and is expected to be nonfunctional (17). No truncated forms of the ADP have been detected with this mutant. The VRX-021 virus, which overexpresses ADP (ADP++), was constructed as described in reference 18 with a shuttle plasmid providing the E3 region that is a hybrid between VRX-006 and VRX-007. The closely related viruses VRX-006 and VRX-007 were described in reference 17. Epithelial cells were plated 24 h before infection and subsequently infected with a multiplicity of infection (MOI) of 10 to 20 infectious units (IU) per cell in serum-free medium. IU levels were determined by a fluorescent-focus-forming assay of infected HeLa cells using a primary antibody (Ab) specific for the E1A proteins. After 2 h, complete medium was added to the cells. For infection of lymphocytes, cells were seeded at least 24 h before infection at a density of 1 × 106 cells per ml. Cells were infected with 100 IU per cell and incubated for 2 h at 37°C. For experiments utilizing “spin infections,” cells were infected with 50 IU per cell and centrifuged at room temperature for 45 min at 1,000 × g. Cells were then incubated at 37°C for 75 min. Following incubation with the virus, the nonadsorbed virus was then washed from the infected cells three times with serum-free medium. Cells were then returned to culture in complete medium at a density of 5 × 105 cells per ml.
RT-PCR analysis of ADP expression.
RNA was purified from the infected cells using an RNA Miniprep kit from Qiagen (catalog no.74104). A 500-ng volume of total RNA was reverse transcribed using a DyNAmo cDNA synthesis kit (Thermo Fisher) according to manufacturer's instructions using a Bio-Rad Thermocycler. Quantitative PCR (qPCR) of the cDNA was then performed in duplicate 25-μl reaction mixtures containing 2× DyNAmo SYBR green master mix (Thermo Fisher), 10 mM (each) forward and reverse primers, and RNase-free water. A minus-reverse transcriptase (RT) negative-control sample was included in quantitative RT-PCR (qRT-PCR) experiments for detection of contaminating DNA. The primers designed for qPCR of the HAdV-C5 ADP were placed in the coding region for the ADP exon (forward primer, 5′-CCGGACTTACATCTACCACAAA-3′ [nucleotides 29516 to 29537]; reverse primer, 5′-CAAACATAAGCGCTATGGAGAAC-3′ [nucleotides 29620 to 29598] [accession number M73260.1]). Hypoxanthine phosphoribosyltransferase 1 (HPRT1) mRNA was found to remain invariant among mock- and adenovirus-infected lymphocytes in previous experiments and was used as an endogenous control. The primers used for HPRT1 were 5′-TGG ACA GGA CTG AAC GTC TTG-3′ and 5′CCA GCA GGT CAG CAA AGA ATT TA-3′. All primers were purchased from Integrated DNA Technologies, Coralville, IA. Cycling conditions were as follows: 15 min at 95°C and 30 cycles of 10 s at 95°C, 30 s at 50°C, and 30 s at 72°C followed by a 1%-increase melting curve gradient from 60°C to 95°C. Relative gene expression levels were determined using the threshold cycle (CT) values for each reaction and using the ΔΔCT method for comparing the relative values of gene expression (19).
Intracellular staining and flow cytometry.
Infected cells were harvested at the indicated times postinfection (pi) and washed with ice-cold phosphate-buffered saline (PBS). The cells were then fixed in the dark with 1% paraformaldehyde obtained from Affymetrix Inc. (catalog no. 19943) and incubated at room temperature for 30 min. The fixed cells were then resuspended in 0.2% Tween 20–PBS (permeabilization buffer) at a density of 106 cells per ml for 15 min at room temperature. This was followed by centrifugation at 1,500 rpm for 5 min at 20°C. The cells were then incubated at room temperature for 30 min with blocking buffer (PBS with 2% bovine serum albumin [BSA]). The cells were centrifuged and resuspended in cell staining buffer obtained from BioLegend (catalog no. 420201). Cell staining buffer contains bovine calf serum and 0.09% sodium azide. The monoclonal antibody (MAb) specific to adenovirus hexon (Chemicon MAb 8051) was used at 10 μg per ml in cell staining buffer. The monoclonal antibody (clone 45.16.4, unknown IgG subclass) to ADP was generated with mice that were immunized with a conjugated peptide corresponding to the HAdV-C5 ADP C-terminal sequence (amino acids 79 to 93). Hybridomas were produced and tested by indirect immunofluorescence. Positive hybridomas were subcloned twice. The concentrated cell culture supernatant fluid containing the monoclonal antibody was used at a 1:50 dilution. Purified mouse IgG1 (BD Pharmigen, catalog no. 557273) was used at 20 μg per ml in cell staining buffer as an isotype control for hexon staining. After incubation with the primary antibody, cells were stained with a fluorescently labeled secondary antibody that was either allophycocyanin (APC)- or fluorescein isothiocyanate (FITC)-labeled goat IgG raised against whole-mouse IgG (H+L) (Life Technologies, catalog no. A-865, F-2761) and used at 20 μg per ml. Cells were washed and resuspended in 0.6 ml of cell staining buffer and acquired by flow cytometry on a BD Fortessa cell analyzer using FACSdiva software.
Trypan blue dye exclusion assay.
The viability of A549, BJAB, Jurkat, and KE37 cells was measured by the exclusion of trypan blue dye as described in reference 2 using a Countess automated cell counter (Invitrogen). A 20-μl volume of cells was diluted with an equal volume of 0.4% trypan blue dye. These experiments were conducted several times with similar results. The data shown represent the averages of the results of several repeat experiments, with error bars representing the standard deviations (SD).
RESULTS
Persistently infected lymphocytes show low levels of ADP.
