

Abstract
In this study, subnucleocapsid nanorings formed by the recombinant nucleoprotein (N) of the respiratory syncytial virus were evaluated as a platform to anchor heterologous antigens. The ectodomain of the influenza virus A matrix protein 2 (M2e) is highly conserved and elicits protective antibodies when it is linked to an immunogenic carrier, making it a promising target to develop universal influenza vaccines. In this context, one or three M2e copies were genetically linked to the C terminus of N to produce N-M2e and N-3M2e chimeric recombinant nanorings. Mice were immunized intranasally with N-M2e or N-3M2e or with M2e or 3M2e control peptides. N-3M2e-vaccinated mice showed the strongest mucosal and systemic antibody responses. These mice presented a reduced viral load and minor weight loss, and all survived upon challenge with influenza virus A/PR8/34 (H1N1) (PR8). We compared the intranasal route to the subcutaneous route of N-3M2e immunization. Only the intranasal route induced a strong local IgA response and led to the protection of mice upon challenge. Finally, we demonstrated that the induction of anti-M2e antibodies by N-3M2e is not impaired by preexisting anti-N immunity. Overall, these results show that the N nanoring is a potent carrier for mucosal delivery of vaccinal antigens.
INTRODUCTION
The respiratory mucosal surfaces are the portal of entry for a variety of pathogens, including viruses. Protective immunity against respiratory viruses requires the induction of mucosal immune effectors that are more efficiently elicited upon mucosal than systemic immunization due to the compartmentalization of the immune system (1). The antigen delivery vehicle also plays a major role, since inert subunit vaccines are poorly immunogenic compared to live vaccines when administered mucosally (2). New vaccinal approaches based on virus-like nanosized particles that could provide sufficient immunogenicity for mucosal vaccination have emerged lately (3). The self-assembly property of one or several viral proteins produced through recombinant technologies results in the formation of subviral particles ranging in size from about 20 to 100 nm (4). These recombinant particles are nonreplicating and thus safe structures. Some viral nanoparticles, like the one formed by hepatitis B virus core (HBc) proteins, will spontaneously encapsidate RNA or DNA fragments, which are natural ligands for pattern recognition receptors (Toll-like receptor 3 [TLR3], TLR7, -8, and -9) and will further enhance nanoparticles' immunogenicity (5, 6).
An original technology was set up in our laboratory to produce and purify a recombinant form of the nucleoprotein (N) of the human respiratory syncytial virus (RSV) assembling as soluble nanometric rings composed of 10 or 11 N monomers bound to random stretches of bacterial RNA (70 bp) (7). These structures, about 15 nm in diameter, were named N subnucleocapsid ring structures (N SRS), and their three-dimensional (3D) structure was solved (8).
We previously documented the immunogenicity of N SRS in BALB/c mice and calves (9, 10). In mice, intranasal (i.n.) vaccination with N SRS elicits strong local and systemic immunity and completely protects mice against an RSV challenge, whereas the same vaccine formulation delivered subcutaneously (s.c.) is only marginally protective (9). The aim of the present study is to demonstrate the efficiency of N SRS as a new mucosal carrier for heterologous viral antigens.
Current influenza virus vaccines are composed of antigenic determinants from hemagglutinin (HA) and neuraminidase (NA) glycoproteins derived from 3 influenza virus strains (A/H1N1, A/H3N2, and B). Viral proteins of human seasonal influenza strains, including HA and NA, evolve gradually through point mutation (drift), allowing the resulting variants to elude host immunity. More rarely, influenza A viruses evolve through segment exchange with other human or animal viruses (shift), possibly resulting in an extensive worldwide epidemic (11). This frequent antigenic drift or shift requires regular updating of the vaccine composition (12). Therefore, several research teams and vaccine manufacturers are focusing on the design of new “universal” vaccine strategies, using the most conserved influenza antigenic motifs like those carried by influenza virus nucleoprotein (NP), M1, the stem domain of HA, and the ectodomain of M2 (M2e) (13).
M2 is a transmembrane protein translated from a spliced RNA derived from the seventh segment of the influenza genome, also coding for M1 (14), that forms a tetrameric ion channel at the surface of the particle. Its ectodomain, M2e, is a minor but evolutionary constant epitope, remarkably conserved between antigenically distant influenza A virus strains of either human or avian origin (15). Indeed, the ability of M2e-based vaccine to provide cross-protective immunity was first demonstrated by Neirynck et al. (16). However, M2e is poorly immunogenic, either during the course of a natural infection or following vaccination with inactivated virions (17, 18). Many vaccinal strategies have been designed to improve M2e immunogenicity, including the use of virus-like particles (VLPs) as an immunogenic platform for M2e (19). Using the highly immunogenic HBc carrier for M2e, it has been shown that antibodies against M2e, while nonneutralizing, mediate viral protection through antibody-dependent cell cytotoxicity (ADCC) and/or complement-dependent cytotoxicity (CDC) (18, 20).
Thus, M2e is an antigen of choice to anchor onto N SRS and thereby demonstrate their potency as immunogenic carriers. M2e-decorated N SRS, intranasally administered, appeared to be strongly immunogenic and induced high levels of anti-M2e antibodies, both in serum and bronchoalveolar lavage fluid (BALf) even when mice had preexisting immunity to the carrier. Moreover, sera from N-3M2e-immunized mice were shown to bind to the native M2 protein at the surface of H1N1-infected cells and in infected-cell lysates. Following challenge with influenza virus A/PuertoRico/8/34 (PR8) H1N1, mice immunized with M2e-decorated N SRS were well protected against clinical signs and viral replication. Interestingly, the choice of the immunization route appeared to be critical since the same vaccine formulation delivered s.c. failed to protect against viral challenge.
MATERIALS AND METHODS
Plasmid constructions and protein expression and purification of N-M2e and N-3M2e SRS.
N-M2e and N-3M2e fusion proteins were obtained by successive insertion of one or multiple M2e epitopes at the C-terminal end of N. First, the plasmid pET22-M2 containing the full-sequence coding for M2 of influenza virus A/PuertoRico/8/34 (H1N1) was mutated by replacing the cysteine residues in positions 17 and 19 of the amino acid sequence of M2 by two serine residues with the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. The mutated plasmid pET22-M2-CC17-19SS was used as a template to amplify M2e sequences containing the double mutation CC17-19SS used to produce N-M2e and N-3M2e fusion protein. M2e and 3M2e epitopes were amplified by overlapping PCR using high-fidelity PfuUltra polymerase (Agilent Technologies, Santa Clara, CA) with gene-specific primers flanked with the appropriate enzymatic restriction sites: SacI/AgeI for M2e and SacI/KpnI, KpnI/EcoRI, and EcoRI/AgeI for 3M2e. Then, we used a pET-N-Sac plasmid derived from pET-N, which contains the full-sequence coding for N from RSV strain Long (9), to subclone the M2e and 3M2e sequences. Briefly, pET-N-Sac was obtained by introducing a SacI restriction site in frame at the end of the sequence coding for N without modifying its amino acid sequence (21). PCR-amplified M2e and 3M2e epitopes were digested by SacI and AgeI and inserted into a pET-N-Sac plasmid that had been identically restricted. The plasmids generated were named pET-N-M2e and pET-N-3M2e. The entire sequences coding for N-M2e and N-3M2e were validated on both strands by DNA sequencing. All sequences and primers are available upon request.
Recombinant N-M2e and N-3M2e expression and purification were performed as previously described by Tran et al. (7). Briefly, the purification of the recombinant proteins is permitted by the specific interaction between the C terminus of P protein (PCT) of RSV (residues 161 to 241) fused to glutathione S-transferase (GST) and N protein. Escherichia coli BL21(DE3) cells were cotransformed with pGEX-PCT and pET-N-M2e or pET-N-3M2e. The transformed cells were cultured at 37°C to an A600 of 0.6 in 1 liter of LB medium containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. To express N SRS, the transformed cells were incubated 15 h at 28°C in 0.33 mM IPTG (isopropyl-β-d-thiogalactopyranoside) LB medium with agitation. The bacteria were harvested and centrifuged for 15 min at 5,000 × g. GST fusion proteins were purified from the bacterial pellets with glutathione-Sepharose 4B beads (Amersham Pharmacia Biotech, Piscataway, NJ) as previously described (21). SRS were separated from the beads by thrombin cleavage. Of note, a small amount of PCT was copurified with N SRS (see Fig. 8A). The purified proteins were subjected to 12.5% SDS-PAGE analysis and detected by Coomassie brilliant blue staining. ProSieve Prestained protein ladders (Lonza, Basel, Switzerland) were used.
FIG 8.

