

Abstract
Autophagy and the effects of its inhibition or induction were investigated during the entire infectious cycle of varicella-zoster virus (VZV), a human herpesvirus. As a baseline, we first enumerated the number of autophagosomes per cell after VZV infection compared with the number after induction of autophagy following serum starvation or treatment with tunicamycin or trehalose. Punctum induction by VZV was similar in degree to punctum induction by trehalose in uninfected cells. Treatment of infected cells with the autophagy inhibitor 3-methyladenine (3-MA) markedly reduced the viral titer, as determined by assays measuring both cell-free virus and infectious foci (P < 0.0001). We next examined a virion-enriched band purified by density gradient sedimentation and observed that treatment with 3-MA decreased the amount of VZV gE, while treatment with trehalose increased the amount of gE in the same band. Because VZV gE is the most abundant glycoprotein, we selected gE as a representative viral glycoprotein. To further investigate the role of autophagy in VZV glycoprotein biosynthesis as well as confirm the results obtained with 3-MA inhibition, we transfected cells with ATG5 small interfering RNA to block autophagosome formation. VZV-induced syncytium formation was markedly reduced by ATG5 knockdown (P < 0.0001). Further, we found that both expression and glycan processing of VZV gE were decreased after ATG5 knockdown, while expression of the nonglycosylated IE62 tegument protein was unchanged. Taken together, our cumulative results not only documented abundant autophagy within VZV-infected cells throughout the infectious cycle but also demonstrated that VZV-induced autophagy facilitated VZV glycoprotein biosynthesis and processing.
INTRODUCTION
Varicella-zoster virus (VZV; human herpesvirus 3) is an alphaherpesviral pathogen that causes primary varicella infection in children (1). A few days after primary infection, a viremia ensues within T lymphocytes, during which infected T cells exit capillaries and infect keratinocytes within the epidermis to cause the characteristic vesicular rash (2). The viral progeny produced in skin lesions migrate in a retrograde manner via sensory neurons into the dorsal root ganglia, where they establish latency. Decades later, the same VZV strain reactivates from these ganglia and travels anterograde to cause the dermatomal disease herpes zoster. Autophagosomes have been identified in the vesicular skin lesions of both varicella and herpes zoster (3, 4).
The response of various viruses to macroautophagy has been a subject of renewed research (5, 6). Macroautophagy (here referred to as autophagy) is a catabolic process by which whole or parts of organelles are sequestered into double-membraned autophagosomes in the cytoplasm and then the cargo is degraded by hydrolases and proteases when the autophagosomes fuse with lysosomes (7, 8). Subsequently, the degradation products are recycled for reuse in other cellular processes. Numerous autophagy-associated (ATG) proteins have been identified, and many other cellular proteins are recruited to autophagy-associated protein complexes (9). Two such complexes relevant to the present study are the phosphoinositide 3-kinase (PI3K) complex and the ATG5-ATG12 complex. Both play roles in the conversion of microtubule-associated protein 1 light chain LC3-I by lipidation to LC3-II (10). LC3-II is a primary structural protein of both the inner and outer membranes of the autophagosome (11). At its core, the PI3K complex is composed of VPS34 (PIK3C3), p150 (PIK3R4), and Beclin-1 (ATG6) (12, 13). VPS34 is a lipid kinase, while p150 is a serine/threonine kinase. The autophagy-associated PI3K complex recruits phosphatidylinositol 3-monophosphate binding proteins (e.g., WIPI1) to the endoplasmic reticulum (ER) membrane (14). In the other complex, ATG5 and ATG12 are conjugated together by an ubiquitin-like conjugation system formed by ATG7 as the E1 ligase and either ATG3 or ATG10 as the E2 ligase (10). The ATG5-ATG12 conjugate then forms a complex with ATG16L, which plays a key role in the elongation of the evolving autophagosome (15). The compound 3-methyladenine (3-MA), a small-molecule inhibitor of class I and III PI3K proteins, is frequently used to study autophagy (16, 17). Likewise, the essential autophagy protein ATG5 is often knocked out in studies of the role of autophagy in disease models (18).
With regard to RNA viruses and retroviruses, autophagy has assumed a wide variety of roles, sometimes considered either proviral or antiviral (19–23). With regard to herpes simplex virus 1 (HSV-1), the majority of publications suggest that autophagy exerts an antiviral effect. One of the earliest papers on herpesvirus-induced autophagy connected protein kinase R (PKR) signaling through the α subunit of eukaryotic initiation factor 2 (eIF2α) with HSV-1 infection and noted that HSV-1 encodes a protein, ICP34.5, that diverts protein phosphatase 1 to dephosphorylate eIF2α (24, 25). Later, it was postulated that HSV induction of autophagy via PKR activation would lead to virus degradation within nascent autophagosomes (26, 27). HSV ICP34.5 was then reported to directly bind Beclin-1 (ATG6 in yeast, such as Saccharomyces cerevisiae), one of the components of the autophagy-specific PI3K complex, and inhibit the formation of autophagosomes in HSV-infected cells (28). Further, viral peptides generated by the degradation process may become available for presentation on the cell surface via the major histocompatibility complex and thereby stimulate adaptive immunity (29–33). It has also been reported that HSV- and Epstein-Barr virus-induced autophagy is cell type specific (34–36) and that simply the presence of viral DNA in the cytoplasm is sufficient to induce autophagy (37, 38). Interestingly, the Kaposi's sarcoma-associated herpesvirus (KSHV) viral FLIP protein modulates the autophagic response in KSHV-infected cells by binding ATG3, the autophagy-associated E2 ubiquitin-like protease (39). Finally, HSV encodes another protein, US11, that is capable of dephosphorylating eIF2α and therefore disarms autophagy (40). In summary, these reports demonstrate that autophagy plays an antiviral role during HSV-1 infection and that this particular herpesvirus has evolved mechanisms capable of antagonizing autophagy.
VZV encodes the smallest genome among the human herpesviruses, lacking homologs of both HSV-1 ICP34.5 and US11 (41). We previously observed that autophagy is a prominent feature in VZV-infected cultured cells and that VZV-induced autophagy was associated with features common to ER stress and the unfolded protein response (UPR) (4, 42), yet VZV-infected cells do not exhibit phosphorylated eIF2α or protein synthesis attenuation (43, 44). On the basis of the findings presented in the above-described papers, our central hypothesis is that VZV-induced autophagy is a cellular response to exaggerated ER stress that acts to prolong the life of an infected cell. In short, this role appears to be very different from that proposed for autophagy during HSV infection (5, 29). This difference is all the more remarkable because HSV and VZV are the 2 most closely related human herpesviruses on the phylogenetic tree (45). On the other hand, among the human betaherpesviruses more distantly related to VZV, cytomegalovirus has been shown to selectively induce ER stress while incorporating components of the UPR (particularly BiP) to form a viral assembly compartment proximal to the ER (46, 47). In the present study, we expand our prior observations and provide experimental evidence that enhanced autophagy in the VZV system is a dynamic process which is evident during the entire infectious cycle. Of great interest, comparisons of results of assays with an inhibitor of autophagy, 3-MA, and results of assays with an inducer of autophagy, trehalose, suggested that VZV-induced autophagy not only facilitates the biosynthesis and maturation of viral glycoproteins but also enhances infectivity, responses not previously documented in a herpesvirus-cell system.
MATERIALS AND METHODS
Viruses and cells.
VZV-32 is a low-passage-number laboratory strain; its genome has been completely sequenced and falls within European clade 1 of VZV genotypes (48). The MRC-5 strain of human fibroblast cells and the MeWo strain of human melanoma cells were grown in tissue culture dishes with and without 12-mm round or 22-mm square coverslips in minimum essential medium (MEM; Gibco, Life Technologies) supplemented with 7% fetal bovine serum (FBS), l-glutamine, nonessential amino acids, and penicillin-streptomycin. When monolayers were nearly confluent, they were inoculated with VZV-infected cells at a ratio of one infected cell to eight uninfected cells by previously described methods (49). Assays for titration of cell-free virus and cell-associated virus (infectious foci) have been described by this laboratory (50). Methods for purification of enveloped virions by density gradient sedimentation have also been described in detail (50).
Primary and secondary antibody reagents.