Species C adenoviruses typically establish a lytic infection in human epithelial cells and produce progeny virus as soon as 12 h postinfection (hpi). During such a lytic infection, cell proliferation ceases and the infected cells die within a few days (2). In contrast, several human lymphocytic cell lines support persistent infections with species C adenoviruses such as the wild-type HAdV-C5 viral strain dl309 (8). Persistently infected BJAB and Ramos cells (B-cell origin) and KE37 cells (T-cell origin) proliferated at the same rate as noninfected cells. Some persistently infected lymphocytic cells maintained the viral genome for over a year in culture (8). However, not all lymphocytic cells support a persistent infection; Jurkat T cells stopped dividing and declined in viability as early as 3 days after infection (8). Thus, infection of Jurkat cells appears to lead to a lytic infection that is similar to infection of epithelial cells.
Because ADP affects cell viability, we compared ADP expression in persistently infected cells (KE37 and BJAB) with that in lytically infected cells (A549 and Jurkat cells). The expression of ADP and a representative late gene (hexon) was evaluated by intracellular staining and flow cytometry in cells that were infected with wild-type HAdV-C5 (dl309) (Fig. 1). Hexon expression was detected in approximately 80% of A549 and Jurkat cells 1 day postinfection (dpi) and in a similar fraction of BJAB and KE37 cells between 3 and 8 dpi. ADP expression, however, was markedly reduced in the persistently infected cells compared to lytically infected cells. In epithelial A549 cells serving as a control, ADP expression was detected in the same fraction of cells as hexon expression. Although the fraction of Jurkat cells positive for ADP was less than the fraction positive for hexon, at least 40% of the Jurkat cells contained detectable levels of ADP on days 1, 3, and 5 postinfection. In contrast, less than 15% of the KE37 cells and less than 20% of the BJAB cells expressed detectable levels of ADP at any time postinfection (Fig. 1, black bars). We note that, as previously published (8, 9), lymphocytic cells require a greater virus inoculum to achieve a level of infection comparable to epithelial cell infection as determined by hexon staining. Although the time courses of hexon gene expression differed among the lytically and persistently infected cells, all cells were well infected and able to direct expression of a representative late gene. This variable time course in late gene expression is evident in the representative flow cytometry analysis of hexon expression (Fig. 1B to D). Five days postinfection, hexon protein was detected in 87% of Jurkat cells (Fig. 1B). At this same time postinfection, we detected hexon in 72% and 63% of KE37 and BJAB cells, respectively. There was some variability in the kinetics of hexon expression between experimental infections of these lymphocytic cell lines; however, it was quite typical for the highest levels of hexon to be detected in BJAB and KE37 cells several days after maximal expression occurred in Jurkat cells infected at the same time (Fig. 1C and D). Hexon expression typically peaked in Jurkat cells within the first few days of infection and was quite high as early as 1 dpi (92%; Fig. 1B). Although both Jurkat and KE37 cells are of T-cell lineage, very little hexon expression could be detected 1 dpi in KE37 cells (13%; Fig. 1C). A similar variation in the timing of ADP expression among the lymphocytic cells was observed. Even though ADP expression was always detected in a fraction of each lymphocytic cell line that was smaller than the fraction of hexon-positive cells, the fraction of ADP-positive cells was always proportionally greater in Jurkat cells than in either KE37 or BJAB cells. Interestingly, the intensity of ADP staining per ADP-positive cell was higher in KE37 cells than in BJAB cells (Fig. 1C and D; bottom panels). KE37 cells appeared to include a population of cells that stained nearly as intensely as A549 cells, which were analyzed in the same experiment as the BJAB cells to serve as a positive control (Fig. 1D). These results show that expression of the ADP gene determined by antibody staining and flow cytometry is markedly reduced compared to expression of the late hexon gene in both cell lines that support a persistent adenovirus infection.
FIG 1.

Expression levels of ADP and the hexon protein differ among cells that sustain a persistent or lytic adenovirus infection. (A) Hexon and ADP levels were determined on the indicated days by flow cytometric analysis of A549, Jurkat, BJAB, and KE37 cells infected with the phenotypically wild-type virus dl309. Lung epithelial tumor cells (A549) were infected with the wild-type virus dl309 at a multiplicity of infection of 20 IU per cell. The lymphocytic cells (Jurkat, KE37, and BJAB) were infected at a multiplicity of infection of 100 IU per cell to achieve similar fractions of infected cells as judged by hexon staining. Monoclonal antibodies specific for ADP or hexon, followed by a fluorescently labeled secondary antibody, were used to stain cells intracellularly. The data are representative of three experiments with similar results. (B to D) Flow cytometry dot plots from a separate experiment are shown for infected Jurkat (B), KE37 (C), and BJAB (D) cells on the indicated dpi. Mock-infected cells and cells stained with an isotype control antibody were used as negative controls. The gate for hexon expression was based on the isotype control antibody stain remaining below 5%. The gate for ADP expression was based on mock-infected cell staining remaining below 5%. As a positive control for ADP staining, A549 cells were infected with the wild-type virus and analyzed 1 dpi, with the results shown in the double-boxed plot. The y axis denotes cell side scatter.
Changes in the viability of infected A549, Jurkat, KE37, and BJAB cells were evaluated by trypan blue dye exclusion (Fig. 2). Mock-infected cells remained viable (>95%) over the course of the experiment. Representative data for mock-infected viability are shown only for A549 cells. Infected A549 epithelial cells displayed the typical cytopathic effect by rounding and detaching from the substrate within 2 dpi (data not shown). After 4 dpi, most A549 cells were fully detached and more than half were nonviable. By 7 dpi, over 95% of the infected A549 cells had died. A similar decrease in cell viability was observed in the infected Jurkat cells. Jurkat cell proliferation ceased after 1 dpi (data not shown), and by 3 dpi, 20% of the cells were dead. Approximately 60% of the infected Jurkat cells died by 7 dpi, with only cellular debris present after that time. The results shown in Fig. 1 indicate that the fraction of Jurkat cells expressing ADP was less than the fraction of A549 cells (see Fig. 1); however, both A549 and Jurkat cells died following infection (Fig. 2). As previously reported, both KE37 cells and BJAB cells remained viable and continued to proliferate during the course of the 10-day experiment. In these studies, expression of ADP appeared to be inversely correlated with cell survival and the ability to establish a persistent infection.
