

Abstract
We previously reported that exosomal transfer of hepatitis C virus (HCV) positive-strand RNA from human Huh-7 hepatoma cells to human plasmacytoid dendritic cells (pDCs) triggers pDC alpha/beta interferon (IFN-α/β) production in a Toll-like receptor 7 (TLR7)-dependent, virus-independent manner. Here we show that human pDCs are also activated by a TLR7-dependent, virus-independent, exosomal RNA transfer mechanism by human and mouse hepatoma and nonhepatoma cells that replicate the negative-strand lymphocytic choriomeningitis virus (LCMV).
TEXT
Interferons (IFNs) are key mediators of the innate immune response to many viruses, including hepatitis C virus (HCV) (1) and the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) (2). Accordingly, HCV and LCMV have evolved mechanisms to block IFN induction in the infected cell (1, 2). Nevertheless, HCV and LCMV infections strongly induce IFN and IFN-stimulated gene (ISG) expression in vivo (3–7). Recently, we reported that Huh-7 cells infected with HCV or containing a subgenomic HCV replicon can trigger alpha/beta IFN (IFN-α/β) production in vitro by exosomal transfer of positive-strand HCV RNA to cocultured human peripheral blood-derived plasmacytoid dendritic cells (pDCs) in a Toll-like receptor 7 (TLR7)-dependent manner without infecting them (1, 8, 9). Here, we have extended those observations to a negative-strand RNA virus. The broad host cell range of LCMV allowed us to show that human pDCs can be activated by a wide variety of infected human and mouse cell lineages, a process that required cocultivation of pDCs and infected cells but no infection of pDCs.
LCMV is a noncytolytic enveloped virus with a bisegmented negative-strand RNA genome (1, 2, 10, 11). LCMV causes a long-term chronic infection in its natural host, the mouse. Human infections occur through mucosal exposure to aerosols or by direct contact of abraded skin with infectious material (3–7, 11). LCMV infection of humans can result in severe disease that in some cases can be fatal (12). LCMV infection of mice is associated with an initial burst of type I interferon produced in large part by infected dendritic cells (DCs) (7, 13–15). However, LCMV nucleoprotein (NP) efficiently blocks interferon regulatory factor 3 (IRF3) activation and thus IFN production in LCMV-infected cells (16). This might explain why only a small fraction of LCMV-infected dendritic cells produce IFN in the infected mice (7). Interestingly, however, IFN production also occurs in pDCs in the spleen in the absence of active LCMV replication, suggesting that pDCs can sense LCMV infection independently of virus production (7). Thus, in this study we asked if pDCs can sense LCMV-infected cells by a mechanism similar to that described for sensing of HCV-infected cells (8, 9).
Blood was collected from healthy adult human volunteers after informed consent was obtained according to procedures approved by the Scripps Research Institute Human Research Committee. In a first set of experiments, we infected Huh-7.5.1c2 cells, a subclone of the human hepatoma Huh-7 cell line that is highly permissive for HCV infection (17), with LCMV (Armstrong strain) (multiplicity of infection [MOI] = 0.1) 3 days before coculture with human peripheral blood-derived pDCs as described previously (9). The supernatant harvested after 24 h of coculturing LCMV-infected Huh-7.5.1c2 cells (2 × 105) with human pDCs (2 × 104) contained up to 100 ng/ml of IFN-α (Fig. 1A, lane 5). This was ≥10-fold higher than the amount of IFN-α produced by pDCs that had been cocultured with Huh-7.5.1c2 cells infected by the cell culture-adapted HCV JFH-1 D183 variant (9, 18) (Fig. 1A, lane 4), which correlated with the relative intracellular viral RNA levels in the HCV- and LCMV-infected cells (Table 1). Interestingly, similar amounts of IFN-α were produced in pDC cocultures with cells infected with a single-cycle