

Abstract
All herpesviruses share a remarkable propensity to establish latent infection. Human Kaposi's sarcoma-associated herpesvirus (KSHV) effectively enters latency after de novo infection, suggesting that KSHV has evolved with strategies to facilitate latent infection. NF-κB activation is imperative for latent infection of gammaherpesviruses. However, how NF-κB is activated during de novo herpesvirus infection is not fully understood. Here, we report that KSHV infection activates the inhibitor of κB kinase β (IKKβ) and the IKK-related kinase epsilon (IKKε) to enable host NF-κB activation and KSHV latent infection. Specifically, KSHV infection activated IKKβ and IKKε that were crucial for latent infection. Knockdown of IKKβ and IKKε caused aberrant lytic gene expression and impaired KSHV latent infection. Biochemical and genetic experiments identified RelA as a key player downstream of IKKβ and IKKε. Remarkably, IKKβ and IKKε were essential for phosphorylation of S536 and S468 of RelA, respectively. Phosphorylation of RelA S536 was required for phosphorylation of S468, which activated NF-κB and promoted KSHV latent infection. Expression of the phosphorylation-resistant RelA S536A increased KSHV lytic gene expression and impaired latent infection. Our findings uncover a scheme wherein NF-κB activation is coordinated by IKKβ and IKKε, which sequentially phosphorylate RelA in a site-specific manner to enable latent infection after KSHV de novo infection.
INTRODUCTION
Human Kaposi's sarcoma-associated herpesvirus (KSHV, also known as human herpesvirus 8, or HHV-8) belongs to lymphotropic gammaherpesvirus 2 family (1). KSHV infection is causatively linked to Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD) (2–5). KS is the leading cause of mortality and morbidity in immunocompromised patients, whereas PEL and MCD are rare lymphomas. Similar to other herpesviruses, KSHV infection has two phases, i.e., lytic replication and latency. Interestingly, cells supporting lytic replication and those carrying latent KSHV genomes are consistently found in KSHV-associated tumors, e.g., KS lesions (6, 7). In human KS lesions, the majority of the tumor comprises KSHV latently infected spindle cells that have been infiltrated with immune cells, displaying characteristics of an excessive inflammatory response (8).
A feature of KSHV is its remarkable propensity to establish latent infection after de novo infection. KSHV infection ex vivo in a variety of cell lines leads to latent infection by default (9, 10). Although latently infected cells can be induced to enter the lytic cycle, biological and chemical reagents are relatively poor in reactivating KSHV and viral yield is low, suggesting tight control of lytic replication (11). By using KSHV latent lymphoma cell lines, KSHV lytic gene expression and productive replication can be induced with exogenous expression of the viral replication transactivator (RTA). Accumulating studies point to RTA as the regulatory node that integrates diverse physiological signaling events to determine the fate of KSHV-infected cells. Thus, RTA-mediated transcription is the paramount regulatory hub during KSHV infection. An array of events that influence RTA expression and RTA-mediated transcription have been reported thus far (12–17). Moreover, RTA-interacting proteins, either of host or viral origin, can alter its activity during viral infection (13, 15). Finally, microRNA and posttranslational events, e.g., phosphorylation and acetylation, can further tune RTA-dependent gene expression (16–19). These findings demonstrate the crucial roles of RTA in determining the fate of KSHV infection and suggest that RTA-mediated transcription is highly suppressed after de novo KSHV infection.
In response to viral infection, innate immune signaling events are immediately initiated to defeat viral replication. Despite the diversity of pattern recognition receptors (PRRs) that sense viral infection, upstream signaling events converge at two kinase complexes, i.e., the inhibitor of κB kinase (IKK), consisting of IKKα, IKKβ, and IKKγ, and the IKK-related TBK1-IKKε complex (20). Activated kinases phosphorylate key transcription factors or inhibitors thereof to upregulate the expression of antiviral genes, thereby establishing an antiviral state (21, 22). As obligate pathogens, viruses have evolved with strategies to evade and exploit host innate immune signaling events. Positive-strand RNA viruses cleave adaptor molecules upstream of IKKαβγ or TBK1-IKKε kinases to disarm the host innate immune defense (23–26). Large DNA herpesviruses dedicate significant portions of their genomes to modulators of host innate and adaptive immune responses. Notable examples are the diverse mechanisms that deregulate the interferon (IFN)-dependent antiviral pathways by various pathogens. Strikingly, our recent studies showed that murine gammaherpesvirus 68 (γHV68), a model herpesvirus for human KSHV and Epstein-Barr virus (EBV), usurped the IKKβ kinase to phosphorylate RTA and promote viral transcriptional activation (27). Loss of IKKβ or components of the same pathway severely impaired γHV68 lytic replication. Moreover, IKKβ was exploited by γHV68 to terminate NF-κB activation and to avoid antiviral cytokine production (28, 29). These findings highlight the dynamic and intricate interactions between viruses and their human host.
Downstream of IKKαβγ or TBK1-IKKε kinases, NF-κB transcription factors are key players in regulating antiviral gene expression, specifically that of inflammatory cytokines and IFNs. Among five members of the NF-κB family, RelA is the most abundantly and ubiquitously expressed (30). Moreover, RelA is the transcriptionally active subunit of the predominant RelA-p50 dimer. Posttranslational modifications of the RelA subunit, e.g., phosphorylation and acetylation, are important means to regulate NF-κB-dependent gene expression (31–34). However, how multiple events are coordinated to achieve regulated gene expression is not clear. Here, we report that KSHV de novo infection activates IKKβ, and IKKε, which enable the phosphorylation of serine 536 (S536) and S468 of RelA, respectively, to promote NF-κB activation and KSHV latent infection. Phosphorylation of S536 of RelA is required for phosphorylation of S468, the latter of which potently inhibited KSHV lytic replication. Conversely, knockdown of IKKβ and IKKε impaired NF-κB activation and elevated KSHV lytic gene expression, resulting in reduced KSHV latent infection. Our findings have uncovered a scheme wherein two closely related kinases are activated to coordinate NF-κB activation in enabling KSHV latent infection and reveal an intimate link between innate immune signaling and viral persistent infection.
MATERIALS AND METHODS
Plasmids.
Unless otherwise specified, IKKα, IKKβ, IKKε, TBK1, RelA, and RelA mutants carrying an S468A, S468E, S536A, S536E, S529A, or S468,536A mutation and Flag-tagged RTA were cloned into pcDNA5/FRT/TO (Invitrogen) and pCDH-puro for transient and stable expression, respectively. All cloned cDNAs were validated by DNA sequencing.
Cells and viruses.
Human ECV endothelial cells and 293T, iSLK-Bac16, and iSLK.219 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 100 U penicillin-streptomycin. iSLK-Bac16 cells were maintained with G418 (250 μg/ml), hygromycin (1.2 mg/ml), and puromycin (1 μg/ml). iSLK.219 cells were maintained with G418 (250 μg/ml), hygromycin (400 μg/ml), and puromycin (10 μg/ml). Recombinant KSHV was induced from iSLK-Bac16 or iSLK.219 cells with doxycycline (1 μg/ml) for up to 96 h, as described previously (35, 36). Recombinant KSHV was concentrated via ultracentrifugation at 32,000 rpm for 2 h at 4°C.
