

Abstract
Influenza virus vaccination strategies are focused upon the elicitation of protective antibody responses through administration of viral protein through either inactivated virions or live attenuated virus. Often overlooked in this strategy is the CD4 T cell response: how it develops into memory, and how it may support future primary B cell responses to heterologous infection. Through the utilization of a peptide-priming regimen, this study describes a strategy for developing CD4 T cell memory with the capacity to robustly expand in the lung-draining lymph node after live influenza virus infection. Not only were frequencies of antigen-specific CD4 T cells enhanced, but these cells also supported an accelerated primary B cell response to influenza virus-derived protein, evidenced by high anti-nucleoprotein (NP) serum antibody titers early, while there is still active viral replication ongoing in the lung. NP-specific antibody-secreting cells and heightened frequencies of germinal center B cells and follicular T helper cells were also readily detectable in the draining lymph node. Surprisingly, a boosted memory CD4 T cell response was not sufficient to provide intermolecular help for antibody responses. Our study demonstrates that CD4 T cell help is selective and limiting to the primary antibody response to influenza virus infection and that preemptive priming of CD4 T cell help can promote effective and rapid conversion of naive B cells to mature antibody-secreting cells.
INTRODUCTION
Ongoing efforts to curtail the ever-present threat of influenza virus infection by either pandemic or more common seasonal strains are largely hinged upon vaccination with trivalent inactivated virus (TIV) or live attenuated influenza virus (LAIV) vaccine (1). While both of these strategies are generally efficacious (2–4), there are often gaps in protection that influenza virus can widen and exploit, such as in the case of a pandemic or antigenic drift. When protection fails, clearance of the virus and recovery from infection are predicated upon the adaptive responses and depend upon the timely expansion of effector CD8 and CD4 T cells as well as helper CD4 T cells and B cells. Though CD4 and CD8 T cell responses can result in viral clearance without a supporting antibody response (5, 6), the primary B cell response has been very closely associated with protection (7–10). Given the important function B cells have in disease amelioration, it follows that the CD4 T helper response is equally essential. Many studies have documented the essential role of helper cells in the establishment of a protective antibody response; without CD4 T cells, B cell responses are suboptimal, leading to delayed clearance of virus (11–13).
CD4 T cells are an obligate participant of the germinal center (GC) response that is necessary to establish high-affinity, class-switched memory B cells and antibody-secreting plasma cells (14–17). CD4 T cells also provide both direct and indirect support to the extrafollicular response that results in a rapid release of influenza virus-specific antibody (15, 18). Central to this provision of B cell help is the follicular T helper (Tfh) cell that is characterized by expression of CXCR5, a chemokine receptor that licenses CD4 T cell access to the B cell zone, where antigen-engaged B cells are positioned, via responsiveness to CXCL13 (14, 19, 20).
Though the primary CD4 T cell response to infection is capable of supporting B cell responses, it is less clear what specific role memory CD4 T cells have in a primary humoral response to a complex pathogen. The issue of whether CD4 frequency is predictive of a B cell response has yet to be well established though recent evidence is accumulating that suggests a close relationship (21, 22). Endogenous or adoptively transferred memory CXCR5+ CD4 T cells can accelerate the B cell response to a model protein antigen (23) and have also been shown to have “superior” functionality in the lymph node (LN) and lung of infected mice (24). In humans, CXCR5-expressing cells in the blood are functionally related to Tfh cells, perhaps representing the memory component of these B cell helpers (25, 26). The B cell-helping capacity of this T cell memory population highlights a potential mechanism for accelerating the primary B cell response to influenza virus infection. Previous studies addressing memory CD4 T cell help for B cells have been somewhat hindered by the difficulty in unlinking development of T cell memory from B cell memory and by limitations on studying help in the context of infection. By selectively priming the CD4 T cell arm of memory, an expanded population of cells with helper capacity could be established, and its role in the primary B cell response to infection can be more clearly defined.
