

Abstract
Posttranscriptional mechanisms are important for regulation of cellular and viral gene expression. The presence of the 5′ cap structure m7G(5′)ppp(5′)Nm is a general feature of mRNAs that provides protection from exoribonuclease digestion and enhances translation. Vaccinia virus and other poxviruses encode enzymes for both cap synthesis and decapping. Decapping is mediated by two related enzymes, D9 and D10, which are synthesized before and after viral DNA replication, respectively. The timing of D10 synthesis correlates better with the shutdown of host gene expression, and deletion of this gene has been shown to cause persistence of host and viral mRNAs in infected cells. Here, we constructed specific mutant viruses in which translation of D10 was prevented by stop codons or activity of D10 was abrogated by catalytic site mutations, without other genomic alterations. Both mutants formed plaques of normal size and replicated to similar extents as the parental virus in monkey epithelial cells and mouse embryonic fibroblasts. The synthesis of viral proteins was slightly delayed, and cellular and viral mRNAs persisted longer in cells infected with the mutants compared to either the parental virus or clonal revertant. Despite the mild effects in vitro, both mutants were more attenuated than the revertants in intranasal and intraperitoneal mouse models, and less infectious virus was recovered from organs. In addition, there was less lung histopathology following intranasal infection with mutant viruses. These data suggest that the D10 decapping enzyme may help restrict antiviral responses by accelerating host mRNA degradation during poxvirus infection.
INTRODUCTION
Posttranscriptional mechanisms are important for the regulation of cellular and viral gene expression at the levels of RNA stability and translation. The presence of a 5′ cap structure m7G(5′)ppp(5′)Nm is a general feature of eukaryotic mRNAs and many viral mRNAs that provides protection from exonuclease digestion and enhances translation (1–6). In eukaryotic cells, mRNA decay begins with shortening of the poly(A) tail and proceeds in either the 5′-to-3′ or 3′-to-5′ direction. The latter is mediated by the cytoplasmic RNA exosome (7–9) and a scavenger enzyme that degrades the cap (8). In the 5′-to-3′ pathway, removal of the cap (10–13) is followed by exoribonuclease Xrn1 digestion (14, 15). Enzymes with nudix hydrolase motifs that decap cytoplasmic mRNA are present in Saccharomyces cerevisiae and mammalian cells and are thought to function in mRNA decay (10–13, 16, 17).
Degradation of cellular mRNA may be advantageous to viruses by decreasing competition for the translational machinery and by reducing the synthesis of factors that contribute to the innate and adaptive immune responses to infection. The ability of viruses to accelerate mRNA decay has been intensively studied with members of the herpesvirus family, which replicate in the nucleus and utilize the transcriptional machinery of the cell (18, 19). For alphaherpesviruses, accelerated mRNA turnover is mediated by the endoribonuclease activity of the FEN1-like viral vhs protein (20, 21). Although deletion of the vhs gene has minimal effect on viral replication in cell culture, such mutants are attenuated in mice (22–24). Enhanced cell mRNA turnover by gammaherpesviruses is mediated by a multifunctional protein that also has DNase activity (18).
Rapid turnover of viral and cellular mRNAs occurs during infection with vaccinia virus (VACV), the prototype poxvirus (25–32). Poxviruses are large DNA viruses that encode enzymes and factors for replication and transcription of their genomes within the cytoplasm (33). Analysis of mRNAs in cells infected with VACV reveals the successive synthesis of early, intermediate, and late stage viral transcripts (32, 34–36). Transcription is mediated by a viral multisubunit DNA-dependent RNA polymerase together with stage-specific transcription factors that recognize cognate promoters. Although VACV encodes a predicted FEN1-like nuclease, it is involved in recombination and double-strand DNA repair, and there is no evidence for associated endo- or exo-RNase activity (37). However, VACV and other poxviruses encode decapping enzymes (38, 39) that may play a role similar to that of the herpesvirus vhs RNase. The two decapping enzymes, D9 and D10, are approximately 25% identical in sequence; both enzymes contain nudix hydrolase motifs that are necessary for liberating m7GDP from capped RNA substrates. D9 is expressed before and D10 is expressed after VACV DNA synthesis suggesting roles throughout virus replication (40). Homologs of D10 are encoded by all poxviruses, whereas D9 is encoded by most chordopoxviruses but not entomopoxviruses. A genome-wide RNA interference (RNAi) screen suggested a role for the 5′-to-3′ exoribonuclease Xrn1 in VACV replication (41), which could act in conjunction with the VACV decapping enzymes to degrade mRNA.
Two approaches have been employed to study the roles of D9 and D10 during VACV infection. Inducible overexpression of D10, and to a lesser extent D9, resulted in inhibition of virus replication accompanied with decreased levels of viral mRNA and proteins (42). Deletion mutants were constructed by replacing the D9R or D10R open reading frames (ORFs) encoding D9 and D10, respectively, with the enhanced green fluorescent protein (EGFP) ORF regulated by a strong promoter (40). Efforts to delete both D9R and D10R simultaneously by this method, however, were unsuccessful. While the D9R mutant showed no observable defect, the D10R mutant made small plaques and the purified virus particles had low infectivity. In addition, deletion of D10R resulted in delayed onset of early and late gene expression, and persistence of viral and cellular mRNAs in BS-C-1 cells (40). There have been no reports on the effects of D9R or D10R deletions on the virulence of poxviruses in animal models.
In considering further in vitro and in vivo studies on the role of D10, we had concerns that the phenotype of the D10R deletion mutant might be affected by transcription of the inserted EGFP ORF regulated by a strong promoter, which could occlude neighboring genes because of inefficient termination. Indeed, this appeared to be the case with an unrelated VACV mutant (43). In addition, there is a possibility that D10 has multiple functions. For the present study, we constructed a null mutant with stop codons that prevented D10 synthesis without perturbing the surrounding genome structure or adding an adventitious gene. In addition, we made catalytic site mutations in D10 to determine whether the phenotype would be as severe as the phenotype of the null mutant. A revertant virus was also constructed to serve as a control in addition to the wild-type (WT) virus. These new mutant viruses allowed us to investigate the phenotypes of D10R mutants in cultured cells and to assess their pathogenicity in a mouse model. Although the mutants replicated well in cell culture, cellular and viral mRNAs of the mutants persisted longer than those of the controls, and the mutants were significantly attenuated in vivo, suggesting an important role for D10 in defense against host antiviral responses.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 and BHK-21 cells were grown in minimum essential medium with Earle's balanced salts (EMEM) supplemented with 2 mM l-glutamine, 100 units penicillin, and 100 μg streptomycin per ml (Quality Biological, Inc.) and containing 10% fetal bovine serum (FBS) (Sigma-Aldrich). C57BL/6 mouse embryonic fibroblasts (MEFs) (ATCC SCRC-1008) were grown in Dulbecco's modified Eagle's medium (DMEM) (Quality Biological, Inc.) supplemented with 4 mM l-glutamine, 15% FBS, 100 units penicillin, and 100 μg streptomycin per ml.