Luciferase reporter assay.
As previously described (37), 293T cells were seeded into 24-well plates (1 × 105 cells/cm2). Sixteen hours later, cells were transfected with a total of 500 ng of plasmid cocktail per well by calcium phosphate precipitation. To determine the effects of human IκBα kinases or RelA on the transcriptional activation of RTA, we transfected 293T cells with a plasmid cocktail that comprised 100 ng of open reading frame 57 (ORF57) or polyadenylated nuclear (PAN) luciferase reporter plasmid, 2 ng of RTA, 200 ng of pCMV-β-galactosidase (β-Gal) plasmid, and 50 ng or 150 ng of IKK or RelA plasmid. At 20 h posttransfection, the activity of firefly luciferase and β-Gal in whole-cell lysates was determined by using a FLUOstar Omega microplate reader (BMG Labtech.). Data from reporter assays represent at least three independent experiments.
Protein expression and purification.
Glutathione S-transferase (GST) and GST-fusion proteins containing the N-terminal region of IκBα or the carboxyl terminus of IFN regulatory factor 3 (IRF3) were expressed after isopropyl-β-d-thiogalactopyranoside induction and purified with glutathione-conjugated Sepharose as previously described (38, 39). Eluted proteins were resuspended in 25% glycerol and stored at −80°C for kinase assays.
Reverse transcript-PCR and qRT-PCR.
To determine the relative level of cellular and viral transcripts, reverse transcription-PCR and quantitative real-time PCR (qRT-PCR) were performed as previously described (27, 28). Briefly, total RNA was extracted from ECV, iSLK.219, or BJAB cells by using TRIzol reagent (Invitrogen). To remove genomic DNA, total RNA was digested with RNase-free DNase I (New England BioLabs) at 37°C for 1 h. DNase I digestion was quenched by heat inactivation at 70°C for 20 min, and total RNA was purified with TRIzol reagent. cDNA was prepared with 1.5 μg total RNA, reverse transcriptase (Invitrogen), and oligo(dT)12–19 primer. RNA was then removed by incubation with RNase H (Epicentre). The abundance levels of cellular and viral mRNAs were assessed by qRT-PCR by using a StepOnePlus real-time PCR system (Applied Biosystems). Human β-actin was used as an internal control. To determine the relative viral genomes in KSHV-infected ECV cells, total genomic DNA was purified by phenol-chloroform extraction and ethanol-sodium acetate precipitation after digestion with proteinase K (Qiagen). The abundance of the KSHV genome in 20 ng total genomic DNA was determined by real-time PCR using primers specific for RTA (also known as ORF50) and ORF9. All primers were synthesized by Integrated DNA Technologies and validated individually.
In vitro kinase assay.
Endogenous IKKβ and IKKε were analyzed in in vitro kinase assays. Briefly, ECV cells were harvested at the indicated time points after KSHV infection. Whole-cell lysates were precipitated with an antibody against IKKγ (also known as NEMO) to obtain the IKKβ kinase complex, or with rabbit anti-IKKε to obtain the IKKε-containing kinase complex. The kinase reaction mixture consisted of 0.5 μg GST or GST-fusion proteins 100 μCi [γ-32P]ATP, and precipitated kinase in 20 μl of kinase buffer. The reaction mixture was incubated at room temperature for 40 min, and denatured proteins were analyzed by SDS-PAGE and autoradiography.
Immunoprecipitation and immunoblotting.
Commercial antibodies used in this study included mouse M2 anti-Flag (Sigma), mouse anti-β-actin (Abcam), rabbit anti-IKKε (Sigma), rabbit anti-RelA (C-20; Santa Cruz Biotech), rabbit anti-RelA S468p (Bethyl), rabbit anti-RelA S536p (Cell Signaling), and rabbit anti-GST (Santa Cruz Biotech) antibodies. Antibodies against IKKβ and IKKγ were kindly provided by E. Zandi, and anti-RTA antibody was a gift from Y. Izumiya (40).
Immunoprecipitation and immunoblotting were carried out as described previously (38, 41, 42). Briefly, cells were harvested and lysed with NP-40 buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 5 mM EDTA) supplemented with a protease inhibitor cocktail. Then, centrifuged cell lysates were precleared with Sepharose 4B beads and incubated with the indicated antibodies and protein A/G-agarose (Thermo Scientific) or antibody-conjugated agarose (Sigma) at 4°C for 4 to 6 h. The agarose beads were washed three times with the corresponding lysis buffer and eluted with 1× SDS sample buffer by boiling at 95°C for 5 min. Immunoblotting analysis was performed with the indicated primary antibodies, and proteins were visualized with IRDye800- or IRDye680-conjugated secondary antibodies (Licor) and an Odyssey infrared imaging system (Licor).
Statistical analysis.
The statistical significance (P value) was calculated by using an unpaired two-tailed Student's t test. A P value of <0.05 was considered statistically significant.
RESULTS
IKKβ and IKKε potently inhibit RTA-mediated transcriptional activation.
We previously showed that IKKβ was usurped by murine gammaherepsvirus 68 (γHV68) to promote viral lytic replication via phosphorylation of RTA (27). We reasoned that IKK and IKK-related kinases likely impact KSHV gene expression driven by RTA as well. To assess the effects of these innate immune kinases on KSHV RTA transcriptional activation, we performed luciferase assays with KSHV lytic promoters, i.e., those of PAN RNA and ORF57, a key player of viral RNA metabolism (43). As shown in Fig. 1A, RTA expression upregulated the gene expression levels driven by PAN and ORF57 promoters by ∼65- and 30-fold, respectively. Exogenous expression of IKKβ and IKKε, but of neither IKKα nor TBK1, potently inhibited the gene expression driven by PAN and ORF57 promoters in a dose-dependent manner. The effect of IKKε was more robust than that of IKKβ, and these two kinases were equally expressed (Fig. 1B). To determine whether the kinase activity of IKKε was required for the inhibition of RTA-dependent transcriptional activation, we utilized the kinase-dead mutant of IKKε, IKKεK38A, for a reporter assay. Compared to wild-type IKKε, the IKKεK38A mutant had a marginal effect on RTA-mediated gene expression (Fig. 1C). Similarly, the kinase-dead mutant of IKKβ, IKKβKD, failed to inhibit RTA-mediated transcription. Thus, IKKβ and IKKε potently inhibit RTA-mediated transcriptional activation in a kinase-dependent manner.
FIG 1.

IKKβ and IKKε inhibit RTA-mediated gene expression. (A and B) 293T cells were transfected with a reporter plasmid cocktail and plasmids containing the indicated genes. At 24 h posttransfection, luciferase activity and β-galactosidase activity in whole-cell lysates were determined (A). Whole-cell lyases were analyzed by immunoblotting with the indicated antibodies (B). (C) Transfection with BJAB cells and luciferase reporter assays were carried out as described for panel A, except a plasmid containing the kinase-dead mutant of IKKε (IKKεK38A) or that of IKKβ (IKKβKD) was included. WT, wild type.
Knockdown of IKKβ and IKKε upregulates KSHV lytic gene expression and impairs KSHV latent infection.
