

Abstract
Progressive multifocal leukoencephalopathy (PML) is a fatal disease with limited treatment options, both clinically and in the research pipeline. Potential therapies would target and neutralize its etiologic agent, JC polyomavirus (JCPyV). The innate immune response to JCPyV infection has not been studied, and little is known about the initial host response to polyomavirus infection. This study examined the ability of a human alpha defensin, HD5, to neutralize JCPyV infection in human fetal glial cells. We show that HD5, by binding to the virion, blocks infection. The JCPyV-HD5 complexes bind to and enter host cells but are reduced in their ability to reach the endoplasmic reticulum (ER), where virions are normally uncoated. Furthermore, HD5 binding to the virion stabilizes the capsid and prevents genome release. Our results show that HD5 neutralizes JCPyV infection at an early postentry step in the viral life cycle by stabilizing the viral capsid and disrupting JCPyV trafficking. This study provides a naturally occurring platform for developing antivirals to treat PML and also expands on the known capabilities of human defensins.
INTRODUCTION
JC polyomavirus (JCPyV) is an important human pathogen that chronically infects the majority of the population (1, 2). Most individuals are infected early in life, resulting in a life-long persistent infection. The infection remains subclinical in immunocompetent individuals, with the host cell immune system controlling infection by incompletely understood mechanisms. Upon onset of immunosuppression, JCPyV replication increases, resulting in dissemination of virus to the central nervous system. There, increased replication of JCPyV in glial cells and cytolytic destruction of the myelin-producing oligodendritic cells leads to the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML) (3, 4). Although it is rare (5% of HIV patients), PML has no cure and no treatments other than reconstitution of the patient's immune system, and it is increasing in incidence (5). PML is also apparent in an increasing number of patients treated with immunomodulatory drugs, including the leukocyte trafficking inhibitor natalizumab (6–10). The recent discovery of 7 new human polyomaviruses, including Merkel cell polyomavirus and trichodysplasia polyomavirus, highlights the need to elucidate pathogenic mechanisms and identify promising treatment targets for this virus family.
JCPyV initially attaches to target cells via a receptor complex containing a sialic acid-containing carbohydrate, lactoseries tetrasaccharide c (LSTc), and the 5HT2a serotonin receptor (11, 12). The virus is internalized via clathrin-mediated endocytosis and enters early endosomes marked by Rab5 (13, 14). Cells expressing dominant negative versions of Rab5 but not Rab7 (late endosome) or Rab11 (recycling endosome) are resistant to infection, suggesting that JCPyV may use redundant pathways to arrive at the endoplasmic reticulum (ER) (14). JCPyV starts to arrive at the ER within 4 to 6 h (15) and uses the same ER proteins as simian virus 40 (SV40) to retrotranslocate to the cytosol (16).
How the host immune system controls JCPyV is poorly understood. It is commonly believed that the major site of JCPyV persistence is the kidney, although other sites of persistent infection have been proposed, including the brain, B cells, bone marrow, tonsil, and salivary gland (17–20). JCPyV does not cause significant disease until the immune system is suppressed, indicating the host immune system plays an important role in controlling the persistent infection. Studies have shown CD4+ and CD8+ T cells as significant contributors to preventing the progression of PML in JCPyV-positive patients, but the innate immune response to JCPyV infection has not been well studied (21, 22).
Defensins are small (18- to 45-amino-acid) cationic peptides that intrinsically possess antimicrobial properties and are key mediators of the innate immune system (reviewed in references 23, 24, and 25). They have characteristic disulfide bonds, and the arrangement of these bonds defines these molecules as alpha or beta defensins. Human defensins are divided into alpha and beta classes and are found in epithelial cells, monocytes, intestinal Paneth cells, NK cells, neutrophils, and also gamma-delta T cells. Their inhibitory effects on viral infection appear to be cell and virus specific and act at different stages of the infection cycle (23–25). Alpha defensins have been shown to exhibit direct antiviral activity against enveloped virus such as herpes simplex (26), influenza, rhabdovirus, and HIV (27, 28). Similar to their antibacterial activity, defensins inhibit enveloped viral infection by damaging the envelope. They have been shown recently to neutralize nonenveloped viral infection as well. Adenovirus infection is neutralized by blocking viral uncoating and subsequent release of the genome (29–31). Alpha defensins have also been shown to block papillomavirus infection by sequestering the virions in their endocytic vesicles (32). The human alpha defensin HD5 inhibits replication of the JCPyV-related polyomavirus BK by blocking binding to host cells (33).
We show here that despite a high degree of sequence identity, JCPyV and BKPyV are neutralized by HD5 by distinct mechanisms. Instead of inhibiting binding to cells, HD5 inhibits JCPyV by stabilizing the viral capsid and inhibiting conformational changes that are critical for infectivity. This study adds to the antiviral capabilities of HD5, suggests multiple modes of neutralization within the same virus family, and highlights the need for continued research in this area to determine conserved mechanisms and motifs for further development as antivirals.
MATERIALS AND METHODS
Cells, virus, antibodies, and defensins.
SVG-A (34) (human fetal glial) cells were maintained in a 37°C CO2 chamber in minimal essential medium (MEM; Gibco) supplemented with 1% penicillin-streptomycin (Gibco) and 10% heat-inactivated fetal bovine serum (Atlanta Biologicals). The Mad1 SVEΔ strain of JCPyV has previously been described (35). Defensins were purchased from Peptides International and reconstituted in water at a concentration of 100 μM. Viral particles used in each experiment and the calculations used to determine these are as follows: for Fig. 2B, 2.2 × 1010 particles = a multiplicity of infection (MOI) of 10,000 particles (not saturating); for Fig. 2C, 2.2 × 108 particles = an MOI of 100 particles (not saturating); for Fig. 3A: 2.2 × 108 particles = an MOI of 100 particles (not saturating); for Fig. 4, an MOI of 5 FFU = 125,000 particles; for Fig. 5, an MOI of 20 = 500,000 particles. Calculations were as follows: 1.7 × 107 viral particles/hemagglutination unit (HAU); 2.2 × 1013 particles/mg; molecular weight of JCPyV = 2.7 × 106; 1.0 HAU = 2.8 × 10−17 M; 1 HAU of a good virus stock = 586 fluorescence-forming units (FFU).
FIG 2.

HD5 directly interacts with JCPyV but does not block binding to cells. (A) HD5 binds to JCPyV. Ninety-six-well plates were coated with saturating concentrations of JCPyV. A 10-μg/ml concentration of HD5 or HBD1 was then added to the plates, and interaction was determined by ELISA. Values are absorbance at 652 nm. JCPyV-488 was also used to rule out a loss of binding ability due to the labeling process. *, P < 0.05 compared to the no-HD5 value. A bar indicates a direct comparison. (B) Effect of HD5 on JCPyV binding to host cells: SVG-A cells were incubated with JCPyV-488–HD5 complexes for 1 h on ice. Cells were then fixed with 4% PFA and analyzed by flow cytometry. Values are the mean fluorescence intensity from 3 independent experiments, and error bars indicate the standard deviations of the mean fluorescence intensities. NS, not significant. A bar indicates a direct comparison. (C) Analysis of JCPyV binding by fluorescence. Mock- and HD5-treated JCPyV-488 was bound to SVG-A cells on ice (to prevent internalization). Images are maximum z-stack projections indicating binding to the plasma membrane. Images are representative. Scale bar = 20 μm. (D) HD5 binds to mutant pentamers. Ninety-six-well plates were coated as described for panel A with the indicated mutant JCPyV VP1 pentamers. HD5 was added, and interaction was determined by ELISA. Values are absorbance at 652 nm (endpoint reading), and error bars indicate the standard deviations from 3 independent experiments.
FIG 3.