FIG 2.

Lymphocytic cells that support a persistent infection remain viable after infection with HAdV-C5. A549 cells were infected with the wild-type virus dl309 at a multiplicity of infection of 20 IU per cell. Jurkat, KE37, and BJAB cells were infected at a multiplicity of infection of 100 IU per cell. Cell viability was measured over the course of 10 days by trypan blue dye exclusion. The dotted line indicates the lower limit of detection. Error bars represent the SD of results from replicates across several different experiments.
We measured levels of the predominant ADP mRNA by quantitative real-time PCR after reverse transcription in order to determine if the amount of ADP measured by antibody staining correlated with the level of ADP mRNA. Total RNA was isolated from infected A549, Jurkat, KE37, and BJAB cells at various times postinfection. ADP mRNA levels were quantified with the use of primers placed in the coding region of the ADP gene and normalized with respect to an invariant, abundant gene, HPRT1. These values were further normalized to the minimal level of ADP mRNA in the lymphocytic cells, which was measured in KE37 cells 5 dpi. Results are expressed as relative changes across the infected cell types in Fig. 3. ADP mRNA was not detected in mock-infected cells, and the PCR primers did not detect signal in samples lacking reverse transcriptase (not shown). The highest levels of ADP mRNA were observed in the lytically infected A549 and Jurkat cells. In replicate experiments, the levels of ADP mRNA in A549 and Jurkat cells were comparable between 1 and 3 dpi. The substantial increase in ADP mRNA levels between 6 and 18 hpi in the A549 cells is consistent with the expression of ADP as a late gene from the major late promoter. Jurkat cells, which contained the largest fraction of ADP-positive cells among the lymphocytic cell lines, also expressed the largest amount of ADP mRNA among the lymphocytic cell lines at similar times (Fig. 3). Jurkat cells were not analyzed at 7 dpi because of the considerable cell death at this time. Although levels of ADP protein appeared low in both KE37 and BJAB cells, ADP mRNA was not absent from either of these cells. Because the levels of ADP mRNA appeared similar between A549 and Jurkat cells at late times of infection but the fraction of cells with detectable ADP was lower in Jurkat cells, these results appear to indicate that the expression of ADP in lymphocytes is not strictly controlled by the level of mRNA.
FIG 3.
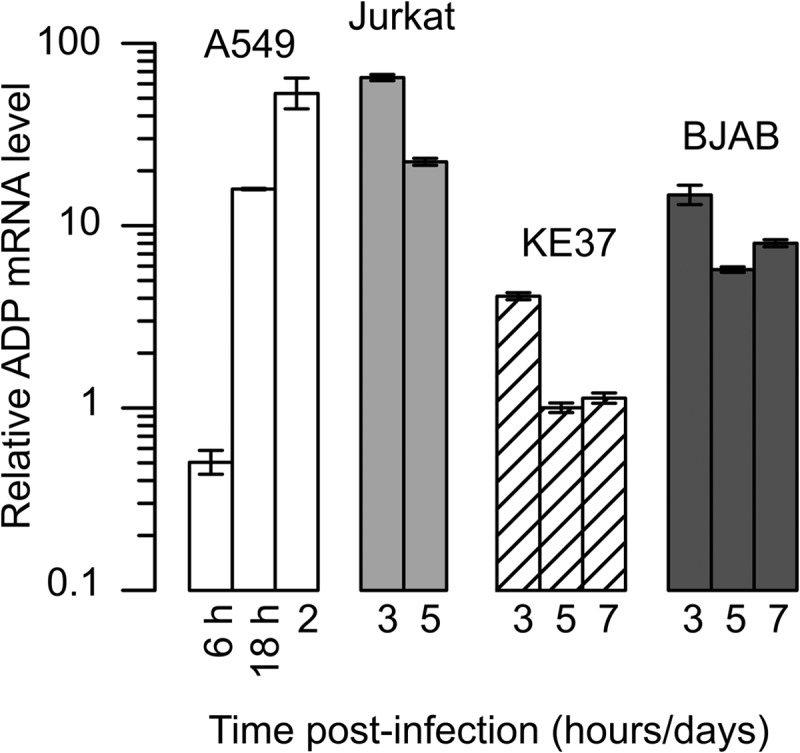
Lymphocytic cells that support a persistent infection contain lower levels of ADP mRNA than cells that support a lytic infection. A549 cells were infected with the wild-type virus dl309 at a multiplicity of infection of 20 IU per cell. Jurkat, KE37, and BJAB cells were infected at a multiplicity of infection of 50 IU per cell using the spin infection method (Materials and Methods). Total cellular RNA was isolated from 3 × 106 cells at the indicated times after infection, and the levels of the viral ADP mRNA and cellular HPRT mRNA were quantified by real-time PCR after reverse transcription. The results were normalized within each preparation to the abundant HPRT transcript as an endogenous cellular gene whose expression was unchanged following infection. The results were further normalized to the level of ADP mRNA measured in KE37 cells at 5 dpi, which was set to 1. Error bars represent the SD of results from technical replicates of a representative experiment.
Rates of cell killing in A549 cells correspond to different levels of ADP expression.
To examine the impact of ADP expression on the survival of infected lymphocytes more closely, mutant adenoviruses that either lack ADP (pm534, identified here as ADP-) (17) or overexpress ADP (VRX-021, identified here as ADP++) (18) were analyzed. Initially, A549 cells were mock infected or infected with the indicated viruses and harvested and levels of hexon and ADP determined (Fig. 4A). Greater than 80% of the infected A549 cells contained detectable levels of hexon protein following infection with each virus. As expected, no ADP protein (<5%) could be detected in mock-infected cells or cells infected with the ADP deletion mutant virus (Fig. 4A, Mock and ADP-). ADP-positive cells were readily detected among A549 cells infected with the ADP-WT virus (Fig. 4A, wild-type) or ADP-overexpressing virus (Fig. 4A, ADP++). On a per cell basis, the mean fluorescent intensity (MFI) indicated that approximately 3-fold more ADP was present in cells infected with the ADP-overexpressing virus (MFI of 379) than in cells infected with ADP-WT (MFI of 113). The viruses VRX-006 and VRX-007, which are closely related to VRX-021, directed the synthesis of an excess of ADP similar to that seen with the wild-type virus as measured by immunoblotting in A549 cells analyzed at 24 and 36 hpi (17).