recombinant LCMV (scrLCMVΔGP/GFP [33]) that cannot produce infectious virus (Fig. 1A, lane 6), suggesting that production of LCMV infectious progeny was not required to trigger IFN-α production by the pDCs. Notably, inoculation of human pDCs with a high dose (MOI = 10) of LCMV for 24 h in the absence of Huh-7 cells did not trigger IFN-α production in the pDCs (Fig. 1A, lane 7). Likewise, pDCs did not produce IFN-α after incubation with the cell culture supernatant (Fig. 1A, lane 8) of the LCMV-infected Huh-7.5.1c2 cells used for the coculture shown in lane 5 of Fig. 1A. These results indicated that production of IFN-α by pDCs did not require that they be infected by LCMV. Human pDCs incubated for 3 days with infectious LCMV were negative for LCMV nucleoprotein (NP) expression by fluorescence-activated cell sorter (FACS) analysis (data not shown), indicating that human pDCs are not likely to be productively infected by LCMV in vitro. It is noteworthy that pDC IFN-α production levels were equally robust when infected Huh-7.5.1c2 cells or infected parental Huh-7 cells were used (Fig. 1A, lane 10). Importantly, neither Huh-7 cells nor Huh-7.5.1c2 cells produced IFN-α themselves either before or after LCMV infection (Fig. 1A, lanes 11 to 13), suggesting that IFN-α production reflects activation of the cocultured human pDCs. This was confirmed by FACS analysis of cocultures of human pDCs and LCMV-infected Huh-7.5.1c2 cells (9) after staining for pDC markers HLA-DR (allophycocyanin [APC]-mouse anti-HLA-DR; eBioscience) and CD123 (phycoerythrin [PE]-Cy7-mouse anti-CD123; Biolegend) and intracellular IFN-α (PE-mouse anti-IFN-α; Miltenyi, Auburn, CA). Approximately 20% of the cocultured HLA-DR-positive (HLA-DR+) pDCs but none of the LCMV-infected HLA-DR− Huh-7.5.1c2 cells produced IFN-α (Fig. 1B) even though all of the Huh-7.5.1c2 cells were infected, as shown by LCMV NP-specific immunofluorescence (IF) analysis using an anti-NP monoclonal antibody (MAb) (1.1.3 [34]) as described previously (9) (Fig. 1B). Levels of infectious virus-independent pDC activation were similarly robust for all four different strains of LCMV tested (Fig. 1C). Together, these results demonstrate that LCMV-infected human Huh-7-derived hepatoma cells are sensed by human pDCs that respond by producing IFN-α even more strongly than when they are stimulated by HCV JFH1-infected cells (9). The lack of IFN-α production by human pDCs directly exposed to infectious LCMV virions strongly suggested that, in similarity to their response to HCV-infected cells, they likely responded to something other than the virus particles themselves.
FIG 1.
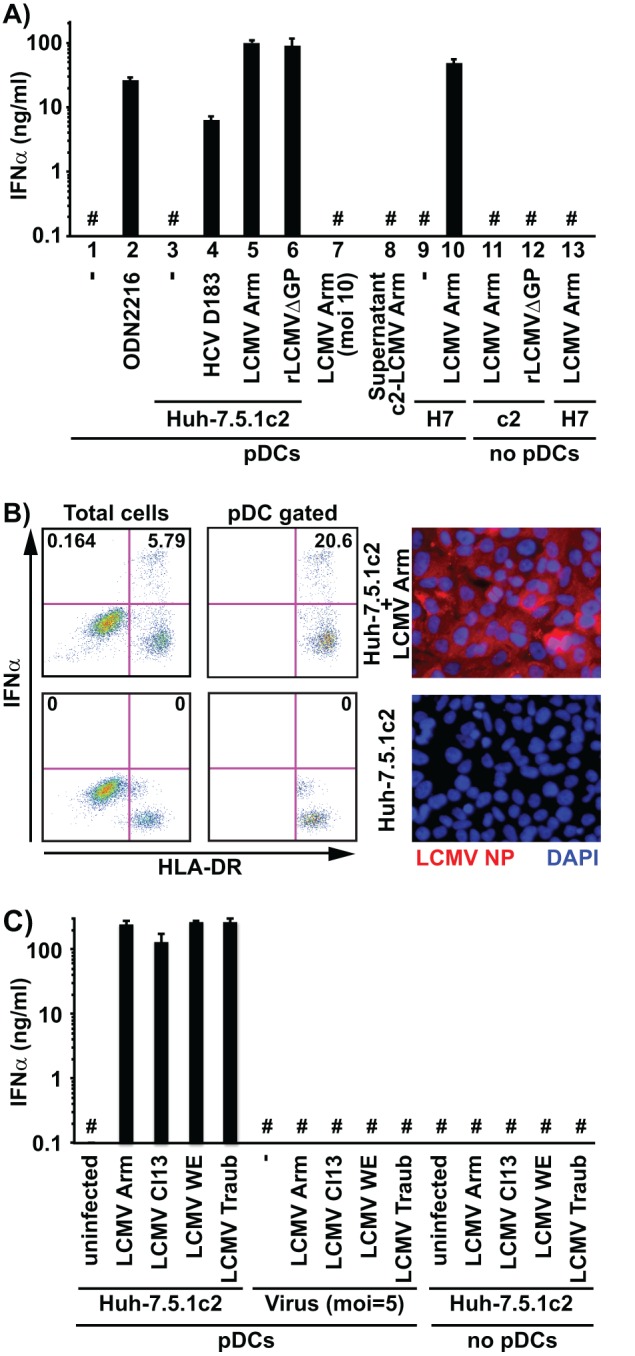
LCMV-infected Huh-7-derived cells trigger IFN-α production in cocultured human peripheral pDCs. (A) Huh-7.5.1c2 or Huh-7 cells (2 × 105) infected with LCMV or HCV D183 (18) at MOI = 0.1 3 days earlier were cocultured with 2 × 104 freshly purified human peripheral pDCs in wells of a 96-well round-bottom plate for 24 h before IFN-α was quantified in the coculture supernatant by enzyme-linked immunosorbent assay (ELISA) as described previously (9). c2, Huh-7.5.1c2 cells; H7, Huh-7 parental cells. (B) Cocultures grown as described for panel A were fixed and analyzed by FACS as described previously (9). All cells are shown in the left panels, and cells gated for HLA-DR and CD123 (pDCs) are shown in the right panels. Huh-7.5.1c2 cells infected with LCMV as described for panel A were analyzed by immunofluorescence as described previously (9) using an anti-NP MAb (1.1.3 [34]) and a secondary Alexa 555-conjugated goat anti-mouse antibody (Invitrogen). (C) IFN-α was quantified in the supernatant of cocultures of Huh-7.5.1c2 cells infected with different LCMV strains (39, 40) with pDCs as described for panel A. #, below the limit of detection of the IFN-α ELISA (36 pg/ml). Error bars represent means ± standard deviations (SD) (n = 3).