Construction of recombinant viruses.
Vaccinia virus (VACV) recombinants were derived from the Western Reserve (WR) strain of VACV (ATCC VR-1354). The vD10mu (v stands for virus, and mu stands for mutant), vD10stop (stop stands for stop codons), and vD10rev (rev stands for revertant) viruses were constructed by replacing the EGFP ORF in vΔD10 (40) with a mutated or wild-type D10R ORF. To generate vD10mu, the EGFP ORF was replaced by D10R containing the active site mutations E144Q and E145Q (38) by homologous recombination with DNA generated by overlap PCR containing the mutated D10R ORF flanked by portions of D9R and D11L. A similar procedure was used to generate vD10stop in which amino acids M81 and K84 of D10R were mutated to produce TGA stop codons and vD10rev in which the WT D10R replaced EGFP. Homologous recombination was carried out by infecting BS-C-1 cells with 2 PFU/cell of vΔD10, followed by transfection of 1.5 μg of D10mu, D10stop, or wild-type D10 PCR products using Lipofectamine 2000 (Life Technologies). After 24 h, cells were subjected to 3 freeze-thaw cycles, and the lysates were diluted and used to infect fresh BS-C-1 cell monolayers. Colorless recombinant plaques were distinguished from the green fluorescent parental plaques and clonally purified 3 times. The inserted regions of the recombinant viruses were PCR amplified and sequenced to confirm their identity.
Purification of virus particles.
Recombinant viruses were grown in 5 × 108 BHK-21 cells for 2 or 3 days, followed by homogenization of the cells in 10 mM Tris-HCl (pH 9.0) with a Dounce homogenizer. The cell lysates were centrifuged through a 36% sucrose cushion, and the pellet was suspended in 10 mM Tris-HCl and purified by a second centrifugation through a 24% to 40% sucrose gradient (44, 45). The milky virus band was isolated, diluted with 1 mM Tris-HCl, and centrifuged to obtain a pellet, which was suspended in 1 mM Tris-HCl (pH 9.0). The number of virus particles was determined with a virus counter (ViroCyt, Denver, CO), and the virus titer was determined by plaque assay.
Plaque assay.
VACV preparations were dispersed in a chilled bath sonicator by pulsing three times for 45 s each time followed by 10-fold serial dilutions in supplemented EMEM with 2.5% FBS. Diluted viruses were distributed to BS-C-1 cell monolayers in 6-well plates. After absorption for 1.5 h, the medium was removed, and the cells were covered by supplemented EMEM containing 2.5% FBS and 0.5% methylcellulose. The cells were incubated at 37°C with 5% CO2 for 2 or 3 days, stained with 0.1% crystal violet in 20% ethanol, and visible plaques were counted. Alternatively, plaques were identified by immunostaining as follows. Infected BS-C-1 cell and MEF monolayers were fixed with methanol-acetone (1:1), washed with tap water, and incubated with rabbit anti-VACV antibody (1:2,000 dilution) for 1 h. The cells were washed again with tap water and incubated with a 1:3,000 dilution of protein A conjugated with peroxidase (Thermo Scientific) for 1 h. The cells were washed and incubated with the substrate dianisidine saturated in ethanol for 5 min. After color formation, the dianisidine solution was removed, and the cells were washed in tap water.
Virus yield determination.
BS-C-1 cells and MEFs were grown in 12-well plates and infected with 5 PFU/cell of VACV in supplemented EMEM with 2.5% FBS for 1.5 h. The cells were washed extensively with the same medium, incubated at 37°C, and harvested at intervals. Harvested cells were lysed by 3 freeze-thaw cycles, and virus titers were determined by plaque assay on BS-C-1 cell monolayers.
Pulse-labeling proteins.
The method for pulse-labeling infected cells was modified from a previous procedure (40). Briefly, BS-C-1 cells or MEFs grown on 12-well plates were infected with 5 PFU/cell of VACV. After 1.5 h, the cells were washed and incubated with fresh medium. At subsequent times, the medium was replaced with methionine- and cysteine-free RPMI 1640 (Sigma-Aldrich) containing 2.5% dialyzed FBS (HyClone); after 30 min, the medium was replaced with fresh methionine- and cysteine-free RPMI 1640 containing 100 μCi/ml of [35S]methionine and [35S]cysteine (PerkinElmer) for 30 min. The cells were harvested, washed, lysed in buffer containing 10 mM Tris (pH 7.5), 10 mM NaCl, 2 mM CaCl2, 0.5% NP-40, and 1× EDTA-free protease inhibitors (Roche) in the presence of 1,000 gel units of micrococcal nuclease (New England BioLabs) at room temperature for 15 min to digest DNA. Lithium dodecyl sulfate sample and reducing buffers (1× NuPAGE; Life Technologies) were added to the lysates, and the resulting lysate mixtures were heated at 70°C for 15 min. Proteins were resolved on 4 to 12% NuPAGE Bis-Tris gels (Life Technologies), dried on Whatman paper, exposed to a PhosphorImager screen, and visualized by a Molecular Dynamics Typhoon 9410 molecular imager (GE Amersham).
Western blotting.
Infected cells were harvested and lysed, and the proteins were resolved by electrophoresis on 4 to 12% NuPAGE Bis-Tris gels (Life Technologies) as described above. The proteins were electrophoretically transferred to a nitrocellulose membrane with an iBlot system (Life Technologies). The membrane was blocked in 5% nonfat milk in phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST) for 1 h, washed with PBST, and incubated with the primary antibody in PBST containing 5% nonfat milk for 1 h. The membrane was washed with PBST and incubated with the secondary antibody conjugated by horseradish peroxidase in PBST with 5% nonfat milk for 1 h. After the membrane was extensively washed, the amount of protein bound by the secondary antibody was visualized by SuperSignal West Femto substrates (Thermo Scientific). The antibodies used and their dilutions are as follows: rabbit polyclonal anti-actin antibody (Sigma-Aldrich) at 1:2,000, mouse anti-E3 monoclonal antibody (MAb) (46) at 1:10, rabbit polyclonal anti-D13 antibody (47) at 1:3,000, and rabbit polyclonal anti-A3 antibody (unpublished) at 1:5,000 dilution. The secondary goat anti-mouse (Thermo Scientific) and goat anti-rabbit (Thermo Scientific) antibodies were both diluted 1:3,000.