We have previously shown that the primary CD4 T cell response to live influenza virus infection is abundant and highly diverse, consisting of more than 100 different epitopes occupying a sizeable portion of the total lymphoid CD4 T cell compartment. Therefore, it was uncertain whether CD4 T cell help is a detectably limiting factor in the B cell response to virus infection. By utilizing a peptide-priming strategy designed to generate influenza virus-specific CD4 T cells without providing epitopes for B cell activation, we generated CD4 memory unlinked to B cell memory. After subcutaneous immunization with influenza virus-derived peptides and subsequent infection, we show that peptide priming leads to the generation of influenza virus-specific memory CD4 T cells that can influence the rate of the primary B cell response to live infection. These accelerated kinetics of the CD4 T cell and B cell responses led to early increased serum titers of virus-specific antibody and increased frequencies of antibody-secreting cells, germinal center B cells, and follicular T helper cells. Surprisingly, examination of the ability of antigen-specific memory CD4 T cells to provide help to B cells of an alternate protein specificity showed that this is likely a very inefficient process that results in a minimal degree of intermolecular help. Taking these observations together, we demonstrate that CD4 T cell help is a limiting factor for the kinetics and magnitude of the early B cell response to infection and that provision of this help is highly selective with regard to antigen specificity, highlighting the role that memory CD4 T cells can play in protective immune responses to novel influenza viruses.
MATERIALS AND METHODS
Influenza virus.
Influenza A/New Caledonia/20/99 virus was prepared in the allantoic cavity of embryonated chicken eggs, as described previously (27). The infectious dose used per mouse was 5 × 104 50% egg infective doses (EID50) in 30 μl of phosphate-buffered saline (PBS) delivered intranasally.
Mice.
Female SJL (I-As) mice were purchased from the National Cancer Institute—Frederick (Frederick, MD). Mice were maintained in a specific-pathogen-free facility at the University of Rochester, according to institutional guidelines specified by the University Committee on Animal Resources, and used at 7 to 16 weeks of age. Mice in all groups were age matched.
Immunization and influenza virus infection of mice.
Mice were immunized in each hind footpad with 50 μl of an influenza virus-derived peptide pool emulsified in Incomplete Freund's Adjuvant (IFA; Sigma-Aldrich) and 0.6 μg/ml lipopolysaccharide (LPS; Sigma-Aldrich). The peptide pool contained either five or six nucleoprotein (NP)-derived or hemagglutinin (HA)-derived immunogenic peptides, respectively, at concentrations of 5 nM. Peptides used in this study include the following: NP 97, YKRVDGKWVRELVLYDK; NP 270, VAHKSCLPACVYGPAVA; NP 342, RVSSFIRGTRVLPRGKL; NP 438, SDMRAEIIKMMESARPE; NP 444, IIKMMESARPEEVSFQG; 6, HA 120, EQLSSVSSFERFEIFPK; HA 126, SSFERFEIFPKESSWPN; HA 132, EIFPKESSWPNHTVTGV; HA 144, TVTGVSASCSHNGKSSF; HA 334, LRNIPSIQSRGLFGAIA; HA 386, NAINGITNKVNSVIEKM. Numbers indicate residue positions within the protein sequences. As a control, a group of mice were immunized with emulsion of IFA and LPS. After 4 weeks, mice were anesthetized by intraperitoneal injection with Avertin (2,2,2-tribromoethanol) at a dose of 200 to 250 μl per mouse. Mice were infected intranasally with 5 × 104 EID50 of A/New Caledonia/20/99 in 30 μl of phosphate-buffered saline (PBS). Mice were euthanized on day 5, day 7, and day 10 postinfection; spleen and popliteal and mediastinal lymph nodes (PLNs and MLNs, respectively) were excised and used as a source of CD4 T cells for in vitro assays, as described below. Serum was collected from individual mice by cardiac puncture or by submandibular bleed.
Synthetic peptides.
Peptides used for enzyme-linked immunosorbent spot (ELISPOT) assays were derived from a set of 17-mer peptides, overlapping by 11 amino acids, that encompassed the entire sequence of hemagglutinin (HA) from influenza A/New Caledonia/20/1999 virus (H1N1) and nucleoprotein (NP) from influenza A/New York/348/2003 virus (H1N1), as previously described (28, 29). The amino acid sequence for NP protein is highly conserved between this virus and A/New Caledonia/20/99. Peptide arrays were obtained from the NIH Biodefense and Emerging Infections Research Repository (NIAID) as follows: NR-2602 for the HA protein of A/New Caledonia/20/1999 and NR-2611 for the NP protein of A/New York/348/2003. Peptides were reconstituted at 10 mM in PBS with or without added dimethyl sulfoxide to increase solubility of hydrophobic peptides and 1 mM dithiothreitol for cysteine-containing peptides. Single peptides were used at a final concentration of 10 μM, and peptide pools were used at a final concentration of 2 μM for each peptide in the pool.