TABLE 1.
Cell lines and viruses triggering IFN-α production by pDCs
| Cell line | Reference(s) | Species | Cell of origin | Virus | Virus strain/genotype | Infection (log GE/μg RNA)a | IFN-α log (ng/ml)b |
|---|---|---|---|---|---|---|---|
| Huh-7.5.1c2 | 17, 19, 20 | Human | Hepatocyte | HCV | JFH1/2a | 7 | 1 |
| LCMV | Armstrong | 8 | 2 | ||||
| Cl13 | 8 | 2 | |||||
| WE | 8 | 2 | |||||
| Traub | 8 | 2 | |||||
| rLCMVΔGFP | 8 | 2 | |||||
| Huh-7 | 20, 21 | Human | Hepatocyte | HCV | JFH1/2a | 7 | 1 |
| LCMV | Armstrong | 8 | 2 | ||||
| HepG2 | 22 | Human | Hepatocyte | LCMV | Armstrong | 8 | 2 |
| HepG2.2.15 | 23 | Human | Hepatocyte | HBV | ayw | 7 | NDf |
| HBV + Resc | ayw/TLR7-agonist | 7 | 1 | ||||
| HBV + LCMV | ayw/Armstrong | 7/8 | 1–2 | ||||
| Hep3B | 22 | Human | Hepatocyte | LCMV | Armstrong | 8 | 2 |
| HeLa | 24 | Human | Cervix | LCMV | Armstrong | 8 | 1 |
| HEK293T | 25, 26 | Human | Embryonic kidney | LCMV | Armstrong | 8 | ND |
| PHHd | Human | Liver | LCMV | Armstrong | 7 | 1 | |
| AML12 | 27 | Mouse | Hepatocyte | LCMV | Armstrong | 8 | 0 |
| NIH 3T3 | 28 | Mouse | Embryonic fibroblast | LCMV | Armstrong | 7 | 1 |
| CV-1 | 29 | Monkey | Kidney | LCMV | Armstrong | nte | ND |
| LMH D2 | 30–32 | Chicken | Hepatocyte | DHBV | DHBV3 | 8 | ND |
| DHBV + Resc | DHBV3/TLR7-agonist | 8 | 1 | ||||
| DHBV + LCMV | DHBV3/Armstrong | 8/8 | 1 |
Approximate magnitude of intracellular viral RNA content in log genome equivalents per μg of total cellular RNA (log GE/μg RNA) at the start of the coculture with human pDCs.
Approximate magnitude of IFN-α production [in log(ng/ml)] after 24 h of coculture of the infected cell line with human pDCs.
Cocultures of HepG2.2.15 and LMH D2 cells and human pDCs were simultaneously treated with 50ng/ml of the TLR7 agonist resiquimod (Res).
Freshly isolated primary human hepatocytes (PHH); Life Technologies, Carlsbad, CA.
nt, not tested.
ND, not detected (<36 pg/ml).