Primary antibodies required for this study include previously described VZV-specific murine monoclonal antibodies (MAbs) 3B3 and 711 (gE; ORF68), 6B5 (gI; ORF67); 158 (gB; ORF31), 233 (gC; ORF14), 258 (gH; ORF37), 5C6 (IE62; ORF62), and 251D9 (pORF41; small capsid protein) (4, 51, 52), as well as one human polyclonal anti-VZV serum (53). Also used were rabbit polyclonal antibodies to MAP1LC3B (sc-28266; Santa Cruz Biotechnology) and beta-tubulin (sc-9104; Santa Cruz Biotechnology), as well as an MAb to ATG5 (sc-133158; Santa Cruz Biotechnology). VZV MAbs were used at a dilution of 1:1,000, rabbit polyclonal antibodies were used at 1:250, and ATG5 MAb was used at 1:500. The secondary antibodies included Alexa 488, 546, and 633 fluoroprobes conjugated to a goat anti-rabbit IgG or goat anti-mouse IgG F(ab′)2 fragment (Invitrogen). These reagents were applied at a dilution of 1:1,250.
Imaging protocols.
Autophagosomes were visualized in samples of infected and uninfected cells by confocal microscopy methods as described previously (54, 55). Briefly, the samples were fixed with 2% paraformaldehyde and permeabilized with 0.02 to 0.05% Triton X-100 in phosphate-buffered saline (PBS; 30 min without agitation) and then blocked in 5% nonfat milk with 2.5% normal goat serum for 2 h at room temperature (RT). The primary antibody was added for 2 h at RT and overnight at 4°C. After washing (3 times for 5 min each time), the samples were incubated with the secondary antibody and the Hoechst 33342 (H33342) double-stranded DNA stain (1:1,000) for 2 h at RT before mounting on slides for viewing. Following preparation, the samples were viewed on a Zeiss 710 confocal fluorescence microscope in the Central Microscopy Research Facility of the University of Iowa. Images were analyzed using Zen 2009 software (Zeiss) and ImageJ software (NIH, Bethesda, MD). Pixel intensity was quantitated using ImageJ, as follows: images were opened in ImageJ in the single color for either gE, gC, IE62, or H33342 and converted to a 32-bit image, after which the Measure tool in the Analysis menu was used to obtain an average pixel intensity. The ratio of the pixel intensity of each viral protein to the H33342 pixel intensity was then calculated for each tile image. The standard error of the mean across each set of images was used to generate error bars. P values were determined by unpaired, two-tailed Student's t tests. Three-dimensional images of VZV-infected cells were reconstructed from large z-stacks of confocal laser scanning microscopic images with Imaris software, version 7.6 (Bitplane Scientific Software), as described by this laboratory (55).
Tunicamycin, 3-MA, and trehalose treatments.
Conditions for the treatment of cultured cells with tunicamycin (10 μg/ml; catalog no. 654380; Calbiochem) have been described in earlier papers investigating VZV glycoprotein biosynthesis (56, 57). For experiments in uninfected cells, tunicamycin was added 24 h after subculturing, and the monolayer was fixed after another 24 h. The compound 3-MA (10 mM; catalog no. M9281; Sigma) was dissolved in sterile MEM per the manufacturer's directions, applied to cultured cells at 6 h postinfection (hpi), and refreshed every 24 h (16). Conditions for examining the effect of the disaccharide trehalose on autophagy have been described previously (58).
Transfection with siRNA.
Uninfected confluent MeWo cell monolayers in T-25 flasks were split 1:3 and placed onto glass coverslips in 6-well dishes in MEM supplemented as described above. Cells were allowed to adhere to the coverslips overnight at 37°C in 5% CO2. Monolayers were transfected on the following day with 50 nM concentrations of small interfering RNA (siRNA) (or an equal volume of RNase-free H2O for the no-siRNA control), using Lipofectamine 2000 (10 μl per well; Invitrogen, Life Technologies) in unsupplemented Opti-MEM medium (Gibco). Monolayers were washed with Opti-MEM at 6 h posttransfection, and medium was replaced with MEM supplemented as described above (but with no penicillin-streptomycin). Medium was changed to fully supplemented MEM at 16 h posttransfection. Transfected cells were incubated for a total of 48 h posttransfection before being infected with VZV-32, as described above. Infection was allowed to proceed for 72 h at 32°C. The following siRNA products were purchased from Santa Cruz Biotechnology: human APG5 (sc-41445), control siRNA-B (sc-44230), or control siRNA-A (fluorescein conjugate; sc-36869). ATG5 siRNA was a pool of 3 target-specific 19- to 25-nucleotide siRNAs designed to knock down expression of the gene for human ATG5.
SDS-PAGE, immunoprecipitation, and immunoblotting.
Methods for SDS-PAGE and immunoblotting have been previously described by this laboratory (56). Electrophoresis was performed in precast 4 to 15% gradient or 7.5% acrylamide gels (Mini-Protean TGX; Bio-Rad). The proteins in the gel were then transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore). The membrane was then blocked with 5% nonfat milk containing 2.5% normal goat serum (Sigma) in Tween 20 in PBS (T-PBS) for 2 h and then incubated in the primary antibody (1:500 in T-PBS) for 2 h at RT and then overnight at 4°C. Following washing (3 times for 5 min each time), the membrane was incubated in goat antirabbit or antimouse secondary horseradish peroxidase (HRP) antibody (catalog numbers A10547 and F21453, respectively; Invitrogen) at 1:10,000 for 2 h at RT and then washed 5 times for 5 min each time. In some immunoblots, TrueBlot Ultra anti-mouse Ig HRP (eBioscience) was substituted for the Invitrogen reagent (1:5,000). Finally, the membrane was incubated in chemiluminescent solution (Super Signal West Pico solution; catalog number P34080; Thermo Scientific) for 5 min at RT. The membrane was then patted dry and exposed to autoradiography film.
RESULTS
Establishing the parameters for assessment of VZV-induced autophagy.
Both starvation and tunicamycin treatment increase autophagy in eukaryotic cells (59, 60). In previously published papers, we have also noted markedly enhanced autophagosome production in VZV-infected cells at single time points (4), yet it is well-known that autophagosome formation can vary depending upon cell type. In order to better validate the procedure for autophagosomal quantitation that was used for multiple experiments described in this report, we first defined the number of LC3-positive puncta under conditions of stress, e.g., serum starvation and tunicamycin treatment. We also added a positive control, trehalose, a newly recognized inducer of autophagy in mammalian cells (61). Trehalose, a disaccharide which is found in high concentrations in many invertebrates, may stabilize lipid membranes (62). Tunicamycin treatment, which inhibits N-linked glycosylation, was included because it is known to induce significant ER stress (63). In general, several images were taken of treated or untreated fibroblast cells immunolabeled with an LC3B antibody, using high-resolution confocal microscopy at a magnification of ×400 (Fig. 1A to F). Untreated cells generally exhibited less than 4 puncta per cell, which is regarded as normal for unstressed eukaryotic cells (64, 65). A minority of cells exhibited more than 4 puncta, perhaps as a consequence of recent cellular division or other cytoskeletal reorganization. Even though our starvation conditions were not extreme, serum-starved fibroblasts exhibited significantly more puncta per cell than the number seen in cells grown in medium with serum (e.g., P < 0.0001). As expected, treatment with both trehalose and tunicamycin induced a wide range of increased LC3-positive puncta, with some cells exhibiting >20 puncta. For example, the stressed cell shown in Fig. 1F exhibited 50 puncta. The LC3-positive puncta in each cell within a given image were enumerated for all conditions to generate a set of statistical distributions (Fig. 1G). Although stress-induced autophagy has been analyzed previously, our confocal microscopy data provided quantities that would allow comparison of the results obtained with this maximal stimulus with those obtained by VZV-induced autophagy in experiments carried out by the same observers in the same laboratory.
FIG 1.