Northern blotting.
Total RNA from infected BS-C-1 cells or MEFs was isolated using a RNeasy minikit (Qiagen). A sample containing 2 μg of total RNA was loaded on a glyoxal gel (Life Technologies); following electrophoresis, the RNA was electrophoretically transferred to a positively charged nylon membrane using an iBlot system (Life Technologies). Subsequent steps were carried out using a Northern Max-Gly kit (Life Technologies) according to the manufacturer's instructions. Double-strand DNA probes labeled with [α-32P]dCTP (PerkinElmer) were made with a Decaprime II random priming kit (Life Technologies) using a 300- to 400-nucleotide PCR fragment amplified from the gene of interest. The probed membrane was exposed to a PhosphorImager screen and visualized by the Molecular Dynamics Typhoon 9410 molecular imager (GE Amersham).
Infection of mice.
Three or five female 6-week-old BALB/c mice (Taconic, Germantown, NY) were kept in one cage for each treatment group. Prior to inoculation, VACV was diluted in PBS containing 0.05% bovine serum albumin (BSA). The virus titers were verified on the day of the procedure by plaque assay. For intranasal (i.n.) inoculation, mice were anesthetized by isoflurane inhalation, and 20 μl of virus was injected into one nostril. For intraperitoneal (i.p.) inoculation, 200 μl of virus was injected into the left or right lower quadrant of the abdomen.
Determination of virulence in mice.
The virulence of vD10stop, vD10mu, and vD10rev were determined by comparing the weight loss and viral titers in the organs. Following i.n. inoculation, mice were weighed daily for 17 days, and their disease symptoms were observed. The mice were euthanized when they lost 30% of their original weight, in compliance with the National Institute of Allergy and Infectious Diseases Animal Care and Use Protocols. For determination of virus titers in organs, mice were sacrificed on day 5 or day 7 following inoculation, and their organs were removed and placed in a solution containing 148 mM NaCl, 5 mM KCl, 1.2 mM MgSO4 · 7H2O, 0.67 mM KH2PO4, 1.18 mM K2HPO4, 2.46 mM CaCl2 · 2H2O, and 10.8 mM HEPES [pH 7.3]). Organs were homogenized, aliquoted, and stored at −80°C. Aliquots of organ homogenates were subjected to 3 freeze-thaw cycles and sonicated, and the virus titers were determined by plaque assay.
Statistical analysis.
Student's t test was used to determine the significance of differences between weights of mice following virus infection. Either Student's t test or Mann-Whitney test was employed to determine the significance of differences in virus titers of organs. Kaplan-Meier plot and log rank test (Mantel-Cox test) were used to analyze the survival rates of mice. Statistical analyses were performed with GraphPad Prism version 6.00 (GraphPad Software, La Jolla, CA, USA).
Histopathology and immunohistochemistry.
For evaluation of histopathology, mice were infected i.n. with vD10stop, vD10mu, or vD10rev at a dose of 104 or 105 PFU and sacrificed on day 7 or 5, respectively. The animals were euthanized by CO2 inhalation and necropsied according to a standard protocol. The lungs and nasal turbinates were fixed by immersion in 10% neutral buffered formalin, embedded in paraffin, cut into 4-μm-thick sections, and stained with hematoxylin and eosin (H&E) for examination by light microscopy. For a control for histopathology, one mouse was sham inoculated with PBS containing 0.05% bovine serum albumin and sacrificed at the same time. A pathologist without knowledge of the treatment group examined the slides.
Serial unstained sections were used for immunohistochemistry. The sections on slides were deparaffinized in xylenes and rehydrated through graded concentrations of ethanol. Heat-mediated antigen retrieval (Diva decloaker; Biocare Medical, Concord, CA) was carried out prior to immunostaining. A polyclonal antibody (48), made by infecting rabbits with VACV WR, was incubated on the tissue sections for 60 min at room temperature. Bound antibody was detected using a biotinylated anti-rabbit secondary antibody followed by a Vectastain standard ABC-AP (ABC stands for avidin-biotinylated enzyme complex, and AP stands for alkaline phosphatase) kit (Vector Laboratories). Vulcan Fast Red (Biocare Medical, Concord, CA) was used as the chromogen, and nuclei were counterstained with hematoxylin.
RESULTS
Construction and replication of D10 mutants.
Starting with vΔD10, in which the EGFP ORF had been substituted for the D10R ORF (40), we made three new constructs by replacing the EGFP ORF with WT D10R or mutated D10R ORFs (Fig. 1A). The revertant vΔD10rev contained the original D10R ORF, the mutant vD10stop had two stop codons near the N terminus of D10, and the mutant vD10mu had two amino acid substitutions in the catalytic site, which were previously shown to completely inactivate decapping activity in vitro (38). The mutants were constructed by homologous recombination and identified by the absence of the green fluorescence exhibited by vΔD10. The viruses were clonally purified by several rounds of plaque purification and analyzed by PCR and DNA sequencing. Since a previously made antibody to D10 had low sensitivity (42), additional D10rev, D10stop, and D10mu viruses were made with a 3×FLAG tag at their C termini. This allowed us to check the efficiency of the stop codons and the stability of the mutated protein. D10stop-3XFLAG could not be detected by Western blotting, whereas the expression of D10mu-3XFLAG was similar to D10rev-3XFLAG (data not shown).
FIG 1.