Next, we asked whether production of IFN-α by human pDCs in response to coculture with LCMV-infected cells was also related to the exosome-mediated mechanism by which they respond to HCV-infected cells (8, 9). Human pDC activation by LCMV-infected Huh-7.5.1c2 cells was inhibited by the TLR7-specific antagonist IRS661 (Fig. 2A), suggesting that activation of pDCs is mediated by TLR7 as we have previously described for HCV (8, 9) and as has also been observed in the spleen of LCMV-infected mice (15). Likewise, as previously described for HCV (9), human pDC activation by LCMV-infected cells was cell-cell contact dependent since cultivation of LCMV-infected Huh-7.5.1c2 cells and human pDCs in transwell chambers did not result in detectable levels of IFN-α production by the pDCs (Fig. 2B). Next, we performed a series of experiments to determine whether LCMV-infected cell-mediated pDC activation might be exosome dependent. The two structurally unrelated exosome release inhibitors GW4869 (10 μM) and spiroepoxide (20 μM) strongly reduced the ability of LCMV-infected Huh-7.5.1c2 cells to trigger IFN-α production by human pDCs (Fig. 2C) without affecting intracellular LCMV RNA levels in the Huh-7.5.1c2 cells (data not shown). Furthermore, cytochalasin D (0.1 μg/ml), an inhibitor of actin-dependent endocytosis (35) that does not affect LCMV infection (36), completely blocked LCMV-infected cell-mediated IFN-α production by pDCs (Fig. 2C). In control experiments, GW4869, spiroepoxide, and cytochalasin D had little or no effect on TLR7 agonist (resiquimod)-triggered IFN-α production by pDCs (Fig. 2C). These results suggest that exosome release from infected cells and active endocytosis by the pDCs are required for pDC stimulation by LCMV-infected cells. These findings, together with the observation that supernatants of scrLCMVΔGP/GFP-infected cells that do not produce infectious virus nevertheless contain membrane-protected LCMV RNA (data not shown), suggest that LCMV RNA is likely to be transferred to pDCs via exosomes as we have previously described for HCV (8). These findings are consistent with the notion that, in similarity to the situation described for HCV (8, 9), human pDCs sensed LCMV-infected hepatoma cells by a short-range exosome-mediated and TLR7-dependent mechanism.
FIG 2.
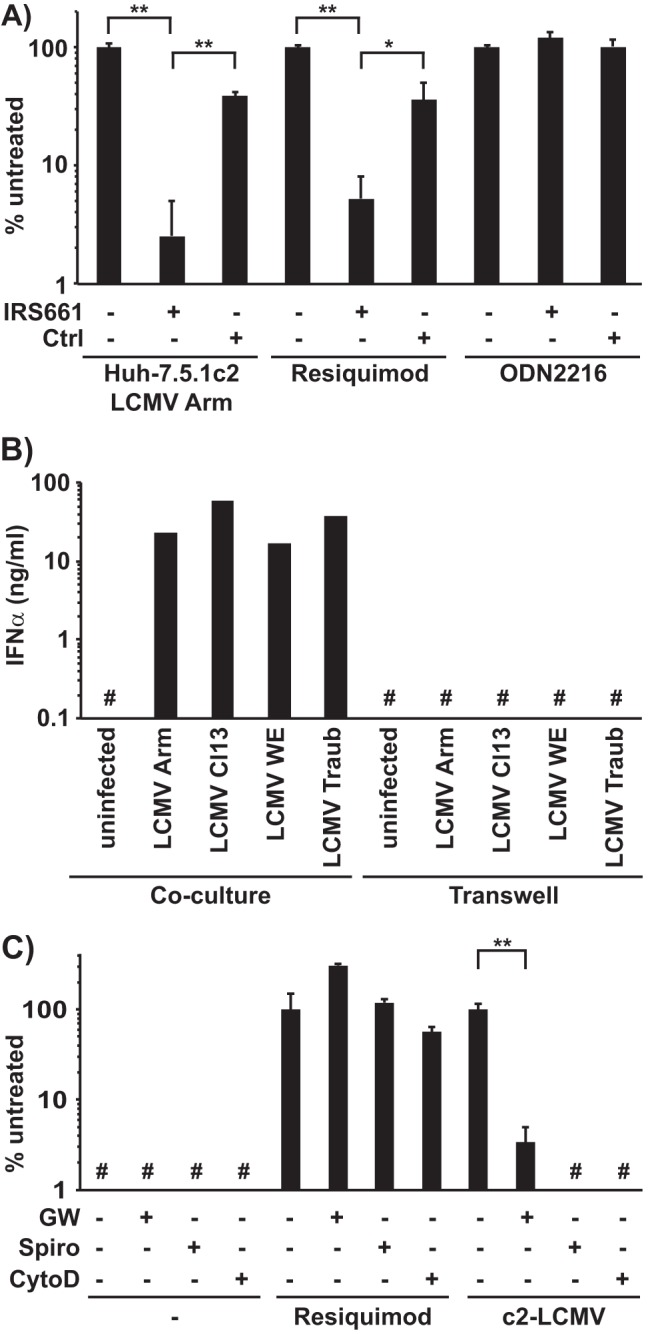
Mechanism of activation of pDCs by LCMV-infected Huh-7.5.1c2 cells. (A) Human peripheral pDCs were cocultured with LCMV Armstrong-infected Huh-7.5.1c2 cells or incubated with a TLR7 (Resiquimod) or TLR9 (ODN2216) ligand and left untreated or treated with a TLR7 antagonist or a control oligonucleotide (Ctrl) exactly as described previously (9). IFN-α production is shown as a percentage of the untreated control level in each group. (B) IFN-α production was quantified by ELISA in the supernatant of cocultures of LCMV-infected Huh-7.5.1c2 cells and pDCs set up exactly as described in the legend to Fig. 1A but either seeded together on top of the membrane of transwell chambers (Coculture) or separated by the membrane (Transwell). Data of individual wells are shown. (C) Human peripheral pDCs (2 × 104) were cocultured with 6.7 × 103 LCMV Armstrong-infected Huh-7.5.1c2 cells or incubated with a TLR7 agonist (Resiquimod) and left untreated or treated with the exosome release inhibitors GW4869 (GW, 10 μM) and spiroepoxide (Spiro, 20 μM) or the endocytosis inhibitor cytochalasin D (CytoD, 0.1 μg/ml) as described previously (8). IFN-α production is shown as a percentage of the untreated control group level. #, below the limit of detection of the IFN-α ELISA (150 pg/ml). Error bars represent means ± SD (n = 3). *, P < 0.05; **, P < 0.01 (paired Student's t test).