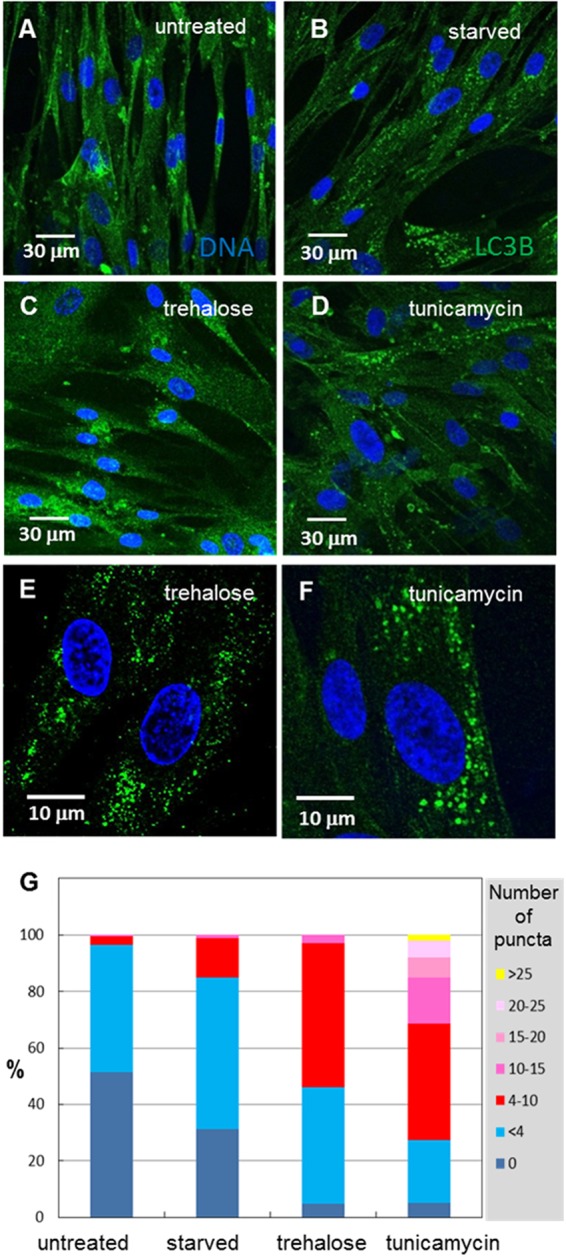
Enumeration of autophagosomes under conditions of cellular stress. MRC-5 fibroblasts were plated onto glass coverslips in tissue culture dishes and then serum starved in plain MEM for 6 h or treated with 100 mM trehalose or 10 μg/ml of tunicamycin for 24 h before fixing, blocking, and labeling for LC3B. Sixteen or more images were taken at a magnification of ×400, and the number of puncta per cell was counted. (A) Untreated fibroblasts showed few LC3B puncta. (B) Serum-starved fibroblasts showed moderately more LC3B puncta. (C) Trehalose-treated fibroblasts exhibited significantly more puncta in each cell. (D) Tunicamycin-treated fibroblasts showed numerous LC3B puncta in one-third or more of the cells. (E, F) Higher magnification (×1,000) of several cells treated with trehalose (E) or with tunicamycin (F); one tunicamycin-treated cell exhibited 49 LC3B puncta. (G) Distribution of the percentage of cells that exhibited a given number of LC3B puncta. Untreated fibroblasts typically showed <4 puncta per cell, while stressed cells showed 10 or more puncta per cell.
VZV-induced autophagosome formation during the entire infectious cycle.
In previous papers, we showed that VZV induces autophagosome formation in infected cultured cells, but we did not enumerate the LC3-positive puncta individually within infected cells over an entire infectious cycle (4, 42). Because of prior data that suggested an early appearance of autophagosomes within the infectious cycle, we previously postulated that autophagy may exert a proviral effect early in VZV infection. Because VZV is renowned for being highly cell associated, calculation of the timing for a single replication cycle is difficult but has ranged from 14 to 18 h (66). Because of the low titer of the inoculum, several replication cycles are required before all cells in a monolayer are infected (50). Another confounder is that VZV is highly fusogenic in certain cells; thus, fusion of one infected cell with a second newly infected cell obscures the end of a replication cycle and the beginning of another (67).
As a marker of the immediate early (IE) kinetic phase of VZV replication, VZV ORF62 (IE62, the homolog of HSV-1 ICP4) is the key viral transcriptional regulator, which is localized to the nucleus early in the replication cycle (68). We took advantage of that property, along with the late appearance of fused cells, to classify cells within an image of a VZV-infected monolayer into (i) bystander uninfected cells, (ii) newly infected cells (with IE62 only in the nucleus), (iii) infected cells (with IE62 in the cytoplasm), and finally, (iv) syncytial cells (illustrated in Fig. 2A and B). After classifying all cells within an image, the LC3-positive puncta per cell or syncytium were enumerated. This procedure was carried out with at least 16 images at a magnification of ×400 at three time points: 24, 36, and 72 hpi. As expected, the number of VZV-induced syncytial cells was the highest at 72 hpi, when over 70% of the cells in the monolayer were infected (Fig. 2C). As a general observation, all VZV-infected cells (newly infected, infected, or syncytial) exhibited more LC3-positive puncta than the uninfected bystander cells (Fig. 2D). When these categories were separately analyzed, newly infected cells (with IE62 only in the nucleus) exhibited more puncta per cell than cells at later phases of infection. In other words, as the infectious cycle progressed within an individual cell, the number of LC3-positive puncta was moderated but remained above 4. Even late in the infectious cycle, when syncytium formation was evident, a constant but elevated level of LC3-positive puncta was observed. Compared with starvation conditions (Fig. 1), VZV infection generally induced more puncta. Compared with tunicamycin treatment (Fig. 1), there was an overlap in the number of puncta, often reaching 20 per cell, although infection rarely induced numbers of puncta as high as 50. Of note, punctum induction by trehalose was similar in degree to that by VZV infection (compare Fig. 1G with Fig. 2D).
FIG 2.

Enumeration of autophagosomes during the entire VZV infectious cycle. MRC-5 fibroblasts were plated onto glass coverslips in tissue culture dishes and then inoculated with VZV-32-infected cells and incubated for several different periods of time before fixing, blocking, and labeling for LC3B and the VZV IE62 protein. Sixteen or more images were taken at a magnification of ×400; cells were classified by their extent of infection: uninfected bystander cells, newly infected cells (IE62 in the nucleus but not in the cytoplasm), infected cells (IE62 in the cytoplasm), and syncytial cells (IE62 in cytoplasm of fused cells). Then, the number of LC3B puncta per cell was counted. (A) The classification of cells by their extent of infection is illustrated by color coding of nuclei: 9 uninfected bystander cells around the periphery, 17 newly infected cells, 7 infected cells, and 11 syncytial cells in a representative image fixed at 36 hpi. (B) VZV IE62 immunolabeling of the cells in panel A. Note the cytoplasmic staining of IE62 in the cells classified as infected and syncytial. (C) The distribution of classified cells from 24 to 72 hpi documented the progression of the infection through the cellular monolayer. un, uninfected. (D) Distributions of classified cells with a given number of LC3B puncta at 24, 36, and 72 hpi. The graphs for uninfected cells show that bystander uninfected cells exhibited slightly more puncta than truly uninfected cells, while newly infected cells showed numerous puncta (4 to 10) early; infected cells and syncytial cells (late infection) showed a wider range of increased numbers of puncta.
Decreased VZV infectivity following treatment with 3-MA.
Tunicamycin initiates cellular stress because it blocks formation of N-linked glycans in the ER (60, 63). We have previously demonstrated that tunicamycin treatment inhibits the formation of infectious VZV particles, because processing of the viral glycoproteins required for final envelopment is disrupted and the misfolded proteins accumulate in the ER (57). As noted above, the compound 3-MA used in the above-described experiments inhibits autophagy. Because autophagy is easily detectable during a productive VZV infection in cultured cells (42), we postulated that treatment with an autophagy-inhibitory compound would also inhibit production of infectious VZV. The first experiment to assay this question required treatment of VZV-infected cells with 3-MA at 6 hpi, with the 3-MA being refreshed every 24 h. Treatment with 3-MA disrupted the typical spread of cell-associated VZV within a monolayer. Rather than undergoing a gradual transition after infection from being newly infected to later syncytium (polykaryon) formation, 3-MA-treated cells were clumped together without the obvious formation of large syncytia (Fig. 3A). The presence of VZV gE indicated that viral replication was not completely blocked. However, 3-MA treatment induced many more VZV-infected cells to become rounded up or pyknotic (Fig. 3B). In contrast, 3-MA treatment of uninfected fibroblasts did not induce the same degree of cytopathology (data not shown). To measure the effect of 3-MA on VZV infectivity, we carried out a plaque assay using cell-free VZV inocula and found that 3-MA profoundly reduced VZV titers (Fig. 3C; P < 0.0001). To confirm that the 3-MA inhibitory effect could also be demonstrated within infected cells that had not been sonically disrupted, we performed an infectious focus assay at 48 and 72 hpi, using treated and untreated infected cells, and found nearly identical results (Fig. 3D). The two sets of titration results with the 3-MA inhibitor of autophagy presented above strongly suggest that autophagy is a critical component during the infectious cycle.