N and N-3M2e SRS are copurified with a small fragment of P (PCT) that induced marginal anti-P antibody response. (A) N-3M2e and N SRS were analyzed by SDS-PAGE and Coomassie blue staining. The identity of each band is indicated on the right. (B) BALB/c mice were immunized twice i.n. at a 2-week interval with 8 μg of N or N-3M2e SRS or 8 μg of P. All antigens were adjuvanted with 5% Montanide gel. The negative-control group received i.n. adjuvant alone (MG). Sera were collected 2 weeks after the boost immunization. P-specific IgG1 and IgG2a were determined individually in sera from each group by indirect ELISA using purified P protein as the capture antigen. The mean (+ SEM) titers for each group are plotted as histograms (n = 5 per experimental group). P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
SDS-PAGE and native PAGE separation.
For SDS-PAGE, samples were prepared in Laemmli buffer (62.5 mM Tris-HCl [pH 6.8], 5% β-mercaptoethanol, 2% SDS, 20% glycerol, 0.01% bromophenol blue) and denatured 5 min at 95°C. Samples were then separated on 12.5% polyacrylamide gel and detected by Coomassie brilliant blue staining. For native PAGE, samples were prepared in buffer containing 50% saccharose, separated on 4% polyacrylamide gel in 0.2× Tris-borate-EDTA (TBE) (pH 8.0), and stained with Coomassie brilliant blue. ProSieve prestained protein ladders (Lonza, Basel, Switzerland) were used.
Western blot assay.
N SRS rings and chimeric N rings (N-M2e and N-3M2e) were loaded and separated with a 12.5% SDS-PAGE. Gels were transferred onto Immobilon-P membranes (Merck Millipore, Billerica, MA) for 1 h at 50 V followed by Western blotting using a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with phosphate-buffered saline (PBS) containing 5% skim milk during 1 h at room temperature and then incubated overnight at 4°C with a rabbit polyclonal antibody against N at a dilution of 1:3,000 or for 1 h at room temperature with a mouse 14C2 monoclonal antibody against M2 (Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:3,000 in PBS with 0.3% Tween 20 and 5% skim milk. Membranes were washed with PBS containing 0.3% Tween 20 and finally incubated 1 h at room temperature with relevant peroxidase-conjugated secondary antibody (P.A.R.I.S. Biotech, Compiègne, France). Proteins were visualized by chemiluminescence with ECL substrate (GE Healthcare, Little Chalfont, United Kingdom). ProSieve prestained protein ladders (Lonza, Basel, Switzerland) were used.
Size measurements by dynamic light scattering.
Size measurements with the Zetasizer Nano series (Malvern instruments, Malvern, United Kingdom), based on the principle of dynamic light scattering, were made at 20°C using a helium-neon laser wavelength of 633 nm and detection angle of 173°. The results were presented as size distribution calculated from the Malvern software.
TEM visualization of N rings.
Electron micrographs were acquired using a CM12 transmission electron microscope (TEM; Royal Philips Electronics, Amsterdam, Netherlands) at 80-kV excitation voltage. Samples of purified N SRS at a concentration of 0.05 mg/ml (N SRS) or 0.025 mg/ml (N-M2e and N-3M2e chimeric SRS) were applied onto an air-glow-discharged carbon-coated 200-mesh copper grid and stained with a 2% uranyl acetate aqueous solution.
Mice and immunization-challenge protocols.
All animal experiments were carried out under the authority of license issued by the “Direction des Services Vétérinaires” (accreditation number 78-27 to S.R.) and approved by the ethics committee COMETHEA (COMité d'ETHique appliqué à l'Expérimentation Animale, INRA Jouy-en-Josas and AgroParisTech; authorization number 12-008). Female BALB/c mice aged 6 to 8 weeks were purchased from the “Centre d'Elevage Janvier” (Le Genest, St Isle, France) and housed under Federation of European Laboratory Animal Science Associations (FELASA) pathogen-free conditions in our animal facilities (IERP, INRA, Jouy-en-Josas, France).
Mice were anesthetized with a solution of ketamine and xylazine (50 and 10 mg/kg of body weight, respectively) and vaccinated twice at a 2-week interval by intranasal (i.n.) instillation of 40 μl of 0.9% endotoxin-free NaCl, containing 2 or 20 μg of M2e, 3M2e, N-M2e, N-3M2e, or N adjuvanted with 5% Montanide gel 01 (a polymeric aqueous adjuvant, based on a dispersion of a high-molecular-weight polyacrylic polymer in water; Seppic, Paris, France). In some experiments, mice were vaccinated s.c. with 20 μg of 3M2e and N-3M2e with 5% Montanide gel. A positive-control group vaccinated s.c. with 2 μg inactivated PR8 in 5% Montanide gel was included in most challenge experiments. Two weeks after the boost injection, mice were either autopsied or anesthetized and inoculated i.n. with 1 × 104 or 5 × 104 PFU of influenza virus A/PuertoRico/8/34 (PR8) in 50 μl of 0.9% endotoxin-free NaCl in biosafety level 2 (BSL2) confinement. Body weight, clinical score, and deaths were monitored daily. We implemented a clinical score scale (0, normal state, no symptoms; 1, slightly ruffled fur; 2, ruffled fur but active mouse; 3, ruffled fur and inactive mouse). Mice that had lost 25% or more of their initial weight were euthanized humanely. For the evaluation of the impact of the preexisting immunity against RSV for N-3M2e immunization, mice were anesthetized and inoculated i.n. with 1.2 × 107 PFU of human RSV (HRSV) strain A2 in 30 μl of 0.9% endotoxin-free NaCl in BSL2 confinement.
Sample collection.
Sera were obtained from blood collected via cheek puncture 14 days after the prime and 14 days after the boost immunizations. Mice were killed by gentle cervical dislocation. Bronchoalveolar lavages (BAL) were performed by flushing the lungs via tracheal puncture four times in and out with 1.5 ml Ca2+- and Mg2+-free PBS supplemented with 1 mM EDTA. BAL fluids (BALf) were centrifuged, and supernatants were stored frozen at −20°C for enzyme-linked immunosorbent assays (ELISAs) of anti-M2 antibodies.
Antigen-specific antibody ELISA.
Individual mouse sera and BALf were assayed for N- or M2-specific antibodies (Ig [H+L], IgG2a, IgG1, or IgA) by ELISA using M2e synthetic peptide and N or P recombinant proteins as coating (200 ng per well) as previously described (9). Endpoint antibody titers were calculated by regression analysis, plotting serum dilution versus the absorbance at 450 nm, using Microcal Origin (OriginLab Corporation, Northampton, MA), with the following regression curve equation: y = (b + cx)/(1 + ax). Endpoint titers were defined as the highest dilution resulting in an absorbance value twice that of blank points (points without immune serum).
Binding of anti-M2e antibodies to infected cells.
Subconfluent monolayers of A549 cells were infected with influenza virus A/WSN/33 (H1N1) (WSN) at a multiplicity of infection of 10. For fluorescence microscopy observations, cells were fixed at 12 h postinfection (p.i.) with PBS containing 4% paraformaldehyde (PFA), permeabilized with PBS containing 0.1% Triton X-100 for 20 min at room temperature, and incubated with PBS containing 0.1 M glycine 10 min at room temperature to quench aldehyde residues. Cells were blocked with PBS containing 3% bovine serum albumin (BSA) and 0.05% Tween 20 for 1 h at room temperature and then incubated 1 h at room temperature with either 14C2 antibody (Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:500, a mouse 9G8 monoclonal antibody against NP (9G8) (Santa Cruz Biotechnology, Dallas, TX) at a dilution of 1:500, or sera collected from mice immunized twice with 8 μg of N-3M2e at a dilution of 1:200 or 1:500 in PBS with 3% BSA and 0.05% Tween 20. Cells were washed with PBS and finally incubated 1 h at room temperature with Alexa Fluor 594-conjugated anti-mouse antibody (Invitrogen, Carlsbad, CA) at a dilution of 1:2,000 in PBS with 3% BSA and 0.05% Tween 20. Cells were observed with an Axio Observer fluorescence microscope (Carl Zeiss, Oberkochen, Germany) using a 40× objective, and images were acquired and processed using AxioVision software (Carl Zeiss, Oberkochen, Germany). For Western blot analysis, cells were collected at 12 h postinfection in Laemmli buffer. Then, cell lysates were loaded and separated on 12.5% SDS-PAGE. Gels were transferred onto Immobilon-P membranes followed by Western blotting (as described above) using 14C2 antibody at a dilution of 1:5,000 or sera collected from mice immunized twice with 8 μg of N-3M2e at a dilution of 1:100 or 1:250.
Determination of pulmonary influenza viral load by plaque assay.