Unlike HCV, LCMV has a broad host cell range in terms of both type and species, which enabled us to determine if the ability to trigger IFN-α production by human pDCs could be extended to other cell types and species. Coculture of human pDCs with either LCMV-infected human cervical epithelium-derived (HeLa) or human hepatoma-derived (HepG2 and Hep3B) cells triggered strong IFN-α production by the human pDCs (Fig. 3A). In contrast, coculture of human pDCs with LCMV-infected human embryonic kidney 293T cells did not result in production of IFN-α, though 293T cells were infected at levels similar to those of all the other cell lines (Fig. 3B). Importantly, human pDCs did not produce IFN-α when cocultured with the uninfected cell lines or when incubated with supernatants of the LCMV-infected cell lines; neither was it produced by LCMV infection of any of the cell lines examined (Fig. 3A). Next, we determined whether freshly prepared cultures of primary human hepatocytes (PHHs) (Life Technologies, Carlsbad, CA) (1.25 × 105 cells per well in 48-well plates) infected with LCMV would also be capable of triggering IFN-α production by cocultured human blood-derived pDCs. As shown in Fig. 4A, 1.25 × 105 PHHs in a 48-well plate (maintained according to the manufacturer's instructions) infected with LCMV Arm for 3 days (d3) or 4 days (d4) triggered IFN-α production by 1 × 105 cocultured human blood-derived pDCs whereas the uninfected PHHs did not. Importantly, the LCMV-infected PHH cells did not produce IFN-α despite containing high levels of LCMV RNA (Fig. 4B) and despite most of the PHH cells being positive for LCMV NP (Fig. 4C), suggesting that pDCs, rather than PHH cells, were the source of IFN-α in the cocultures (Fig. 4A). These results demonstrated that sensing of virus-infected cells by human pDCs is not restricted to Huh-7-derived cells but also extends to cells of nonhepatic human origin (i.e., HeLa cells), to other human hepatoma-derived cell lines (HepG2 and Hep3B), and, most importantly, to primary human hepatocytes.
FIG 3.
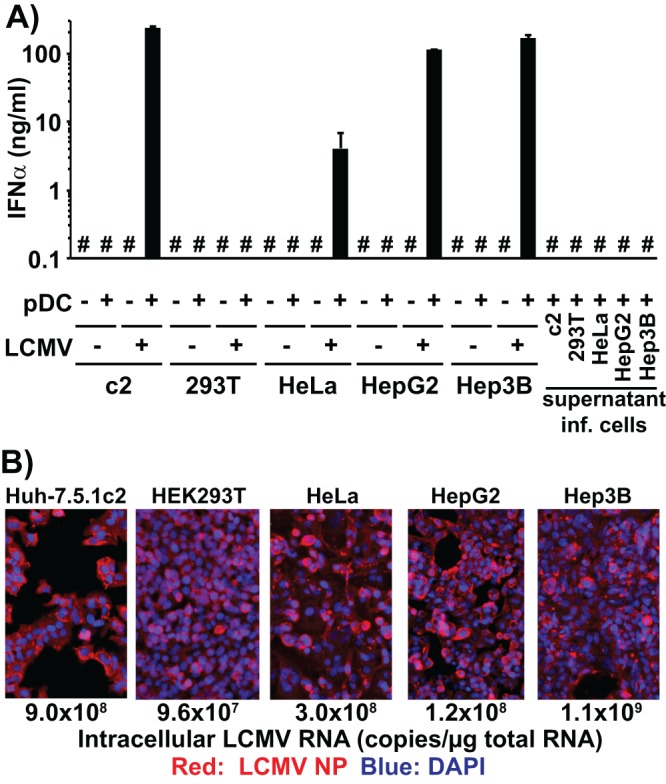
LCMV-infected human cell lines other than Huh-7 trigger IFN-α production in cocultured pDCs. (A) Quantification of IFN-α production in cell culture supernatants of uninfected or LCMV Armstrong-infected cells (LCMV +/−) cocultured or not with human peripheral pDCs (pDC +/−) set up as described for Fig. 1A. Alternatively, 2 × 104 pDCs were incubated with the supernatant of LCMV-infected cells collected 3 days after LCMV inoculation. #, below the limit of detection of the IFN-α ELISA (36 pg/ml); inf, infected. Error bars represent means ± SD (n = 3). (B) Analysis of LCMV infection 3 days after LCMV Armstrong inoculation (MOI = 0.1). LCMV-infected cells were visualized by LCMV NP-specific immunofluorescence. Intracellular LCMV RNA levels were determined by LCMV-specific RT-qPCR and normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA levels as described previously (9).