FIG 4.

Adenovirus variants direct the accumulation of ADP to differing levels and kill A549 cells at different rates. A549 epithelial cells were mock infected or infected with HAdV-C5 variants that differ in their ability to express ADP. The virus pm534 contains two nonsense mutations that prevent ADP expression (ADP-). VRX-021 overexpresses the ADP gene (ADP++). The phenotypically wild-type virus dl309 expresses normal levels of ADP. (A) Cells were infected with the indicated viruses at a multiplicity of infection of 20 IU per cell, harvested after 48 h, fixed, and processed for intracellular staining of the hexon and ADP proteins. The stained cells were analyzed by flow cytometry where the indicated gates identifying positively stained cells were compared to those identifying mock-infected cells. The y axis denotes cell side scatter. MFI = mean fluorescence intensity. (B) A549 cells infected with the indicated viruses at a multiplicity of infection of 20 IU per cell were evaluated for viability by trypan blue dye exclusion over the course of 10 days following infection with the indicated viruses. Results from a representative experiment are shown.
The viability of A549 cells infected with the different ADP-expressing viruses was evaluated over time (Fig. 4B). Mock-infected cells remained viable through the experiment, with decreased viability due to overcrowding noted at 10 dpi. In contrast, the infected cells ceased proliferating shortly after infection (data not shown). As expected, cells infected with the ADP-deleted virus remained viable for the longest time, with cell death observed only after 5 dpi. The wild-type virus killed A549 cells at the expected rate, with approximately 20% dead cells at 5 dpi. In contrast, the ADP-overexpressing virus killed A549 cells much sooner and at a greater rate. By 7 dpi, virtually no viable cells were detected following infection with the ADP-overexpressing virus. Thus, in the epithelial A549 cell line, the level of ADP expression correlated with the rate of cell killing.
The survival of lymphocytes able to sustain a persistent infection is not affected by infection with an ADP-overexpressing virus.
Lymphocytic cells known to establish persistent infections were infected with the dl309 (wild-type) or VRX-021 (ADP++) virus, and cell viability was determined on 1, 3, 7, and 10 dpi (Fig. 5A). The genotype of the infecting virus had no impact on the viability of the infected KE37 or BJAB cells. In replicate experiments, both KE37 and BJAB cells remained viable (85% to 95%) over the course of the infection. Additionally, the viability of these cells infected with the ADP-deletion virus pm534 did not increase (data not shown). The failure to detect a change in cell viability after infection of KE37 or BJAB cells with the ADP-overexpressing virus may reflect a limited increase in ADP protein expression directed by the ADP++ virus in these cells. In this regard, the percentage of ADP++ virus-infected BJAB and KE37 cells expressing ADP remained low (Fig. 5B) and this remained true even when hexon expression could be detected in over 90% of the infected cells (Fig. 5B; BJAB). To increase the efficiency of infection of KE37 cells, we performed additional experiments using the spin infection method (Materials and Methods). KE37 and BJAB cells infected by this method were analyzed at 3 and 5 dpi, respectively. Hexon expression was detected in over 70% of infected KE37 cells and over 90% of infected BJAB cells (Fig. 5C and D, respectively). Nonetheless, only a small fraction of cells infected with wild-type virus contained detectable levels of ADP (<5%). Under these infection conditions, the fraction of ADP-positive cells infected with the ADP++ virus increased over that seen in wild-type virus-infected cells; however, these values (14% of KE37 cells and 10% of BJAB cells) remained much lower than the fraction of hexon-positive cells at that time postinfection. It should be noted that the gating strategy used for ADP expression in these experiments was based on ADP- virus-infected cells remaining below 5% positive for ADP. In contrast, the ADP expression depicted in Fig. 1 (which did not include the ADP- virus) was based on mock-infected cells remaining below 5% and resulted in more-inclusive gating for what was considered the ADP-positive population of cells. Curiously, ADP expression directed by the ADP++ virus did not increase cell death in either KE37 or BJAB cells as it did in the ADP++ virus-infected A549 cells (Fig. 4). These results suggest the possibility that the expression of ADP and its ability to promote cell death proceed by mechanisms in A549 cells different from those in lymphocytic cells.
FIG 5.

Cell death and ADP protein levels remain low in lymphocytic cells infected with the ADP-overexpressing adenovirus. KE37 and BJAB cells were infected with the wild-type virus or ADP-overexpressing virus VRX-021 (ADP++). (A) Cell viability was evaluated by trypan blue dye exclusion at the indicated time postinfection with 50 IU of virus per cell. Error bars represent the SD of results from replicates performed on at least three different days. (B) On the indicated days postinfection, KE37 and BJAB cells were harvested, fixed, and processed for intracellular staining of the hexon and ADP proteins as described in Materials and Methods. The dotted line indicates the limited of detection, set by cells infected with the ADP- virus. (C and D) Representative flow cytometry dot plots from a separate experiment are shown for infected KE37 cells (C) and BJAB cells (D) on the indicated day after spin infection with each virus. Cells stained with an isotype control antibody were used as negative controls for hexon staining. The gate for ADP expression was based on that of cells infected with the ADP- virus remaining below 5%. The y axis denotes cell side scatter.