Lungs were crushed in 700 μl of modified Eagle medium (MEM) with a Precellys 24 bead grinder homogenizer (Bertin Technologies, St Quentin en Yvelines, France) 2 × 15 s at 4 m/s. Lung homogenates were clarified by centrifugation 10 min at 2,000 × g and titrated on MDCK cells using a plaque assay procedure adapted from Matrosovich et al. (22). Briefly, serial dilutions of lung homogenates in MEM were added to a confluent monolayer of MDCK cells in a 12-well plate and incubated 60 min at 37°C under a 5% CO2 atmosphere. Then, 1.5 ml of a gelled medium composed of 1 mM glutamine, 0.25% sodium bicarbonate, 0.5 μg/ml l-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK) trypsine, and 1.2% Avicel (FMC Biopolymer, Philadelphia, PA) was added to each well. Three days later, plates were fixed with a 4% formaldehyde solution and colored by crystal violet.
Determination of RSV pulmonary viral load by qRT-PCR.
Individual viral loads were measured from lung homogenate by quantitative real-time (qRT)-PCR. Briefly, lungs were crushed, and total RNA was extracted from lung homogenates using the RNeasy minikit (Qiagen, Venlo, Netherlands) exactly as previously described (23). Four micrograms of individual RNA samples was reverse transcribed, and individual cDNAs were mixed with SYBR green PCR Master Mix (Life Technologies, Carlsbad, CA) with primers targeting a conserved region of the N gene or primers targeting the murine HPRT gene (23). Real-time PCR was run on the ABI Prism 7900HT Sequence Detector (PerkinElmer, Waltham, MA) as described earlier. Fluorescence curves were analyzed using the Sequence Detector System software (PerkinElmer, Waltham, MA) to determine the cycle threshold (CT) values for each gene. Individual data were normalized to HPRT by calculating the ΔCT value [CT(N) median − CT(HPRT) median]; the ΔΔCT (sample ΔCT − mean ΔCT of control mice) was calculated for each sample. The relative quantity of N transcripts, normalized to HPRT, is expressed as the percentage of the negative-control group (MG).
May-Grünwald-Giemsa staining.
BAL fluid cells from individual mice were enumerated, spread on duplicate microscope Superfrost slides (Thermo Scientific, Waltham, MA) by cytocentrifugation (5 min, 700 rpm) using Shandon Cytospin 5 (Thermo Scientific, Waltham, MA), and stained with May-Grünwald and Giemsa stain. At least 400 leukocytes were counted blindly for each sample.
Statistical data analysis.
Data were expressed as arithmetic means ± standard errors of the means (SEM). The nonparametric Mann-Whitney or unpaired t test (two groups), one-way analysis of variance (ANOVA) Tukey's multiple-comparison test, or log rank Mantel-Cox test (>2 groups) was used to compare unpaired values (GraphPad software, San Diego, CA). P values of <0.05 were considered significant; levels of significance are indicated on the graphs by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
RESULTS
Design and characterization of N SRS carrying M2e peptide in single or tandem repeats.
To investigate the potential of N SRS as an immunogenic carrier for M2e peptide, we constructed two types of N SRS chimeric proteins by genetically linking one or three M2e copies (named N-M2e and N-3M2e, respectively). The repetition of the M2e motif in the 3M2e version of the fusion protein was made in an attempt to increase the antigen-specific immune response as was done to optimize the M2e-HBc construct (24). The C-terminal end of N, exposed at the surface of the ring, was chosen to fuse single or triple M2e epitopes according to the recently determined tridimensional structure of N SRS (Fig. 1A) (8). The two cysteine residues at positions 17 and 19 were replaced by two serine residues in order to avoid a possible formation of disulfide bonds that could disturb the assembly of the SRS. The resulting N-M2e and N-3M2e fusion proteins were expressed in E. coli and purified to homogeneity according to the procedure previously described for N SRS (7). To demonstrate that the fusion of one or three M2e peptide sequences did not interfere with the formation of SRS, purified N-M2e, N-3M2e, and N as control were analyzed by native PAGE and by dynamic light scattering (DLS) (Fig. 1B and C). The native PAGE showed two predominant bands corresponding to nanorings of 10 and 11 monomers of either N, N-M2e, or N-3M2e, as was shown for N SRS by Tran et al. (7). DLS plots showed a major peak for both N-M2e and N-3M2e indicative of homogeneous population of rings with a hydrodynamic radius compatible with the one of N SRS in solution (7). To determine the protein composition of these rings, N-M2e and N-3M2e were subjected to SDS-PAGE followed by Coomassie blue staining (Fig. 1D). A band was obtained for each fusion protein at the expected size. Furthermore, the analysis of the protein content of the chimeric rings was carried out by Western blot analysis using anti-N and anti-M2 14C2 antibodies (Fig. 1E), validating the presence of the M2e epitopes. Although no degradation was observed in the SDS-PAGE analysis, a fuzzy band corresponding to the same apparent molecular weight as that of N-M2e was observed in the lane of N-3M2e, suggesting that the rings were slightly susceptible to degradation. However, this band revealed both by anti-N and by 14C2 antibodies indicated that at least one M2e peptide remained associated to N. Finally, the observation by electron microscopy revealed that N-M2e and N-3M2e chimeric proteins assembled as ringlike structures with a less regular shape than SRS containing N only (Fig. 1F). Overall, these results clearly demonstrated that the fusion of one or three M2e domains on each monomer of N had no impact on the formation and the integrity of SRS and that we were able to produce and purify to homogeneity these chimeric nanoparticles.
FIG 1.

Design of chimeric N SRS fused to M2e epitopes and biochemical and biophysical characterization. N protein was fused at its C-terminal end (Cter) to either one (N-M2e) or three (N-3M2e) M2e peptides from A/PuertoRico/8/34 (H1N1). (A) Schematic representation of one N-3M2e ring based on the known 3D structure of N SRS seen from the top (8). The sequence of the C terminus of N protein is in green, M2e epitopes are in red, and the 2-amino-acid (aa) linkers are in black. N SRS (empty carrier) and chimeric N SRS (N-M2e, N-3M2e) were produced and purified as described in Materials and Methods and then analyzed by native PAGE (B), SDS-PAGE (D), and Coomassie blue staining. The hydrodynamic radius of chimeric N rings was also estimated by dynamic light scattering (DLS) (C). Western blotting (E) with anti-N (left panel) or 14C2 anti-M2 (right panel) antibodies confirmed the presence of M2e epitopes on N SRS. The relative molecular mass (kDa) is indicated on the left. (F) Purified chimeric rings were negatively stained and observed by transmission electronic microscopy (TEM). The structure integrity of chimeric rings was preserved upon the addition of 1 (middle panel) or 3 (right panel) M2e peptides. For comparison, a photograph of native N rings is shown in the left panel.
The antibody response to M2e is enhanced when M2e is linked to N SRS.
First, the immunogenicity of the M2e synthetic peptide was compared to the immunogenicity of N-M2e. BALB/c mice were vaccinated twice i.n. at a 2-week interval with either M2e (20 μg) or N-M2e (2 μg) with 5% Montanide gel as adjuvant. This polymeric aqueous adjuvant has been extensively tested in several animal species and presents a good safety profile, including for mucosal administration (25). Control groups were vaccinated with 20 μg of N SRS (i.n.) or 2 μg of inactivated A/PuertoRico/8/34 (iPR8) virions (s.c.). Although M2e did not induce detectable antibody responses at a 20-μg dose, N-M2e at a 2-μg dose elicited a significant antibody response composed of both IgG1 and IgG2a subtypes (Fig. 2A). It was previously reported that M2e induces stronger specific antibody responses when administered as a synthetic multiply antigenic peptide (26, 27, 28). Thus, we compared the synthetic tripeptide (3M2e) with N-3M2e at two doses (2 and 20 μg) for their capacity to induce systemic antibody response (Fig. 2B and C). Nasal immunization with the 3M2e peptide (2 μg) was able to elicit a specific serum antibody response, mostly of IgG1 isotype, which was further increased after the boost (Fig. 2B). In contrast, the same dose of N-3M2e gave rise to a stronger M2e-specific antibody response, with IgG1 detectable upon the first immunization and IgG2a following the boost immunization (Fig. 2B). Larger doses of both 3M2e and N-3M2e antigens (20 μg) elicited the same pattern of antibody responses but at higher levels (Fig. 2C). Then, we evaluated the capacity of N-3M2e to induce a local antibody response in BAL fluids, compared to M2e or 3M2e peptides (Fig. 3). Nasal immunization with N-3M2e induced a specific IgG1, IgG2a, and IgA antibody response in BALf that was significantly stronger than the one elicited by M2e (not detectable) and better than the one elicited by 3M2e (significantly for IgG2a). From these results, we conclude that N SRS is a highly efficient immunogenic carrier for mucosal vaccination, with the highest antibody responses obtained when three copies of M2e are fused to each monomer of N.
FIG 2.