FIG 3.

Inhibition of VZV infectivity following 3-MA treatment. VZV-32-infected monolayers were prepared as described in the legend to Fig. 2. At 6 hpi, half of the wells were treated with 10 mM 3-MA, which was refreshed every 24 h. At 72 hpi, the cells were fixed and labeled with antibodies against VZV gE and LC3B. (A) Confocal fluorescent images of 3-MA-treated versus untreated VZV-infected cells. 3-MA treatment inhibited the progression of syncytial cytopathology. (B) Effect of 3-MA treatment on the infected cell phenotype. Treatment with 3-MA increased the number of rolled up or pyknotic cells within a given image. (C) Titers of cell-free VZV measured by plaque assay. (D) Titers of VZV infectious foci measured by infectious focus assay. Both titration assays showed significantly reduced VZV titers following 3-MA treatment (P < 0.0001 at 72 hpi with either inoculum; panel C, n = 18; panel D, n = 12).
Decreased VZV gE production within the infectivity fraction after 3-MA treatment and density gradient sedimentation.
We have previously defined methods for density gradient sedimentation and fractionation to obtain a single infectivity fraction containing mainly infectious VZV particles (50). These methods required considerable experimentation because VZV is renowned for its low titer in cultured cells as well as the fragility of its viral envelope when subjected to gradient sedimentation. Based on the results presented above, we postulated that the decreased titers after 3-MA treatment may be a consequence of diminished viral glycoprotein production. Since VZV gE is the best characterized of the VZV envelope proteins, we next investigated gE as a representative viral glycoprotein. To examine and compare the amounts of gE within an infectivity fraction, we subjected treated and untreated infected cultures to one potassium tartrate-glycerol density gradient sedimentation, followed by analysis of the different fractions by SDS-PAGE with immunoblotting for the easily detectable gE glycoprotein (Fig. 4A to E). In the first experiment, we repeated the above-described experiment in which cultures were treated with 3-MA. When the control and treated cultures were harvested and analyzed, we showed that the infectivity fraction had decreased in width in the 3-MA-treated culture and had become less dense (compare fraction 4a in the 3-MA gradient with fractions 4a and 4b in the control gradient). When the infectivity fractions from each gradient were analyzed by SDS-PAGE and immunoblotting, the gE band was decreased in intensity in the treated fraction; also, the gE dimeric forms (at about 200 kDa, marked by black circles in Fig. 4A to E) were virtually absent from the treated fraction. When gE is produced in abundance in infected cells, there is a broad range of mature forms, including dimers and possible trimers (69, 70). The ORF41 blot was performed as a positive control for the infectivity fraction, since ORF41 is a small capsid protein that is not found within other fractions after density gradient sedimentation (71).
FIG 4.

Diminished VZV gE in the infectivity fraction following 3-MA treatment. VZV-infected monolayers were incubated with complete MEM with or without 3-MA. At 72 hpi, the monolayers were harvested, sonically disrupted, and subjected to glycerol-potassium tartrate density sedimentation. (A) Six fractions were collected from each density gradient. The fractions were diluted in PBS and centrifuged at 100 × g for 1 h. The pellets were resuspended in PBS and mixed with SDS-PAGE sample buffer. The infectivity fractions (fractions 4a and 4b) are outlined by a bracket on the right. (B to E) Each fraction was probed by immunoblotting with antibodies against VZV gE and VZV ORF41 protein. ORF41 was found in the infectivity fraction, while gE was found both in the infectivity fraction and in light particles (envelopes without capsids) that sediment throughout the less dense fractions, fractions 1 to 3. Results for four exposure times, 1 s (B), 15 s (C), 1 min (D), and 30 min (E), of each Western blot are shown. The expected bands of each protein are noted by black arrows. The fraction number is indicated below each lane. Molecular mass markers (kDa) are indicated in the left margin of each blot. Note the higher gE forms (200 kDa) in fractions 4a and 4b (black circles).
Increased gE production within the infectivity fraction after trehalose treatment and density gradient sedimentation.
Based on the trehalose data shown in Fig. 1, we postulated that this inducer of autophagy would increase gE production, in contradistinction to the effect of 3-MA shown in Fig. 4. To this end, we repeated the VZV infections in cultured cells, treating one set of cultures with trehalose and leaving the second set as a control (Fig. 5). Thereafter, we processed the infected cells for gradient sedimentation and for SDS-PAGE and immunoblotting. The results showed a marked increase in gE production in the infectivity fraction, following trehalose treatment (Fig. 5B, right, trehalose, lane 4). The overly abundant amounts of the gE protein facilitated detection of higher-molecular-mass gE forms (Fig. 5B, right, trehalose, lane 4).
FIG 5.

Increased VZV gE in the infectivity fraction following trehalose treatment. VZV-infected monolayers were incubated with complete MEM with or without 100 mM trehalose. At 72 hpi, the monolayers were harvested and sonically disrupted. The sonicated samples were sedimented on glycerol-potassium tartrate density gradients. (A) Six fractions were collected from each density gradient. The fractions were diluted in PBS and centrifuged at 100 × g for 1 h. The pellets were resuspended in PBS and mixed with SDS-PAGE sample buffer. Infectivity fraction 4 is outlined by a bracket on the right. (B) Each fraction was probed by immunoblotting with antibodies against VZV gE and VZV ORF41 protein. The expected band of each protein is marked by a black arrow. The fraction number is indicated below each lane. Molecular mass markers (kDa) are indicated in the left margin of each blot. Note the dramatic increase in VZV gE in the trehalose-treated virion fraction (lane 4), visible after a 2-s exposure.
Reduced cytopathic effect following 3-MA treatment of VZV-infected melanoma cells.
Because of the above-described dramatic effects of inhibitors and enhancers of autophagy on VZV glycoprotein biosynthesis, we pursued further confirmatory studies of the related effects of autophagy in our virus-cell systems (49). VZV glycoprotein production is especially prolific in the ER/Golgi apparatus of melanoma cells, more so than in the ER/Golgi apparatus of fibroblast cells, and leads to extensive syncytium formation (72). In fact, very little biosynthesis of cellular glycoproteins secondary to the abundant viral glycoprotein biosynthesis is detectable within 24 hpi (72). Because of these older observations, we again postulated that inhibition of autophagy by 3-MA in VZV-infected melanoma cells would reduce biosynthesis of the viral glycoproteins and thereby reduce the glycoprotein-dependent cytopathic effect (CPE). To test this hypothesis, we performed two experiments to examine VZV glycoprotein biosynthesis under conditions of 3-MA treatment. In the first set of experiments, we captured images of the CPE in infected and treated cultures (Fig. 6A). The VZV-induced CPE in melanoma cells was very distinctive, with the formation of large syncytia eventually occurring in the entire monolayer. These syncytia were induced by the viral fusogenic gH-gL and gB complexes, although viral spread preceding syncytium formation requires the gE-gI complex (73, 74). As can be seen in the micrographs, cultures treated with 3-MA had virtually no syncytium formation even late in infection (Fig. 6A; compare the two panels for 96 hpi). To delineate the amount of glycoprotein biosynthesis between treated and untreated infected cultures, we examined infected cells by confocal fluorescence microscopy and found that less gE was expressed when cells were treated with 3-MA (Fig. 6B). To observe the diminished glycoprotein biosynthesis following 3-MA treatment within an entire cell in more detail, we created three-dimensional (3D) animations of control and treated cells using Imaris software; representative two-dimensional (2D) images are shown in Fig. 6C. The diminished syncytium size after 3-MA treatment was also evident in the 3D animations (Fig. 6D). Finally, the gE pixel intensity over an entire monolayer, as quantitated using the Measure tool of ImageJ, was statistically significantly less in 3-MA-treated monolayers (Fig. 6E; P < 0.001).
FIG 6.