Construction and in vitro replication of D10R mutants. (A) Representation of D10R and flanking ORFs of WT and mutant viruses. The D10 deletion mutant (vΔD10) was constructed by replacing the D10R ORF of WT virus with EGFP regulated by the VACV P11 promoter (40). The revertant virus (vD10rev) containing the intact D10R ORF, catalytic site mutant (vD10mu), and stop codon mutant (vD10stop) were derived from vΔD10 by homologous recombination. The crosses in vD10mu denote the point mutations in the D10 nudix catalytic domain. The signs in vD10stop indicate the locations of the stop codons. (B) Plaque formation. Monolayers of BS-C-1 cells and MEFs were infected with the indicated VACV strains for 48 h. The cells were washed, incubated with anti-VACV polyclonal rabbit antibody, washed again, and incubated with protein A conjugated with peroxidase followed by the substrate dianisidine for visualization of plaques. (C) Growth curves. BS-C-1 cells and MEFs were grown in 12-well plates and infected with 5 PFU/cell of VACV strains. BS-C-1 cells were harvested at 3, 6, 13, 26, and 38 h postinfection, and MEFs were harvested at 2, 12.5, 26, and 38 h postinfection. Harvested cells were lysed and diluted, and virus titers were determined by plaque assay on BS-C-1 cells. The MEF titers are the averages of two independent experiments.
Replication of D10 mutants.
To avoid a potential effect of the epitope tag on function, all subsequent studies were carried out with the untagged mutants. The plaques formed by WT VACV, vΔD10, and the three new viruses in BS-C-1 cells, which are commonly used with VACV, were visualized by staining with crystal violet (not shown) and antibody to VACV followed by a secondary antibody conjugated to peroxidase (Fig. 1B). Since mouse studies were planned, we also analyzed plaque formation in MEFs (Fig. 1B). The plaques formed by WT VACV, vD10rev, vD10stop, and vD10mu were similar in size and appearance. However, the plaques formed by vΔD10 were slightly smaller than the others. The viruses all replicated to similar levels in BS-C-1 cells and MEFs (Fig. 1C). In some experiments (not shown), the yields of vΔD10 in BS-C-1 cells were less than those of the other viruses; however, when the data were combined, this was not statistically significant. A discrepancy between plaque size and virus yield can have multiple reasons that were not explored here.
Virus particles were purified from cell lysates by sedimentation through a sucrose cushion and gradient. Infectivity was determined by plaque assay, and the number of virions was ascertained by using a virus counter, which is a specialized flow cytometer that scores particles that contain nucleic acids and proteins. When propagated in HeLa cells, vΔD10 had a higher particle/PFU ratio than WT VACV did, as previously reported (40). However, we discovered that the calculated particle/PFU ratios of the purified mutant and revertant viruses grown in BHK-21 cells varied from 30 to 60, which was considered to be within the error of the analytical procedures. Therefore, multiplicities for infections were determined from the plaque titers.
Effects of D10 mutations on protein synthesis.
VACV gene expression is divided into early, intermediate, and late stages. Early proteins may be detected by 3 h and intermediate and late proteins may be detected by 6 h with the exact times and relative amounts depending on the multiplicity and synchronicity of infection and the cell type. The progression of host cell and viral protein synthesis can be monitored by autoradiography after pulse-labeling infected cells with [35S]methionine and [35S]cysteine. At 3 h, there were only subtle differences between the band patterns from mock- and VACV-infected cells (Fig. 2A). At 6 h, there were distinct differences between the mock infection and each of the VACV infections. At 9 h, the virus-specific postreplicative protein bands were evident in each of the samples but were most intense in the WT. However, the intensities of the virus-induced proteins from the different virus infections became more similar to each other at 12 to 24 h. The bands that were predominant in the mock-infected sample were sharply reduced in all infected samples by 12 h. A similar progression was seen in MEFs (Fig. 2B).
FIG 2.

Time course of viral protein synthesis determined by pulse-labeling with radioactive amino acids. (A and B) BS-C-1 cells (A) and MEFs (B) were mock infected or infected with 5 PFU/cell of WT VACV or vD10rev, vΔD10, vD10mu, and vD10stop mutant viruses. At the indicated time after infection (shown in hours), the cells were pulse-labeled with [35S]methionine and [35S]cysteine for 30 min. The cells were harvested and lysed, and proteins were resolved by SDS-polyacrylamide gel electrophoresis and exposed to a PhosphorImager for visualization of newly synthesized proteins. The positions and masses (in kilodaltons) of marker proteins are indicated to the left of the gels.
Viral protein synthesis was also evaluated by Western blotting using antibodies to representative early (E3), intermediate (D13), and late (A3) VACV proteins. In BS-C-1 cells, viral intermediate and late protein synthesis appeared to be slightly delayed in the D10 mutants compared to the WT (Fig. 3A). No consistent differences in viral protein synthesis between the viruses were discerned in MEFs (Fig. 3B).
FIG 3.

Time course of synthesis of specific early, intermediate, and late viral proteins. (A and B) BS-C-1 cells (A) and MEFs (B) were infected with 5 PFU/cell of WT VACV or vD10rev, vΔD10, vD10mu, or vD10stop and harvested at the indicated time (shown in hours). The lysates were analyzed by Western blotting using antibodies to the E3 early protein, the D13 intermediate protein, and the A3 late protein. Antibody to actin served as a loading control. One set of cells was infected in the presence of 1-β-d-arabinofuranosylcytosine (AraC) to inhibit viral DNA replication and confirm the stage of expression of the viral proteins.
Effects of D10 mutations on mRNA accumulation.
Northern blotting was used to compare the amounts of representative cellular and viral mRNAs. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a representative cell mRNA, decreased more rapidly in BS-C-1 cells infected with the WT virus or vD10rev than in cells infected with vΔD10, vD10stop, or vD10mu during the first 9 h, but by 12 h the RNA was barely detected in all samples (Fig. 4A). The patterns of the two VACV early mRNAs examined, C11 and E3, were similar to the pattern of the GAPDH mRNA: more rapid degradation in BS-C-1 cells infected with the WT and revertant than in the D10 mutants (Fig. 4A). F17 was selected as an example of a late mRNA because it migrates as a discrete band in contrast to most other late mRNAs. F17 mRNA was more abundant at 6 h and 9 h in the cells infected with the D10 mutants than in cells infected with the WT or revertant (Fig. 4A). Similarly, C11 and F17 mRNAs were more abundant at 6 h and 9 h in MEFs infected with D10 mutants than in MEFs infected with the revertant (Fig. 4B).
FIG 4.

Levels of viral and cellular mRNAs following infection with WT and mutant viruses. (A and B) BS-C-1 cells (A) and MEFs (B) were infected with WT VACV, vD10rev, vΔD10, vD10mu, or vD10stop and harvested at the indicated time (in hours). Total RNAs were isolated and resolved on a glyoxal RNA gel, and the C11, E3, and F17 viral mRNAs and GAPDH cell mRNA were analyzed by Northern blotting.