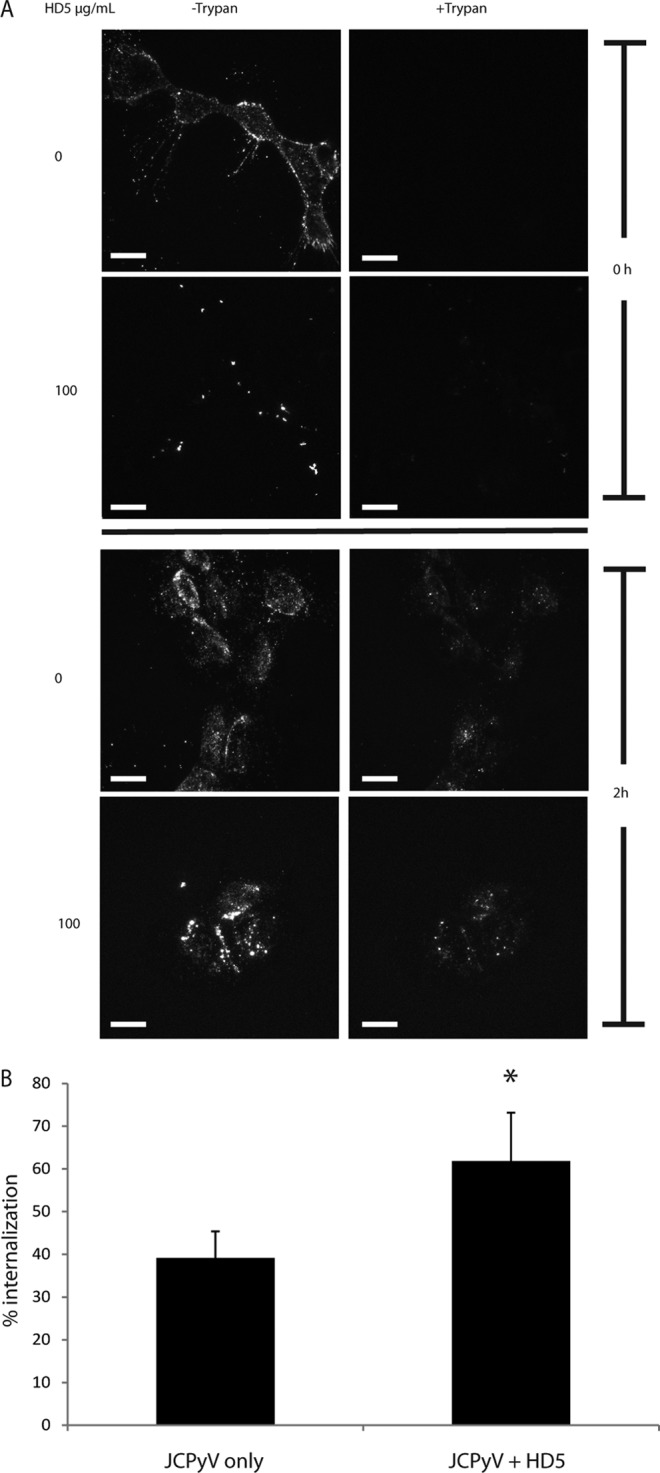
HD5 does not inhibit JCPyV internalization. (A) Analysis of internalization. Prechilled SVG-A cells were incubated with either JCPyV-488 or HD5-treated JCPyV-488 for 1 h on ice to prevent internalization. The top four panels were taken at 0 hpi and the bottom four at 2 hpi (allowing for internalization). A 1:20 dilution of trypan blue was added directly to the field, and the images are z-stack projections with and without trypan blue. Images are representative of 3 independent experiments. Scale bar = 20 μm. (B) Quantification of internalization. Total fluorescence was calculated per cell in the pre-trypan blue maximum-projection z-stack images. The same process was done for the post-trypan blue images. Images are the projection of the entire cell. Values are the average remaining fluorescence of 5 to 10 cells per condition per experiment (total, 15 to 30 cells per condition) 2 hpi divided by the total fluorescence before the addition of trypan blue. Error bars indicate standard deviations. *, P < 0.05.
FIG 4.

HD5 reduces JCPyV traffic to the ER. (A) Colocalization analysis. SVG-A cells were infected with JCPyV-633 at an MOI of ∼5 for 10 h. JCPyV was either mock treated with water, treated with 100 μg/ml HD5 or HBD1, or added to cells pretreated with 10 ng/ml of brefeldin A (used as an ER trafficking inhibitor control). At 10 hpi, cells were fixed and stained for PDI. Images are merged z-stack slices from a representative plane (a plane with a significant amount of virus). Inserts are enlarged sections of the image indicated by the white squares. JCPyV-633 is red, PDI is green, and colocalization is yellow. Images show representative cells, and 40 to 50 cells were analyzed per condition. Scale bar = 10 μm. (B) Quantification of colocalization between JCPyV-633 and ER proteins. Colocalization analysis was performed on the set of images described above using Mander's coefficient of colocalization with M1 indicating amount of virus channel overlapping PDI channel. Box plots indicate minimum, 1st-quartile, mean, 3rd-quartile, and maximum values. *, P < 0.0001 compared to JCPyV alone. A bar indicates a direct comparison.
FIG 5.

HD5 reduces exposure of VP2 at a time point consistent with ER arrival. (A) Imaging of VP2 exposure. Cells were infected with JCPyV that had been treated with 100 μg/ml HD5 at an MOI of 20 for 10 h. At 10 hpi, cells were fixed and stained for VP2 and nuclei. Nuclei are pseudocolored blue, and VP2 is green. VP2 exposure results in a punctate, perinuclear staining pattern, as seen in the top left image. A 100-μg/ml concentration of HBD1 and a 10-ng/ml concentration of brefeldin A (BFA) were used as controls as described in the text. Scale bar = 50 μm. (B) Quantification of VP2 exposure. A total of 2,500 cells were imaged per condition and counted using the counter in ImageJ. VP2-positive cells were counted in the same way and normalized to the total amount of cells per condition. Values are the percent positive VP2 cells, and error bars indicate standard deviations. *, P < 0.05 compared to JCPyV alone. (C) Visualization of virus, VP2, and PDI at once. Viruses and cells were treated as for panel A, and cells were infected at an MOI of 5. At 10 hpi, cells were fixed and stained for VP2. Additionally, after VP2 staining, cells were stained for PDI overnight. PDI is pseudocolored red, virus green, and VP2 blue. Areas of colocalization appear white and are indicated by yellow arrows. White boxes indicate areas enlarged in the panels immediately below. Scale bar = 20 μm.
Virus propagation and labeling.
JCPyV was propagated and purified as previously described (36). Briefly, 10 1,700-cm2 roller bottles were seeded with SVG-A cells and infected with JCPyV (MOI = 0.01 FFU per cell) for 14 days. Lysates were frozen and thawed three times. The lysate was then sonicated three times on ice using a Fisher 150E sonicator (1 min sonication, power setting of 3, 50% amplitude, duty cycle of 50%). Deoxycholate was added to a final concentration of 0.25% (vol/vol) and incubated for 30 min at 37°C. Lysates were then pelleted for 30 min at 4°C and 15,000 rpm, and the supernatant collected. This supernatant was overlaid onto a 20% sucrose column and centrifuged in an SW40 Ti rotor at 35,000 rpm for 3 h at 15°C (Beckman-Coulter). The resulting pellet was suspended in buffer A (10 mM Tris, 50 mM NaCl, 0.1 mM CaCl2, 0.01% Triton X-100, 0.05% sodium azide) and loaded on a cesium chloride (CsCl) step gradient (1.23, 1.29, 1.33, and 1.35 g/ml CsCl). This was centrifuged to equilibrium in an SW55 Ti rotor at 33,000 rpm for 18 h at 4°C. The band corresponding to virions was pulled and extensively dialyzed against buffer A. Concentrations were determined by A280 values. JCPyV was labeled with Alexa Fluor 488 or 633 according to the manufacturer's instructions (Invitrogen). Briefly, purified JCPyV was dialyzed extensively against 0.1 M carbonate buffer, pH 8.3, and then labeled with Alexa Fluor 488 or 633 at a molar ratio of 200:1 for 1 h at 21°C. Labeled JCPyV was then dialyzed against buffer A, aliquoted, and stored at −80°C until use. Infectivity measurements were performed on this labeled virus, and it was found that the labeling process slightly reduced the infectivity compared to that of unlabeled virus.
VP1 pentamer and mutant production.