Compared to human umbilical vein endothelial cells (HUVEC), ECV endothelial cells express high levels of IKKβ and IKKε and can be easily amplified for biochemical studies. Thus, we employed ECV cells to investigate innate immune signaling in KSHV latent infection. To probe the roles of IKKβ and IKKε in KSHV infection, we knocked down their expression with short hairpin RNA (shRNA) and assessed KSHV infection. Two separate shRNAs, expressed via lentivirus infection, reduced the IKKε protein level in human ECV endothelial cells without affecting the IKKβ protein level (Fig. 2A). After infection with shRNA-expressing lentivirus, ECV cells were then infected with recombinant KSHV.219 (rKSHV.219), which carries green fluorescent protein (GFP) as an infection marker and red fluorescent protein (RFP; under the control of the PAN promoter) as a lytic replication marker (44). rKSHV.219 infection in ECV cells expressing shRNA27 yielded cells with higher GFP intensity, likely due to viral genome replication. In support of this, knockdown of IKKε greatly elevated RFP+ cells as well, suggesting more robust lytic gene expression when IKKε expression was suppressed (Fig. 2B). shRNA36 had a mild effect on KSHV lytic gene expression, as judged by the number of RFP+ cells (data not shown). Indeed, qRT-PCR analysis using primers specific for KSHV lytic genes, including RTA, PAN, ORF57, ORF21, and K8.1, showed that knockdown of IKKε increased viral lytic transcripts by ∼5-fold for RTA, ORF57, ORF21, and K8.1, whereas PAN RNA was elevated by more than 10-fold at 72 h postinfection (hpi) (Fig. 2C). Consistent with increased viral gene expression, the viral genome copy number gradually increased in ECV cells in which IKKε was knocked down, whereas it gradually decreased in control ECV cells (Fig. 2D). By 72 hpi, the copy number of the KSHV genome in IKKε knockdown cells was approximately 6-fold higher than that in control ECV cells. To quantitatively measure KSHV latent infection, we used recombinant KSHV derived from BAC16, which permits selection with hygromycin, accommodating the puromycin resistance conferred by shRNA expression vectors. ECV cells were infected with lentivirus expressing IKKε shRNA and selected with puromycin. Cells were then infected with recombinant KSHV generated from BAC16 (hygromycin resistant) and selected with puromycin and hygromycin for latently infected KSHV episomes. When ECV cells were selected with puromycin and hygromycin for latently infected cells, knockdown of IKKε in ECV cells reduced KSHV latently infected cells by more than 95% for shRNA27 and by ∼75% for shRNA36 (Fig. 2E).
FIG 2.

Knockdown of IKKε increases KSHV lytic gene expression and impairs latent infection. ECV cells were infected with lentivirus expressing control (CTL) shRNA or shRNA27 and shRNA36 of IKKε and then selected with puromycin. (A) Whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (B) ECV cells were infected with KSHV at a multiplicity of infection (MOI) of 2 and monitored by fluorescence microscopy at 48 and 72 h postinfection. (C and D) Total RNA (C) or genomic DNA (D) was extracted from ECV cells at the indicated time points after KSHV infection. The mRNA abundance of selected viral genes (C) or the copy number of viral episomes (D) was analyzed by qRT-PCR. (E) ECV cells were infected with rKSHV derived from BAC16 at an MOI of 0.5. At 72 hpi, cells were selected with hygromycin and colonies were counted. (F, G, and H) ECV cells were infected with control lentivirus or lentivirus containing Flag-IKKεK38A. Whole-cell lysates were prepared and analyzed by immunoblotting with the indicated antibodies (F). ECV cells were infected with rKSHV.219 at an MOI of 0.5. Lytic replication was analyzed by fluorescence microscopy (G) and quantitative real-time PCR with total RNA (H).
We further examined the effects of the kinase-dead IKKεK38A mutant on KSHV latent infection. When IKKεK38A was expressed in ECV cells (Fig. 2F), we observed an increase in RFP+ cells at 72 hpi (Fig. 2G). qRT-PCR analysis, using primers specific for KSHV lytic genes, showed that the expression of IKKεK38A increased the abundance of various lytic transcripts, although its effects at 24 or 48 hpi were marginal (Fig. 2H). The effects of IKKε knockdown and exogenously expressed IKKεK38A on KSHV lytic gene expression were prominent at late time points during KSHV infection, i.e., 72 hpi, implying its temporal effect during KSHV infection. Taken together, these results indicate that IKKε is crucial for KSHV latent infection.
Similarly, we examined the roles of IKKβ in KSHV latent infection with shRNA-mediated knockdown. ECV cells were infected with lentivirus expressing shRNAs specific for IKKβ. Of two shRNAs, one caused diminished IKKβ protein expression, as determined by immunoblotting analysis (Fig. 3A). qRT-PCR analysis showed that IKKβ knockdown resulted in an increase of the mRNAs for ORF57 and RTA by ∼2- to 3-fold (Fig. 3B). The effect of IKKβ knockdown on KSHV gene expression was not as robust as that of IKKε knockdown, in agreement with the lower inhibition of IKKβ than that of IKKε for RTA-mediated transcriptional activation in reporter assays. Furthermore, the increased RTA mRNA abundance correlated with a higher protein level in ECV cells infected with KSHV (Fig. 3C). Finally, we examined the outcome of IKKβ knockdown on KSHV latent infection. Knockdown of IKKβ diminished cells latently infected with KSHV, as assessed by fluorescence microscopy. Semiquantitative measurement showed that shRNA knockdown of IKKβ reduced KSHV latent infection by ∼65%, indicating that IKKβ is necessary for KSHV latent infection (Fig. 3D).
FIG 3.
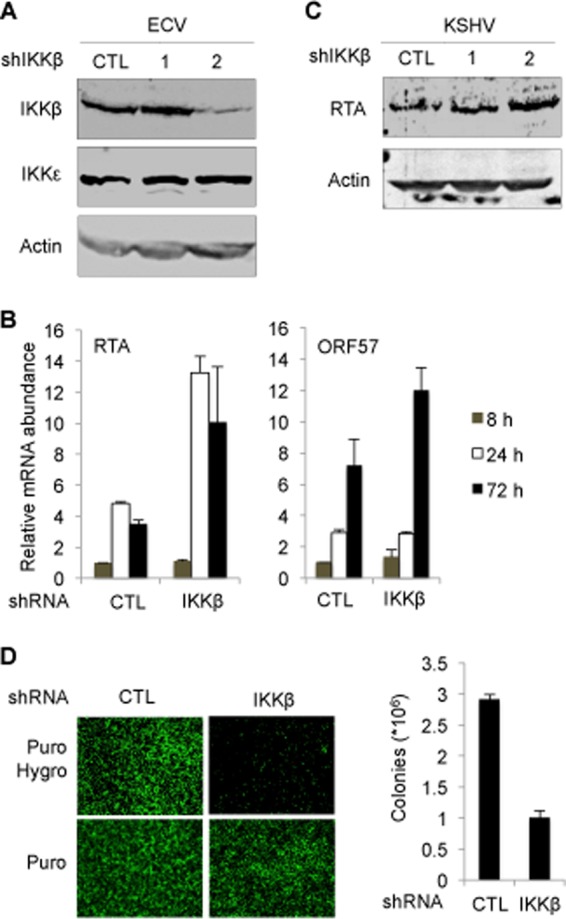
Depletion of IKKβ promotes KSHV lytic gene expression and impairs latent infection. ECV cells were infected with lentivirus expressing control (CTL) shRNA or shRNA1 and shRNA2 of IKKβ and selected with puromycin. (A) Whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (B and C) ECV cells were infected with KSHV at a multiplicity of infection (MOI) of 2. Total RNA was extracted and analyzed by qRT-PCR (B). Whole-cell lysates were prepared at 48 h postinfection and analyzed by immunoblotting with anti-RTA and anti-β-actin antibodies (C). (D) ECV cells were infected with KSHV at an MOI of 0.5. At 72 hpi, cells were selected with hygromycin, fluorescence of surviving cells was recorded by using a fluorescence microscope (left), and colonies were counted (right).