ELISPOT assays for cytokine-secreting cells.
As described previously (28), CD4 T cells were analyzed for abundance and specificity using cytokine ELISPOT assays. Briefly, mice were euthanized at the indicated times postinfection (Fig. 1A and throughout Results), and single-cell suspensions from spleen, mediastinal lymph nodes, and popliteal lymph nodes were collected and processed in culture medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 10% fetal bovine serum [FBS]). Splenocytes were depleted of red blood cells by treatment with ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA in H2O, pH 7.2 to 7.4) for 5 min at room temperature and then washed and enriched for CD4 T cells by antibody and complement-mediated lysis. Monoclonal antibody-producing cell lines obtained from the American Type Culture Collection included 3.155 (anti-CD8), RA3/3A1/6.1 (anti B220), and 10.2.16 (anti-I-As) for SJL mice. After incubation for 30 min at 4°C with the monoclonal antibodies at 2 × 107 cells/ml, cells were washed and resuspended in a 1:20 dilution of guinea pig complement (Low Tox M; Cedarlane Laboratories, Burlington, NC) at a concentration of 2 × 107 cells/ml and incubated at 37°C for 30 min. Viable cells were purified by density gradient centrifugation with Lympholyte-M (Cedarlane Laboratories). Ninety-six-well MultiScreen HTS filter plates (Millipore, Billerica, MA) were coated with 2 μg/ml of purified rat anti-mouse gamma interferon (IFN-γ) (clone AN18; BD Bioscience, San Jose, CA) in PBS at room temperature for 2 h or overnight at 4°C. Plates were washed and blocked for 1 h at room temperature with culture medium. CD4 T cells (various graded doses up to 300,000 cells to maximize plate spot readability) were cocultured with syngeneic splenocytes as antigen-presenting cells (APC; 500,000 cells) and recall peptides in a total volume of 200 μl for 16 to 18 h at 37°C and 5% CO2. Plates were washed and developed using a Vector Blue Substrate Kit III (Vector Laboratories, Burlingame, CA) prepared in 100 mM Tris, pH 8.2. After being dried, the plates were processed for spot counting using an Immunospot Reader Series 2A with Immunospot software, version 3.2 (Cellular Technology, Ltd., Cleveland, OH). Data were calculated and presented as spots per million CD4 T cells, with background values subtracted.
FIG 1.
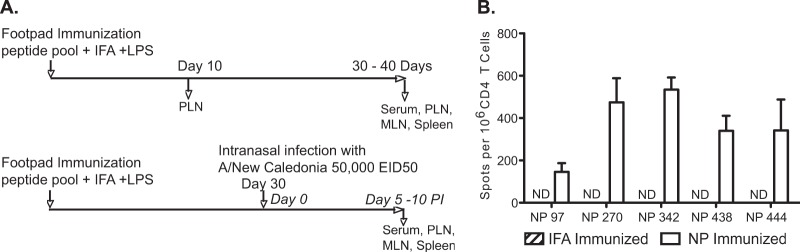
Immunodominant influenza virus epitopes prime a robust CD4 T cell response. (A) Experimental protocol used throughout this study. To study the effects of memory on the response to subsequent infection, SJL mice were immunized in the footpad with an emulsion of peptides in IFA and LPS, as described in Materials and Methods, and were either tested directly 10 days postimmunization or rested for 4 weeks for studies on infection. One cohort of these mice was not infected, and a second cohort was infected intranasally with 5 × 104 EID50 A/New Caledonia/20/99 at 30 days postimmunization. (B) To determine the primary response to peptides used for priming, CD4 T cells were isolated from the draining popliteal lymph nodes (PLN) 10 days after immunization. CD4 T cells from NP- and control IFA-immunized mice were recalled with NP peptides used in immunization. The data are presented as mean spots per million CD4 T cells from three individual mice and are representative of three experiments. ND, no detectable response. Error bars indicate standard deviations.
Antibody ELISAs.