VP1 pentamers were produced as previously described (12). Briefly, cDNA coding for amino acids 22 to 289 of the Mad-1 strain of JCPyV VP1 was cloned into the pET15b expression vector (Novagen). Mutations were introduced using Phusion mutagenesis (NEB) according to the manufacturer's instructions and confirmed by sequencing. Proteins were overexpressed in Escherichia coli and purified using nickel affinity chromatography.
Infectivity assays.
SVG-A cells were seeded overnight in 96-well plates so that each well would be 50% confluent. Labeled or unlabeled JCPyV at an MOI of 0.5 was preincubated with defensin (0.01 to 200 μg/ml) in 20-μl reaction mixtures in MEM containing 2% fetal bovine serum (FBS) for 1 h on ice. This mixture was then overlaid onto SVG-A cells and incubated at 37°C for 1 h, followed by addition of 100 μl per well of complete medium (MEM with 10% FBS). At 72 h postinfection (hpi), the cells were washed once with phosphate-buffered saline (PBS) and fixed with 100% ice-cold methanol for 30 min at −20°C. Infected nuclei were visualized by indirect immunofluorescence, using an antibody to VP1 (PAB597) and an Alexa Fluor 488-labeled secondary antibody (Invitrogen). Infected cells per well were scored and normalized to a virus-only positive control. The time course experiment was performed by adding 50 μg/ml HD5 to the cells at the indicated time points postinfection and assaying for infected cells 72 hpi. The pretreatment experiment was done by adding 50 μg/ml HD5 to the cell culture medium for 1 or 2 h before infection in 2% MEM in 20 μl. HD5 was not kept in the medium for the 72 h after initial infection in this experiment. Infected cells were scored as mentioned above.
Cell viability assay.
SVG-A cells were seeded as in the infectivity assays. Medium was aspirated and replaced with 100 μl complete medium containing 100 μg/ml of defensin. For the positive control, medium with no defensin was added, and for the negative control, medium was replaced with 100 μl PBS. At 72 hpi, 20 μl of CellTiter 96 AQOne solution (Promega) was added directly to the culture medium. This reagent measures the amount of soluble formazan, which is directly proportional to the number of viable cells. The plate was incubated in a humidified 37°C incubator for 2 h. Absorbance was then read at 490 nm.
ELISA.
Binding of HD5 or HBD1 to JC virions or VP1 pentamers was determined by enzyme-linked immunosorbent assay (ELISA). Saturating amounts (10 μg/ml) of JCPyV or pentamers (Alexa Fluor 488 labeled or unlabeled) were bound to 96-well plates in PBS supplemented with 0.1% sodium azide for 24 h at 4°C (Corning). After being washed three times with PBS for 5 min each, plates were blocked with PBS-Tween (PBST) plus 0.25% bovine serum albumin (BSA) for 1 h at room temperature and then washed three times again. A 10-μg/ml concentration of HD5 or HBD1 in 50 μl was added and incubated for 2 h at 4°C. HD5 was detected using a murine-generated monoclonal antibody to human HD5 (1:100; Santa Cruz) and a goat anti-mouse IgG secondary antibody conjugated to horseradish peroxidase (HRP; 1:10,000; Invitrogen). HBD1 was captured using a polyclonal antibody generated in rabbits (1:100; Santa Cruz) and a goat anti-rabbit IgG secondary antibody conjugated to HRP (1:10,000; Invitrogen). TMB Ultra solution was added, and absorbance was read kinetically every minute for 45 min at 652 nm. The time point shown in the results is after 15 min of TMB Ultra incubation.
Flow cytometry.
SVG-A cells were seeded in 6-well dishes at a density of 5 × 105 cells per well the night before the experiment, resulting in 1 × 106 cells on the day of the experiment. Cells were removed from the dish using Cellstripper (Cellgro) and washed twice with PBS. Meanwhile, 100 μg/ml HD5 was preincubated with ∼1 μg of Alexa Fluor 488-labeled JCPyV (JCPyV-488) on ice for 1 h in 100 μl PBS. The virus-HD5 complexes were then bound to cells for 2 h on ice. Cells were then washed and fixed with 1% paraformaldehyde (PFA), and mean fluorescence intensities determined by flow cytometry (FACSCanto II; Becton Dickinson). Experiments were done in triplicate and analyzed with FlowJo software (Treestar, Ashland, OR).
Live-cell microscopy.
Labeled JCPyV (5 to 10 ng) was premixed with HD5 (100 μg/ml) for 1 h on ice in 100 μl 2% MEM. SVG-A cells were plated into 60-mm optical dishes (World Precision Instruments, Inc.) the night before infection at a density of 200,000 cells/dish. These cells were prechilled at 4°C for 30 min and then overlaid with the JCPyV-HD5 complexes in a volume of 100 μl for 2 h. After unbound virus had been washed out with medium, to analyze time zero, cells were immediately brought to the Zeiss 710 laser scanning microscope for analysis. To analyze later time points, cells bound with complex were washed and then incubated at 37°C, and the Zeiss LSM 710 was prewarmed to 37°C on both the stage and objective collar. Cells were imaged at a ×63 magnification.
Internalization assay.
The internalization assay method was adapted from reference 37. Briefly, cells were treated with labeled JCPyV complexes as in live cell microscopy. z-stack images were taken and then a 1:20 dilution of trypan blue was added directly to the imaged area (trypan blue addition results in a quenching of fluorescence at 488 nm). A z-stack image was taken after the quenching, and total fluorescence was computed and compared between “before” and “after” images. Cell outlines were drawn using the bright-field images (not shown), and the sum of the intensities inside the cell using the maximum projection pre-z stack was divided by the sum of the intensities of the maximum-projection post-z stack and converted to percentages.
Endoplasmic reticulum colocalization.
Labeled JCPyV (JCPyV-633) was premixed with 100 μg/ml of either HD5 or HBD1 for 1 h on ice in a 20-μl reaction mixture in 2% MEM. SVG-A cells seeded in a 96-well glass-bottom plate were then infected at an MOI of ∼5. A subset of cells was pretreated with 10 ng/ml brefeldin A (BFA) for 1 h during the defensin-virus incubations. BFA was kept in those wells for the remainder of the experiment. At 10 hpi, cells were fixed with 4% PFA for 10 min at room temperature, permeabilized with 0.2% Triton-X for 10 min at 21°C, blocked for 1 h at 37°C with 10% goat serum in PBS containing 1% BSA, and incubated with a 1:100 dilution of a rabbit monoclonal antibody to protein disulfide isomerase (PDI; Abcam) and a mouse monoclonal to human HD5 overnight with gentle shaking at 4°C. The next day, cells were washed in PBS and incubated at room temperature for 1 h with a 1:500 dilution of goat anti-rabbit AF488 and goat anti-mouse AF405. Cells were imaged on a Zeiss 710 laser scanning confocal microscope. Colocalization analysis was done per field of view according to reference 38 using ImageJ software with the JACoP plugin and Mander's coefficient for colocalization. Briefly, images were 3D hybrid-median filtered and thresholded using the JACoP plugin. Virus channel and PDI channel thresholds were chosen to reflect a conservative and appropriate (i.e., no saturation of pixels) amount of signal and kept constant over the course of an experimental replicate of 10 images (each containing 1 to 3 cells) per condition. Mander's M1 coefficient represents the amount of virus channel overlapping the PDI channel. For image representation, images were fast Fourier transform band pass filtered to better define the ER boundaries. The whole depth of the cell was used for colocalization quantification, but the images shown are from a plane in the cell with significant amounts of virus for visualization purposes. The results are from 3 fully independent experiments and ∼50 total cells per treatment per experiment.
VP2 exposure.