KSHV de novo infection activates IKKβ and IKKε.
IKK and IKK-related kinases are key signaling molecules in innate immune responses against viral infections (22, 45). We reasoned that KSHV de novo infection activates IKKβ and IKKε, which in turn impinge on KSHV lytic gene expression and latent infection. To test this hypothesis, we monitored the kinase activities of IKKβ and IKKε after KSHV infection in an in vitro kinase assay. Human ECV endothelial cells were infected with rKSHV.219, and the IKKβ kinase complex was precipitated with anti-IKKγ. An in vitro kinase assay showed that KSHV infection gradually increased IKKβ kinase activity by up to ∼3-fold at 4 hpi (Fig. 4A). Interestingly, we found that the kinase activity of IKKε was much more robustly induced by KSHV infection, with an ∼5- to 10-fold increase in its ability to phosphorylate IRF3C (Fig. 4B). To corroborate the activation of IKKβ and IKKε in in vitro kinase assays, we carried out qRT-PCR analysis and assessed the expression of host inflammatory genes, represented by IFN-α1, IFN-α2, IFIT3, CCL5, interleukin-8 (IL-8), and IFN-γ-inducible protein 10 (IP10). Interestingly, IFNα1, IFNα2, IFIT3, and CCL5 shared similar expression patterns (Fig. 4C). The levels of these cytokine mRNAs were reduced at 4 hpi and then induced at 8 hpi by ∼2- to 5-fold. At 24 hpi, these mRNAs returned to levels below those for mock-infected cells. By contrast, IL-8 and IP10 were gradually induced up to 6- and 4-fold at 24 hpi, respectively. Evidently, the first four cytokines are the faster responders upon KSHV infection, and the other two chemokines, i.e., IL-8 and IP10, perhaps represent the slower responders. When viral lytic transcripts, i.e., RTA and ORF57, were examined by qRT-PCR, we observed a gradual and robust increase in viral gene expression, indicating the progression of viral infection (Fig. 4D). Notably, both lytic and latent genes are expressed during the first 24 h postinfection to facilitate the establishment of KSHV latent infection (46). These results collectively indicate that KSHV de novo infection activates both IKKβ and, more potently, IKKε.
FIG 4.
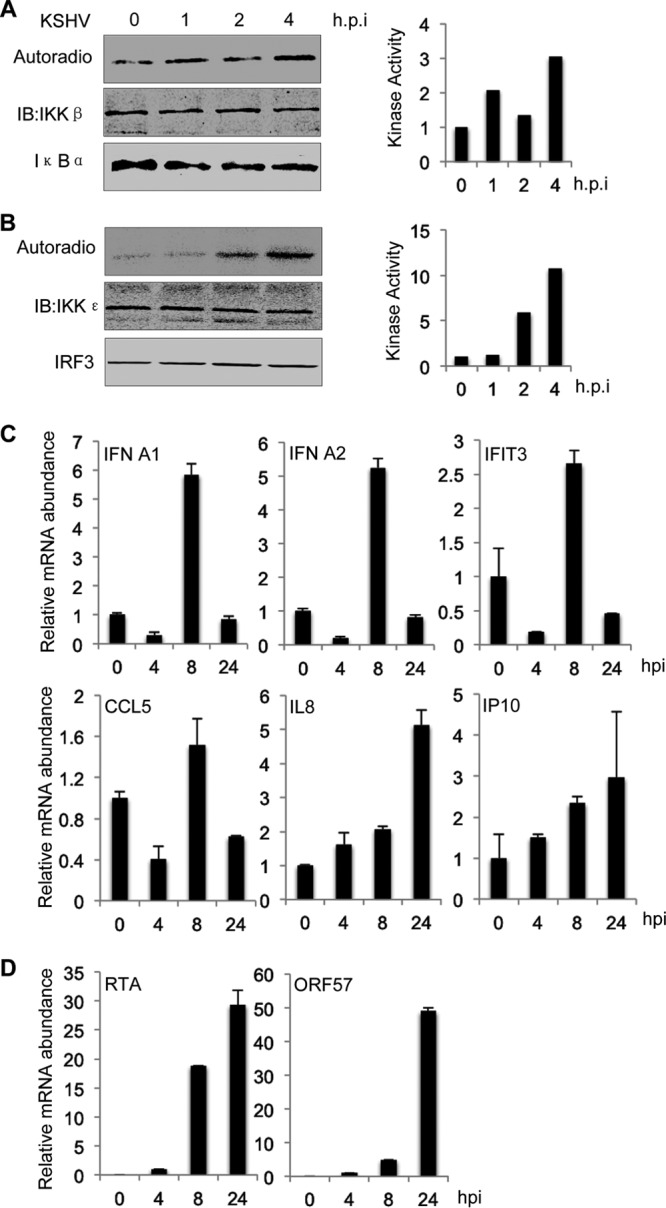
KSHV de novo infection activates IKKβ and IKKε. (A and B) IKKβ and IKKε were precipitated from ECV cells at the indicated time points after KSHV infection. GST fusion proteins carrying the N terminus of IκBα or the C terminus of IRF3 (IRF3C) were added to precipitated IKKβ (A) and IKKε (B), respectively. Phosphorylation of GST fusion proteins and other proteins were analyzed by autoradiography and immunoblotting (IB), respectively. Phosphorylation was also quantified with densitometry; results are shown in the graphs on the right. Data represent three independent experiments. (C and D) Total RNA was extracted from ECV cells at the indicated time points after KSHV infection and analyzed by qRT-PCR for mRNA abundance of selected host (C) and viral (D) genes.
RelA is an effector downstream of IKKβ and IKKε that inhibits RTA-mediated transcriptional activation.
We previously reported that IKKβ can phosphorylate γHV68 RTA to promote viral transcriptional activation (27). To this end, we extensively analyzed RTA phosphorylation by IKKβ and IKKε in an in vitro kinase assay and via mass spectrometry. Although the in vitro kinase assay identified three phosphorylation sites within the carboxy terminus of RTA, the ability of IKKε to phosphorylate these sites was not confirmed by mass spectrometry analysis when we used RTA purified from transfected 293T cells (data not shown). Moreover, RTA mutants carrying phosphorylation-resistant mutations demonstrated a similar ability to activate gene expression in reporter assays and in iSLK.219 cells that ectopically expressed RTA (data not shown). These results suggest that RTA phosphorylation by IKKβ and IKKε does not contribute to suppression of KSHV lytic replication by these two kinases.