ADP transcript levels in lymphocytes infected with the ADP-overexpressing virus were quantified and compared to levels measured in wild-type virus-infected cells. As seen with the wild-type virus, A549 epithelial cells and Jurkat T cells that were infected with VRX-021 (ADP++) contained the greatest amount of ADP mRNA at late times postinfection (Fig. 6A). Also as seen before, KE37 and BJAB cells typically contained less ADP mRNA than Jurkat cells on any given day postinfection. Interestingly, all cells infected with the ADP-overexpressing virus contained between 20- and 100-fold more ADP mRNA than cells infected with the wild-type virus (Fig. 6B). This substantial increase in ADP mRNA was greater than the observed increase in levels of ADP protein at comparable times postinfection. This result suggests that the ADP++ virus is able to direct increased accumulation of ADP mRNA in both epithelial and lymphocytic cells but that the fraction of cells containing detectable levels of ADP protein remains low (<20%) in the lymphocytic cells that can be persistently infected.
FIG 6.

Levels of ADP mRNA are greater in cells infected with the ADP-overexpressing virus than in cells infected with the wild-type ADP-expressing virus. A549 cells were infected with the ADP-overexpressing virus VRX-021 at a multiplicity of infection of 20 IU per cell. Lymphocytic cells were infected with 50 IU per cell of VRX-021 using the spin infection method (Materials and Methods). The levels of mRNA for HPRT and ADP were quantified as described in the legend for Fig. 3. (A) Total RNA was isolated from equal numbers of cells at the indicated day or hour postinfection. HPRT and ADP mRNA levels were quantified and normalized as described in the legend to Fig. 3. The relative amounts of ADP mRNA are shown in reference to the relative amount observed in KE37 cells 5 dpi with the wild-type virus. Error bars represent the SD of results from technical replicates of a representative experiment. (B) The level of ADP mRNA in VRX-021-infected cells was expressed as a fold increase (log10 scale) over the amount of ADP mRNA measured in wild-type virus-infected cells on the same day. The geometric mean of the fold change is shown in parentheses. In this panel, A549 cells were analyzed at 18 hpi and 2 dpi, Jurkat cells at 3 and 5 dpi, and KE37 and BJAB cells on 3, 5, and 7 dpi; the earliest times are represented by the upper bar.
Deletion of ADP converts a lytic infection to a persistent infection phenotype in lymphocytes.
Jurkat cells were infected with viruses that express ADP to differing levels, and cell viability was measured 1, 3, 7, and 10 dpi. Jurkat cells infected with the wild-type or ADP-overexpressing (ADP++) viruses were dead by 10 dpi. We saw no difference in the rates of cell death for cells infected with the wild-type and ADP-overexpressing virus in replicate experiments. In contrast, Jurkat cells infected with the ADP-deletion mutant pm534 (ADP-) remained viable (Fig. 7A). Immunostaining for hexon and ADP confirmed that the cells were productively infected by each virus (Fig. 7B). As before, the fraction of ADP-positive cells identified by flow cytometry was less than the hexon-positive fraction. However, these values were greater than the ADP-positive fraction measured in either BJAB or KE37 cells infected with the same viruses (see Fig. 5). Notably, ADP expression was detected in approximately 50% of Jurkat cells infected with either the wild-type virus or ADP++ virus (Fig. 7B). The Jurkat T-cell line infected with the ADP- virus contained a remarkably high percentage of viable cells (>70%) at 10 dpi. Additionally, these infected cells continued to proliferate in culture for at least 45 days (data not shown). Indeed, after 33 days, high levels of viral DNA remained in these cells (4.2 × 108 viral genomes per 107 cells), suggesting that the virus did indeed persist in these cells. These results suggest that the ability of adenovirus to establish a lytic infection in the Jurkat cell line requires ADP expression. Together with the low level of ADP expression observed in the persistently infected lymphocytic cells, these data suggest that downregulation of ADP expression can convert a lytic infection in lymphocytes to a persistent infection.
FIG 7.

Jurkat cells remain viable after infection with the ADP-deletion virus. Jurkat cells were infected with the wild-type virus dl309 (wild-type), ADP-mutant virus pm534 (ADP-), or the ADP-overexpressing virus VRX-012 (VRX++). (A) Cell viability was determined by trypan blue dye exclusion at the indicated days postinfection. Error bars represent the SD of results from replicates of at least three experiments. (B) The expression of hexon and ADP was determined by flow cytometry as described previously. The dotted lines indicate the limits of detection, set by cells infected with the ADP- virus.
DISCUSSION
Species C adenoviruses typically proceed with a lytic infection in human cells of epithelial or fibroblast origin. The lysis and death of lytically infected cells are accelerated by the adenovirus death protein (ADP) (2, 20). However, species C adenoviruses are also able to infect some lymphocytic and macrophage cell lines without proceeding through a lytic infection (11, 13, 21, 22). We recently found that adenovirus types 2 and 5 are able to persist in several lymphocytic cell lines for over a year in culture (8). Moreover, species C adenoviruses appear to persist in T cells recovered from the adenoids and tonsils removed from children (6). The mechanism that allows adenovirus to persist in these cells remains unknown. This study explored the role of ADP in determining whether infected lymphocytes enter the lytic or persistent pathway. We found that reduced expression of ADP permitted the establishment of a persistent infection.