FIG 4.
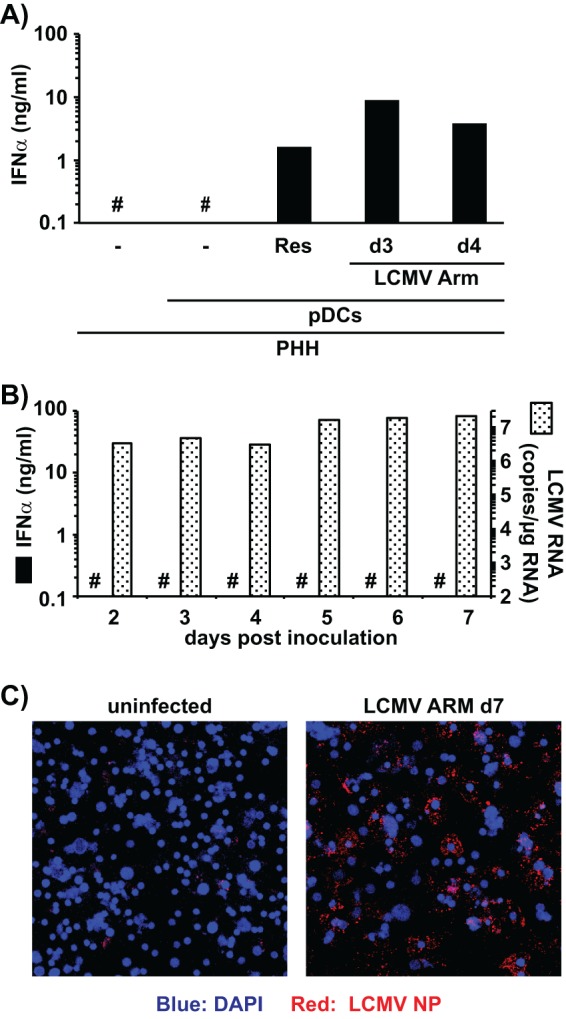
LCMV-infected primary human hepatocytes (PHHs) trigger IFN-α production in cocultured pDCs. (A) Quantification of IFN-α production in cell culture supernatants of uninfected PHHs cocultured (20 h) or not with human peripheral pDCs and cocultures of human pDCs with PHHs infected with LCMV (MOI = 0.1) for 3 days (d3) or 4 days (d4). #, below the limit of detection of the IFN-α ELISA (36 pg/ml); Res, resiquimod. Single wells were analyzed. (B) Analysis of LCMV infection and IFN-α production of PHHs at different time points after LCMV Armstrong inoculation (MOI = 0.1). Intracellular LCMV RNA levels were determined by LCMV-specific RT-qPCR and normalized to GAPDH mRNA levels as described previously (9). #, below the limit of detection of the IFN-α ELISA (36 pg/ml). (C) LCMV-infected PHHs were visualized by LCMV NP-specific immunofluorescence 7 days postinoculation.
Since mice are the natural host of LCMV, we asked if LCMV-infected mouse cell lines would trigger IFN-α production by human pDCs. Both LCMV-infected murine hepatoma (AML12) and fibroblast (NIH 3T3) cells triggered IFN-α production by cocultured human pDCs, while the corresponding uninfected cells did not (Fig. 5A). Furthermore, the LCMV-infected cells themselves did not produce IFN-α, indicating that the pDCs in the cocultures were the source of the IFN-α (Fig. 5A). Interestingly, we observed substantial differences in the magnitude of IFN-α production by pDCs depending on the LCMV-infected cell type used in the coculture, although the levels of LCMV infection were similar in all cell lines tested (Fig. 5B) as determined by LCMV-NP-specific immunofluorescence and reverse transcription-quantitative PCR (RT-qPCR) using LCMV NP-specific primers (LCMV_NP-up [GTTGCGCATTGAAGAGGTCGG] and LCMV_NP-lo [CCAACCACAGAACGGGCAGT]). This suggested that there may be cell type- and species-specific differences in the efficiency of viral RNA transfer to pDCs.
FIG 5.