Because IKKβ and IKKε are important for NF-κB activation which, in turn, enables the latent infection of gammaherpesvirus, we surmised that NF-κB activation is a major player downstream of IKKβ and IKKε. To test this hypothesis, we examined the inhibitory effects of IKKβ and IKKε on RTA-mediated transcriptional activation in reporter assays in which the three transcriptionally active subunits of NF-κB, i.e., RelA, RelB, and c-Rel, were knocked down with shRNAs. We found that knockdown of RelA, but not that of RelB or c-Rel, diminished the inhibition of RTA-dependent gene expression by IKKβ (Fig. 5A). Similar results were obtained for the inhibition of RTA-mediated transcriptional activation by IKKε. These results identified RelA as a key player downstream of IKKβ and IKKε for inhibition of RTA-mediated transcription. The efficiencies of knockdown of all three Rel family members were validated by qRT-PCR analysis, the results of which demonstrated a reduction of gene expression of >60% (Fig. 5B). To further corroborate the role of RelA downstream of IKKβ and IKKε, we performed an electrophoresis mobility shift assay. KSHV infection elevated the nuclear binding activity of an NF-κB probe, indicative of NF-κB activation (Fig. 5C). Moreover, an antibody against RelA further retarded the migration of the NF-κB–DNA complex, indicating that KSHV-induced NF-κB contains RelA (Fig. 5C). These results collectively support that RelA is an important effector downstream of IKKβ and IKKε.
FIG 5.

RelA is an effector downstream of IKKβ and IKKε. 293T cells were infected with lentivirus expressing control (Ctrl) shRNA or shRNA specific for RelA, RelB, or c-Rel. (A) 293T cells were used to determine the inhibition of IKKβ (left) and IKKε (right) on RTA-dependent transcriptional activation in a luciferase reporter assay. (B) Total RNA was extracted for qRT-PCR analysis using primers specific for RelA, RelB, and c-Rel. (C) Nuclear extract was incubated with a [γ-32P]ATP and analyzed by polyacrylamide gel electrophoresis. (D) Whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. Numbers below the blots indicate the remaining RelA, which was determined by densitometry analysis. (E and F) ECV cells were infected with lentivirus expressing control (CTL) or RelA-specific shRNA, selected with puromycin, and infected with KSHV at a multiplicity of infection of 2. Viral lytic gene expression was analyzed by qRT-PCR (E) and fluorescence microscopy (F).
To examine the role of RelA in KSHV latent infection, we knocked down RelA expression in ECV cells and examined KSHV infection. While shRNA1 reduced the RelA protein level by ∼38.8%, shRNA2 nearly abolished the RelA protein (Fig. 5D). Moreover, knockdown of RelA resulted in an increase in viral lytic transcripts, including PAN, ORF57, and ORF21, by ∼2- to 3-fold (Fig. 5E). By using rKSHV.219, which expresses RFP as a lytic marker, we found that knockdown of RelA elevated lytic replicating cells by ∼5- to 10-fold at 72 hpi. Whereas the RFP+ cells were not detected at 48 hpi in ECV cells expressing control shRNA, RFP+ ECV cells were prominent in those cells expressing RelA shRNA (Fig. 5F). Finally, quantitative measurement of KSHV latently infected cells indicated that RelA knockdown reduced KSHV latent infection by ∼80% (Fig. 5F). These results indicate that RelA, an effector downstream of IKKβ and IKKε, is critical for suppressing KSHV lytic gene expression, thereby enabling KSHV latent infection.
IKKε and IKKβ are required for phosphorylation of S468 and S536 of RelA, respectively, to inhibit RTA-mediated transcriptional activation.
Although IKKβ and IKKε were shown to phosphorylate the inhibitor of κB (IκB, e.g., IκBα), these kinases can directly phosphorylate NF-κB subunits such as RelA, representing a regulatory step likely functioning at post-nuclear translocation. In fact, RelA phosphorylation of S536 and S468 are implicated in distinct outcomes of NF-κB, depending on cellular conditions (31, 47). Considering that IKKβ and IKKε remained highly active at 4 hpi, we reasoned that events after NF-κB subunits released from IκB, e.g., RelA phosphorylation by IKKβ and IKKε, are likely important to influence NF-κB activation and KSHV latent infection. We thus examined RelA phosphorylation of S468 (S468p) and S536 (S536p), both of which are important for NF-κB activation. Upon KSHV infection, the level of RelA S468p gradually increased within the first 4 hpi, whereas that of RelA S536p increased at 1 and 2 hpi and then declined to levels below that of mock-infected cells at 4 hpi (Fig. 6A). To test whether RelA phosphorylation of S468 and S536 is important for the inhibitory effect of RelA, we overexpressed RelA or its mutants and examined RTA-mediated transcriptional activation in reporter assays. In agreement with a previous report (48), RelA expression potently inhibited RTA-mediated transcriptional activation on the ORF57 promoter (Fig. 6B). Interestingly, RelA S468A failed to inhibit gene expression driven by the RTA-dependent ORF57 promoter, whereas RelA S468E, a phosphorylation mimetic mutant, was as potent as wild-type RelA. Moreover, a dose-dependent expression of RelA wild type and RelA S468E showed that RelA S468E was not more potent than RelA wild type, suggesting that RelA phosphorylated by endogenous kinases is sufficient to inhibit RTA-mediated transcription (Fig. 6C). Interestingly, RelA S536A greatly increased RTA-dependent transcription, potentially exerting a dominant negative effect on endogenous RelA. The S536E mutation reduced the inhibitory effect of RelA, although RelA S536E increased the basal promoter activity of ORF57 (Fig. 6B). These results suggest that RelA S536 phosphorylation is required, but not sufficient, for RelA-mediated inhibition. On the other hand, RelA S468 phosphorylation is sufficient to inhibit RTA-dependent transcription.
FIG 6.

Phosphorylation of RelA is coordinated by IKKβ and IKKε during KSHV infection. (A) ECV cells were infected with KSHV and harvested at the indicated time points postinfection. Whole-cell lysates were analyzed for RelA phosphorylation of S468 and S536 by immunoblotting with the indicated antibodies. (B) RTA activation was determined in a luciferase reporter assay in 293T cells with plasmids carrying RelA wild type (WT) or mutants, as indicated. (C) Reporter assays were carried out as described for panel B, except with an increasing amount of plasmid containing RelA WT or RelA S468E. (D) ECV cells were infected with lentivirus expressing control shRNA or shRNA specific for IKKε and IKKβ and selected with puromycin. At various time points after KSHV infection, RelA phosphorylation was analyzed by immunoblotting with the indicated antibodies. (E) IKKε-mediated (top) or RelA-mediated (bottom) repression of RTA-dependent transcriptional activation in control or IKK knockdown ECV cells was analyzed in luciferase reporter assays. (F) ECV cells expressing RelA WT, RelA S468A, or RelA S536A were infected with KSHV, and whole-cell lysates were analyzed by immunoblotting with the indicated antibodies.