Linking M2e in one or three repeats onto N SRS strongly increased the antigen-specific serum antibody response. BALB/c mice were immunized i.n. twice at a 2-week interval with 20 μg of M2e synthetic peptide or 2 μg of N-M2e SRS (A), or with 2 μg of 3M2e synthetic peptide or N-3M2e SRS (B), or with 20 μg of 3M2e synthetic peptide or N-3M2e SRS (C). As a control, mice were immunized i.n. with 20 μg of N or s.c. with 2 μg of inactivated PR8 virus (iPR8). All immunogens were adjuvanted with 5% Montanide gel. Sera of vaccinated mice were collected 2 weeks after the prime immunization (1st) or 2 weeks after the boost immunization (2nd). M2e-specific Ig (H+L), IgG1, and IgG2a titers were determined individually in sera by indirect ELISA using M2e synthetic peptide as the capture antigen. Data are means + SEM of antibody titers for each group (n = 5 to 33 per experimental group; 1 to 5 independent experiments). P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 3.
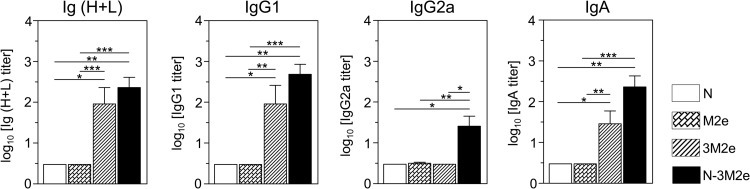
Linking M2e in one or three repeats onto N SRS strongly increased the antigen-specific mucosal IgA antibody response. BALB/c mice were immunized i.n. with 20 μg of M2e or 3M2e synthetic peptides or 20 μg of N-3M2e chimeric rings. As a negative control, mice were immunized i.n. with 20 μg of N. All immunogens were adjuvanted with 5% Montanide gel. Bronchoalveolar lavage fluids (BALf) of vaccinated mice were collected 2 weeks after the boost immunization. M2e-specific Ig (H+L), IgG1, IgG2a, and IgA titers were determined individually by indirect ELISA using M2e synthetic peptide as the capture antigen. Data are means + SEM of antibody titers for each group (n = 4 to 10 per experimental group, 1 to 2 independent experiments). P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
The anti-M2e antibodies induced in mice upon N-3M2e immunization efficiently bind to native M2 protein at the surface of influenza virus-infected cells.
Previous studies suggested that the binding of anti-M2e antibodies to the M2 protein expressed at the surface of infected cells could contribute to virus clearance by promoting ADCC (18, 20). Thus, we investigated whether anti-M2e antibodies contained in the sera of N-3M2e-immunized mice were able to bind to influenza virus-infected cells. To that end, A549 cells were infected with WSN virus and subjected to immunofluorescence staining using sera collected from N-3M2e-immunized mice or, as positive controls, anti-M2 (14C2) and anti-NP monoclonal antibodies (Fig. 4A, B, C, D, and E). A strong cytoplasmic staining was observed using anti-NP antibodies, validating the efficacy of infection (Fig. 4A). Moreover, a strong specific staining was observed using sera from N-3M2e vaccinated mice at dilutions of 1:250 and 1:500 compared to an absence of staining of noninfected cells (Fig. 4C, D, and E). This pattern of M2 staining was very similar to the one observed with 14C2 antibody (Fig. 4B). Furthermore, infected cell lysates were analyzed by Western blotting (Fig. 4F). An intense and unique band was obtained at 17 kDa using sera collected from N-3M2e-vaccinated mice at a dilution of 1:100 or 1:250 or using the 14C2 antibody. These results demonstrated that anti-M2e antibodies induced by N-3M2e efficiently bind to the M2 protein in virus-infected cells. Thus, the substitution of the cysteine residues in positions 17 and 19 of fused M2e peptides did not impair the conformational recognition of native M2 antigen. Moreover, these results also indicated that antibodies induced in mice by N-3M2e immunization and directed to the M2e of PR8 virus are cross-reactive with the M2 protein of WSN virus.
FIG 4.

Anti-M2e antibodies induced in mice by N-3M2e immunization bind to the M2 protein expressed by influenza virus-infected cells. Subconfluent monolayer A549 cells were infected with influenza virus WSN at a multiplicity of infection of 10. At 12 h p.i., cells were fixed in PBS–4% PFA and stained as described in Materials and Methods with anti-NP monoclonal antibody (A), 14C2 anti-M2 antibody (B), or sera collected from i.n. N-3M2e immunized mice (C, D, E) at a dilution of 1:200 (C, E) or 1:500 (D). (E) Negative control for staining of noninfected cells. Samples were observed by fluorescence microscopy using a 40× objective. Alternatively, infected (WSN) or noninfected (NI) cell extracts were analyzed by Western blotting using 14C2 antibody or sera collected from i.n. N-3M2e-immunized mice at a dilution of 1:100 or 1:250 (F).
Nasal vaccination with N-3M2e rings confers clinical and virological protection against influenza challenge.
To investigate the level of protection conferred by N-3M2e or N-M2e immunization, mice were vaccinated i.n. with 20 μg of M2e or 3M2e versus 2 μg of N-M2e or N-3M2e SRS. A group of mice was immunized s.c. with 2 μg of iPR8 virus as a positive control for protection. All antigens were adjuvanted with 5% Montanide gel. Two weeks after the boost immunization, mice were challenged i.n. with 5 × 104 PFU of PR8 virus and monitored daily for weight loss, clinical score, and mortality (Fig. 5). All the mice immunized with N-3M2e or iPR8 virus survived the challenge with little weight loss (mean percentages of initial weight of 94% and 99% at day 5 p.i. for N-3M2e- and iPR8-vaccinated mice, respectively) and low clinical scores. In contrast, mice immunized with 3M2e or N-M2e were only partially protected (survival rates of 78% and 75%, respectively) with moderate weight loss (mean percentage of initial weight of 91% for N-M2e at day 5 p.i.) and higher clinical scores. Finally, M2e-immunized mice presented a severe weight loss between day 0 and day 5 (mean percentage of initial weight of 80% at day 5 p.i.) together with the highest clinical scores and mortality (survival rate of 22%).
FIG 5.

Nasal immunization with N-3M2e SRS protected against a homologous virus challenge. BALB/c mice were immunized twice i.n. at a 2-week interval with 20 μg of M2e or 3M2e synthetic peptides or 2 μg of N-M2e or N-3M2e SRS. The positive-control group received s.c. 2 μg of iPR8. All antigens were adjuvanted with 5% Montanide gel. Two weeks after the boost immunization, mice were lightly anesthetized and inoculated i.n. with 5 × 104 PFU of PR8 virus. (A) Survival curves of infected mice are expressed as the percentages of surviving mice. (B) Weight curves of infected mice. Values represent the mean weights of mice expressed as a percentage of initial weight at day of inoculation (100%). (C) Clinical scores of infected mice were rated from 0 to 3 as described in Materials and Methods. Plotted data are means ± SEM (B, C). Curves are identified in panel A. The significance of the differences observed between N-3M2e curves and each of the other curves was evaluated according to the one-way ANOVA Tukey's multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) (statistical test was performed between day 2 and day 5 p.i. for weight and clinical score curves). The log rank Mantel-Cox test was used to compare survival curves (n = 8 to 11 per experimental group).
To evaluate to what extent the clinical protection was related to a reduction of pulmonary viral load, mice immunized with two doses (2 or 20 μg) of N-3M2e or 3M2e were challenged with either 1 × 104 or 5 × 104 PFU of PR8 virus and sacrificed 4 days after infection (Fig. 6). Negative-control groups were immunized with N SRS or diluent (0.9% NaCl) and positive-control groups with iPR8. Mice vaccinated with 20 μg N-3M2e exhibited significantly reduced pulmonary viral load compared to mice vaccinated with 3M2e or N SRS, at both doses of challenge virus (Fig. 6A and C). This reduction of viral load was correlated with a significant protection from weight loss compared to mock-vaccinated mice (Fig. 6B and D). However, when a lower dose of vaccine antigen (2 μg) and a stronger dose of challenge virus (5 × 104 PFU) were tested, no significant reduction of lung viral load was observed for N-3M2e-vaccinated mice compared to mock-vaccinated mice (Fig. 6E). Nevertheless, N-3M2e-vaccinated mice remained significantly protected from weight loss compared to mock-vaccinated mice (Fig. 6F). Finally, under all conditions tested, mice vaccinated with iPR8 had lower viral loads than mice vaccinated with N-3M2e (Fig. 6A, C, and E), but both groups were equally protected from weight loss (Fig. 6B, D, and F). In conclusion, N-3M2e i.n. vaccination efficiently prevented weight loss and mortality upon viral challenge, despite a partial (or delayed) viral clearance.
FIG 6.