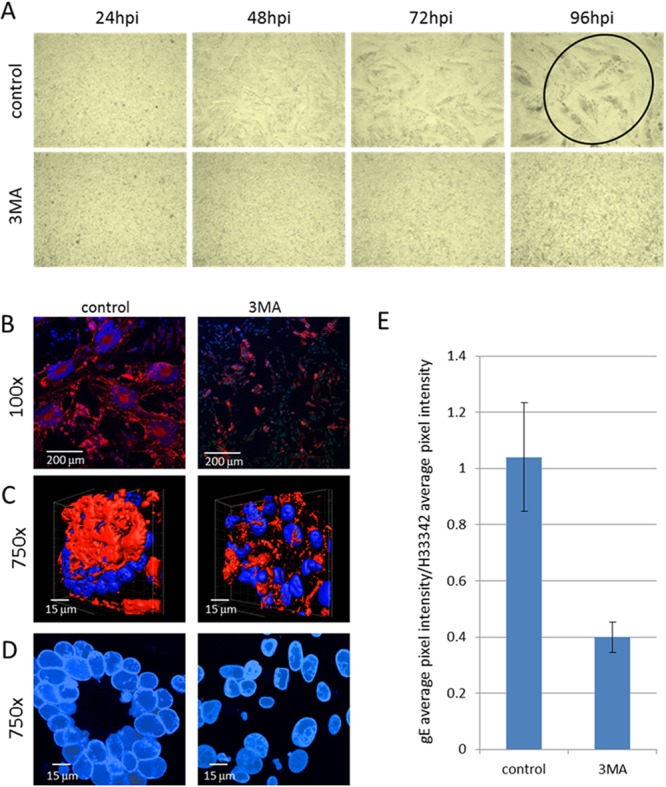
Reduced VZV gE expression and syncytium size following 3-MA treatment. VZV-infected melanoma cell monolayers were incubated with MEM with or without 3-MA. (A) Images of live cells were collected with a light microscope at 24, 48, 72, and 96 hpi. Magnification, ×40. Note the complete CPE at 96 hpi in the control infected sample (circled), while the corresponding treated sample had a minimal CPE. (B) Confocal microscopy. At 72 hpi, the cells were fixed and stained with an anti-VZV gE MAb and Hoechst 33342 DNA stain. Images of the fixed cells were collected on a laser scanning confocal microscope. Magnification, ×100. (C) 3D animations. In order to show the decreased gE production within entire cells, stacks of confocal images were converted into 3D animations. Single images from the 3D animations are shown in panel C. Nuclei are blue, and total gE expression is shown in red. (D) Syncytium sizes. 2D images from the animations were collected in order to show the differences in syncytium sizes (polykaryons) between control and treated cells. (E) Pixel intensities of VZV gE. ImageJ software was used to obtain the average pixel intensities of gE and Hoechst 33342 labeling in confocal images. Magnification, ×100. The ratio of the gE average pixel intensity over the H33342 average pixel intensity is shown in panel E (P < 0.001; n = 4 images).
Decreased syncytium size after knockdown of ATG5 by siRNA in VZV-infected cells.
In order to further investigate the role of autophagy during the VZV infectious cycle, we postulated that inhibition of autophagy with siRNA would provide an alternative strategy by which to confirm the autophagy results obtained with 3-MA treatment. Because the ATG5 protein is essential for the maturation and closure of the autophagosome, we selected ATG5 siRNA for these experiments (75). As noted in Materials and Methods, MeWo cell monolayers were transfected for a total of 48 h with either ATG5 siRNA or a control (nontargeting/scrambled) siRNA and then inoculated with VZV-32-infected cells and incubated for a further 72 h; a set of infected but nontransfected control monolayers was also included (Fig. 7). In order to confirm successful knockdown of the ATG5 protein after siRNA transfections, samples were analyzed for ATG5 protein by Western blotting; we detected >50% knockdown of the ATG5-ATG12 protein complex by Western blotting, whereas no significant knockdown was detected in either the no-siRNA control or the nontargeting siRNA control (Fig. 7E).
FIG 7.

Reduced VZV cytopathic effect following ATG5 knockdown. Melanoma cells were plated onto coverslips in a 6-well dish. At 60% confluence, the wells were transfected with control B (ctrl B), ATG5, or mock siRNA and then incubated for 48 h before VZV infection. At 72 hpi, the cells were fixed and stained with the Hoechst 33342 DNA stain and viewed on a Zeiss 710 confocal microscope. The nuclei in more than 20 polykaryons in 15 images were enumerated for each condition using ImageJ software. (A to C) Representative images of the nuclei of VZV-infected cells that were transfected with no siRNA (A), ATG5 siRNA (B), or control siRNA (C). Magnifications, ×400. (D) Number of nuclei per syncytium, with error bars representing SEMs. Infected cells with ATG5 knockdown exhibited smaller syncytia with significantly fewer nuclei. ***, P < 0.001 (n = 24 syncytia). (E) Western blot of ATG5. Immunoblotting was performed to confirm the knockdown of the ATG5 protein after siRNA transfections. Molecular mass markers (kDa) are included; beta-tubulin was used as a protein loading control.
In cells transfected with ATG5 siRNA and then infected with VZV-32, we observed a decrease in the overall size of the syncytia that formed in the monolayer (Fig. 7A to C). Again, this decrease in cell-to-cell fusion suggested decreased formation of fusogenic VZV glycoproteins (76). To quantitate this decrease in polykaryon size, we counted the number of nuclei included in each syncytium, inspecting more than 15 images under each condition at either ×200 or ×400 magnification. Under conditions of ATG5 knockdown, VZV infection induced smaller polykaryons with significantly fewer nuclei per syncytium (Fig. 7D); ATG5 siRNA versus no siRNA, P < 0.0001; ATG5 siRNA versus control siRNA, P < 0.0001. In other words, this assay confirmed that inhibition of ATG5 by siRNA inhibited VZV cell-to-cell spread by disrupting syncytium formation.
Reduced VZV glycoprotein expression after knockdown of ATG5 by siRNA in VZV-infected cells.
Based on our hypothesis that autophagy was required for efficient glycoprotein biosynthesis, we further investigated the effects of ATG5 siRNA knockdown on VZV infection using confocal microscopy to immunodetect individual viral proteins in monolayers that had been transfected. In other words, if autophagy facilitated the stress response of the infected cell to accommodate early and abundant glycoprotein-processing events within the ER, inhibition of autophagy would diminish viral glycoprotein production within the ER and subsequently within the Golgi apparatus. As a marker of VZV glycoprotein biosynthesis, we selected the predominant VZV gE-gI complex for further investigation. VZV gE, in particular, is heavily modified by both N- and O-linked glycans during its maturation; within the Golgi apparatus, gI also binds gE, and this complex formation modifies the final glycosylation of gE. When expression of the predominant gE glycoprotein was analyzed in transfected and infected cells, we observed a decrease in the amount and brightness of gE immunolabeling after ATG5 siRNA transfection compared with those of gE expression in the no-siRNA or nontargeting siRNA control transfections and infections (Fig. 8A1 to A3). After visual inspection of the images from these three sets of experiments, we concluded that VZV spread throughout a monolayer of cells was limited by ATG5 siRNA transfection prior to infection.
FIG 8.