KE37 and BJAB cells are lymphocytic cells that support a persistent adenoviral infection. During a persistent infection, these cells maintain the viral genome and divide at the same rate as noninfected cells (8). We reasoned that expression of the death-promoting ADP must be limited in order for the persistently infected cells to survive. Accordingly, ADP was detected in a smaller fraction of KE37 and BJAB cells than in the Jurkat T-cell line and A549 epithelial cell line, both of which support a lytic infection. The frequency of ADP-positive cells (<20%) was much lower than the frequency of hexon-positive cells (80%) in the cell lines that support a persistent infection. The difference in ADP-positive and hexon-positive cells was less substantial for Jurkat cells (50% ADP-positive cells versus 90% hexon-positive cells) and nonexistent for A549 cells, both of which support a lytic infection (Fig. 1). Because BJAB and KE37 cells contained less ADP mRNA than Jurkat or A549 cells (Fig. 3), the absence of ADP detected by intracellular staining and flow cytometry may have been due in part to the low level of ADP mRNA. However, even though the levels of ADP mRNA in Jurkat and A549 cells were similar, fewer ADP-positive Jurkat cells were detected than hexon-positive cells. Consequently, other mechanisms must account for the apparent absence of ADP-positive lymphocytic cells. For example, the steady-state level of ADP protein can be regulated by posttranscriptional means. ADP is N- and O-glycosylated (23) and palmitoylated on the C terminus (24). ADP variants with mutations in the lumenal domain were found to be unstable or no longer able to accelerate cell lysis (25). It is possible, therefore, that lymphocyte cell-specific modifications that perturb the localization or stability of ADP impair the function of the protein. Alternatively, a cell type-specific modification or cellular factor could mask the ADP epitope recognized by the monoclonal antibody used in this study. It should be noted, however, that this epitope is not present in the ADP of Ad2 and Ad6. The apparent lack of selective pressure to conserve this sequence implies that it is unlikely to participate in the binding of a key regulatory protein. Nonetheless, it is conceivable that either of these mechanisms could impair the function of the ADP protein in the lymphocytic cell.
We tested directly the hypothesis that ADP levels are relevant to the establishment or maintenance of a persistent infection using viruses that either overexpressed or failed to express ADP. In accordance with published studies (17, 26), the rate of epithelial cell killing correlated directly with the level of ADP. In A549 cells, increased cell death was associated with a 2- to 3-fold increase in ADP protein levels as determined by antibody staining (Fig. 4). Although we measured increases in levels of both mRNA (Fig. 6) and protein in KE37 and BJAB cells infected with the ADP++ virus, no increase in cell death was detected (Fig. 5A). This suggests either that the levels of ADP were not sufficient to induce cell death or that the death-promoting activity of ADP is inhibited in these cells.
While the increased level of ADP expression achieved here failed to convert a persistent infection of some lymphocyte cells into a lytic infection, the absence of ADP expression from the infecting virus converted Jurkat cells from lytic to persistent infection. Jurkat cells infected with the wild-type virus died within 7 to 10 days, while Jurkat cells infected with the ADP-null virus survived in culture for more than 40 days. Greater than 80% of these cells expressed hexon shortly after infection. Additionally, these persistently infected Jurkat cells maintained a high level of viral DNA (more than 4 × 108 genomes per 107 total cells) for over 1 month after the initial infection. These results indicate that downregulation of ADP could allow the virus to persist in some cells that typically exhibit a lytic infection phenotype. Because A549 cells did not become persistently infected with the ADP-deleted virus, it seems likely that additional factors contribute to the switch between a lytic infection and a persistent infection. It will be of interest to determine if this molecular decision can occur in cells other than those of lymphocytic origin. Additional experiments should determine if latently infected cells can be reactivated without ADP expression. By this means, infectious virus could be formed in a cell with the potential to persist and traffic and spread to distal locations.
In epithelial cells, ADP facilitates the efficient release of progeny virions without affecting the final yield of progeny virus (2, 20). Because the impact of ADP on virus yield from infected lymphocytes has not been described, we compared the yields of virus among Jurkat and KE37 lymphocytic cells as well as A549 epithelial cells that were infected with the three ADP variant viruses studied here. Although the yields of virus differed between the cell types, we found that ADP expression had no impact on the total virus yield from a given cell type (data not shown). Our experiments are consistent with the suggestion that this property of ADP occurs similarly in lymphocytes and epithelial cells and that ADP expression does not impact the total yield of progeny virus. Experiments measuring extracellular virus are under way to determine if ADP impacts the virus released from infected lymphocytes in a manner similar to that seen with epithelial cells. It would be of interest to determine the nature of any signals that can overcome the restraint imposed on ADP expression, as these signals would trigger the release of infectious virus and may play an important role in the reactivation and spread of the virus.
Viral infection can lead to either the lytic death of the cell or the long-term survival of the persistently or latently infected cell. Viruses that establish latent or persistent infections typically express products that suppress programmed cell death. The alphaherpesviruses can be cytolytic in epithelial cells while being noncytolytic in sensory neurons in which a latent infection is established (27). The latency-associated transcript (LAT) of herpes simplex virus promotes survival of the infected neurons by many potential mechanisms, including the suppression of apoptosis (28, 29) and contribution of regulatory small RNA molecules that restrict viral gene expression (30, 31). Although varicella-zoster virus does not encode a homolog to the LAT, this virus expresses the ORF63 protein that provides an antiapoptotic function in neuronal cells (32). The adenovirus E1B-19K protein is a potent anti-apoptotic Bcl-2 homolog (33) that can suppress cell death (34). However, in the studies reported here, the presence of the E1B-19K protein did not prevent death promoted by ADP.
Our studies suggest that downregulated expression of the ADP may contribute to the switch from a lytic to a persistent adenovirus infection. This may be the first report describing the regulated expression of a death-promoting viral gene as a mechanism of establishing persistence. Nonetheless, other viruses that establish long-term infections could proceed by a similar mechanism. For example, hepatitis C (HCV) virus establishes a chronic infection in the liver. Viral persistence is due in part to the ability of this virus to defeat host immune mechanisms (35). However, the p7 protein of HCV is a death-promoting viral product (36) that induces apoptosis in the Huh7.5 liver cell line. Lymphoid cells, which have been suggested to be a reservoir of HCV (37), can support a persistent HCV infection (38). Intriguingly, peripheral memory B cells were reported to resist HCV-induced apoptosis (39). Perhaps downregulation of the p7 protein in cells of lymphoid origin contributes to the persistence of HCV in these cells. Parvovirus B19 is another example of a virus whose pathogenic nature stems from its ability to replicate in and destroy erythroid precursor cells (40). The 11-kDa nonstructural protein of B19 was shown to contribute to apoptosis in these cells (41). In contrast, the frequent detection of B19 DNA in the skin (42) and other tissue samples from solid organs (43) of healthy individuals suggests that the virus may be able to persist in other cells or tissue. It remains to be determined if cell type-specific differences in the patterns of B19 gene expression contribute to the persistence of this virus in certain tissues.