Reduction of pulmonary viral load in BALB/c mice immunized with N-3M2e SRS after homologous challenge. BALB/c mice were immunized twice i.n. at a 2-week interval with either 2 μg (E and F) or 20 μg (A, B, C, D) of 3M2e synthetic peptide or N-3M2e SRS or s.c. with 2 μg of iPR8 as a positive control for protection. The negative-control groups received i.n. either 2 μg (E and F) or 20 μg (A, B, C, D) of N SRS or the adjuvant alone in 0.9% NaCl (phy S). All immunogens were adjuvanted with 5% Montanide gel. Two weeks after the boost immunization, mice were lightly anesthetized and inoculated with either 1 × 104 (A, B) or 5 × 104 (C, D, E, F) PFU of PR8 virus. (A, C, E) Four days after challenge, mice were sacrificed and their lungs were collected. Titers of infectious virus in lung homogenates were measured by plaque assay on MDCK cells. Individual values are represented for each experimental group. The mean value is represented by a horizontal bar, SEM by a vertical bar, and the limit of detection by a dotted line (n = 3 to 6 per experimental group, 1 or 2 independent experiments). The doses of vaccine antigens and challenge virus are indicated above each panel. P values were determined according to the unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (B, D, F) Weight curves of infected mice. Values represent the mean weights of mice expressed as a percentage of initial weight at day of inoculation (100%). Plotted data are means ± SEM (n = 3 to 6 per experimental group, 1 or 2 independent experiments). Curves are identified in panel B. The significance of the differences observed between N-3M2e curves and each of the other curves was evaluated according to the one-way ANOVA Tukey's multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) (statistical test was performed between day 3 and day 4 p.i.).
The immunization route is critical for the induction of a local anti-M2e IgA response and subsequent protection against viral challenge.
Previous studies have revealed that the efficacy of protective anti-M2 antibody responses varies depending on the immunization route (27, 29). Thus, we evaluated the outcome of i.n. versus s.c. routes of immunization with N-3M2e (20 μg) on the nature of the mucosal and systemic anti-M2e antibody responses and the degree of protection against an influenza challenge. Both immunization routes elicited potent anti-M2e IgG1 and IgG2a antibody responses in serum and BALf (Fig. 7A and B). The s.c. immunization gave rise to the strongest anti-M2e IgG2a and IgG1 antibody titers in serum following the first immunization, whereas following the boost immunization, the s.c. and i.n. routes elicited similar anti-M2e antibody titers (Fig. 7A). Remarkably, only the i.n. route was able to induce anti-M2e in serum and BALf (Fig. 7A and B). As a positive control for immunization, we evaluated the antibody response to the carrier protein (N) and showed that the same patterns of IgG1, IgG2a, and IgA antibodies are found in serum and BAL fluid, notably for the restriction of antigen-specific IgA to the i.n. route of immunization (Fig. 7C and D). We have also evaluated the immunogenicity of the P C-terminal fragment (PCT) copurified with, but not linked to, SRS (see Materials and Methods) (Fig. 8A). Upon i.n. immunization, PCT appeared to be poorly immunogenic and induced a low anti-P antibody response by comparison with the entire P protein (Fig. 8B).
FIG 7.

Only the i.n. delivery route of N-3M2e SRS gave rise to specific IgA. BALB/c mice were immunized twice i.n. or s.c. at a 2-week interval with 20 μg of N-3M2e SRS adjuvanted with 5% Montanide gel. As a negative control, mice received i.n. adjuvant alone (MG [i.n.]). Sera of vaccinated mice were collected 2 weeks after the prime immunization (1st) and 2 weeks after the boost immunization (2nd), and BALf were collected 2 weeks after the boost immunization. M2e-specific (A, B) or N-specific (C, D) Ig (H+L), IgG1, IgG2a, and IgA titers were determined individually in sera (A, C) or BALf (B, D) from each group by indirect ELISA using M2e synthetic peptide or N rings as the capture antigen. Data are means + SEM of antibody titers for each group (n = 4 to 10 per experimental group). Bars are identified at the top right of the panel A. P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Then, the mice immunized as described above were challenged 2 weeks after the boost with 5 × 104 PFU of PR8 virus. The best protection was obtained when N-3M2e was administered i.n. (green circles), with a survival rate of 80%, whereas only 30% of the mice that received N-3M2e by the s.c. route (red squares) survived, similarly to the control groups (Fig. 9A). Moreover, s.c.-immunized mice presented a more important weight loss and higher clinical scores than mice immunized i.n. (Fig. 9B and C). To get more insights into the difference between the two vaccination routes, mice were vaccinated as described above. As an additional control, mice were immunized i.n. or s.c. with 20 μg of 3M2e. Then, mice were challenged with 5 × 104 PFU of PR8 and sacrificed at day 8 p.i. The percentages of granulocytes (neutrophils and eosinophils) were found to be significantly increased in BALf of mice vaccinated by the s.c. route compared to the i.n. route (40% ± 11% neutrophils and 3% ± 0.1% eosinophils for the s.c. route versus 10% ± 2% neutrophils and 1% ± 1% eosinophils for the i.n. route for N-3M2e-vaccinated mice; 45% ± 19% neutrophils and 3% ± 3% eosinophils for the s.c. route versus 17% ± 12% neutrophils and 1.8% ± 1% eosinophils for the i.n. route for 3M2e-vaccinated mice) (Fig. 9D). Viral loads measured at day 8 p.i. revealed a complete clearance of the virus when mice were immunized i.n. with N-3M2e (mean viral titer, <1.2 log10 PFU/lung), unlike mice vaccinated s.c. with N-3M2e (mean viral titer, 4.7 ± 0.1 log10 PFU/lung), mice vaccinated i.n. or s.c. with 3M2e (mean viral titers, 3.8 ± 1.8 log10 and 4.8 ± 0.6 PFU/lung, respectively), or control mice (mean viral titer, 5.2 ± 0.3 log10 PFU/lung) (Fig. 9E). These results show that the i.n. route is by far better than the s.c. route for N-3M2e SRS vaccination and suggest that mucosal anti-M2e IgA, which was induced only upon i.n. delivery of N-3M2e, might play a front role in the protection of vaccinated mice against influenza infection.
FIG 9.

The efficacy of the N-3M2e vaccine against virus challenge depended upon the immunization route. (A, B, C) BALB/c mice were immunized twice i.n. or s.c. at a 2-week interval with 2 μg of N-3M2e SRS adjuvanted with 5% Montanide gel. As a negative control, mice received i.n. or s.c. adjuvant alone (MG [IN] or MG [SC], respectively). Two weeks after the boost immunization, mice were lightly anesthetized and inoculated with 5 × 104 PFU of PR8 virus. (A) Survival curves of infected mice are expressed as the percentages of surviving mice. (B) Weight curves of infected mice. Values represent the mean weights of surviving mice expressed as a percentage of initial weight at day of infection (100%). (C) Clinical status of infected mice was rated from 0 to 3 as described in Materials and Methods. Values represent the mean scores, and SEM is represented by vertical bars. Keys to curve identification are in panel A. The significance of the differences observed between N-3M2e [IN] curves and each one of the other curves was evaluated according to the one-way ANOVA Tukey's multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) (statistical test was performed between day 2 and day 5 p.i. for weight curves and between day 2 and day 9 p.i. for clinical score curves). The log rank Mantel-Cox test was used to compare survival curves (n = 10 or 11 per experimental group). (D, E) BALB/c mice were immunized twice i.n. or s.c. at a 2-week interval with 20 μg of N-3M2e or 3M2e adjuvanted with 5% Montanide gel. The negative-control group received i.n. adjuvant alone (MG). Two weeks after the boost immunization, mice were lightly anesthetized and inoculated with 5 × 104 PFU of PR8 virus. Eight days after the infection, surviving mice were sacrificed and BALf and lung were collected. (D) BAL fluid cells from individual mice were spread on slides and labeled by May-Grünwald-Giemsa, and leukocytes were counted (neutro, neutrophils; macro, macrophages; lympho, lymphocytes; eosino, eosinophils). The percentage of each subset is represented in a pie chart for each immunized group. Percentages are indicated on the chart, and color keys are shown. (E) Titers of infectious virus in lung homogenate were measured by plaque assay on MDCK cells. The values obtained for each individual mouse are represented by a dot. Mean value is represented by a horizontal bar, SEM by a vertical bar, and the limit of detection by a dotted line. P values were determined according to the unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) (n = 4 or 5 per experimental group).
Preexisting anti-N immunity induced by RSV infection or N SRS immunization does not impede the anti-M2e antibody response upon N-3M2e immunization.
An important issue when developing a new vaccine carrier is the risk of interference with preexisting immunity toward the carrier itself, in our case, antibodies against N of RSV. To clarify this important point, mice previously primed by RSV infection or N SRS vaccination were immunized with N-3M2e by the i.n. route. As expected, all primed mice acquired high levels of anti-N antibodies 2 weeks after RSV infection or N SRS immunization (Fig. 10A). However, the levels of serum anti-M2e Ig(H+L) and IgG1 elicited after the first or the second immunization were not significantly different between N-primed and nonprimed mice (Fig. 10B). Interestingly, anti-M2e IgG2a titers were even higher for RSV-primed mice after the first immunization and for RSV- or N SRS-primed mice after the second immunization (Fig. 10B). Similar results were obtained with BALf, for which we notably recorded an increase of anti-M2e IgA and IgG2a in primed mice (Fig. 10C). Overall, these results demonstrated that the induction of the anti-M2e antibody response by N-3M2e in mice was not impaired by preexisting anti-N immunity and in fact could even be enhanced by it.