To probe the roles of IKKβ and IKKε in phosphorylating S468 and S536 of RelA, we knocked down IKKβ and IKKε and examined RelA phosphorylation after KSHV infection. In control ECV cells, KSHV infection resulted in an increase of RelA S536p by 50% and a more robust increase of RelA S468p (Fig. 6D). IKKε knockdown in ECV cells diminished RelA S468p and increased RelA S536p, indicating that IKKε is necessary for RelA S468 phosphorylation (Fig. 6D). The increase in RelA S536p upon IKKε depletion suggested that RelA S536p is subsequently phosphorylated and targeted for degradation by IKKε. Surprisingly, IKKβ knockdown resulted in low levels of both RelA S536p and RelA S468p (Fig. 6D). This result indicates that IKKβ is necessary for phosphorylation of both S468 and S536 of RelA. Taken together, these results suggest a possibility that RelA S536 phosphorylation is necessary for RelA S468 phosphorylation. Under this scenario, IKKβ is responsible for phosphorylation of S536, whereas IKKε is responsible for phosphorylating RelA at S468. We thus surmised that the inhibition of IKKε on RTA-mediated transcriptional activation depends, at least partly, on IKKβ. To test this hypothesis, we knocked down IKKβ and examined RTA-dependent transcription when IKKε was expressed. As expected, knockdown of IKKβ partly restored the transcriptional activity of RTA that was inhibited by IKKε (Fig. 6E). When IKKβ or IKKε was knocked down with shRNA, RelA inhibition of RTA-mediated transcriptional activation was partially restored (Fig. 6E). Finally, we constructed a RelA mutant carrying the S536A or S468A mutation and examined the phosphorylation of S468 and S536 in ECV cells infected with KSHV. We found that KSHV infection robustly elevated the S536 phosphorylation of RelA wild type and RelA S468A, but not the phosphorylation of S468 of the RelA S536A mutant (Fig. 6F). These results further confirmed that S536 and its phosphorylation are required for the phosphorylation of S468.
NF-κB activation enables KSHV latent infection.
To determine the roles of NF-κB activation on KSHV latent infection, we utilized the loss-of-function and dominant negative mutant of RelA, RelA S536A, to examine the effect of NF-κB activation on KSHV latent infection. Although RelA S468A lost its ability to inhibit RTA-mediated transcriptional activation, it did not have a dominant negative effect. Thus, we used RelA S468E to probe the phosphorylation of S468 in KSHV lytic gene expression and latent infection. ECV cells stably expressing RelA S536A, RelA S468E, or RelA S468,536A were established with lentivirus infection (Fig. 7A) and infected with rKSHV.219. Fluorescence microscopy analysis indicated that expression of RelA S536A and RelA S468,536A, but not that of RelA wild type or RelA S468E, increased RFP+ cells, which also correlated with elevated GFP fluorescence (Fig. 7B). We then examined viral lytic gene expression by qRT-PCR and found that RelA S536A expression increased viral mRNA transcripts of RTA, ORF57, and K8.1 by ∼5-fold. A similar effect on KSHV lytic gene expression was observed for the RelA S468,536A mutant. By contrast, expression of RelA S468E or RelA wild type had minimal effects on KSHV lytic gene expression, suggesting that endogenous RelA is sufficient to inhibit KSHV lytic gene expression (Fig. 7C). This result indicated that NF-κB activation driven by RelA is a potent inhibitor of KSHV lytic replication. We further examined the effects of these two RelA mutants on KSHV latent infection. Using hygromycin to select for stable episomes of KSHV in ECV cells, we found that the expression of RelA S536A and RelA S468,536A reduced KSHV latently infected cells by ∼75% after de novo infection (Fig. 7D). In contrast, the expression of the RelA S468E mutant or RelA wild type had a marginal effect on KSHV colony formation. The fact that RelA S468,536A and RelA S536A demonstrated the same level of inhibition on KSHV latent infection and promotion on KSHV lytic gene expression supports the conclusion that phosphorylation of RelA S536 is a critical step for RelA-mediated inhibition. Collectively, these results indicate that RelA S536A impairs KSHV latent infection by promoting lytic gene expression.
FIG 7.

RelA S536A promotes KSHV lytic gene expression and impairs latent infection. ECV cells were infected with lentiviruses expressing RelA wild type (WT), RelA S468E, RelA S536A, or RelA S468,536A (AA). (A) Whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. (B) Cells were infected with rKSHV.219, and fluorescence was determined by using a fluorescence microscope at 72 hpi. (C) Total RNA was extracted and analyzed by qRT-PCR with primers specific to the indicated genes. (D) Cells were selected with hygromycin at 72 hpi, and colonies were counted.
DISCUSSION
NF-κB activation is crucial for a plethora of biological processes, ranging from fundamental development to highly diseased clinical conditions (49). Not surprisingly, NF-κB is a key determinant of and crucially required for the latent infection of gammaherpesviruses. Specifically, NF-κB activation is likely exploited by gammaherpesviruses to suppress viral lytic gene expression during latent phase (50–53). For human KSHV and murine γHV68, NF-κB activation is sufficient to inhibit RTA-dependent transcriptional activation (48). Conversely, RTAs of KSHV and γHV68 were also shown to induce RelA degradation and terminate NF-κB activation (29, 54). This likely contributed to the efficient lytic replication of γHV68 via evasion of antiviral cytokine production, although the significance of RelA degradation by KSHV RTA remains less clear. In fact, γHV68 hijacks MAVS and IKKβ to induce RelA degradation, in conjunction with RTA serving as an E3 ligase to ubiquitinate RelA (29). These findings highlight the dynamic regulation of NF-κB as being important for the outcome of KSHV and γHV68 infection. Given the propensity of KSHV to establish latent infection, we have addressed the importance of NF-κB activation during KSHV infection. In determining the roles of IKKβ and IKKε in KSHV infection, we found that these two kinases are coordinated to promote NF-κB activation which, in turn, enables KSHV latent infection by suppressing viral lytic gene expression.
The IKK complex is composed of two kinase subunits, IKKα and IKKβ, and a scaffold subunit, IKKγ. IKKε is an IKK-related kinase, and its function remains less understood, despite its presumed redundant function with TBK1 in phosphorylating interferon regulatory factors (55, 56). However, IKKε is largely dispensable for proinflammatory cytokine production in response to viral infection (57). Instead, IKKε was shown to regulate IFN-mediated signal transduction downstream of interferon receptors (58, 59). Upon viral infection, these innate immune kinases are activated to provoke antiviral cytokine production, which acts to defeat viral infection. Our recent studies of murine γHV68 suggested the possibility that these immune kinases directly phosphorylate KSHV RTA to influence viral transcription (27). Indeed, IKKβ and IKKε potently phosphorylated KSHV RTA in an in vitro kinase assay (data not shown). However, we were unable to confirm RTA phosphorylation in cells and to establish the biological significance of RTA phosphorylation when we used recombinant KSHV (data not shown). Thus, we determined that NF-κB activation is an effector downstream of IKKβ and IKKε. By employing shRNA-mediated knockdown, we showed that RelA, but neither RelB nor c-RelA, contributed to the inhibition of IKKβ and IKKε on RTA-mediated transcriptional activation and KSHV lytic gene expression. The viral lytic gene expression also conversely correlated with latent infection of KSHV. However, knockdown of RelA only partly restored the inhibition of IKKβ and IKKε on RTA-dependent transcriptional activation, implying that additional cellular or viral factors are inhibited by IKKβ and IKKε during KSHV infection. This was further supported by the observation that a fraction of the NF-κB–DNA complex was shifted by an anti-RelA antibody.