Decreased VZV gE expression and processing following ATG5 knockdown. Cells were plated onto coverslips in a 6-well dish. At 60% confluence, the wells were transfected with either control B, ATG5, or mock siRNA and then incubated for 48 h before VZV infection. At 72 hpi, the cells were fixed and labeled with an MAb to VZV gE, gC, or IE62 (in separate wells). After washing, the cells were labeled with fluorescent secondary antibodies and the Hoechst 33342 DNA stain. After mounting on slides, the samples were viewed on a Zeiss 710 confocal microscope and a number of images were taken. Magnifications, ×200. (A1 to A3) Representative images of VZV gE expression in cells with no siRNA (A1), ATG5 siRNA (A2), and control siRNA (A3); (A4) the brightness of gE fluorescence versus the DNA was measured for each of the three conditions using ImageJ and graphed (**, P = 0.0004; ***, P = 0.0025; n = 15 images). (B1 to B3) Representative images of VZV gC expression in cells with no siRNA (B1), ATG5 siRNA (B2), and control siRNA (B3); (B4) the brightness of gC fluorescence versus that of the DNA fluorescence was measured for each of the three conditions using ImageJ and graphed (***, P < 0.0001; n = 15 images). (C1 to C3) Representative images of VZV IE62 expression in cells with no siRNA (C1), ATG5 siRNA (C2), and control siRNA (C3). (C4) The brightness of IE62 fluorescence versus that of the DNA fluorescence was measured for each of the three conditions using ImageJ and graphed (for no siRNA versus ATG5, P = 0.14; for ATG5 versus control siRNA, P = 0.18; n = 9 images). (D, E) Detection of VZV gE after precipitation with MAb 3B3 and immunoblotting with MAb 3B3 for a 5-s exposure (D) and a 10-s exposure (E). (F, G) Detection of VZV gE from lysates by Western blotting with anti-gE MAb 711. MeWo cells transfected with the indicated siRNA were then infected with VZV-32 and lysed at 72 hpi. Lysates were reduced by boiling in sample buffer containing dithiothreitol. Equal amounts of protein were loaded in each well, as determined by Bio-Rad DC protein assay, and separated by 7.5% SDS-PAGE; blots were probed with MAb 711. Note the presence of the gE dimer in ATG5 siRNA-treated MeWo cells. (F) A 10-s exposure; (G) a 20-s exposure. (H) Detection of the VZV gE-gI complex after precipitation with anti-gI MAb 6B5. Lysates were precipitated with MAb 6B5 and blotted with high-titer anti-VZV human serum. (I, J) Quantitation of Western blot band intensities for VZV gE (I) and IE62 (J) proteins. Melanoma cells were transfected and infected as described at the beginning of this legend, and lysates were reduced by boiling in dithiothreitol. Equal amounts of protein were loaded in each well, as determined by Bio-Rad DC protein assay. Lysates were separated by 7.5% SDS-PAGE and probed with MAb 3B3 (against gE) or MAb 5C6 (against IE62). Western blot films were developed and scanned, and the Measure tool from ImageJ was used to determine the intensity of the bands. The intensities of the no-siRNA bands were set to 1, and the values for ATG5 and control B are shown relative to those for no siRNA. Error bars represent SEMs. Statistical significance, determined by Student's t test, is noted for gE (***, P < 0.0001; **, P < 0.01; n = 5 blots); there was no statistically significant difference for IE62 (n = 3 blots).
To verify that impression among the three sets of images, we used ImageJ software to quantify the decrease in gE staining by measuring the pixel intensity and comparing it to the pixel intensity of DNA (from H33342 staining) (Fig. 8A4). MAb 3B3 is a particularly good probe for this pixel analysis, because of its high affinity of binding to the defined epitope on the gE N terminus (77). After measuring more than 15 images for each condition, we calculated a statistically significant decrease in the relative pixel intensity of gE immunolabeling following ATG5 knockdown compared to the gE pixel intensity under conditions with no siRNA (Fig. 8A4; P = 0.0004) or the nontargeting siRNA control (Fig. 8A4; P = 0.0025). Another glycoprotein, gC, is produced late in VZV infection and therefore is a marker for the end of the infectious cycle (Fig. 8B1 to B4). We also observed a decrease in gC expression after ATG5 siRNA transfection in VZV-infected cells (Fig. 8B4; ATG5 versus no siRNA, P < 0.0001; ATG5 versus control siRNA, P < 0.0001).
Finally, we examined the level of expression for a nonglycoprotein viral product, IE62, to determine if the knockdown of ATG5 led to a global decrease in VZV gene products or if it was specific to glycoproteins (Fig. 8C1 to C4). Knockdown of ATG5 did not lead to a significant decrease in IE62 expression after VZV infection (Fig. 8C4; ATG5 siRNA versus no siRNA, P = 0.14; ATG5 siRNA versus control siRNA, P = 0.18). To confirm the results presented above, we repeated the quantitation by immunoblotting for both the gE and the IE62 proteins under the same 3 experimental conditions. As shown in Fig. 8I and J, the production of gE was significantly less during ATG5 knockdown (ATG5 versus no siRNA, P < 0.001; ATG5 versus control siRNA, P < 0.01), while IE62 production was not affected.
Impaired VZV gE processing during ATG5 knockdown.
Because the above-described differences were confined to glycoproteins, we next postulated that diminished autophagy compromised the later stages of VZV glycoprotein processing in the Golgi apparatus. The gE glycoprotein is the most completely characterized VZV glycoprotein; it is heavily modified during its transit through the ER/Golgi apparatus (69, 78). As with many other type 1 herpesviral glycoproteins, the gE ectodomain contains high-mannose glycans that are subsequently processed into complex-type sialylated glycans in the mid- to trans-Golgi apparatus. O-linked glycans are also added in the Golgi apparatus. All of the above-described modifications led to a mature gE glycoprotein monomer with a molecular mass of 98 kDa. As shown in Fig. 8D and E, the molecular mass of the gE glycoprotein was diminished in the presence of ATG5 treatment. On the basis of numerous prior gE analyses, this result suggests diminished processing of high mannose to complex-type glycans.
Further, gE also exists as a dimer (70). The dimer is rarely seen for two reasons: it is not produced in abundance, and it is not easily detected by most anti-gE MAb probes. The presence of the gE dimer was best documented when gE expression experiments were carried out in a baculovirus system (79). Insect cells facilitate the steps required for addition of high-mannose glycans to gE but not those required for processing to complex-type glycans. Since the dimer was more easily detected in insect cells, the data indicated that gE dimerization was enhanced when gE existed in its less mature high-mannose form. As shown in Fig. 8F and G, more gE dimer production occurred under conditions of ATG5 treatment. This result strongly supports the data in Fig. 8D and E and again implies that ATG5 knockdown leads to less complete gE processing.
We could not evaluate the detailed effects of ATG5 knockdown on gC processing, because the glycosylation and intermediary gC forms have not been sufficiently well characterized (80). Instead, we postulated that the better-delineated processing steps of VZV gI (ORF 67), the type 1 glycoprotein partner of gE, would also be impaired under conditions that diminished gE processing (69). To this end, we coprecipitated the gE-gI complex with anti-gI MAb 6B5 and subjected the complex to SDS-PAGE under denaturing conditions, before immunoblotting with a polyclonal anti-VZV antiserum. As shown in Fig. 8H, the molecular masses of both gE and gI were lower in the ATG5 knockdown lane. Thus, the processing of the gI glycoprotein was also impaired by inhibition of autophagy.
DISCUSSION
In this report, we expand upon the proviral role of autophagy during the VZV infectious cycle in cultured cells, as noted in two earlier publications (4, 42). When we previously studied VZV-induced autophagy in both fibroblasts and melanoma cells, we observed that autophagosome formation occurred earlier after infection in melanoma cells than in fibroblasts but the totality of the response was similar. Now we present the following observations: (i) compared with known inducers of autophagy, serum starvation and tunicamycin, VZV-induced autophagy, as assayed by enumeration of LC3-positve puncta, was greater than that induced by serum starvation and less than that induced by tunicamycin; (ii) inhibition of VZV-induced autophagy reduced infectivity; (iii) inhibition of autophagy reduced syncytium formation in infected monolayers; and (iv) inhibition of autophagy reduced the glycosylation and maturation of the VZV gE-gI complex. The above-described effects following inhibition of autophagy within the VZV system fit with an evolving consensus that the core components required for formation of the preautophagosome (phagophore) are recruited to the ER (81); in other words, adverse effects on autophagy in the ER also impair glycoprotein processing. We also investigated the effects of the disaccharide trehalose, a newly discovered inducer of autophagy (61), and confirmed (i) that treatment with trehalose induced abundant autophagosome formation in uninfected cells, while (ii) similar treatment of VZV-infected cells led to enhanced VZV gE biosynthesis and maturation. Both VZV glycoprotein gE and its partner, gI, harbor tyrosine-based motifs in their endodomains, allowing them to undergo endocytosis and recycling after they traffic to the outer cell membrane (82). Thus, the findings of the current study support a suspected intersection between autophagy and glycoprotein trafficking pathways within a cell (81).
We had previously investigated autophagic flux by measurement of the level of polyubiquitin-binding protein p62, the protein product of the sequestosome 1 (SQSTM1) gene found on human chromosome 5 (42). Polymerization of p62 forms protein aggregates that are degraded over time within autophagosomes (83). Compared with uninfected cells, p62 levels were lower in VZV-infected cells when measured by immunoblotting, a result that suggested degradation of p62 as part of autophagic flux during VZV infection. Treatment of a VZV-infected monolayer with 3-MA should halt this degradation process, a result that points to autophagy as a major explanation for protein degradation in infected cells (54).