FIG 10.

Preexisting anti-N immunity induced by RSV infection or vaccination with N SRS did not interfere with the induction of anti-M2e antibody response upon N-3M2e immunization. BALB/c mice were primed i.n. with 1.2 × 107 PFU of HRSV-A2 virus (RSV primed) or 20 μg of N SRS adjuvanted with 5% Montanide gel (N primed). A nonprimed group was also added as a control (no prime). Two weeks after, the 3 groups were immunized twice i.n. at a 2-week interval with 2 μg of N-3M2e adjuvanted with 5% Montanide gel. Sera of the vaccinated mice were collected before (NI, not immunized) and 2 weeks after (prime) the prime and 2 weeks after the first (1st) and the second (2nd) immunizations with N-3M2e. BALf were collected 2 weeks after the second immunization. N-specific Ig (H+L) (A) or M2e-specific Ig (H+L), IgG1, IgG2a, or IgA (B, C) were determined individually in sera (A, B) or BALf (C) from each group by indirect ELISA using N rings or M2e synthetic peptide as the capture antigen. The means + SEM of titers for each group are plotted as histograms (n = 5 per experimental group). Bar identifications are indicated at the top left of panel B. P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Nasal vaccination with N-3M2e rings confers protection against RSV challenge in mice.
RSV and influenza virus infection are two major causes of infantile lower respiratory tract infections. Therefore, the development of a bivalent vaccine efficient against both RSV and influenza would be of great interest for an optimal prevention of bronchiolitis in infants. As mentioned in the introduction, we have previously demonstrated that N SRS vaccination is protective against RSV (9). To validate that N-3M2e is also effective for the control of RSV infection, mice were immunized with 8 μg of N or N-3M2e SRS and challenged with RSV. N and N-3M2e immunizations induced similar levels of antibody responses against N (Fig. 11A). Viral loads measured at day 5 postinfection revealed that both N- and N-3M2e-vaccinated mice were efficiently protected against RSV replication in lungs compared to the nonimmunized control group (more than 95% reduction of viral RNA in lungs) (Fig. 11B). These results suggest that N-3M2e could be used as a bivalent vaccine efficient against both RSV and influenza virus.
FIG 11.

Nasal immunization with N-3M2e SRS protected mice against viral replication in the lung after an RSV challenge. BALB/c mice were immunized twice i.n. at a 2-week interval with 8 μg of N or N-3M2e SRS. The negative-control group received i.n. adjuvant alone (MG). All antigens were adjuvanted with 5% Montanide gel. (A) Sera were collected before (NI, not immunized) and 2 weeks after (1st) the prime immunization and 2 weeks after the boost immunization (2nd), and N-specific Ig (H+L) levels were determined individually in sera from each group by ELISA. (B) Two weeks after the boost immunization, mice were lightly anesthetized and inoculated i.n. with 1.2 × 107 PFU of HRSV-A2 virus. Five days after challenge, mice were sacrificed and their lungs were collected. Individual viral loads were measured from lung homogenate by qRT-PCR. The relative quantity of N transcripts, normalized to HPRT, is expressed as the percentage of the negative-control group (MG). P values were determined according to the Mann-Whitney test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) (n = 5 per experimental group).
DISCUSSION
The highly conserved ectodomain of the M2 protein (M2e) is, to date, one of the most promising targets for the development of cross-protective or “universal” anti-influenza vaccines (19, 30). However, M2e is poorly immunogenic per se, and the use of M2e as a vaccine candidate requires the implementation of a strategy to improve its immunogenicity (17). In the present study, we made the proof of concept for a new and original immunogenic carrier for M2e peptides: subnucleocapsid ring structures (SRS) formed by the nucleoprotein of RSV. Using chimeric N SRS fused to 1 (N-M2e) or 3 (N-3M2e) M2e epitopes, we succeeded in inducing in BALB/c mice a systemic and local antibody response against M2e that was stronger than the response elicited upon immunization with M2e or 3M2e synthetic peptides or inactivated influenza virus (iPR8). Of note, 1 μg of synthetic M2e peptides contains more M2e epitopes than 1 μg of chimeric N SRS/M2e (molecular ratio of 1/17 for N-M2e/M2e and 1/6 for N-3M2e/3M2e). Thus, the efficiency of N SRS as a vaccinal carrier was certainly underrated in our experiments. We are currently investigating what makes the SRS nanoparticle an immunogenic carrier. Two main features can be involved: the fragment of bacterial RNA and the nanoring structure. So far, we have not been able to remove the fragment of RNA encapsidated in the SRS without destroying their architecture. Nevertheless, our preliminary results suggest that the SRS nanostructure is critical for immunogenicity.
We also did some preliminary investigations of memory T cell responses against M2e, and we found no difference between the M2e peptide alone and M2e fused to N SRS in their capacity to elicit antigen-specific gamma interferon (IFN-γ)-producing cells in spleen (our unpublished data). Others have also shown that M2e immunization of BALB/c mice using multiple antigenic peptides induces M2e-specific T cell responses (28). However, the role of memory T cells in M2e-mediated immune protection has been ruled out by studies showing that T CD8/CD4 depletion in HBc-M2e-vaccinated mice does not impede vaccine protection whereas immune T CD4/CD8 transfer prior to challenge fails to confer protection (18). Conversely, passive immunoglobulin transfer from M2e-immune mice to naive mice is protective against viral challenge (16). Thus, we have chosen to focus the present study on the analysis of the antibody response to M2e.
Intranasal immunization with N-3M2e efficiently protected mice against mortality and clinical signs following influenza virus A challenge and significantly reduced pulmonary viral load at day 4 p.i. However, the reduction of viral load induced by N-3M2e was only partial compared to the reduction of viral load observed in mice immunized with iPR8. A partial reduction of pulmonary viral load was also recorded in several studies evaluating the efficacy of M2e-based vaccine strategies, regardless of the antigen carrier and the dose of challenge virus (16, 27, 31–35). In most cases, mice are efficiently protected against mortality even when the pulmonary viral load remains high. Indeed, anti-M2e antibodies are not neutralizing, in contrast to the anti-HA antibodies induced by the conventional vaccination with inactivated virus, which impede the attachment of the viral particle on its receptor at the surface of target cells, thus providing a sterilizing immunity (36). Nevertheless, we have seen a complete elimination of the virus at day 8 p.i. in mice immunized i.n. with N-3M2e. This demonstrated that the viral clearance occurred but was delayed in N-3M2e-vaccinated mice compared to mice immunized with inactivated virus. Indeed, it has been shown that anti-M2e antibodies are not able to bind the viral particle, probably due to HA and NA steric hindrance, but are able to bind to infected cells that profusely express M2 protein at their surface (18). In agreement with these results, we have shown that anti-M2e antibodies induced in N-3M2e-immunized mice efficiently bind to the M2 protein at the surface of infected cells. Likewise, anti-M2e antibodies could prevent the viral budding that is M2 mediated and reduce viral diffusion, as it was suggested using plaque reduction assays in vitro (37, 38), or activate lysis of infected cells after opsonization by antibody-dependent cell cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), or phagocytosis by alveolar macrophages (18, 20). Moreover, the low level of viral replication until day 8 p.i. could allow a primary immune response against determinants carried by the challenge virus itself, including the induction of specific neutralizing anti-HA antibodies, contributing to viral clearance.
We have seen that the i.n. route was clearly the best one for N-3M2e immunization, stimulating a strong local IgA response and offering the best protection rate compared to the s.c. route. The i.n. route was chosen according to our previous studies showing that it is more efficient than the s.c. route for N SRS immunization and subsequent protection against pulmonary RSV challenge (9). Previous studies using multiple M2e peptides have compared the i.n. and s.c. routes in mice (27, 29). For instance, Wolf et al. demonstrated that only the i.n. route is able to induce anti-M2e IgA antibodies in BALf, which is in agreement with our findings (27). However, these studies have shown a limited advantage of the i.n. route for the protection of vaccinated mice against a lethal influenza challenge in terms of pulmonary viral load. In 1999, Neirynck et al. compared the i.n. route to the intraperitoneal (i.p.) route using M2e fused to HBc protein. They also showed that i.n. is the best route of immunization, given that it is as efficient as the i.p. route to protect mice against viral challenge and induced a much higher antibody response, without requiring adjuvant (16). In our present study, we have shown a much higher protection of mice immunized by the i.n. route than by the s.c. route. Moreover, our results suggest that the immune responses elicited upon s.c. delivery could even exacerbate pulmonary inflammation after infection and thus enhance clinical signs and weight loss. Since only the i.n. route of immunization with N-3M2e elicited a strong IgA secretion in BALf, we suggest that these local antibodies could play a major role in the protection of vaccinated mice against virus challenge.