SVG-A cells (5 × 105) were seeded in a 96-well glass bottom plate and infected at an MOI of 20 with mock-treated or 100 μg/ml HD5- or HBD1-treated JCPyV in a volume of 20 μl in 2% MEM. After 1 h, complete medium was added. HD5 preincubation was done for 1 h on ice. A subset of cells was pretreated with 10 ng/ml brefeldin A for 1 h during the defensin-virus incubations. BFA was kept in those wells for the remainder of the experiment. After 10 hpi, cells were fixed and stained for the major viral protein 2 (VP2) using a polyclonal rabbit antibody to the SV40 VP2/3 (Abcam), which cross-reacts with JCPyV VP2. Nuclei were counterstained with Hoechst dye (1:10,000). Imaging was done on a Zeiss 200 M Axiovert fluorescence microscope at a ×20 magnification. Ten fields of view were imaged per experimental condition, resulting in quantification of ∼2,500 cells per treatment. Cells were considered to have exposed VP2 by showing a perinuclear punctate staining pattern. VP2+ cells were counted in ImageJ and normalized to the number of total cells. To analyze VP2, PDI, and JCPyV all at once, cells were treated with defensins at 100 μg/ml, infected at an MOI of 5 (same as ER colocalization), fixed, and stained first for VP2 using the antibody described above. After the primary and secondary (Alexa Fluor 405) antibodies for VP2 were incubated and washed away, a rabbit monoclonal to PDI was added overnight at 4°C with gentle shaking. The following day, PDI was detected using an Alexa Fluor 488 secondary antibody. Images shown are fast Fourier transform (FFT) band pass filtered, and PDI is pseudocolored red, VP2 blue, and JCPyV-633 green. All Alexa Fluor secondary antibodies were used at 1:500.
DNase protection assay.
Purified JC virions at a concentration of 1 μg/μl (5 μl used) were preincubated with 100 μg/ml HD5 for 1 h on ice and/or microsomes (3.3 μg/ml) for 3 h at 37°C after HD5 treatment in 20-μl reaction mixtures (PBS). Microsomes were prepared as described in reference 39 and stored at −80°C at a concentration of 1 mg/ml. Samples were analyzed for DNA content using a NanoDrop (Thermo Scientific) spectrophotometer. To degrade DNA not protected from the viral capsids, samples were then incubated with DNase (1 U/μg DNA) for 15 min at room temperature. Twenty microliters of 25 mM EDTA was added to stop the DNase reaction and incubated at 65°C for 10 min (1 μl of 25 mM EDTA inactivates 1 U of DNase). To release viral DNA from the capsids, proteinase K was used as described in the Qiagen blood and tissue kit (Qiagen, Inc.). Viral DNA was purified using the Qiagen column per the manufacturer's instructions, and the DNA was then eluted in a new microcentrifuge tube with 40 μl double-distilled water (ddH2O).
PCR.
Conventional PCR was used to amplify JC viral genomes. One microliter of eluted DNA was mixed with 1 μg of each custom primer (5′-TGA GAC TTG GGA AGA GCA TTG TG-3′ and 5′-TGA AGA TGT AAA AGG GAC AGG AGC-3′; Invitrogen) and reagents in a standard PCR kit. Reaction conditions were 94°C for 4 min once, followed by 30 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, finishing at 4°C. Real-time PCR (RT-PCR) was conducted using the Bio-Rad iQ5 system. All samples were investigated in triplicate in a 25-μl reaction using 12.5 μl SYBR green master mix (Applied Biosystems). One microliter of template and 400 nM forward and reverse primers specific to JCPyV T antigen were used.
Electron microscopy.
One μg of purified JCPyV was preincubated with 100 μg/ml of defensin for 1 h on ice in 20-μl reactions (PBS) and then treated with water or 3.3 μg/ml microsomes for 3 h at 37°C. Complexes were then overlaid onto carbon-coated Formvar grids (Electron Microscopy Sciences) for 30 s, blotted to remove excess, and stained with NanoW, a tungsten-based stain reagent, for 30 s (Nanoprobes). Excess stain was blotted, and grids were visualized using a Phillips 410 transmission electron microscope.
Statistical analysis.
Unless otherwise indicated, experiments were performed in triplicate 3 independent times. Means were compared using an unpaired Student's t test, and comparisons are indicated in the figure legends. Values were considered significant if the P value was below 0.05.
RESULTS
HD5 neutralizes JCPyV in a dose-dependent manner and inhibits early steps in the viral life cycle.
In order to determine the effect of defensins on JCPyV infection, we first tested the ability of a panel of alpha and beta defensins to neutralize JCPyV. Preincubation of purified JCPyV with 100 μg/ml of each defensin resulted in no decrease in infectivity for most defensins, while the alpha defensin HD5 and the beta defensin HBD3 were significantly neutralizing at this concentration (Fig. 1A). To eliminate the possibility that the neutralization of infection was due to cytotoxicity, cell viabilities were determined after exposure to different defensins. HBD3 demonstrated levels of cytotoxicity comparable to those of our cell death control, and we therefore attribute the lack of infectivity of HBD3-treated JCPyV to cytotoxicity (Fig. 1B). We focused on HD5, since this was the only defensin to inhibit JCPyV infection without significant cytotoxicity. HD5 inhibited JCPyV infection in a dose-dependent manner, with 25 μg/ml HD5 reducing infection by ∼50% (Fig. 1C). Since Alexa Fluor-labeled JCPyV virions (JCPyV-488 and 633) were used in later experiments, we also determined that HD5 neutralized JCPyV-488 with similar efficiencies. We next sought to determine the kinetics of HD5-mediated neutralization. Cells were infected with JCPyV, and at various times postinfection, medium was removed and replaced with medium containing a neutralizing concentration of HD5 (50 μg/ml). HD5 efficiently inhibited JCPyV infection at early time points with a half-life (t1/2) of approximately 6 hpi. Since JCPyV undergoes rapid endocytosis (16), this result suggests that HD5 interferes with early steps in JCPyV entry and trafficking in host cells (Fig. 1D). Defensins are also known to stimulate antiviral innate immune signals in cells, and this response can inhibit viral replication (40, 41). To test whether neutralization of JCPyV by HD5 is due to promotion of a cellular antiviral state instead of directly neutralizing viral capsids, we pretreated cells with 100 μg/ml of HD5 for 1 or 2 h. HD5 was then washed from these samples, and cells were inoculated with JCPyV and scored for VP1 production at 72 hpi. When HD5 was added at 1 or 2 h prior to inoculation, more than 70% of JCPyV infectivity remained, suggesting that a cellular response is not the primary mechanism by which HD5 neutralizes JCPyV (Fig. 1E). Adding HD5 at the time of infection and allowing HD5 to remain in these cultures efficiently inhibited JCPyV infection, consistent with our time-of-addition experiments (Fig. 1D).
FIG 1.

Neutralization of JCPyV infection by HD5. (A) Testing the efficacy of a defensin panel. A 100-μg/ml concentration of the indicated defensins was preincubated with JCPyV (MOI, 0.5) on ice for 1 h and then overlaid on SVG-A cells. After 1 h, complete medium was added, and the infected cells were incubated for 72 h. At 72 hpi, cells were fixed and stained for VP1. Error bars indicate the standard deviations from 3 independent experiments. *, P < 0.05 compared to no defensin. (B) Cytotoxic effect of defensins. A 100-μg/ml concentration of each defensin was added to SVG-A cells and remained in the medium for 72 h to mimic one replication cycle of JCPyV. At 72 hpi, cells were assayed for viability using a commercial MTS assay. “(+)ive” indicates cells with no defensin, and “(-)ive” indicates cells in PBS for 72 h. Error bars indicate the standard deviations from 3 independent experiments. *, P < 0.05 compared to the positive control. A bar indicates a direct comparison. (C) Dose-response curve for HD5. Approximately 5 to 10 ng of either WT JCPyV or JCPyV488 was preincubated with the indicated concentrations of HD5 for 1 h at 4°C. The complexes were then overlaid onto SVG-A cells for 1 h and stained for the presence of VP1 in the nucleus 72 hpi. Values are percentages of the value for the positive control (no HD5 plus virus). Error bars indicate standard deviations from 3 independent experiments. *, P < 0.05 compared to the virus control. (D) HD5 time course. A 50-μg/ml concentration of HD5 (a neutralizing concentration, as seen in panel C) was added to SVG-A cells at the indicated time points after infection with JCPyV (MOI, 0.5). Values are percentages of the value for the positive control after staining for VP1 72 hpi. (E) Effect of HD5 pretreatment. A 50-μg/ml concentration of HD5 was added to SVG-A cells directly in a 20-μl reaction for either 2 h before, 1 h before, or at the point of infection (+ive) with ∼5 to 10 ng of JCPyV. The pretreatment reactions were carried out in the presence of 2% FBS. Additional experiments were performed in PBS to rule out the effect of serum in the pretreatment (data not shown). Values are the percent VP1-positive cells 72 hpi. Error bars indicate standard deviations from 3 independent experiments. *, P < 0.05 compared to the positive control. A bar indicates a direct comparison.