In subsequent experiments, we observed that 3-MA treatment diminished the appearance of a cytopathic effect (CPE) in infected monolayers. Since a CPE is heavily dependent on the presence of fusogenic viral glycoproteins in the outer cell membrane (76), we probed for viral glycoproteins in the presence of 3-MA treatment and documented less viral glycoprotein biosynthesis. To test our hypothesis that 3-MA treatment reduced VZV titers, thereby reducing the CPE, we titrated the amount of infectious virus at increasing intervals postinfection in treated and untreated VZV-infected monolayers and observed a significantly lower titer in the 3-MA-treated cells. Further, we examined the viral glycoprotein profile of the infectivity fraction purified by density gradient sedimentation after 3-MA treatment and confirmed that production of the representative gE glycoprotein was diminished.
Based on the above-described observations, we postulated that directed inhibition of autophagosome formation should impair viral glycoprotein biosynthesis. As noted, the targeting of LC3-II to phagophores requires another conjugation system, in which ATG12-ATG5 complexes are noncovalently linked with ATG16L to form a multimeric complex (84, 85). In our experiments with ATG5 siRNA, we mainly investigated gE (ORF68), considered to be an early late protein, because gE is also one of the most abundantly produced viral glycoproteins; therefore, gE is invariably easy to detect and any reduction would be apparent (69). Both the biosynthesis and processing of VZV gE were diminished under conditions of autophagy inhibition. Further, the ATG5 knockdown results supported our argument that the 3-MA data accurately reflected a reduction in autophagy.
Taken together, the totality of our results strongly support our hypothesis that autophagy within the VZV system promotes cell survival by relieving the ER stress generated, at least in part, by the abundant biosynthesis of VZV glycoproteins. When ER stress reaches a sufficiently heightened level, autophagy provides an auxiliary pathway for the relief of ER stress (86, 87). A similar concept of ER stress secondary to abundant accumulation of viral structural glycoproteins has recently been documented during the life cycle of canine distemper virus (CDV) (88). This enveloped RNA virus, which is closely related to measles virus, is strongly cell associated during its replication cycle, similar to VZV. The CDV investigators delineated markers of an enlarged ER accompanied by ER stress both in infected cells and after transient expression of the two CDV glycoproteins, but they did not study the effects on autophagy.
ACKNOWLEDGMENTS
We acknowledge the University of Iowa Central Microscopy Research Facility for use of equipment.
The research was supported by NIH grant AI89716.
Footnotes
Published ahead of print 6 November 2013
REFERENCES
- 1.Weller TH. 1983. Varicella and herpes zoster. Changing concepts of the natural history, control, and importance of a not-so-benign virus. N. Engl. J. Med. 309:1434–1440 [DOI] [PubMed] [Google Scholar]
- 2.Ku CC, Besser J, Abendroth A, Grose C, Arvin AM. 2005. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J. Virol. 79:2651–2658. 10.1128/JVI.79.5.2651-2658.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grose C. 2010. Autophagy during common bacterial and viral infections of children. Pediatr. Infect. Dis. J. 29:1040–1042. 10.1097/INF.0b013e3181e77f43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter JE, Jackson W, Benetti L, Grose C. 2011. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 85:9414–9424. 10.1128/JVI.00281-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudchodkar SB, Levine B. 2009. Viruses and autophagy. Rev. Med. Virol. 19:359–378. 10.1002/rmv.630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, Green KY, Lopez-Otin C, Xavier RJ, Thackray LB, Virgin HW. 2012. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 11:397–409. 10.1016/j.chom.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Klionsky DJ. 2009. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335:1–32. 10.1007/978-3-642-00302-8_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147:728–741. 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 9.Kiel JA. 2010. Autophagy in unicellular eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365:819–830. 10.1098/rstb.2009.0237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McEwan DG, Dikic I. 2011. The Three Musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 21:195–201. 10.1016/j.tcb.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. 2004. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117:2805–2812. 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- 12.Furuya N, Yu J, Byfield M, Pattingre S, Levine B. 2005. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1:46–52. 10.4161/auto.1.1.1542 [DOI] [PubMed] [Google Scholar]
- 13.Funderburk SF, Wang QJ, Yue Z. 2010. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 20:355–362. 10.1016/j.tcb.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskaran S, Ragusa MJ, Hurley JH. 2012. How Atg18 and the WIPIs sense phosphatidylinositol 3-phosphate. Autophagy 8:1851–1852. 10.4161/auto.22077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. 2003. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116:1679–1688. 10.1242/jcs.00381 [DOI] [PubMed] [Google Scholar]
- 16.Seglen PO, Gordon PB. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 79:1889–1892. 10.1073/pnas.79.6.1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275:992–998. 10.1074/jbc.275.2.992 [DOI] [PubMed] [Google Scholar]
- 18.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. 2005. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25:1025–1040. 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051. 10.1073/pnas.0907344106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi X, Zhang X, Wang B, Wang T, Wang J, Huang H, Wang J, Jin Q, Zhao Z. 2013. The interplays between autophagy and apoptosis induced by enterovirus 71. PLoS One 8:e56966. 10.1371/journal.pone.0056966 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Alirezaei M, Flynn CT, Whitton JL. 2012. Interactions between enteroviruses and autophagy in vivo. Autophagy 8:973–975. 10.4161/auto.20160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang SW, Ducroux A, Jeang KT, Neuveut C. 2012. Impact of cellular autophagy on viruses: insights from hepatitis B virus and human retroviruses. J. Biomed. Sci. 19:92. 10.1186/1423-0127-19-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B. 2013. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. 10.1038/nature11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talloczy Z, Jiang W, Virgin HW, IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190–195. 10.1073/pnas.012485299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. 2007. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81:12128–12134. 10.1128/JVI.01356-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexander DE, Leib DA. 2008. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 4:101–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talloczy Z, Virgin HW, IV, Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29 [DOI] [PubMed] [Google Scholar]
- 28.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- 29.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippe R, Desjardins M. 2009. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10:480–487. 10.1038/ni.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S. 2005. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U. S. A. 102:7922–7927. 10.1073/pnas.0501190102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549. 10.1016/j.chom.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. 2009. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 83:12164–12171. 10.1128/JVI.01676-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tey SK, Khanna R. 2012. Host immune system strikes back: autophagy-mediated antigen presentation bypasses viral blockade of the classic MHC class I processing pathway. Autophagy 8:1839–1841. 10.4161/auto.21860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gobeil PA, Leib DA. 2012. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 3(5):e00267–12. 10.1128/mBio.00267-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. 2012. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12:334–345. 10.1016/j.chom.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valencia SM, Hutt-Fletcher LM. 2012. Important but differential roles for actin in trafficking of Epstein-Barr virus in B cells and epithelial cells. J. Virol. 86:2–10. 10.1128/JVI.05883-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasmussen SB, Horan KA, Holm CK, Stranks AJ, Mettenleiter TC, Simon AK, Jensen SB, Rixon FJ, He B, Paludan SR. 2011. Activation of autophagy by alpha-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J. Immunol. 187:5268–5276. 10.4049/jimmunol.1100949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 85:4212–4221. 10.1128/JVI.02435-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. 2012. Subversion of autophagy by Kaposi's sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 11:167–180. 10.1016/j.chom.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 40.Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. 2013. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 87:859–871. 10.1128/JVI.01158-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759–1816. 10.1099/0022-1317-67-9-1759 [DOI] [PubMed] [Google Scholar]