De Filette et al. addressed whether preexisting anti-HBc immunity can interfere with the protective immune response induced by M2e-HBc vaccination (39). They have shown that anti-HBc immunity does not influence the anti-M2e response. RSV is the main virus isolated in respiratory infection (40), and anti-N antibodies are abundantly found in the sera of infected patients (41, 42). In the perspective of a clinical evaluation of N SRS as a new vaccine carrier for heterologous antigens, it is critical to investigate whether preexisting anti-N immunity induced by RSV infection may interfere with the specific adaptive anti-M2e response induced by chimeric M2e fused to N SRS. In this context, we have shown that preexisting immunity against N, induced by either RSV infection or vaccination with N SRS did not impair immune response against M2e. Yet, we did not test whether N-primed mice immunized with N-3M2e were protected against influenza challenge. Surprisingly, anti-M2e antibody titers (IgG2a in serum and IgA in BALf) were slightly higher in N-primed mice than in nonprimed mice. Interestingly, the same phenomenon was observed to a lesser extent by De Filette et al. concerning anti HBc preimmunity (39). In our case, it could suggest that the formation of immune complexes composed of N-3M2e and anti-N preexisting antibodies are more efficiently captured and processed by antigen-presenting cells via Fc receptors, leading to a better T helper cell priming (43). Overall, our results demonstrate that anti-M2e immunity could be induced by chimeric M2e/N SRS in RSV-immune individuals.
To conclude, we have made the proof of concept that N SRS can be used as a vaccine carrier for heterologous viral antigens like M2e and are particularly well suited for induction of IgA antibodies upon mucosal vaccination. We are currently investigating whether conserved epitopes from the stalk of influenza virus A HA (44, 45) can be successfully linked to N SRS and preserve their conformational display. Then, it would be interesting to investigate the potential of N SRS fused to these HA epitopes or to M2e to induce a cross-protective or heterosubtypic immunity against different strains of influenza virus A (including highly pathogenic influenza viruses such as H5N1). It is also conceivable to link to the N protein 3 different M2e sequences covering most of their diversity among avian and human influenza viruses, as it was previously done using replication-defective adenovirus vectors or VLPs (46, 47). Furthermore, the i.n. administration of live or inactivated influenza vaccines is known to favor cross-protection against heterotypic virus strains in relationship with the induction of secretory IgA (sIgA) in the respiratory tract (48, 49). Accordingly, influenza VLPs formed by HA, NA, and M1 from the 1918 H1N1 strain were shown to confer cross-protection against H5N1 virus only when delivered i.n., in relation with the induction of local IgA (50). The capacity of N SRS to induce a strong local IgA response when administered i.n. is therefore of great interest.
In the present study, we have also validated that N-3M2e-vaccinated mice were protected against RSV challenge just as efficiently as N SRS-vaccinated mice. Thus, we propose that N-3M2e chimeric rings could be used as a bivalent vaccine efficient against two respiratory viruses, influenza virus and RSV.
ACKNOWLEDGMENTS
Pierre-Louis Hervé has benefited from funding granted by the Ile de France region (DIM Malinf), and Mariam Raliou benefited from funding granted by the INRA.
We thank Jean Lepault from the Laboratoire de Virologie Moléculaire et Structurale, CNRS (UPR 3296) (Gif sur Yvette, France), for microscopic analysis of N SRS. We thank Sébastien Deville from Seppic, who provided us with Montanide gel adjuvant. We also thank Bernard Charley and Isabelle Schwartz-Cornil for the final read-through of the manuscript.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Belyakov IM, Ahlers JD. 2009. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J. Immunol. 183:6883–6892. 10.4049/jimmunol.0901466 [DOI] [PubMed] [Google Scholar]
- 2.Lycke N. 2012. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 12:592–605. 10.1038/nri3251 [DOI] [PubMed] [Google Scholar]
- 3.Schneider-Ohrum K, Ross TM. 2012. Virus-like particles for antigen delivery at mucosal surfaces. Curr. Top. Microbiol. Immunol. 354:53–73. 10.1007/82_2011_135 [DOI] [PubMed] [Google Scholar]
- 4.Scheerlinck J-PY, Greenwood DLV. 2008. Virus-sized vaccine delivery systems. Drug Discov. Today 13:882–887. 10.1016/j.drudis.2008.06.016 [DOI] [PubMed] [Google Scholar]
- 5.Bachmann MF, Jennings GT. 2010. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 10:787–796. 10.1038/nri2868 [DOI] [PubMed] [Google Scholar]
- 6.Vanlandschoot P, Cao T, Leroux-Roels G. 2003. The nucleocapsid of the hepatitis B virus: a remarkable immunogenic structure. Antiviral Res. 60:67–74. 10.1016/j.antiviral.2003.08.011 [DOI] [PubMed] [Google Scholar]
- 7.Tran T-L, Castagné N, Bhella D, Varela PF, Bernard J, Chilmonczyk S, Berkenkamp S, Benhamo V, Grznarova K, Grosclaude J, Nespoulos C, Rey FA, Eléouët J-F. 2007. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J. Gen. Virol. 88:196–206. 10.1099/vir.0.82282-0 [DOI] [PubMed] [Google Scholar]
- 8.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagné N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eléouët J-F, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. 10.1126/science.1177634 [DOI] [PubMed] [Google Scholar]
- 9.Roux X, Dubuquoy C, Durand G, Tran-Tolla T-L, Castagné N, Bernard J, Petit-Camurdan A, Eléouët J-F, Riffault S. 2008. Sub-nucleocapsid nanoparticles: a nasal vaccine against respiratory syncytial virus. PLoS One 3:e1766. 10.1371/journal.pone.0001766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riffault S, Meyer G, Deplanche M, Dubuquoy C, Durand G, Soulestin M, Castagné N, Bernard J, Bernardet P, Dubosclard V, Bernex F, Petit-Camurdan A, Deville S, Schwartz-Cornil I, Eléouët J-F. 2010. A new subunit vaccine based on nucleoprotein nanoparticles confers partial clinical and virological protection in calves against bovine respiratory syncytial virus. Vaccine 28:3722–3734. 10.1016/j.vaccine.2010.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elderfield R, Barclay W. 2011. Influenza pandemics. Adv. Exp. Med. Biol. 719:81–103. 10.1007/978-1-4614-0204-6_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boni M. 2008. Vaccination and antigenic drift in influenza. Vaccine 26:C8–C14. 10.1016/j.vaccine.2008.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staneková Z, Varečková E. 2010. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol. J. 7:351. 10.1186/1743-422X-7-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamb RA, Lai C-J. 1980. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 21:475–485. 10.1016/0092-8674(80)90484-5 [DOI] [PubMed] [Google Scholar]
- 15.Fiers W, De Filette M, Birkett A, Neirynck S, Min Jou W. 2004. A “universal” human influenza A vaccine. Virus Res. 103:173–176. 10.1016/j.virusres.2004.02.030 [DOI] [PubMed] [Google Scholar]
- 16.Neirynck S, Deroo T, Saelens X. 1999. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 5:1157–1163. 10.1038/13484 [DOI] [PubMed] [Google Scholar]
- 17.Feng J, Zhang M, Mozdzanowska K, Zharikova D, Hoff H, Wunner W, Couch RB, Gerhard W. 2006. Influenza A virus infection engenders a poor antibody response against the ectodomain of matrix protein 2. Virol. J. 3:102. 10.1186/1743-422X-3-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jegerlehner A, Schmitz N, Storni T, Bachmann MF. 2004. Influenza A vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J. Immunol. 172:5598–5605 [DOI] [PubMed] [Google Scholar]
- 19.Kang SM, Song JM, Compans RW. 2011. Novel vaccines against influenza viruses. Virus Res. 162:31–38. 10.1016/j.virusres.2011.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X. 2011. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 186:1022–1031. 10.4049/jimmunol.0902147 [DOI] [PubMed] [Google Scholar]
- 21.Castagne N, Barbier A, Bernard J, Rezaei H, Huet J, Henry C, Da Costa B, Eléouët J. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J. Gen. Virol. 85:1643–1653. 10.1099/vir.0.79830-0 [DOI] [PubMed] [Google Scholar]