A key component downstream of IKKβ and IKKε is NF-κB, which is activated by phosphorylation and degradation of the inhibitor of κB (IκB). Unleashed from IκBs, NF-κB dimers translocate into the nucleus to upregulate expression of diverse cellular inflammatory genes. Notably, posttranslational modifications, e.g., phosphorylation, further influence NF-κB activation and impinge on the outcome of infection by gammaherpesviruses. Among the multiple phosphorylated forms of RelA identified thus far, phosphorylations of S536 and S468 have been relatively well defined. RelA S536p was reported to enable NF-κB activation via recruitment of coactivators, such as p300, to promote targeted gene expression (31). However, phosphorylation of RelA S468 was originally identified for its role in priming RelA for degradation by the ubiquitin/proteasome system (60). In agreement with this, we previously showed that murine γHV68 induced RelA S468 phosphorylation to promote its degradation (28). In this study, we showed that RelA phosphorylation of S468 is necessary for NF-κB activation and KSHV latent infection. These findings support the conclusion that the outcome of RelA phosphorylation is context dependent. Nevertheless, we found that RelA S468A failed to inhibit RTA-mediated transcriptional activation, implying that S468 phosphorylation is necessary for RelA-mediated inhibition. Recently, we also found that the G protein-coupled receptor of KSHV enables NF-κB activation via phosphorylation of S468, which is relayed by IKKε (61). These results collectively support the corollary that S468 phosphorylation marks RelA for activation and perhaps undergoes immediate degradation, thereby coupling RelA degradation to its activation. This notion is consistent with the observation that many transcriptional factors are kept at low expression levels via the coupling of degradation to transcriptional activation. In support of this, depletion of IKKε elevated the levels of RelA S536p, which presumably accumulated due to lack of phosphorylation at S468 and impaired degradation thereof.
In this study, we report that both IKKβ and IKKε are critical for NF-κB activation and that these two kinases are coordinated to phosphorylate S536 and S468 of RelA, respectively. Moreover, RelA S536 phosphorylation is necessary for subsequent phosphorylation of S468, highlighting the sequential actions of IKKβ and IKKε in activating NF-κB. This is the first example wherein two closely related kinases are activated by a pathogen to enable NF-κB activation via site-specific phosphorylation. It is not clear how these closely related kinases achieve site-specific phosphorylation in cells. When purified, IKKβ and IKKε did not display selectivity to phosphorylate S536 and S468 of RelA in vitro (data not shown), suggesting that other cellular factors are required for the specific phosphorylation of RelA by IKKβ and IKKε in cells. Alternatively, it is also possible that specific phosphorylation is mediated by other kinases, which are selectively activated by IKKβ and IKKε. Surprisingly, IKKs, IKK-related kinases, and IRAK1 were previously reported to phosphorylate S536 or S468 of RelA under various physiological conditions (28, 60–63). Our recent studies further validate these phosphorylation events for RelA that occur in KSHV-infected cells. Notably, the KSHV G protein-coupled receptor (kGPCR) also activates IKKε, which promotes the phosphorylation of S468 of RelA to activate NF-κB, and NF-κB activation by IKKε is critical for kGPCR tumorigenesis (61). Here, we found that IKKε also phosphorylates RelA S468 to enable NF-κB activation and inhibition of KSHV lytic gene expression, thereby promoting KSHV latent infection. These studies support the possibility that RelA S468p is an activated form of NF-κB, and our results also suggest that IKKε is an important signaling molecule in activating NF-κB under diverse physiological conditions.
The assembly of the IKK kinase complex and subsequent activation by phosphorylation of a serine residue within the so-called activation loop defines a prototypical activation mechanism of IKKβ. Surprisingly, the S → E mutation within the equivalent activation loop of IKKε resulted in reduced kinase activity (64–66), suggesting a new mechanism of kinase activation distinct from those of IKKα and IKKβ. Our recent studies involving KSHV infection have provided compelling evidence that IKKε is critical for NF-κB activation, and the findings suggest that KSHV deploys viral factors, e.g., kGPCR, to activate IKKε. The identification of viral activators (e.g., kGPCR and others) will offer useful tools to dissect IKKε activation and determine its role in fundamental biological processes.
ACKNOWLEDGMENTS
We thank Y. Izumiya and E. Zandi for providing antibodies to RTA and IKKγ, respectively. We also thank Y. Nguyen (rotation student, UT Southwestern Medical Center) for assistance in the in vitro kinase assay using purified RTA.
This work is supported by grants from NIH (DE021445 and CA134421 to P. Feng; CA082057, CA31363, and CA115284 to J. U. Jung) and ACS (RSG-11-162-01-MPC to P. Feng).
We declare no conflict of interest.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 4.Nador RG, Cesarman E, Knowles DM, Said JW. 1995. Herpes-like DNA sequences in a body-cavity-based lymphoma in an HIV-negative patient. N. Engl. J. Med. 333:943. [DOI] [PubMed] [Google Scholar]
- 5.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 6.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiou CJ, Poole LJ, Kim PS, Ciufo DM, Cannon JS, ap Rhys CM, Alcendor DJ, Zong JC, Ambinder RF, Hayward GS. 2002. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:3421–3439. 10.1128/JVI.76.7.3421-3439.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273–296. 10.1146/annurevpathol.1.110304.100133 [DOI] [PubMed] [Google Scholar]
- 9.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185–6196. 10.1128/JVI.76.12.6185-6196.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. 10.1016/j.chom.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220. 10.1128/JVI.77.7.4205-4220.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaber T, Yuan Y. 2013. A virally encoded small peptide regulates RTA stability and facilitates Kaposi's sarcoma-associated herpesvirus lytic replication. J. Virol. 87:3461–3470. 10.1128/JVI.02746-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y, He Z, Liang D, Zhang Q, Zhang H, Deng Q, Robertson ES, Lan K. 2012. Carboxyl-terminal amino acids 1052 to 1082 of the latency-associated nuclear antigen (LANA) interact with RBP-Jκ and are responsible for LANA-mediated RTA repression. J. Virol. 86:4956–4969. 10.1128/JVI.06788-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6:570–575. 10.1016/j.chom.2009.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Y, Ganem D. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. U. S. A. 100:8490–8495. 10.1073/pnas.1432843100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gwack Y, Nakamura H, Lee SH, Souvlis J, Yustein JT, Gygi S, Kung HJ, Jung JU. 2003. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 23:8282–8294. 10.1128/MCB.23.22.8282-8294.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gwack Y, Baek HJ, Nakamura H, Lee SH, Meisterernst M, Roeder RG, Jung JU. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055–2067. 10.1128/MCB.23.6.2055-2067.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West JT, Wood C. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150–5163. 10.1038/sj.onc.1206555 [DOI] [PubMed] [Google Scholar]
- 19.Staudt MR, Dittmer DP. 2007. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr. Top. Microbiol. Immunol. 312:71–100. 10.1007/978-3-540-34344-8_3 [DOI] [PubMed] [Google Scholar]
- 20.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. 1997. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 91:243–252. 10.1016/S0092-8674(00)80406-7 [DOI] [PubMed] [Google Scholar]