HD5 does not inhibit JCPyV binding to host cells.
Since HD5 interferes with early steps in JCPyV replication, we asked if HD5 binds the JCPyV capsid and prevents binding to host cells. To determine whether HD5 binds to JCPyV, we used an enzyme-linked immunosorbent assay (ELISA); virions were adsorbed to the plate and probed for any bound HD5 with a monoclonal antibody to HD5. Using this assay, we showed that HD5 binds to JCPyV and JCPyV-488 equivalently (Fig. 2A). We also tested the ability of a nonneutralizing beta defensin, HBD1, to bind virions. As shown in Fig. 2A, HBD1 does not bind to either JCPyV or JCPyV-488, which further suggests that neutralization correlates with direct capsid binding by defensins. The HBD1 ELISA was validated by adsorbing HBD1 to the plate instead of virions and detecting HBD1 using a commercial antibody to HBD1 (data not shown). The HD5 binding is not due to affinity with BSA (the blocking reagent), as shown in the no-virus control. We also tested a panel of VP1 pentamer mutants for the ability to bind HD5 by ELISA (Fig. 2D). VP1 pentamers are the functional capsid protein responsible for host cell receptor engagement; they recapitulate early JCPyV trafficking events and serve as a tool for polyomavirus studies (16). Surprisingly, HD5 was able to bind to all mutants tested, including 3 residues that are vital for LSTc recognition (S266, S268, and N123) (12). To examine whether HD5 prevents JCPyV binding to host cells, we preincubated JCPyV-488 and HD5 for 1 h and then added this complex to SVG-A cells on ice for 2 h. Cells were washed and fixed in PFA and analyzed by flow cytometry. HD5-treated JCPyV-488 bound SVG-A cells as efficiently as JCPyV-488 alone (Fig. 2B). We also examined binding by fluorescence microscopy. HD5-virus complexes were bound to chilled SVG-A cells (to prevent internalization) and imaged (Fig. 2C). Mock- and HD5-treated virus both bound to cells, but the HD5-treated virus bound in clumps, suggesting that HD5 induces virion aggregation.
HD5 does not neutralize JCPyV infection by blocking internalization.
Since HD5 caused aggregation of virions but did not block binding to cells, we hypothesized that HD5-mediated aggregation of virions would prevent viral endocytosis. To examine this, we used a fluorescence-quenching assay to distinguish extracellular virions from those that had entered cells (37).
SVG-A cells were chilled to prevent endocytosis and were inoculated with JCPyV-488. After virions had been allowed to bind to cells for 1 h, the cells were washed and either imaged immediately or warmed to 37°C to allow sufficient time for JCPyV endocytosis (2 h). To evaluate internalization, trypan blue was added to quench any labeled virus remaining on the plasma membrane or attached to the extracellular matrix. Since trypan blue is a membrane-impermeative dye, it does not quench the fluorescence from virions that have been internalized (Fig. 3A). Quantifying the images resulted in no decrease in internalization compared to virus-only controls at the 2-hpi time point (Fig. 3B). In fact, pretreatment of JCPyV with HD5 led to a slight increase in internalization, presented as a percentage of total fluorescence prequenching.
HD5 reduces JCPyV traffic to the endoplasmic reticulum.
After internalization, JCPyV enters early endosomes and traffics to the endoplasmic reticulum (ER) within several hours (16). Since HD5 did not prevent viral internalization, we sought to determine if HD5 binding to JCPyV inhibited retrograde ER transport of virions. SVG-A cells were infected with JCPyV-633 and stained after 10 h for PDI, an ER marker. To mimic disruption of ER traffic, the COP-1 vesicle inhibitor brefeldin A was used as a positive control. Brefeldin A has been shown to disrupt other polyomavirus trafficking to the ER (42). The nonneutralizing beta defensin HBD1 was used as a negative control as in other experiments to show that the mechanism of action was specific to HD5. JCPyV-633 mock treated with water colocalizes with PDI 10 hpi with Mander's coefficient of colocalization, consistent with previously published data (16) (Fig. 4A). Upon treatment with HD5, colocalization is reduced. Quantifying the colocalization using Mander's coefficient resulted in no difference between HD5-treated JCPyV-633 and brefeldin A-treated JCPyV-633 (Fig. 4B), indicating a significant reduction in ER traffic. HBD1 did not affect colocalization with PDI (Fig. 4A, third row).
HD5 reduces exposure of VP2.
In order to confirm reduced ER trafficking of HD5-treated JCPyV, we asked whether HD5 prevented the exposure of VP2. VP2 is exposed upon arrival at the ER after exposure to the ER host chaperones and isomerases and is necessary for viral retrotranslocation to the cytosol (43–45). We hypothesized that less VP2 would be exposed, since virions are hindered from exposure to these proteins encountered inside the ER. SVG-A cells were infected with JCPyV at an MOI of 20 and incubated for 10 h. At this time point, the majority of the virions should have arrived at the ER (16, 46). At 10 hpi, untreated JCPyV shows a perinuclear punctate staining pattern, indicative of VP2 exposure in the ER (Fig. 5A, top left). Both HD5 and BFA significantly reduced exposure of VP2, but HBD1 had no effect (Fig. 5A and B). To rule out the possibility that the reduction in VP2 exposure was due to fewer virions being present due to some degradative process, we visualized labeled virus (mock or HD5/HBD1 treated), VP2, and PDI in a single experiment. Labeled virus colocalized with PDI in all cases, and the amount of colocalization was reduced when HD5 was present, consistent with reduced trafficking of HD5-treated virions to the ER. HD5-treated virions that did colocalize to the ER showed no evidence of VP2 exposure, suggesting that HD5 may also stabilize virions that do the reach the ER from host cell chaperones that drive the unfolding reaction. There is precedent in the literature for this, as other viruses have been shown to be stabilized by alpha defensins (29, 47).
To test this possibility, we asked whether HD5 treatment would stabilize JCPyV virions against either heat or exposure to ER extracts (48). Treatment of JCPyV with ER extracts resulted in an altered virion morphology, characterized by partially unfolded capsids of nonuniform diameter (Fig. 6A, middle). Pretreatment of the virions with HD5 shows the typical aggregation (compare to Fig. 2B), and after subsequent treatment with ER extract, the HD5-treated JCPyV virions remained qualitatively intact and uniform. This suggests that HD5 could interfere with important capsid rearrangements necessary for a productive infection. HD5 also protected the virions from disassembly following incubation at 95C for 30 (Fig. 6A, bottom). To further demonstrate the ability of HD5 to stabilize the viral capsid, DNase protection assays were used to determine if HD5 blocks access to the genome by DNase I. JCPyV and HD5-treated JCPyV were exposed to microsomes or heat as in the transmission electron microscopy (TEM) experiments. The samples were then treated with DNase to digest all exposed DNA, followed by proteinase K to break down the viral capsid. The viral genome was amplified with specific primers. Figure 6B shows that the non-HD5 samples treated with either microsomes or high heat contained much less DNA content after DNase digestion, as evidenced by an increase in the threshold cycle (CT) needed to reach the user-defined threshold. Samples treated with HD5 and those not exposed to microsomes or heat contained the same amount of DNA after digestion, indicating that the microsomes and heat treatments were unable to efficiently expose the genome. This is seen clearly in Fig. 6C, where the average CT values for each sample were converted to fold changes using the ΔΔCT method between DNase treatment and no DNase. Without HD5 treatment, viral DNA was decreased 1,000- to 10,000-fold compared to unmanipulated JCPyV. Upon treatment with HD5, relatively equivalent amounts of DNA remained before and after ER extraction and high-heat treatment, indicating that the capsid was not rearranged and the genome was protected. Plasmid DNA incubated with each reagent without the protection of a viral capsid was used as a control to show that none of the reagents interfered with the DNase reaction and that the DNase degraded available genome. The amount of EDTA used in the control plasmid reaction (and correspondingly in the microsome reaction) is much less than the amount used to eventually stop the DNase reaction.
FIG 6.