Although we were able to convert a lytic viral infection of Jurkat cells into a persistent infection by eliminating ADP expression, we were unable to convert a persistent infection of KE37 cells into a lytic infection by overexpressing ADP. Because lymphocytes infected with the ADP-overexpressing virus accumulated significantly more ADP mRNA but showed only a small increase in the fraction of infected cells with detectable ADP, we view these results as inconclusive. It will be necessary to impose higher levels of ADP expression in order to determine if persistently infected lymphocytes can be converted to a lytic infection by this means. This may prove to be an experimental challenge because ADP expression may be regulated differently in epithelial cells and lymphocytes. The mechanisms underlying this differential regulation remain unknown and are under investigation. Altered expression of viral genes between lymphocytes and epithelial cells has been reported previously (9). Although delayed expression of both early and late genes was seen among infected lymphocytes by McNees et al., the selective reduction of a single viral gene over other viral genes was not observed, making the regulation of ADP expression in lymphocytes unique among the adenoviral genes thus evaluated. It was noted that certain adenoviral gene promoters are sensitive to T-cell activation signals (44). It will be interesting to see if these activation signals also affect ADP expression at the level of transcription or at a posttranscriptional step during reactivation of persistently infected cells. Given the unique interaction of adenoviruses with lymphocytes, it is important to fully understand the dynamics of adenoviral infections in lymphocytes.
ACKNOWLEDGMENTS
This research was supported by an internal Research Initiation Grant from Georgia State University and R01 CA127621 from the National Cancer Institute.
C. Garnett-Benson previously published under the name C. T. Garnett.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Wold WSM, Ison MG. 2013. Chapter 56. Adenoviruses, p 1732–1767 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. 1996. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology 220:152–162. 10.1006/viro.1996.0295 [DOI] [PubMed] [Google Scholar]
- 3.Goodrum FD, Ornelles DA. 1998. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J. Virol. 72:9479–9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox JP, Brandt CD, Wassermann FE, Hall CE, Spigland I, Kogon A, Elveback LR. 1969. The virus watch program: a continuing surveillance of viral infections in metropolitan New York families. VI. Observations of adenovirus infections: virus excretion patterns, antibody response, efficiency of surveillance, patterns of infections, and relation to illness. Am. J. Epidemiol. 89:25–50 [DOI] [PubMed] [Google Scholar]
- 5.Fox JP, Hall CE, Cooney MK. 1977. The Seattle Virus Watch. VII. Observations of adenovirus infections. Am. J. Epidemiol. 105:362–386 [DOI] [PubMed] [Google Scholar]
- 6.Garnett CT, Erdman D, Xu W, Gooding LR. 2002. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J. Virol. 76:10608–10616. 10.1128/JVI.76.21.10608-10616.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. 2009. Latent species C adenoviruses in human tonsil tissues. J. Virol. 83:2417–2428. 10.1128/JVI.02392-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Huang W, Ornelles DA, Gooding LR. 2010. Modeling adenovirus latency in human lymphocyte cell lines. J. Virol. 84:8799–8810. 10.1128/JVI.00562-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNees AL, Mahr JA, Ornelles D, Gooding LR. 2004. Postinternalization inhibition of adenovirus gene expression and infectious virus production in human T-cell lines. J. Virol. 78:6955–6966. 10.1128/JVI.78.13.6955-6966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colin M, Renaut L, Mailly L, D'Halluin JC. 2004. Factors involved in the sensitivity of different hematopoietic cell lines to infection by subgroup C adenovirus: implication for gene therapy of human lymphocytic malignancies. Virology 320:23–39. 10.1016/j.virol.2003.09.043 [DOI] [PubMed] [Google Scholar]
- 11.Lavery D, Fu SM, Lufkin T, Chen-Kiang S. 1987. Productive infection of cultured human lymphoid cells by adenovirus. J. Virol. 61:1466–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meeker TC, Lay LT, Wroblewski JM, Turturro F, Li Z, Seth P. 1997. Adenoviral vectors efficiently target cell lines derived from selected lymphocytic malignancies, including anaplastic large cell lymphoma and Hodgkin's disease. Clin. Cancer Res. 3:357–364 [PubMed] [Google Scholar]
- 13.Silver L, Anderson CW. 1988. Interaction of human adenovirus serotype 2 with human lymphoid cells. Virology 165:377–387. 10.1016/0042-6822(88)90582-X [DOI] [PubMed] [Google Scholar]
- 14.Ginsberg HS, Lundholm-Beauchamp U, Horswood RL, Pernis B, Wold WS, Chanock RM, Prince GA. 1989. Role of early region 3 (E3) in pathogenesis of adenovirus disease. Proc. Natl. Acad. Sci. U. S. A. 86:3823–3827. 10.1073/pnas.86.10.3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nevins JR. 1987. Regulation of early adenovirus gene expression. Microbiol. Rev. 51:419–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones N, Shenk T. 1979. Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells. Cell 17:683–689. 10.1016/0092-8674(79)90275-7 [DOI] [PubMed] [Google Scholar]
- 17.Doronin K, Toth K, Kuppuswamy M, Krajcsi P, Tollefson AE, Wold WS. 2003. Overexpression of the ADP (E3-11.6K) protein increases cell lysis and spread of adenovirus. Virology 305:378–387. 10.1006/viro.2002.1772 [DOI] [PubMed] [Google Scholar]