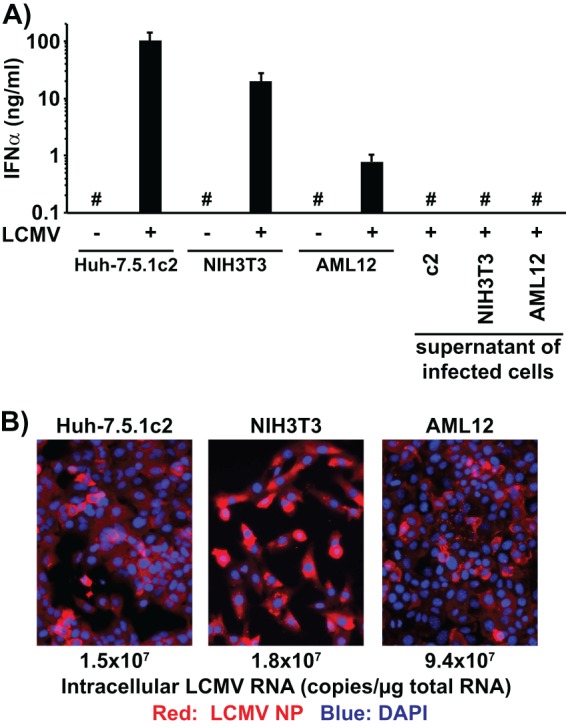
LCMV-infected murine cell lines trigger IFN-α production in cocultured human peripheral pDCs. (A) IFN-α production in cell culture supernatants of uninfected or LCMV Armstrong-infected cells (LCMV +/−) cocultured with human peripheral pDCs (pDC +/−) set up exactly as described for Fig. 1A. Alternatively, pDCs were incubated with the supernatants of LCMV-infected cells collected 3 days after LCMV inoculation and set up exactly as described for Fig. 1A. #, below the limit of detection of the IFN-α ELISA (36 pg/ml). Error bars represent means ± SD (n = 3). (B) Analysis of LCMV infection 3 days after LCMV Armstrong inoculation (MOI = 0.1). LCMV-infected cells were visualized by LCMV NP-specific immunofluorescence. Intracellular LCMV RNA levels were determined by LCMV-specific RT-qPCR and normalized to GAPDH mRNA levels as described previously (9).
Table 1 shows a complete list of the IFN-α responses shown by human pDCs during coculture with LCMV-infected cells of different origins and species. Interestingly, human hepatoma cells (HepG2.2.15 [37]) that replicate hepatitis B virus (HBV) and secrete infectious virions did not trigger IFN-α production by pDCs, and the same was true for chicken hepatocyte-derived cells (LMH D2 [38]) that replicate the duck hepatitis B virus (DHBV) (Table 1). For both systems, however, the failure to trigger pDC activation seemed to be virus specific, since superinfection of the same cells with LCMV resulted in strong IFN-α production by human pDCs. Furthermore, pDCs cocultured with HBV- and DHBV-producing HepG2.2.15 and LMH D2 cells were fully able to produce IFN-α in response to the TLR7 agonist resiquimod (Table 1) compared to resiquimod stimulation of human pDCs only (data not shown), suggesting that neither HBV- nor DHBV-infected cells impaired the ability of pDCs to produce IFN-α in response to TLR7 ligation.
While we were unable to detect IFN-α production in in vitro cocultures of murine splenic pDCs and LCMV-infected mouse or human cell lines (data not shown), it is well documented that LCMV infection triggers IFN-α production by pDCs in vivo in mice (7, 15). Interestingly, it has recently been shown in vivo that the majority of IFN-α-producing pDCs in the mouse spleen early after LCMV Cl13 infection are not productively infected and it was suggested that those pDCs might sense infected cells by a mechanism that is independent of intrinsic virus replication in pDCs, e.g., by the sensing of LCMV-infected cells (7). The results reported here might explain how those pDCs sense LCMV infection in vivo in the mouse spleen. Together with our previous studies employing analysis of the responsiveness of human pDCs to activation by HCV-infected cells (8, 9), the results presented here support the concept that the ability of noninfected pDCs to direct a strong IFN-α response upon sensing infected cells might be a general mechanism by which the host can circumvent the ability of viruses to block innate signaling in productively infected cells and thus mount efficient innate immune responses that have the potential to control viral infection.
ACKNOWLEDGMENTS
We thank Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for providing the infectious JFH-1 molecular clone and Charles M. Rice (Rockefeller University, New York, NY) for providing the Huh-7.5 cells from which the Huh-7.5.1c2 cells were derived.
This work was supported by grants from the National Institutes of Health to F.V.C. (AI079043 and AI088778) and J.-C.D.L.T. (AI077719).
Footnotes
Published ahead of print 23 October 2013
This is manuscript no. 24077 from The Scripps Research Institute.