- 42.Takahashi MN, Jackson W, Laird DT, Culp TD, Grose C, Haynes JI, II, Benetti L. 2009. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J. Virol. 83:5466–5476. 10.1128/JVI.02670-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Desloges N, Rahaus M, Wolff MH. 2005. Role of the protein kinase PKR in the inhibition of varicella-zoster virus replication by beta interferon and gamma interferon. J. Gen. Virol. 86:1–6. 10.1099/vir.0.80466-0 [DOI] [PubMed] [Google Scholar]
- 44.Ambagala AP, Cohen JI. 2007. Varicella-zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J. Virol. 81:7844–7851. 10.1128/JVI.00325-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGeoch DJ, Rixon FJ, Davison AJ. 2006. Topics in herpesvirus genomics and evolution. Virus Res. 117:90–104. 10.1016/j.virusres.2006.01.002 [DOI] [PubMed] [Google Scholar]
- 46.Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890–6899. 10.1128/JVI.79.11.6890-6899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the HCMV assembly compartment. J. Virol. 83:11421–11428. 10.1128/JVI.00762-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters GA, Tyler SD, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. 2006. A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J. Virol. 80:9850–9860. 10.1128/JVI.00715-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grose C, Brunel PA. 1978. Varicella-zoster virus: isolation and propagation in human melanoma cells at 36 and 32 degrees C. Infect. Immun. 19:199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grose C, Perrotta DM, Brunell PA, Smith GC. 1979. Cell-free varicella-zoster virus in cultured human melanoma cells. J. Gen. Virol. 43:15–27. 10.1099/0022-1317-43-1-15 [DOI] [PubMed] [Google Scholar]
- 51.Storlie J, Carpenter JE, Jackson W, Grose C. 2008. Discordant varicella-zoster virus glycoprotein C expression and localization between cultured cells and human skin vesicles. Virology 382:171–181. 10.1016/j.virol.2008.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grose C, Edwards DP, Weigle KA, Friedrichs WE, McGuire WL. 1984. Varicella-zoster virus-specific gp140: a highly immunogenic and disulfide-linked structural glycoprotein. Virology 132:138–146. 10.1016/0042-6822(84)90098-9 [DOI] [PubMed] [Google Scholar]
- 53.Grose C, Friedrichs WE. 1982. Immunoprecipitable polypeptides specified by varicella-zoster virus. Virology 118:86–95. 10.1016/0042-6822(82)90322-1 [DOI] [PubMed] [Google Scholar]
- 54.Klionsky DJ, Cuervo AM, Seglen PO. 2007. Methods for monitoring autophagy from yeast to human. Autophagy 3:181–206 [DOI] [PubMed] [Google Scholar]
- 55.Jackson W, Yamada M, Moninger T, Grose C. 2013. Visualization and quantitation of abundant macroautophagy in virus-infected cells by confocal three-dimensional fluorescence imaging. J. Virol. Methods 193:244–250. 10.1016/j.jviromet.2013.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carpenter JE, Jackson W, de Souza GA, Haarr L, Grose C. 2010. Insulin-degrading enzyme binds to the nonglycosylated precursor of varicella-zoster virus gE protein found in the endoplasmic reticulum. J. Virol. 84:847–855. 10.1128/JVI.01801-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montalvo EA, Parmley RT, Grose C. 1985. Structural analysis of the varicella-zoster virus gp98-gp62 complex: posttranslational addition of N-linked and O-linked oligosaccharide moieties. J. Virol. 53:761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. 2007. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 282:5641–5652. 10.1074/jbc.M609532200 [DOI] [PubMed] [Google Scholar]
- 59.Abeliovich H, Klionsky DJ. 2001. Autophagy in yeast: mechanistic insights and physiological function. Microbiol. Mol. Biol. Rev. 65:463–479. 10.1128/MMBR.65.3.463-479.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. 2006. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281:30299–30304. 10.1074/jbc.M607007200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suarez AL, Kong R, George T, He L, Yue Z, van Dyk LF. 2011. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J. Virol. 85:6293–6308. 10.1128/JVI.00001-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jain NK, Roy I. 2009. Effect of trehalose on protein structure. Protein Sci. 18:24–36. 10.1002/pro.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tkacz JS, Lampen O. 1975. Tunicamycin inhibition of polyisoprenyl N-acetylglucosaminyl pyrophosphate formation in calf-liver microsomes. Biochem. Biophys. Res. Commun. 65:248–257. 10.1016/S0006-291X(75)80086-6 [DOI] [PubMed] [Google Scholar]
- 64.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tra T, Gong L, Kao LP, Li XL, Grandela C, Devenish RJ, Wolvetang E, Prescott M. 2011. Autophagy in human embryonic stem cells. PLoS One 6:e27485. 10.1371/journal.pone.0027485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 83:3904–3918. 10.1128/JVI.02137-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harson R, Grose C. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eisfeld AJ, Turse SE, Jackson SA, Lerner EC, Kinchington PR. 2006. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J. Virol. 80:1710–1723. 10.1128/JVI.80.4.1710-1723.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grose C. 2002. The predominant varicella-zoster virus gE and gI glycoprotein complex, p 195–223 In Holzenburg A, Bogner E. (ed), Structure-function relationships of human pathogenic viruses. Kluwer Academic Press, New York, NY [Google Scholar]
- 70.Grose C, Vleck S, Karlsen OA, Montalvo EA. 2011. Structure-function profiles of the nine varicella-zoster virus glycoproteins, p 153–174 In Weller SK. (ed), Alphaherpesviruses. Molecular virology. Caister Press, Norfolk, United Kingdom [Google Scholar]
- 71.Friedrichs WE, Grose C. 1986. Varicella-zoster virus p32/p36 complex is present in both the viral capsid and the nuclear matrix of the infected cell. J. Virol. 57:155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grose C. 1980. The synthesis of glycoproteins in human melanoma cells infected with varicella-zoster virus. Virology 101:1–9. 10.1016/0042-6822(80)90478-X [DOI] [PubMed] [Google Scholar]
- 73.Cole NL, Grose C. 2003. Membrane fusion mediated by herpesvirus glycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13:207–222. 10.1002/rmv.377 [DOI] [PubMed] [Google Scholar]
- 74.Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:1911–1916. 10.1073/pnas.1216985110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668. 10.1083/jcb.152.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duus KM, Hatfield C, Grose C. 1995. Cell surface expression and fusion by the varicella-zoster virus gH:gL glycoprotein complex: analysis by laser scanning confocal microscopy. Virology 210:429–440. 10.1006/viro.1995.1359 [DOI] [PubMed] [Google Scholar]
- 77.Santos RA, Hatfield CC, Cole NL, Padilla JA, Moffat JF, Arvin AM, Ruyechan WT, Hay J, Grose C. 2000. Varicella-zoster virus gE escape mutant VZV-MSP exhibits an accelerated cell-to-cell spread phenotype in both infected cell cultures and SCID-hu mice. Virology 275:306–317. 10.1006/viro.2000.0507 [DOI] [PubMed] [Google Scholar]
- 78.Grose C. 1990. Glycoproteins encoded by varicella-zoster virus: biosynthesis, phosphorylation, and intracellular trafficking. Annu. Rev. Microbiol. 44:59–80. 10.1146/annurev.mi.44.100190.000423 [DOI] [PubMed] [Google Scholar]
- 79.Olson JK, Bishop GA, Grose C. 1997. Varicella-zoster virus Fc receptor gE glycoprotein: serine/threonine and tyrosine phosphorylation of monomeric and dimeric forms. J. Virol. 71:110–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grose C, Carpenter JE, Jackson W, Duus KM. 2010. Overview of varicella-zoster virus glycoproteins gC, gH and gL. Curr. Top. Microbiol. Immunol. 342:113–128. 10.1007/82_2009_4 [DOI] [PubMed] [Google Scholar]
- 81.Lamb CA, Dooley HC, Tooze SA. 2013. Endocytosis and autophagy: shared machinery for degradation. Bioessays 35:34–45. 10.1002/bies.201200130 [DOI] [PubMed] [Google Scholar]
- 82.Olson JK, Grose C. 1998. Complex formation facilitates endocytosis of the varicella-zoster virus gE:gI Fc receptor. J. Virol. 72:1542–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614. 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. 2007. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282:37298–37302. 10.1074/jbc.C700195200 [DOI] [PubMed] [Google Scholar]
- 85.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. 2008. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19:2092–2100. 10.1091/mbc.E07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yorimitsu T, Klionsky DJ. 2007. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3:160–162 [DOI] [PubMed] [Google Scholar]
- 87.He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93. 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brunner JM, Plattet P, Doucey MA, Rosso L, Curie T, Montagner A, Wittek R, Vandelvelde M, Zurbriggen A, Hirling H, Desvergne B. 2012. Morbillivirus glycoprotein expression induces ER stress, alters Ca2+ homeostasis and results in the release of vasostatin. PLoS One 7:e32803. 10.1371/journal.pone.0032803 [DOI] [PMC free article] [PubMed] [Google Scholar]