Attenuation of D10 mutants in a mouse model.
We excluded vΔD10 for animal studies because of the expression of EGFP and the smaller plaques formed by this virus. vD10rev was preferred to WT VACV as a control because it was derived from the same clonally purified parent as vD10stop and vD10mu. The mouse i.n. model has been extensively used for VACV pathogenesis studies. Morbidity, which can be followed by weight loss, results from replication of virus in the lungs, followed by viremia and dissemination to other organs (49). Groups of BALB/c mice were inoculated i.n. with 104, 105, and 106 PFU of vD10rev, vD10stop, or vD10mu and monitored for 17 days. At the low dose of 104 PFU, only mice inoculated with vD10rev had weight loss, and there were no deaths (Fig. 5A). The weight loss between mice infected with vD10rev and the two mutant viruses was significant (P < 0.05) from day 4 through day 15 and highly significant (P < 0.001) from days 6 to 9. At 105 PFU, all infected mice lost weight, but there was greater weight loss in those infected with vD10rev than with either of the D10 mutants (Fig. 5B). The weight loss between vD10rev and the two mutant viruses was significant from days 5 through 17 (except for day 9) and highly significant for days 13 to 17. Furthermore, mice inoculated with vD10stop and vD10mu survived, whereas 70% of those receiving vD10rev died or were sacrificed because of >30% weight loss (Fig. 5C). Statistical significance determined by a Kaplan-Meier plot and a log rank test (Mantel-Cox test) gave P values of <0.0013 for the values for mice infected with the vD10rev virus and with either the vD10mu or vD10stop virus or uninfected weight control.
FIG 5.
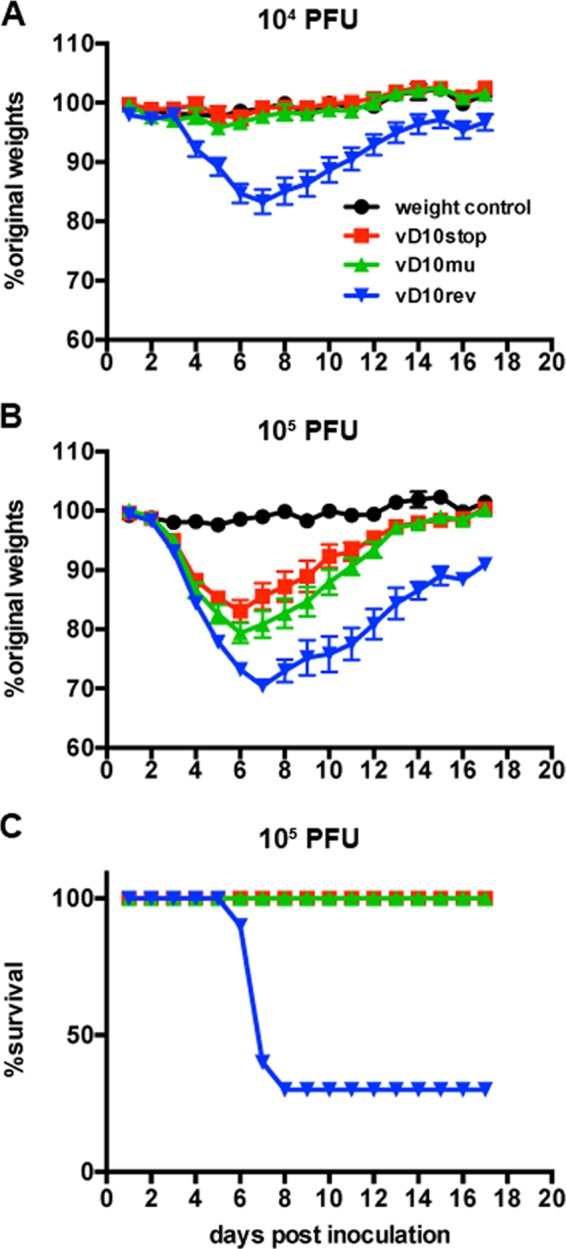
Morbidity and mortality of mice following infection with control and mutant viruses. (A and B) BALB/c mice were inoculated i.n. with 104 PFU (A) or 105 PFU (B) of vD10stop, vD10mu, or vD10rev or with PBS containing 0.05% BSA as a control. The weights of the mice were measured and recorded daily. The daily weights collected from two independent experiments (n = 5 in each experiment) were combined and plotted against the percentage of the original weights prior to inoculation. (C) Survival rates of mice inoculated with 105 PFU.
The virus titers in the nasal turbinates, lungs, abdominal organs, and brain were determined on day 7 after intranasal inoculation with 104 PFU of vD10rev, vD10stop, and vD10mu (Fig. 6A). Significantly higher titers of vD10rev than that of either vD10stop or vD10mu were found in the nasal turbinates (P < 0.04) and lungs (P < 0.02), though the larger amount of virus in the spleens of vD10rev-infected mice did not reach statistical significance (P < 0.06). In contrast, the differences between the virus titers of vD10stop and vD10mu were not statistically significant. In addition, virus was recovered from the livers and brains of mice infected with vD10rev but not from those organs of mice infected with vD10stop or vD10mu (Fig. 6A).
FIG 6.