REFERENCES
- 1.Heim MH. 2013. Innate immunity and HCV. J. Hepatol. 58:564–574. 10.1002/hep.26227, [DOI] [PubMed] [Google Scholar]
- 2.Borrow P, Martínez-Sobrido L, de la Torre JC. 2010. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2:2443–2480. 10.3390/v2112443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, Lemon SM, Lanford RE. 2004. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 78:13779–13792. 10.1128/JVI.78.24.13779-13792.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bigger CB, Brasky KM, Lanford RE. 2001. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 75:7059–7066. 10.1128/JVI.75.15.7059-7066.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. 2002. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:15669–15674. 10.1073/pnas.202608199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee LN, Burke S, Montoya M, Borrow P. 2009. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J. Immunol. 182:7178–7189. 10.4049/jimmunol.0802526 [DOI] [PubMed] [Google Scholar]
- 7.Macal M, Lewis GM, Kunz S, Flavell R, Harker JA, Zuniga EI. 2012. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe 11:617–630. 10.1016/j.chom.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dreux M, Garaigorta U, Boyd B, Décembre E, Chung J, Whitten-Bauer C, Wieland S, Chisari FV. 2012. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12:558–570. 10.1016/j.chom.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. 2010. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U. S. A. 107:7431–7436. 10.1073/pnas.1002301107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer BJ, de la Torre JC, Southern PJ. 2002. Arenaviruses: genomic RNAs, transcription, and replication. Curr. Top. Microbiol. Immunol. 262:139–157. 10.1007/978-3-642-56029-3_6 [DOI] [PubMed] [Google Scholar]
- 11.Buchmeier MJ, De La Torre JC, Peters CJ. 2007. Arenaviridae: the viruses and their replication, p 1791–1827 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5 ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 12.Peters CJ. 2006. Lymphocytic choriomeningitis virus—an old enemy up to new tricks. N. Engl. J. Med. 354:2208–2211. 10.1056/NEJMp068021 [DOI] [PubMed] [Google Scholar]
- 13.Merigan TC, Oldstone MB, Welsh RM. 1977. Interferon production during lymphocytic choriomeningitis virus infection of nude and normal mice. Nature 268:67–68. 10.1038/268067a0 [DOI] [PubMed] [Google Scholar]
- 14.Zuniga EI, Liou L-Y, Mack L, Mendoza M, Oldstone MBA. 2008. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 4:374–386. 10.1016/j.chom.2008.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, Akira S. 2008. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82:196–206. 10.1128/JVI.01640-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martínez-Sobrido L, Zúñiga EI, Rosario D, García-Sastre A, de la Torre JC. 2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 80:9192–9199. 10.1128/JVI.00555-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922. 10.1038/nature06205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082–11093. 10.1128/JVI.01307-;06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014. 10.1128/JVI.76.24.13001-13014.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858–3863 [PubMed] [Google Scholar]
- 21.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. 1979. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282:615–616 [DOI] [PubMed] [Google Scholar]
- 23.Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. U. S. A. 84:1005–1009. 10.1073/pnas.84.4.1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scherer WF, Syverton JT, Gey GO. 1953. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix.. J. Exp. Med. 97:695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–74. 10.1099/0022-1317-36-1-59 [DOI] [PubMed] [Google Scholar]
- 27.Wu JC, Merlino G, Fausto N. 1994. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. U. S. A. 91:674–678. 10.1073/pnas.91.2.674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Todaro GJ, Green H. 1963. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17:299–313. 10.1083/jcb.17.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen FC, Girardi AJ, Gilden RV, Koprowski H. 1964. Infection of human and simian tissue cultures with rous sarcoma virus. Proc. Natl. Acad. Sci. U. S. A. 52:53–59. 10.1073/pnas.52.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawaguchi T, Nomura K, Hirayama Y, Kitagawa T. 1987. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res. 47:4460–4464 [PubMed] [Google Scholar]
- 31.Condreay LD, Aldrich CE, Coates L, Mason WS, Wu TT. 1990. Efficient duck hepatitis B virus production by an avian liver tumor cell line. J. Virol. 64:3249–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong SS, Jensen AD, Chang CJ, Rogler CE. 1999. Double-stranded linear duck hepatitis B virus (DHBV) stably integrates at a higher frequency than wild-type DHBV in LMH chicken hepatoma cells. J. Virol. 73:1492–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodrigo WW, de la Torre JC, Martínez-Sobrido L. 2011. Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. J. Virol. 85:1684–1695. 10.1128/JVI.02229-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emonet SF, Garidou L, McGavern DB, de la Torre JC. 2009. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc. Natl. Acad. Sci. U. S. A. 106:3473–3478. 10.1073/pnas.0900088106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. 1993. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J. Cell Biol. 120:695–710. 10.1083/jcb.120.3.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borrow P, Oldstone MB. 1994. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 198:1–9. 10.1006/viro.1994.1001 [DOI] [PubMed] [Google Scholar]
- 37.Sells MA, Zelent AZ, Shvartsman M, Acs G. 1988. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J. Virol. 62:2836–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong SS, Jensen AD, Wang H, Rogler CE. 1995. Duck hepatitis B virus integrations in LMH chicken hepatoma cells: identification and characterization of new episomally derived integrations. J. Virol. 69:8102–8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dutko FJ, Oldstone MB. 1983. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 64(Pt 8):1689–1698. 10.1099/0022-1317-64-8-1689 [DOI] [PubMed] [Google Scholar]
- 40.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521–540 [DOI] [PMC free article] [PubMed] [Google Scholar]