HD5 stabilizes the viral capsid in the presence of either high heat or microsome treatment. (A) Morphological changes in JCPyV after harsh treatments. Mock- and HD5-treated JCPyV was subjected to treatment with either high heat (95°C for 10 min) or microsomes (ER extracts with chaperones and proteins that would begin to manipulate the viral capsid). Ultrastructural analysis using TEM showed that treatment with heat (middle left) and microsomes (bottom left) resulted in misshaped and incomplete capsids, indicated by the arrows. JCPyV that was treated with 100 μg/ml HD5, however, remained aggregated and shows a morphology very similar to that of mock-treated virions. Scale bar = 100 nm. Magnification, ×54,800. (B and C) Protection from DNase digestion. Mock-treated JCPyV and JCPyV treated with 100 μg/ml HD5 were treated as indicated for panel A and then subjected to DNase treatment to degrade any exposed genome. After the DNase reaction was stopped, capsids were digested with proteinase K, and DNA was isolated and purified. Each condition had a reaction with DNase and without DNase to control for the amount of genome available. Viral genomes were amplified and quantified using real-time PCR. Values indicate threshold cycle (or CT value) (B) or the log fold change in viral DNA content normalized to the no-DNase reactions using the mean DNA content (as determined by CT value) of 3 independent experiments (C). The last 4 samples indicate viral plasmid combined with each reagent in the DNase reaction mixture to ensure that no reagent interfered with DNase activity by itself. *, P < 0.05. NTC, no-template control for the PCR. “Everything” indicates plasmid DNA plus each reagent listed (EDTA, HD5, and microsomes). A bar indicates a direct comparison.
DISCUSSION
Evidence of defensin-mediated neutralization of viral infection has greatly increased in the literature in recent years (reviewed in references 23 and 24). Defensins act on multiple classes of viruses by diverse mechanisms. They have been shown to directly interact with virions and also stimulate immune signaling. This study elucidates the mechanism of HD5 (an alpha defensin)-mediated neutralization of JCPyV infection. We show that the mechanism of neutralization is different from that of neutralization of the highly related polyomavirus BK, affecting distinct stages in the polyomavirus life cycle. Additionally, HD5 stabilizes JCPyV capsids and prevents uncoating.
Previously, we showed that HD5 neutralizes BKPyV by preventing attachment to host cells (33). The data presented here demonstrate that HD5 neutralizes JCPyV infection at a later step in the viral life cycle, after internalization. Viral attachment to host cells is unaffected. The reason for two divergent mechanisms of neutralization of two highly similar viruses by the same defensin may be due to the different receptor complexes utilized by JCPyV and BKPyV to attach to cells. BKPyV attaches to the gangliosides GT1b and GD1b and primarily infects the kidney. The JCPyV receptor complex consists of a sialic acid-containing carbohydrate, LSTc, and the 5HT2a serotonin receptor (11, 12, 49). It is possible that HD5, which associates directly with both viruses, occludes the binding site for gangliosides in the context of a BKPyV infection, whereas the binding site for the sialic acid-containing receptor complex is still available in a JCPyV infection. The data for HD5 binding to mutant VP1 pentamer are consistent with this possibility, as HD5 can still bind to pentamer mutants that abolish JCPyV infection due to a lack of LSTc engagement (50). Thus, it does not appear that HD5 specifically competes with LSTc on the viral capsid, driving the virus to bind a different receptor and trafficking pathway. The relatively low binding signal in Fig. 2A can be attributed to the fact that a large fraction of the HD5 surface area would be occluded by interaction with the virion. This occlusion appears to be less apparent in binding to pentamers (Fig. 2D).
The molecular epitopes driving HD5-mediated neutralization of JCPyV are not known. Residues conferring hydrophobicity and supporting multimerization have been shown to be vital for HD5's antiviral properties in other viral systems (51–53). Several arginine residues in HD5 have been shown to be critical in its ability to neutralize adenovirus (53). The conformation also seems to be necessary as the linearized form of HD5 (reduction of its 3 characteristic disulfide bonds) loses its antiviral capabilities (31). It is still unclear what motifs drive HD5-mediated neutralization of polyomaviruses. Future studies examining the binding motif of HD5 and JCPyV or BKPyV will greatly increase our understanding of how this small molecule inhibits polyomavirus infection.
We have shown that HD5 neutralizes JCPyV up to 3 hpi. Interestingly, the kinetics of JCPyV entry indicates that the majority of the virus is internalized 2 hpi (13). This suggests that HD5 is internalized and inhibits JCPyV at a postendocytic step. Defensins are known to be internalized irrespective of virus, as was shown in the context of herpes simplex virus infection (54). Thus, HD5 could seemingly “catch up” to JCPyV at an early time point in the JCPyV life cycle. Interestingly, our data show that the neutralization capabilities of HD5 start to decrease after 6 h, a time point consistent with the beginning of ER arrival. ELISA and TEM analysis showed that HD5 directly interacted with both labeled and unlabeled virions. HD5-mediated JCPyV complexes are internalized into SVG-A cells within 2 h. Thus, HD5-treated JCPyV is internalized within a time frame similar to that of mock-treated JCPyV even though the complexes are much larger than cargo traditionally internalized by clathrin-mediated endocytosis (reviewed in reference 55). However, studies on vesicular stomatitis virus (VSV) internalization have demonstrated that clathrin-mediated endocytosis is able to internalize large cargo molecules. In those studies, clathrin buckets enlarge, incompletely coating the pit. Actin is then recruited to the pits, but clathrin still proves vital for VSV endocytosis, suggesting that clathrin-mediated endocytosis can accommodate a range of cargos (56).
Pretreatment of cells with HD5 modestly neutralized infection, suggesting that HD5 also has potential indirect effects on the target cell promoting an antiviral response. Nuclear factor of activated T cells (NFAT) and NF-κB are transcription factors that are known to coordinate immune responses and are vital for JCPyV infection (57–59). Alpha defensin genes have NFAT and NF-κB binding sites, so the modest reduction in infection could be attributed to the beginning of a cellular response initiated by these factors due to exogenous HD5 addition. One study showed that addition of HD5 upregulates cell survival genes and inflammatory genes in an NF-κB-mediated fashion (60). Future work will be focused on elucidating the relationship between defensins and these transcription factors in the context of polyomavirus infection. Moreover, the effect of JCPyV infection of kidney and glia-derived cell lines on defensin induction is currently being studied in our laboratory.
Our results are consistent with previously published data, which demonstrate that defensins can alter viral trafficking. Defensins block papillomavirus infection by sequestering the virions in endosomes (32). Our studies show that HD5-treated JCPyV is significantly prevented from arriving at the ER, a step critical for productive infection (44). It is likely that a reduction in ER transport of JCPyV eventually results in targeting of virions to a degradative pathway. This study demonstrates that HD5-mediated neutralization of JCPyV can occur at multiple stages in the viral life cycle. Although the techniques used were different, the reduction in VP2 exposure was much more pronounced after HD5 treatment than the reduction in ER traffic. The MOI used in the VP2 experiment was four times higher than that in the ER colocalization experiment, thus making the effect even greater. A higher MOI was necessary due to the microscopy used (fluorescence versus confocal). Moreover, we costained for virus, PDI, and VP2 in the same confocal experiment to further link the ER colocalization and VP2 exposure experiments. The reduction in VP2 is not due to fewer virions or acute increased degradation, as a large amount of labeled virus was still present at 10 hpi inside the cell near to, or colocalized with, PDI. Thus, we aimed to show what happened to HD5-treated virions that still arrived at the ER. We show here that HD5 also stabilizes the viral capsid, so that even if the virion arrives at the ER, the critical unfolding step necessary to expose VP2 and subsequent retrotranslocation to the cytosol is blocked. VP2 exposure is blocked and the genome is protected from DNase digestion after treatment with ER chaperone proteins. As a result, HD5 acts to prevent a normal JCPyV life cycle but also serves as a potential building block for a JCPyV antiviral molecule due to its direct effects on the capsid.