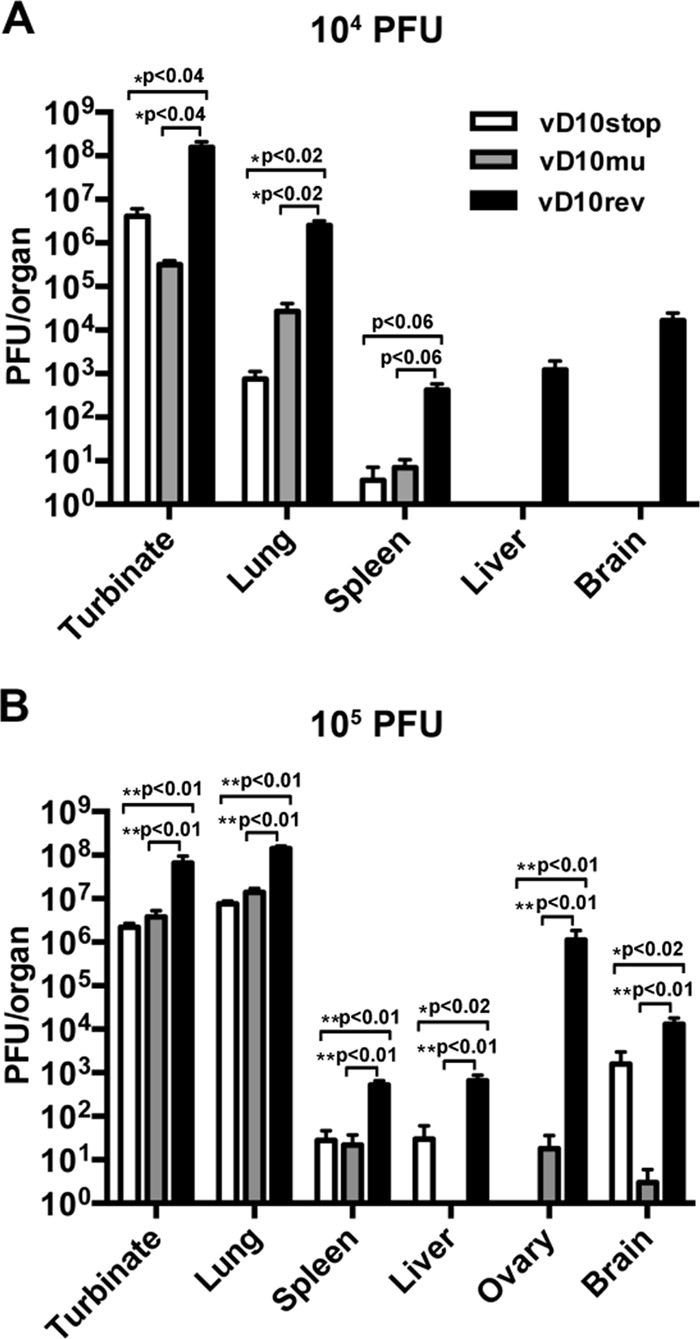
Virus titers in organs of mice following i.n. infection with control and mutant VACV. Groups of BALB/c mice were inoculated i.n. with virus in two independent experiments. (A) The mice were inoculated with 104 PFU and sacrificed on day 7 (n = 3). (B) The mice were inoculated with 105 PFU and sacrificed on day 5 (n = 5). The organs were removed and homogenized, and the virus titers of homogenates were determined by plaque assay on BS-C-1 cells. Statistical significance was determined by Student's t test for panel A and by Mann-Whitney test for panel B.
The same pattern was found on day 5 following inoculation with 105 PFU in an independent experiment (Fig. 6B). Significantly higher virus titers were found in the turbinates (P < 0.01), lungs (P < 0.01), spleens (P < 0.01), livers (P < 0.02), ovaries (P < 0.01), and brains (P < 0.02) of mice infected with vD10rev than in mice infected with either vD10stop or vD10mu. Again, differences between vD10stop and vD10mu were not statistically significant (Fig. 6B).
To evaluate the virulence of D10 mutants in vivo, tissue sections from the upper and lower respiratory tracts were examined by routine H&E staining and by VACV immunohistochemistry. There was evidence of efficient replication in the nasal epithelium and the underlying glands at 7 days after i.n. infection with 104 PFU of each virus (Fig. 7). Histologically, there was hyperplasia and necrosis of nasal epithelial cells often with abundant fluid and cellular debris in the nasal passages. In the lungs, lesions were generally mild and ranged from minimal alveolitis to multifocal bronchiolar epithelial necrosis with extension into the surrounding alveoli. The extent and severity of the pathology and level of virus infection in both the upper and lower respiratory tracts were highest in mice infected with vD10rev compared with mice infected with vD10stop or vD10mu. Irrespective of the virus inoculum, the degree of histopathology and virus replication was higher in the nasal turbinates than in the lung for each animal. We also examined the upper and lower respiratory tissues of mice on day 5 after infection with 105 PFU of each virus. There was extensive lung pathology in each case, making comparisons difficult (data not shown).
FIG 7.

Histology of nasal turbinates and lungs of mice following i.n. inoculations with control and mutant VACV. Groups (n = 3) of BALB/c mice were inoculated i.n. with 104 PFU of vD10rev, vD10stop, or vD10mu. Mice were sacrificed on day 7, and their organs were removed, fixed, embedded in paraffin, and sectioned. The sections were stained with hematoxylin and eosin (H&E) and with anti-VACV antibody for immunohistochemistry (IHC).
The i.p. route has also been used to study VACV infection of mice. However, even with the pathogenic WR strain of VACV, little or no weight loss is observed and doses of >108 PFU are required for lethality. At 5 days after infection with 106 PFU of revertant and mutant viruses, the mice were sacrificed, and the virus titers in abdominal organs, lungs, brains, and nasal turbinates were determined (Fig. 8). The titers of vD10rev were significantly higher than vD10stop and vD10mu in the liver (P < 0.02), spleen (P < 0.04), and kidney (P < 0.02). Virus was obtained only from the turbinates and brains of animals infected with vD10rev (Fig. 8). Differences between the vD10stop and vD10rev values were not statistically significant.
FIG 8.
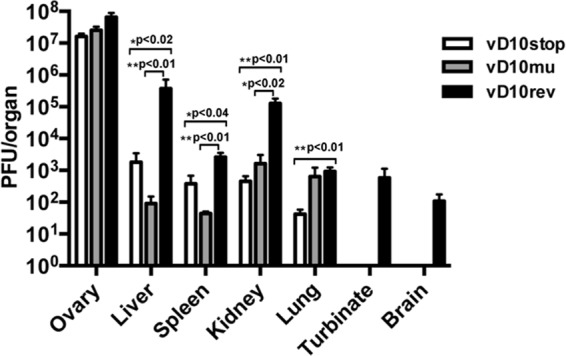
Virus titers in organs of mice following intraperitoneal infection with control and mutant VACV. Mice (n = 5 per group) were inoculated with 106 PFU of vD10rev, vD10stop, or vD10mu and sacrificed on day 5. Their organs were homogenized, and virus titers were determined by plaque assay on BS-C-1 cells. Statistical significance was determined by the Mann-Whitney test.