- 18.Tollefson AE, Ying B, Doronin K, Sidor PD, Wold WS. 2007. Identification of a new human adenovirus protein encoded by a novel late l-strand transcription unit. J. Virol. 81:12918–12926. 10.1128/JVI.01531-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 20.Tollefson AE, Scaria A, Hermiston TW, Ryerse JS, Wold LJ, Wold WS. 1996. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 70:2296–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McManus TE, Marley AM, Baxter N, Christie SN, Elborn JS, Heaney LG, Coyle PV, Kidney JC. 2007. Acute and latent adenovirus in COPD. Respir. Med. 101:2084–2090. 10.1016/j.rmed.2007.05.015 [DOI] [PubMed] [Google Scholar]
- 22.Chu Y, Sperber K, Mayer L, Hsu MT. 1992. Persistent infection of human adenovirus type 5 in human monocyte cell lines. Virology 188:793–800. 10.1016/0042-6822(92)90534-V [DOI] [PubMed] [Google Scholar]
- 23.Scaria A, Tollefson AE, Saha SK, Wold WS. 1992. The E3-11.6K protein of adenovirus is an Asn-glycosylated integral membrane protein that localizes to the nuclear membrane. Virology 191:743–753. 10.1016/0042-6822(92)90250-S [DOI] [PubMed] [Google Scholar]
- 24.Hausmann J, Ortmann D, Witt E, Veit M, Seidel W. 1998. Adenovirus death protein, a transmembrane protein encoded in the E3 region, is palmitoylated at the cytoplasmic tail. Virology 244:343–351. 10.1006/viro.1998.9135 [DOI] [PubMed] [Google Scholar]
- 25.Tollefson AE, Scaria A, Ying B, Wold WS. 2003. Mutations within the ADP (E3-11.6K) protein alter processing and localization of ADP and the kinetics of cell lysis of adenovirus-infected cells. J. Virol. 77:7764–7778. 10.1128/JVI.77.14.7764-7778.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou A, Atencio I, Huang WM, Horn M, Ramachandra M. 2004. Overexpression of adenovirus E3-11.6K protein induces cell killing by both caspase-dependent and caspase-independent mechanisms. Virology 326:240–249. 10.1016/j.virol.2004.06.007 [DOI] [PubMed] [Google Scholar]
- 27.Nicoll MP, Proenca JT, Efstathiou S. 2012. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 36:684–705. 10.1111/j.1574-6976.2011.00320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C. 2004. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J. Neurovirol. 10:260–265. 10.1080/13550280490468690 [DOI] [PubMed] [Google Scholar]
- 29.Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. 2006. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 442:82–85. 10.1038/nature04836 [DOI] [PubMed] [Google Scholar]
- 30.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. U. S. A. 102:16055–16059. 10.1073/pnas.0505850102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roizman B, Zhou G, Du T. 2011. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J. Neurovirol. 17:512–517. 10.1007/s13365-011-0058-x [DOI] [PubMed] [Google Scholar]
- 32.Hood C, Cunningham AL, Slobedman B, Arvin AM, Sommer MH, Kinchington PR, Abendroth A. 2006. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 80:1025–1031. 10.1128/JVI.80.2.1025-1031.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiou SK, Tseng CC, Rao L, White E. 1994. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 68:6553–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White E, Sabbatini P, Debbas M, Wold WS, Kusher DI, Gooding LR. 1992. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol. Cell. Biol. 12:2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thimme R, Binder M, Bartenschlager R. 2012. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 36:663–683. 10.1111/j.1574-6976.2011.00319.x [DOI] [PubMed] [Google Scholar]
- 36.Aweya JJ, Mak TM, Lim SG, Tan YJ. 2013. The p7 protein of the hepatitis C virus induces cell death differently from the influenza A virus viroporin M2. Virus Res. 172:24–34. 10.1016/j.virusres.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo Y, Ueno Y, Shimosegawa T. 2012. Biological significance of HCV in various kinds of lymphoid cells. Int. J. Microbiol. 2012:647581 10.1155/2012/647581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serafino A, Valli MB, Alessandrini A, Ponzetto A, Carloni G, Bertolini L. 1997. Ultrastructural observations of viral particles within hepatitis C virus-infected human B lymphoblastoid cell line. Res. Virol. 148:153–159. 10.1016/S0923-2516(97)89902-5 [DOI] [PubMed] [Google Scholar]
- 39.Mizuochi T, Ito M, Takai K, Yamaguchi K. 2011. Peripheral blood memory B cells are resistant to apoptosis in chronic hepatitis C patients. Virus Res. 155:349–351. 10.1016/j.virusres.2010.09.017 [DOI] [PubMed] [Google Scholar]
- 40.Broliden K, Tolfvenstam T, Norbeck O. 2006. Clinical aspects of parvovirus B19 infection. J. Intern. Med. 260:285–304. 10.1111/j.1365-2796.2006.01697.x [DOI] [PubMed] [Google Scholar]
- 41.Chen AY, Zhang EY, Guan W, Cheng F, Kleiboeker S, Yankee TM, Qiu J. 2010. The small 11 kDa nonstructural protein of human parvovirus B19 plays a key role in inducing apoptosis during B19 virus infection of primary erythroid progenitor cells. Blood 115:1070–1080. 10.1182/blood-2009-04-215756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonvicini F, La Placa M, Manaresi E, Gallinella G, Gentilomi GA, Zerbini M, Musiani M. 2010. Parvovirus b19 DNA is commonly harboured in human skin. Dermatology 220:138–142. 10.1159/000277431 [DOI] [PubMed] [Google Scholar]
- 43.Corcioli F, Zakrzewska K, Rinieri A, Fanci R, Innocenti M, Civinini R, De Giorgi V, Di Lollo S, Azzi A. 2008. Tissue persistence of parvovirus B19 genotypes in asymptomatic persons. J. Med. Virol. 80:2005–2011. 10.1002/jmv.21289 [DOI] [PubMed] [Google Scholar]
- 44.Mahr JA, Boss JM, Gooding LR. 2003. The adenovirus e3 promoter is sensitive to activation signals in human T cells. J. Virol. 77:1112–1119. 10.1128/JVI.77.2.1112-1119.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]