Viral capsid stabilization has been described as an antiviral mechanism for other viruses. For example, chemical compounds produced by Sterling-Winthrop, Inc. (WIN) have been demonstrated to inhibit “breathing” of picornaviruses, resulting in an inability of the virus to transiently expose the N terminus of its capsid protein, thus preventing infection (61, 62). Other groups have shown that defensins neutralize adenovirus infection by stabilizing the viral capsid. Adenovirus escapes from endosomes after exposure of pVI and subsequent puncturing of the endosomal membrane (63). HD5 blocks exposure of pVI and results in an accumulation in endosomes and lysosomes (29, 31). These data suggest that HD5 is acting like “glue,” keeping the capsid intact and preventing genome release to the nucleus. This stabilization effect works in concert with JCPyV aggregation, thus preventing critical rearrangement of the viral capsid and altering trafficking, rather than preventing binding to the host cell. This work builds upon the capsid stabilization literature and shows that defensins can potentially be used either to treat polyomavirus-induced disease or to provide a rationale for the development of a polyomavirus-blocking peptide.
It is entirely possible that JCPyV would be exposed to HD5 in the course of an infection in vivo. Since JCPyV is hypothesized to be transmitted via the fecal-oral route, it is likely that the virus encounters defensins in the tonsillar tissue, the gut, and the kidney epithelium. HD5 is primarily produced in the intestinal Paneth cells, but interestingly, a recent study showed basal levels of HD5 expression in the human kidney and urinary tract that increased in patients with pyelonephritis (64). It was originally thought that only beta defensins were expressed in the kidney. It is interesting that only HD5 significantly reduced JCPyV infection in the panel of defensins tested. Detailed structural studies would be beneficial, since defensins are highly similar in secondary structure.
The concentrations used in this study are physiologically relevant. Physiologic concentrations of defensins can range from 400 ng/ml in plasma of healthy individuals to >10 mg/ml in neutrophils of infected patients. The concentrations vary with the defensin, the defensin-producing cell, and the infection (65, 66). With regard to HD5, lavages from urethral sections of men with gonorrhea have shown HD5 levels to be significantly greater than 10 μg/ml.
Hosts with variant expression levels of defensin genes show susceptibilities to autoimmune diseases such as Crohn's disease as well as HIV infection (67, 68). In addition, not all patients with AIDS develop PML, and the majority of the population is seropositive for JCPyV. It is possible that having higher defensin expression can result in decreased susceptibility to persistent infections and/or autoimmune diseases.
In summary, this study adds to our knowledge of the antiviral capabilities of human defensins and shows that HD5 can act in distinct mechanisms within the same virus family. This has implications for future studies that aim to find a conserved mechanism of action or motif attributed to defensins. This study also provides a platform and rationale for developing a small-molecule inhibitor of JCPyV infection that might be used to treat patients at risk for PML. Finally, this work provides clues as to how JCPyV might be controlled in healthy patients, since there is evidence HD5 is expressed in sites of initial infection.
ACKNOWLEDGMENTS
We thank all members of the Atwood lab for their helpful discussions.
We have no conflicts of interest to declare.
Work in our laboratory was supported by P01NS065719 (W.J.A.), R01CA071878 (W.J.A.), and R01NS043097 (W.J.A.) and by Ruth L. Kirschstein National Research Service Awards F32NS070687 (C.D.S.N), F31NS080625 (S.R.Z.), and F32NS064870 (M.S.M.) from the National Institute of Neurological Disorders and Stroke. Confocal microscopy analysis was completed in the Leduc Bioimaging Facility at Brown University. Immunoblot analysis was performed in the Center for Genomics and Proteomics at Brown University, which is supported by P30GM103410 (W.J.A.).
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Johne R, Buck CB, Allander T, Atwood WJ, Garcea RL, Imperiale MJ, Major EO, Ramqvist T, Norkin LC. 2011. Taxonomical developments in the family Polyomaviridae. Arch. Virol. 156:1627–1634. 10.1007/s00705-011-1008-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. 1971. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet i:1257–1260 [DOI] [PubMed] [Google Scholar]
- 3.Kean JM, Rao S, Wang M, Garcea RL. 2009. Seroepidemiology of human polyomaviruses. PLoS Pathog. 5:e1000363. 10.1371/journal.ppat.1000363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan CS, Koralnik IJ. 2010. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 9:425–437. 10.1016/S1474-4422(10)70040-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. 1992. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 5:49–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 353:369–374. 10.1056/NEJMoa051782 [DOI] [PubMed] [Google Scholar]
- 7.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 353:375–381. 10.1056/NEJMoa051847 [DOI] [PubMed] [Google Scholar]
- 8.Major EO. 2010. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 61:35–47. 10.1146/annurev.med.080708.082655 [DOI] [PubMed] [Google Scholar]
- 9.Tyler KL, Khalili K. 2005. Natalizumab and progressive multifocal leukoencephalopathy: highlights of the International Workshop on JC Virus/PML and Multiple Sclerosis, June 3–4, 2005, Philadelphia PA. Rev. Neurol. Dis. 2:144–149 [PubMed] [Google Scholar]
- 10.Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353:362–368. 10.1056/NEJMoa051586 [DOI] [PubMed] [Google Scholar]
- 11.Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. 2004. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380–1383. 10.1126/science.1103492 [DOI] [PubMed] [Google Scholar]
- 12.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. 10.1016/j.chom.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pho MT, Ashok A, Atwood WJ. 2000. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 74:2288–2292. 10.1128/JVI.74.5.2288-2292.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Querbes W, O'Hara BA, Williams G, Atwood WJ. 2006. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J. Virol. 80:9402–9413. 10.1128/JVI.01086-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A. 2007. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131:516–529. 10.1016/j.cell.2007.09.038 [DOI] [PubMed] [Google Scholar]
- 16.Nelson CD, Derdowski A, Maginnis MS, O'Hara BA, Atwood WJ. 2012. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 428:30–40. 10.1016/j.virol.2012.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chesters PM, Heritage J, McCance DJ. 1983. Persistence of DNA sequences of BK virus and JC virus in normal human tissues and in diseased tissues. J. Infect. Dis. 147:676–684. 10.1093/infdis/147.4.676 [DOI] [PubMed] [Google Scholar]
- 18.Chapagain ML, Nerurkar VR. 2010. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J. Infect. Dis. 202:184–191. 10.1086/653823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei G, Liu CK, Atwood WJ. 2000. JC virus binds to primary human glial cells, tonsillar stromal cells, and B-lymphocytes, but not to T lymphocytes. J. Neurovirol. 6:127–136. 10.3109/13550280009013156 [DOI] [PubMed] [Google Scholar]
- 20.Atwood WJ, Amemiya K, Traub R, Harms J, Major EO. 1992. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology 190:716–723. 10.1016/0042-6822(92)90909-9 [DOI] [PubMed] [Google Scholar]
- 21.Gasnault J, Kahraman M, de Goer de Herve MG, Durali D, Delfraissy JF, Taoufik Y. 2003. Critical role of JC virus-specific CD4 T-cell responses in preventing progressive multifocal leukoencephalopathy. AIDS 17:1443–1449. 10.1097/00002030-200307040-00004 [DOI] [PubMed] [Google Scholar]
- 22.Koralnik IJ. 2002. Overview of the cellular immunity against JC virus in progressive multifocal leukoencephalopathy. J. Neurovirol 8(Suppl 2):59–65. 10.1080/13550280290167894 [DOI] [PubMed] [Google Scholar]
- 23.Ding J, Chou YY, Chang TL. 2009. Defensins in viral infections. J. Innate Immun. 1:413–420. 10.1159/000226256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klotman ME, Chang TL. 2006. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 6:447–456. 10.1038/nri1860 [DOI] [PubMed] [Google Scholar]
- 25.Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. 2004. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu. Rev. Immunol. 22:181–215. 10.1146/annurev.immunol.22.012703.104603 [DOI] [PubMed] [Google Scholar]