DISCUSSION
Our previous studies indicated that deletion of the D10R ORF resulted in persistence of cellular and viral mRNAs over time compared to the WT virus, which could be attributed to the reduction in decapping activity (38, 40). However, the mutant virus was constructed by replacing the D10R ORF with EGFP regulated by a strong promoter, which contributed to impaired replication of an unrelated VACV mutant (43). Therefore, we constructed a new set of mutants without foreign genes or genome rearrangements for the present study. Starting from the original cloned deletion mutant, we made a revertant in which the EGFP ORF was replaced by the original D10R ORF, a mutant with D10R containing stop codons near the N terminus, and a mutant with D10R containing catalytic site mutations that had previously been shown to eliminate D10 decapping activity in vitro (38). Western blotting confirmed the efficacy of the stop codons and the expression of the catalytic site mutant. The new mutants and the revertant made plaques similar in size to those of the WT virus, whereas the original deletion mutant made slightly smaller plaques, suggesting that the EGFP insertion was indeed detrimental.
The two new mutant viruses and the revertant replicated to similar titers in BS-C-1 cells and primary MEFs. We used MEFs in addition to BS-C-1 cells because our plan was to carry out experiments in mice to determine whether loss of decapping activity would affect virulence. In BS-C-1 cells, early and late viral mRNAs and cellular GAPDH persisted longer in cells infected with the mutant viruses than in cells infected with the revertant. In addition, viral protein synthesis was slightly delayed in BS-C-1 cells infected with the mutants compared to cells infected with the revertant. Persistence of viral mRNAs also occurred in MEFs infected with mutants compared to the revertant, although the time course of viral protein synthesis was similar. Thus, both the virus with the stop codon and virus with catalytic site mutations replicated normally in cultured cells, and the major defect with each appeared to be greater stability of mRNAs. Further studies with WT and D10 mutants are planned to determine whether there is specificity in decapping and degradation of cellular mRNAs. Interestingly, global host protein shutoff occurred at late times despite the absence of the D10 protein, indicating that additional factors are responsible. In our previous study (40), host shutoff was unaffected by D9 deletion, and the early synthesis of this protein does not correlate with the late shutoff of host protein synthesis. Nevertheless, it is possible that the host shutoff is determined by the combined action of the two decapping enzymes. Since previous efforts to make a double D9/D10 deletion mutant failed (40), we are working on alternative approaches to achieve this goal. However, other mechanisms, including the sequestering of translation factors in virus factories, may contribute to inhibition of host protein synthesis (50, 51).
In contrast to the mild effects in vitro, the stop codon and active site mutant viruses caused less weight loss and mortality than the revertant virus following i.n. infection of mice. Moreover, the titers of the revertant virus were significantly higher than those of the mutants in the nasal turbinates, lungs, livers, spleens, ovaries, and brains of mice infected with virus in independent experiments carried out with different doses of virus. Histological examination of the lungs confirmed less extensive pathology in mice infected with the mutants. Significantly higher organ titers of revertant virus compared to mutant viruses were also recorded after i.p. infection of mice. The similar in vitro and in vivo results obtained with the null mutant and the active site mutant indicated that the attenuation is due to decreased decapping activity, rather than another unknown activity of D10. The more dramatic effect of the VACV decapping mutants in mice compared to cultured cells parallels that found for herpes simplex virus (HSV) vhs mutants. There is accumulating evidence that by accelerating mRNA decay, vhs counters innate and adaptive immune responses (24, 52–54) and is important for pathogenicity (22, 23, 55, 56). Although VACV accelerates host mRNA decay by a different mechanism than HSV, the end result for both may be to diminish the magnitude of antiviral responses.
ACKNOWLEDGMENTS
We thank Catherine Cotter for help in preparing cells, Tatiana Koonin (Senkevich), and Liliana Maruri-Avidal for their suggestions on recombinant virus construction, Robin Kastenmayer and Jeffery Americo for assisting organ harvesting and virus inoculation of mice, Patricia Earl for suggestions regarding statistical analysis, and the NIAID Comparative Medicine Branch for maintaining and weighing the mice.
The research was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Wei CM, Moss B. 1975. Methylated nucleotides block 5′-terminus of vaccinia virus mRNA. Proc. Natl. Acad. Sci. U. S. A. 72:318–322. 10.1073/pnas.72.1.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furuichi Y, Morgan M, Muthukrishnan S, Shatkin AJ. 1975. Reovirus messenger RNA contains a methylated, blocked 5′-terminal structure: m7G(5′)ppp(5′)GmpCp. Proc. Natl. Acad. Sci. U. S. A. 72:362–366. 10.1073/pnas.72.1.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei CM, Gershowitz A, Moss B. 1975. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4:379–386. 10.1016/0092-8674(75)90158-0 [DOI] [PubMed] [Google Scholar]
- 4.Furuichi Y, Morgan M, Shatkin AJ, Jelinek W, Salditt-Georgieff M, Darnell JE. 1975. Methylated, blocked 5 termini in HeLa cell mRNA. Proc. Natl. Acad. Sci. U. S. A. 72:1904–1908. 10.1073/pnas.72.5.1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muthukrishnan S, Morgan M, Banerjee AK, Shatkin AJ. 1976. Influence of 5′-terminal m7G and 2′-O-methylated residues on messenger ribonucleic acid binding to ribosomes. Biochemistry 15:5761–5768. 10.1021/bi00671a012 [DOI] [PubMed] [Google Scholar]
- 6.Shuman S. 2001. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acids Res. Mol. Biol. 66:1–40. 10.1016/S0079-6603(00)66025-7 [DOI] [PubMed] [Google Scholar]
- 7.Anderson JS, Parker RP. 1998. The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J. 17:1497–1506. 10.1093/emboj/17.5.1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu H, Rodgers ND, Jiao X, Kiledjian M. 2002. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 21:4699–4708. 10.1093/emboj/cdf448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q, Greimann JC, Lima CD. 2006. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127:1223–1237. 10.1016/j.cell.2006.10.037 [DOI] [PubMed] [Google Scholar]
- 10.Dunckley T, Parker R. 1999. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J. 18:5411–5422. 10.1093/emboj/18.19.5411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. 2002. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc. Natl. Acad. Sci. U. S. A. 99:12663–12668. 10.1073/pnas.192445599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21:6915–6924. 10.1093/emboj/cdf678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lykke-Andersen J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 22:8114–8121. 10.1128/MCB.22.23.8114-8121.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Decker CJ, Parker R. 1993. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 7:1632–1643. 10.1101/gad.7.8.1632 [DOI] [PubMed] [Google Scholar]