- 22.Matrosovich M, Matrosovich T, Garten W, Klenk H-D. 2006. New low-viscosity overlay medium for viral plaque assays. Virol. J. 3:63. 10.1186/1743-422X-3-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Remot A, Roux X, Dubuquoy C, Fix J, Bouet S, Moudjou M, Eléouët J-F, Riffault S, Petit-Camurdan A. 2012. Nucleoprotein nanostructures combined with adjuvants adapted to the neonatal immune context: a candidate mucosal RSV vaccine. PLoS One 7:e37722. 10.1371/journal.pone.0037722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Filette M, Min Jou W, Birkett A, Lyons K, Schultz B, Tonkyro A, Resch S, Fiers W. 2005. Universal influenza A vaccine: optimization of M2-based constructs. Virology 337:149–161. 10.1016/j.virol.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 25.Parker R, Deville S, Dupuis L, Bertrand F, Aucouturier J. 2009. Adjuvant formulation for veterinary vaccines: MontanideTM Gel safety profile. Procedia Vaccinol. 1:140–147. 10.1016/j.provac.2009.07.026 [DOI] [Google Scholar]
- 26.Shim B-S, Choi YK, Yun C-H, Lee E-G, Jeon YS, Park S-M, Cheon IS, Joo D-H, Cho CH, Song M-S, Seo S-U, Byun Y-H, Park H-J, Poo H, Seong BL, Kim JO, Nguyen HH, Stadler K, Kim DW, Hong K-J, Czerkinsky C, Song MK. 2011. Sublingual immunization with M2-based vaccine induces broad protective immunity against influenza. PLoS One 6:e27953. 10.1371/journal.pone.0027953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf AI, Mozdzanowska K, Williams KL, Singer D, Richter M, Hoffmann R, Caton AJ, Otvos L, Erikson J. 2011. Vaccination with M2e-based multiple antigenic peptides: characterization of the B cell response and protection efficacy in inbred and outbred mice. PLoS One 6:e28445. 10.1371/journal.pone.0028445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mozdzanowska K, Feng J, Eid M, Kragol G, Cudic M, Otvos L, Gerhard W. 2003. Induction of influenza type A virus-specific resistance by immunization of mice with a synthetic multiple antigenic peptide vaccine that contains ectodomains of matrix protein 2. Vaccine 21:2616–2626. 10.1016/S0264-410X(03)00040-9 [DOI] [PubMed] [Google Scholar]
- 29.Mozdzanowska K, Zharikova D, Cudic M, Otvos L, Gerhard W. 2007. Roles of adjuvant and route of vaccination in antibody response and protection engendered by a synthetic matrix protein 2-based influenza A virus vaccine in the mouse. Virol. J. 4:118. 10.1186/1743-422X-4-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiers W, De Filette M, El Bakkouri K, Schepens B, Roose K, Schotsaert M, Birkett A, Saelens X. 2009. M2e-based universal influenza A vaccine. Vaccine 27:6280–6283. 10.1016/j.vaccine.2009.07.007 [DOI] [PubMed] [Google Scholar]
- 31.Hashemi H, Pouyanfard S, Bandehpour M, Noroozbabaei Z, Kazemi B, Saelens X, Mokhtari-Azad T. 2012. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza A virus challenge. PLoS One 7:e45765. 10.1371/journal.pone.0045765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersson A-MC, Håkansson KO, Jensen BAH, Christensen D, Andersen P, Thomsen AR, Christensen JP. 2012. Increased immunogenicity and protective efficacy of influenza M2e fused to a tetramerizing protein. PLoS One 7:e46395. 10.1371/journal.pone.0046395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song J-M, Wang B-Z, Park K-M, Van Rooijen N, Quan F-S, Kim M-C, Jin H-T, Pekosz A, Compans RW, Kang S-M. 2011. Influenza virus-like particles containing M2 induce broadly cross protective immunity. PLoS One 6:e14538. 10.1371/journal.pone.0014538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jazi MHZ, Dabaghian M, Tebianian M, Gharagozlou MJ, Ebrahimi SM. 2012. In vivo electroporation enhances immunogenicity and protection against influenza A virus challenge of an M2e-HSP70c DNA vaccine. Virus Res. 167:219–225. 10.1016/j.virusres.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 35.Denis J, Acosta-Ramirez E, Zhao Y, Hamelin M-E, Koukavica I, Baz M, Abed Y, Savard C, Pare C, Lopez Macias C, Boivin G, Leclerc D. 2008. Development of a universal influenza A vaccine based on the M2e peptide fused to the papaya mosaic virus (PapMV) vaccine platform. Vaccine 26:3395–3403. 10.1016/j.vaccine.2008.04.052 [DOI] [PubMed] [Google Scholar]
- 36.Johansson BE, Moran TM, Kilbourne ED. 1987. Antigen-presenting B cells and helper T cells cooperatively mediate intravirionic antigenic competition between influenza A virus surface glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 84:6869–6873. 10.1073/pnas.84.19.6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabbard J, Velappan N, Di Niro R, Schmidt J, Jones CA, Tompkins SM, Bradbury AR. 2009. A humanized anti-M2 scFv shows protective in vitro activity against influenza. Protein Eng. Des. Sel. 22:189–198. 10.1093/protein/gzn070 [DOI] [PubMed] [Google Scholar]
- 38.Muto NA, Yoshida R, Suzuki T, Kobayashi S, Ozaki H, Fujikura D, Manzoor R, Muramatsu M, Takada A, Kimura T, Sawa H. 2012. Inhibitory effects of an M2-specific monoclonal antibody on different strains of influenza A virus. Jpn. J. Vet. Res. 60:71–83 http://hdl.handle.net/2115/50096 [PubMed] [Google Scholar]
- 39.De Filette M, Martens W, Smet A, Schotsaert M, Birkett A, Londoño-Arcila P, Fiers W, Saelens X. 2008. Universal influenza A M2e-HBc vaccine protects against disease even in the presence of pre-existing anti-HBc antibodies. Vaccine 26:6503–6507. 10.1016/j.vaccine.2008.09.038 [DOI] [PubMed] [Google Scholar]
- 40.Simoes EA. 1999. Respiratory syncytial virus infection. Lancet 354:847–852. 10.1016/S0140-6736(99)80040-3 [DOI] [PubMed] [Google Scholar]
- 41.Gimenez HB, Keir HM, Cash P. 1987. Immunoblot analysis of the human antibody response to respiratory syncytial virus infection. J. Gen. Virol. 68(Part 5):1267–1275 [DOI] [PubMed] [Google Scholar]
- 42.Levine S, Dajani A, Klaiber-Franco R. 1988. The response of infants with bronchiolitis to the proteins of respiratory syncytial virus. J. Gen. Virol. 69(Part 6):1229–1239 [DOI] [PubMed] [Google Scholar]
- 43.Manca F, Fenoglio D, Li Pira G, Kunkl A, Celada F. 1991. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. J. Exp. Med. 173:37–48. 10.1084/jem.173.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ekiert DC, Bhabha G, Elsliger M, Friesen RHE, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. 10.1126/science.1171491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prabhu N, Prabakaran M, Ho H-T, Velumani S, Qiang J, Goutama M, Kwang J. 2009. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J. Virol. 83:2553–2562. 10.1128/JVI.02165-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou D, Wu T-L, Lasaro MO, Latimer BP, Parzych EM, Bian A, Li Y, Li H, Erikson J, Xiang Z, Ertl HCJ. 2010. A universal influenza A vaccine based on adenovirus expressing matrix-2 ectodomain and nucleoprotein protects mice from lethal challenge. Mol. Ther. 18:2182–2189. 10.1038/mt.2010.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim M-C, Song J-M, O E, Kwon Y-M, Lee Y-J, Compans RW, Kang S-M. 2013. Virus-like particles containing multiple M2 extracellular domains confer improved cross-protection against various subtypes of influenza virus. Mol. Ther. 21:485–492. 10.1038/mt.2012.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asahi-Ozaki Y, Yoshikawa T, Iwakura Y, Suzuki Y, Tamura S-I, Kurata T, Sata T. 2004. Secretory IgA antibodies provide cross-protection against infection with different strains of influenza B virus. J. Med. Virol. 74:328–335. 10.1002/jmv.20173 [DOI] [PubMed] [Google Scholar]
- 49.Asahi Y, Yoshikawa T, Watanabe I, Iwasaki T, Hasegawa H, Sato Y, Shimada S, Nanno M, Matsuoka Y, Ohwaki M, Iwakura Y, Suzuki Y, Aizawa C, Sata T, Kurata T, Tamura S. 2002. Protection against influenza virus infection in polymeric Ig receptor knockout mice immunized intranasally with adjuvant-combined vaccines. J. Immunol. 168:2930–2938 [DOI] [PubMed] [Google Scholar]
- 50.Perrone LA, Ahmad A, Veguilla V, Lu X, Smith G, Katz JM, Pushko P, Tumpey TM. 2009. Intranasal vaccination with 1918 influenza virus-like particles protects mice and ferrets from lethal 1918 and H5N1 influenza virus challenge. J. Virol. 83:5726–5734. 10.1128/JVI.00207-09 [DOI] [PMC free article] [PubMed] [Google Scholar]