- 21.Chen ZJ, Parent L, Maniatis T. 1996. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 84:853–862. 10.1016/S0092-8674(00)81064-8 [DOI] [PubMed] [Google Scholar]
- 22.Hacker H, Karin M. 2006. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006:re13. 10.1126/stke.3572006re13 [DOI] [PubMed] [Google Scholar]
- 23.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 24.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102:2992–2997. 10.1073/pnas.0408824102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U. S. A. 102:17717–17722. 10.1073/pnas.0508531102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 104:7253–7258. 10.1073/pnas.0611506104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong X, Feng H, Sun Q, Li H, Wu TT, Sun R, Tibbetts SA, Chen ZJ, Feng P. 2010. Murine gamma-herpesvirus 68 hijacks MAVS and IKKβ to initiate lytic replication. PLoS Pathog. 6(7):e1001001. 10.1371/journal.ppat.1001001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong X, Feng P. 2011. Murine gamma herpesvirus 68 hijacks MAVS and IKKβ to abrogate NFκB activation and antiviral cytokine production. PLoS Pathog. 7(11):e1002336. 10.1371/journal.ppat.1002336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong X, He Z, Durakoglugil D, Arneson L, Shen Y, Feng P. 2012. Murine gammaherpesvirus 68 evades host cytokine production via replication transactivator-induced RelA degradation. J. Virol. 86:1930–1941. 10.1128/JVI.06127-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkins ND. 2007. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49–62. 10.1038/nrm2083 [DOI] [PubMed] [Google Scholar]
- 31.Zhong H, Voll RE, Ghosh S. 1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1:661–671 [DOI] [PubMed] [Google Scholar]
- 32.Neumann M, Naumann M. 2007. Beyond IκBs: alternative regulation of NF-κB activity. FASEB J. 21:2642–2654. 10.1096/fj.06-7615rev [DOI] [PubMed] [Google Scholar]
- 33.Huang B, Yang XD, Lamb A, Chen LF. 2010. Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell. Signal. 22:1282–1290. 10.1016/j.cellsig.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. 2005. NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 25:7966–7975. 10.1128/MCB.25.18.7966-7975.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21. 10.1016/jviromet.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng H, Dong X, Negaard A, Feng P. 2008. Kaposi's sarcoma-associated herpesvirus K7 induces viral G protein-coupled receptor degradation and reduces its tumorigenicity. PLoS Pathog. 4(9):e1000157. 10.1371/journal.ppat.1000157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng P, Scott CW, Cho NH, Nakamura H, Chung YH, Monteiro MJ, Jung JU. 2004. Kaposi's sarcoma-associated herpesvirus K7 protein targets a ubiquitin-like/ubiquitin-associated domain-containing protein to promote protein degradation. Mol. Cell. Biol. 24:3938–3948. 10.1128/MCB.24.9.3938-3947.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng P, Everly DN, Jr, Read GS. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272–10280. 10.1128/JVI.75.21.10272-10280.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. 2009. NF-κB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jκ coactivator. J. Virol. 83:4435–4446. 10.1128/JVI.01999-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng P, Park J, Lee BS, Lee SH, Bram RJ, Jung JU. 2002. Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 76:11491–11504. 10.1128/JVI.76.22.11491-11504.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng P, Liang C, Shin YC, Xiaofei E, Zhang W, Gravel R, Wu TT, Sun R, Usherwood E, Jung JU. 2007. A novel inhibitory mechanism of mitochondrion-dependent apoptosis by a herpesviral protein. PLoS Pathog. 3(12):e174. 10.1371/journal.ppat.0030174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahin BB, Patel D, Conrad NK. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 6(3):e1000799. 10.1371/journal.ppat.1000799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240. 10.1016/j.virol.2004.03.049 [DOI] [PubMed] [Google Scholar]
- 45.Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. 2008. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3:352–363. 10.1016/j.chom.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 46.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620. 10.1128/JVI.78.7.3601-3620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. 2005. IKKalpha limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 434:1138–1143. 10.1038/nature03491 [DOI] [PubMed] [Google Scholar]
- 48.Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. 2003. NF-κB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540. 10.1128/JVI.77.15.8532-8540.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 50.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. 2006. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene 25:6844–6867. 10.1038/sj.onc.1209941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krug LT, Moser JM, Dickerson SM, Speck SH. 2007. Inhibition of NF-κB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3(1):e11. 10.1371/journal.ppat.0030011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968. 10.1128/JVI.02333-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guasparri I, Keller SA, Cesarman E. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199:993–1003. 10.1084/jem.20031467 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Yu Y, Wang SE, Hayward GS. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59–70. 10.1016/j.immuni.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 55.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 56.Sharma S, ten Oever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. 10.1126/science.1081315 [DOI] [PubMed] [Google Scholar]
- 57.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. 2004. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199:1641–1650. 10.1084/jem.20040520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. 2007. Multiple functions of the IKK-related kinase IKKε in interferon-mediated antiviral immunity. Science 315:1274–1278. 10.1126/science.1136567 [DOI] [PubMed] [Google Scholar]
- 59.Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T. 2011. IκB kinase epsilon (IKKε) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. U. S. A. 108:21170–21175. 10.1073/pnas.1119137109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, Repaka A, Mayo MW, Burstein E. 2009. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev. 23:849–861. 10.1101/gad.1748409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Lu X, Zhu L, Shen Y, Chengedza S, Feng H, Wang L, Jung JU, Gutkind JS, Feng P. 2013. IKK epsilon kinase is crucial for viral G protein-coupled receptor tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 110:11139–11144. 10.1073/pnas.1219829110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song YJ, Jen KY, Soni V, Kieff E, Cahir-McFarland E. 2006. IL-1 receptor-associated kinase 1 is critical for latent membrane protein 1-induced p65/RelA serine 536 phosphorylation and NF-κB activation. Proc. Natl. Acad. Sci. U. S. A. 103:2689–2694. 10.1073/pnas.0511096103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adli M, Baldwin AS. 2006. IKK-i/IKKε controls constitutive, cancer cell-associated NF-κB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 281:26976–26984. 10.1074/jbc.M603133200 [DOI] [PubMed] [Google Scholar]
- 64.Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, Boddupalli H, Combs RG, Kretzmer K, Tripp CS. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277:13840–13847. 10.1074/jbc.M110474200 [DOI] [PubMed] [Google Scholar]
- 65.Huynh QK, Kishore N, Mathialagan S, Donnelly AM, Tripp CS. 2002. Kinetic mechanisms of IκB-related kinases (IKK) inducible IKK and TBK-1 differ from IKK-1/IKK-2 heterodimer. J. Biol. Chem. 277:12550–12558. 10.1074/jbc.M111526200 [DOI] [PubMed] [Google Scholar]
- 66.Peters RT, Liao SM, Maniatis T. 2000. IKKε is part of a novel PMA-inducible IκB kinase complex. Mol. Cell 5:513–522. 10.1016/S1097-2765(00)80445-1 [DOI] [PubMed] [Google Scholar]