- 15.Hsu CL, Stevens A. 1993. Yeast cells lacking 5′→3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol. Cell. Biol. 13:4826–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Song M, Kiledjian M. 2011. Differential utilization of decapping enzymes in mammalian mRNA decay pathways. RNA 17:419–428. 10.1261/rna.2439811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song MG, Li Y, Kiledjian M. 2010. Multiple mRNA decapping enzymes in mammalian cells. Mol. Cell 40:423–432. 10.1016/j.molcel.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glaunsinger BA, Ganem DE. 2006. Messenger RNA turnover and its regulation in herpesviral infection. Adv. Virus Res. 66:337–394. 10.1016/S0065-3527(06)66007-7 [DOI] [PubMed] [Google Scholar]
- 19.Gaglia MM, Covarrubias S, Wong W, Glaunsinger BA. 2012. A common strategy for host RNA degradation by divergent viruses. J. Virol. 86:9527–9530. 10.1128/JVI.01230-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwong AD, Frenkel N. 1989. The herpes simplex virus virion host shutoff function. J. Virol. 63:4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taddeo B, Roizman B. 2006. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 80:9341–9345. 10.1128/JVI.01008-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strelow LI, Leib DA. 1995. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 69:6779–6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strelow L, Smith T, Leib D. 1997. The virion host shutoff function of herpes simplex virus type 1 plays a role in corneal invasion and functions independently of the cell cycle. Virology 231:28–34. 10.1006/viro.1997.8497 [DOI] [PubMed] [Google Scholar]
- 24.Smiley JR. 2004. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 78:1063–1068. 10.1128/JVI.78.3.1063-1068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oda K, Joklik WK. 1967. Hybridization and sedimentation studies on “early” and “late” vaccinia messenger RNA. J. Mol. Biol. 27:395–419. 10.1016/0022-2836(67)90047-2 [DOI] [PubMed] [Google Scholar]
- 26.Sebring ED, Salzman NP. 1967. Metabolic properties of early and late vaccinia messenger ribonucleic acid. J. Virol. 1:550–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boone RF, Moss B. 1978. Sequence complexity and relative abundance of vaccinia virus mRNA's synthesized in vivo and in vitro. J. Virol. 26:554–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice AP, Roberts BE. 1983. Vaccinia virus induces cellular mRNA degradation. J. Virol. 47:529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jindal S, Young RA. 1992. Vaccinia virus infection induces a stress response that leads to association of hsp70 with viral proteins. J. Virol. 66:5357–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brum LM, Lopez MC, Varela JC, Baker HV, Moyer RW. 2003. Microarray analysis of A549 cells infected with rabbitpox virus (RPV): a comparison of wild-type RPV and RPV deleted for the host range gene, SPI-1. Virology 315:322–334. 10.1016/S0042-6822(03)00532-4 [DOI] [PubMed] [Google Scholar]
- 31.Guerra S, Lopez-Fernandez LA, Conde R, Pascual-Montano A, Harshman K, Esteban M. 2004. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J. Virol. 78:5820–5834. 10.1128/JVI.78.11.5820-5834.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:11513–11518. 10.1073/pnas.1006594107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss B. 2013. Poxviridae, p 2129–2159 In Knipe DM, Howley PM. (ed), Fields virology, 6th ed, vol 2 Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 34.Baldick CJ, Jr, Moss B. 1993. Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate stage genes. J. Virol. 67:3515–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Assarsson E, Greenbaum JA, Sundstrom M, Schaffer L, Hammond JA, Pasquetto V, Oseroff C, Hendrickson RC, Lefkowitz EJ, Tscharke DC, Sidney J, Grey HM, Head SR, Peters B, Sette A. 2008. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. U. S. A. 105:2140–2145. 10.1073/pnas.0711573105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908. 10.1128/JVI.05446-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senkevich TG, Koonin EV, Moss B. 2009. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc. Natl. Acad. Sci. U. S. A. 106:17921–17926. 10.1073/pnas.0909529106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parrish S, Resch W, Moss B. 2007. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. U. S. A. 104:2139–2144. 10.1073/pnas.0611685104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parrish S, Moss B. 2007. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 81:12973–12978. 10.1128/JVI.01668-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parrish S, Moss B. 2006. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J. Virol. 80:553–561. 10.1128/JVI.80.2.553-561.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sivan G, Martin SE, Myers TG, Buehler E, Szymczyk KH, Ormanoglu P, Moss B. 2013. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:3519–3524. 10.1073/pnas.1300708110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shors T, Keck JG, Moss B. 1999. Down regulation of gene expression by the vaccinia virus D10 protein. J. Virol. 73:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Senkevich TG, Wyatt LS, Weisberg AS, Koonin EV, Moss B. 2008. A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture. Virology 374:506–514. 10.1016/j.virol.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr. Protoc. Mol. Biol. Chapter 16:Unit 16.17. 10.1002/0471142727.mb1617s43 [DOI] [PubMed] [Google Scholar]
- 45.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 2001. Preparation of cell cultures and vaccinia virus stocks. Curr. Protoc. Mol. Biol. Chapter 16:Unit 16.16. 10.1002/0471142727.mb1616s43 [DOI] [PubMed] [Google Scholar]
- 46.Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B. 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3l gene. Virology 195:732–744. 10.1006/viro.1993.1424 [DOI] [PubMed] [Google Scholar]
- 47.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies DH, Wyatt LS, Newman FK, Earl PL, Chun S, Hernandez JE, Molina DM, Hirst S, Moss B, Frey SE, Felgner PL. 2008. Antibody profiling by proteome microarray reveals the immunogenicity of the attenuated smallpox vaccine modified vaccinia virus Ankara is comparable to that of Dryvax. J. Virol. 82:652–663. 10.1128/JVI.01706-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. 1990. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 71:2761–2767. 10.1099/0022-1317-71-11-2761 [DOI] [PubMed] [Google Scholar]
- 50.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. 10.1016/j.chom.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Mohr I. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 28:2648–2658. 10.1128/MCB.01631-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cotter CR, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2010. The virion host shut-off (vhs) protein blocks a TLR-independent pathway of herpes simplex virus type 1 recognition in human and mouse dendritic cells. PLoS One 5:e8684. 10.1371/journal.pone.0008684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cotter CR, Kim WK, Nguyen ML, Yount JS, Lopez CB, Blaho JA, Moran TM. 2011. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J. Virol. 85:12662–12672. 10.1128/JVI.05557-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J. Virol. 87:3271–3276. 10.1128/JVI.03049-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663–672. 10.1084/jem.189.4.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy JA, Duerst RJ, Smith TJ, Morrison LA. 2003. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 77:9337–9345. 10.1128/JVI.77.17.9337-9345.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]