- 26.Sinha S, Cheshenko N, Lehrer RI, Herold BC. 2003. NP-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrob. Agents Chemother. 47:494–500. 10.1128/AAC.47.2.494-500.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daher KA, Selsted ME, Lehrer RI. 1986. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 60:1068–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Yu W, He T, Yu J, Caffrey RE, Dalmasso EA, Fu S, Pham T, Mei J, Ho JJ, Zhang W, Lopez P, Ho DD. 2002. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 298:995–1000. 10.1126/science.1076185 [DOI] [PubMed] [Google Scholar]
- 29.Nguyen EK, Nemerow GR, Smith JG. 2010. Direct evidence from single-cell analysis that human α-defensins block adenovirus uncoating to neutralize infection. J. Virol. 84:4041–4049. 10.1128/JVI.02471-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith JG, Nemerow GR. 2008. Mechanism of adenovirus neutralization by human alpha-defensins. Cell Host Microbe 3:11–19. 10.1016/j.chom.2007.12.001 [DOI] [PubMed] [Google Scholar]
- 31.Smith JG, Silvestry M, Lindert S, Lu W, Nemerow GR, Stewart PL. 2010. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog. 6:e1000959. 10.1371/journal.ppat.1000959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. 2006. Human alpha-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. U. S. A. 103:1516–1521. 10.1073/pnas.0508033103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dugan AS, Maginnis MS, Jordan JA, Gasparovic ML, Manley K, Page R, Williams G, Porter E, O'Hara BA, Atwood WJ. 2008. Human alpha-defensins inhibit BK virus infection by aggregating virions and blocking binding to host cells. J. Biol. Chem. 283:31125–31132. 10.1074/jbc.M805902200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. U. S. A. 82:1257–1261. 10.1073/pnas.82.4.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vacante DA, Traub R, Major EO. 1989. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170:353–361. 10.1016/0042-6822(89)90425-X [DOI] [PubMed] [Google Scholar]
- 36.Shen PS, Enderlein D, Nelson CD, Carter WS, Kawano M, Xing L, Swenson RD, Olson NH, Baker TS, Cheng RH, Atwood WJ, Johne R, Belnap DM. 2011. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology 411:142–152. 10.1016/j.virol.2010.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J. Virol. 85:4198–4211. 10.1128/JVI.02179-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. Oxford 224:213–232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 39.Magnuson B, Rainey EK, Benjamin T, Baryshev M, Mkrtchian S, Tsai B. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 20:289–300. 10.1016/j.molcel.2005.08.034 [DOI] [PubMed] [Google Scholar]
- 40.Territo MC, Ganz T, Selsted ME, Lehrer R. 1989. Monocyte-chemotactic activity of defensins from human neutrophils. J. Clin. Invest. 84:2017–2020. 10.1172/JCI114394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinstraesser L, Kraneburg U, Jacobsen F, Al-Benna S. 2011. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 216:322–333. 10.1016/j.imbio.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 42.Inoue T, Tsai B. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 7:e1002037. 10.1371/journal.ppat.1002037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniels R, Rusan NM, Wadsworth P, Hebert DN. 2006. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol. Cell 24:955–966. 10.1016/j.molcel.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 44.Gasparovic ML, Gee GV, Atwood WJ. 2006. JC virus minor capsid proteins Vp2 and Vp3 are essential for virus propagation. J. Virol. 80:10858–10861. 10.1128/JVI.01298-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A. 2011. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 13:1305–1314. 10.1038/ncb2339 [DOI] [PubMed] [Google Scholar]
- 46.Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards AP, Van Goor KE, Paton AW, Paton JC, Atwood WJ, Tsai B, DiMaio D. 2011. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2:e0010–11. 10.1128/mBio.00101-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snijder J, Reddy VS, May ER, Roos WH, Nemerow GR, Wuite GJ. 2013. Integrin and defensin modulate the mechanical properties of adenovirus. J. Virol. 87:2756–2766. 10.1128/JVI.02516-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawano MA, Xing L, Tsukamoto H, Inoue T, Handa H, Cheng RH. 2009. Calcium bridge triggers capsid disassembly in the cell entry process of simian virus 40. J. Biol. Chem. 284:34703–34712. 10.1074/jbc.M109.015107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gee GV, Dugan AS, Tsomaia N, Mierke DF, Atwood WJ. 2006. The role of sialic acid in human polyomavirus infections. Glycoconj. J. 23:19–26. 10.1007/s10719-006-5434-z [DOI] [PubMed] [Google Scholar]
- 50.Maginnis MS, Stroh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. 2013. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 4:e00247–13. 10.1128/mBio.00247-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Leeuw E, Rajabi M, Zou GZ, Pazgier M, Lu WY. 2009. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett. 583:2507–2512. 10.1016/j.febslet.2009.06.051 [DOI] [PubMed] [Google Scholar]
- 52.Rajabi M, Ericksen B, Wu XJ, de Leeuw E, Zhao L, Pazgier M, Lu WY. 2012. Functional determinants of human enteric alpha-defensin HD5. Crucial role for hydrophobicity at dimer interface. J. Biol. Chem. 287:21615–21627. 10.1074/jbc.M112.367995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gounder AP, Wiens ME, Wilson SS, Lu W, Smith JG. 2012. Critical determinants of human alpha-defensin 5 activity against non-enveloped viruses. J. Biol. Chem. 287:24554–24562. 10.1074/jbc.M112.354068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hazrati E, Galen B, Lu W, Wang W, Ouyang Y, Keller MJ, Lehrer RI, Herold BC. 2006. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 177:8658–8666 http://www.jimmunol.org/content/177/12/8658.long [DOI] [PubMed] [Google Scholar]
- 55.McMahon HT, Boucrot E. 2011. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12:517–533. 10.1038/nrm3151 [DOI] [PubMed] [Google Scholar]
- 56.Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SPJ. 2009. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 5:e1000394. 10.1371/journal.ppat.1000394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jordan JA, Manley K, Dugan AS, O'Hara BA, Atwood WJ. 2010. Transcriptional regulation of BK virus by nuclear factor of activated T cells. J. Virol. 84:1722–1730. 10.1128/JVI.01918-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manley K, O'Hara BA, Gee GV, Simkevich CP, Sedivy JM, Atwood WJ. 2006. NFAT4 is required for JC virus infection of glial cells. J. Virol. 80:12079–12085. 10.1128/JVI.01456-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manley K, O'Hara BA, Atwood WJ. 2008. Nuclear factor of activated T-cells (NFAT) plays a role in SV40 infection. Virology 372:48–55. 10.1016/j.virol.2007.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu W, de Leeuw E. 2013. Pro-inflammatory and pro-apoptotic properties of human defensin 5. Biochem. Biophys. Res. Commun. 436:557–562. 10.1016/j.bbrc.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phelps DK, Post CB. 1995. A novel basis of capsid stabilization by antiviral compounds. J. Mol. Biol. 254:544–551. 10.1006/jmbi.1995.0637 [DOI] [PubMed] [Google Scholar]
- 62.Reisdorph N, Thomas JJ, Katpally U, Chase E, Harris K, Siuzdak G, Smith TJ. 2003. Human rhinovirus capsid dynamics is controlled by canyon flexibility. Virology 314:34–44. 10.1016/S0042-6822(03)00452-5 [DOI] [PubMed] [Google Scholar]
- 63.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 79:1992–2000. 10.1128/JVI.79.4.1992-2000.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spencer JD, Hains DS, Porter E, Bevins CL, DiRosario J, Becknell B, Wang H, Schwaderer AL. 2012. Human alpha defensin 5 expression in the human kidney and urinary tract. PLoS One 7:e31712. 10.1371/journal.pone.0031712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ihi T, Nakazato M, Mukae H, Matsukura S. 1997. Elevated concentrations of human neutrophil peptides in plasma, blood, and body fluids from patients with infections. Clin. Infect. Dis. 25:1134–1140. 10.1086/516075 [DOI] [PubMed] [Google Scholar]
- 66.Lehrer RI. 1997. Questions and answers about defensins. Clin. Infect. Dis. 25:1141–1142. 10.1086/516082 [DOI] [PubMed] [Google Scholar]
- 67.Aldred PM, Hollox EJ, Armour JA. 2005. Copy number polymorphism and expression level variation of the human alpha-defensin genes DEFA1 and DEFA3. Hum. Mol. Genet. 14:2045–2052. 10.1093/hmg/ddi209 [DOI] [PubMed] [Google Scholar]
- 68.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H, Jr, Fellermann K, Ganz T, Stange EF, Bevins CL. 2005. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc. Natl. Acad. Sci. U. S. A. 102:18129–18134. 10.1073/pnas.0505256102 [DOI] [PMC free article] [PubMed] [Google Scholar]