Mouse sera were collected from individual mice, and NP protein- and HA protein-specific antibodies were determined by an enzyme-linked immunosorbent assay (ELISA) using recombinant NP protein derived from A/New Caledonia/20/99 as previously described (28) or recombinant HA protein derived from A/New Caledonia/20/99 (Protein Sciences Corporation, Meriden, CT). Briefly, 96-well polystyrene flat-bottom plates (Costar) were coated overnight at 4°C with 200 ng/100 μl of purified NP or HA protein per well. Wells were rinsed with wash buffer (0.05% Tween 20 [Sigma-Aldrich] in PBS) and then incubated with blocking buffer (3% bovine serum albumin [BSA] in PBS) for 1 h at room temperature. Blocking buffer was removed, and serial 3-fold sample dilutions (in 0.5% BSA-PBS) were added to the plates and incubated for 2 to 3 h at room temperature. The wells were washed with PBS and incubated for 1 h at room temperature with 100 μl/well alkaline phosphatase-conjugated goat anti-mouse secondary antibody specific for IgG (Southern Biotech, Birmingham, AL) diluted in 1% BSA-PBS at a 1/1,000 dilution. One p-nitrophenyl phosphate substrate tablet (5 mg/tablet; Sigma) was dissolved in 15 ml of diethanolamine substrate buffer, pH 9.8 (9.7% [vol/vol] diethanolamine, 0.02% [wt/vol] sodium azide, 0.5 mM MgCl2). Subsequently, wells were washed with wash buffer, and 100 μl of substrate per well was added and developed at room temperature (25 to 40 min.). Absorbance at 405 nm was read using SoftMax Pro software and a VMax plate reader.
Flow cytometry.
Cells prepared as a single-cell suspension, as described above, were stained first in a suspension of Fc Block (BD Biosciences) for 20 min at 4°C, followed by addition of the indicated antibodies to detect Tfh cells and various B cell subsets. Cells were incubated with antibody for an additional 25 min at 4°C and protected from light; they were then washed one time in fluorescence-activated cell sorting (FACS) buffer (Dulbecco's PBS [DPBS] plus 2% FBS), resuspended in a secondary stain if necessary, and incubated for 25 min at 4°C, protected from light. Cells were then washed two times in FACS buffer and resuspended for flow cytometry data acquisition. Immediately prior to acquisition, 7-aminoactinomycin D (7-AAD) reagent (BD Biosciences) was added to each sample for delineation of live cells. Samples were acquired on a BD FACSCanto II system with 488-nm, 633-nm, and 405-nm lasers using FACS Diva software. Data files were analyzed using FlowJo, version 8.8.6, software (Tree Star, Inc.). Antibodies and reagents, purchased from BD Biosciences unless otherwise noted, were as follows: B220 (RA3-6B2), CD4 (RM4-5), CD44 (IM7), CD95 (Jo2), CXCR5 (2G8), PD-1 (J43; eBiosciences), T and B cell activation antigen (GL-7), and streptavidin-phycoerythrin (PE).
Statistical analyses.
Statistical significance was evaluated using an unpaired Student's t test and by Pearson correlation assuming Gaussian distribution. A P value of <0.05 was considered statistically significant. Prism (GraphPad Software, CA) was used for all statistical tests.
RESULTS
Empirically defined immunodominant influenza virus peptide epitopes prime a robust CD4 T cell response.
The murine CD4 T cell response to a recently circulating strain of influenza virus, A/New Caledonia/20/99, has been well characterized by our laboratory (27, 30–33), with many CD4 T cell epitopes recognized in the primary response to infection in the context of many different class II haplotypes having been identified. Nucleoprotein (NP) was chosen for the current study because, as noted in the aforementioned studies, CD4 T cells with NP specificity are elicited in multiple MHC haplotypic backgrounds. Additionally, NP is genetically conserved across multiple influenza virus strains, and the NP protein can be readily used to enumerate virus protein-specific B cells.
Initially, peptides were selected based on prior identification as robust epitopes elicited by infection (30) and were evaluated as immunogens for peptide priming. Our experimental strategy (Fig. 1A) involved subcutaneous priming of mice with NP-derived peptides emulsified in Incomplete Freund's Adjuvant (IFA) with lipopolysaccharide (LPS), or IFA/LPS alone as a control. The primary CD4 T cell response was evaluated in the draining popliteal lymph node (PLN) after 10 days and again at memory 30 days postimmunization. We then assessed responses 7 days after challenge with live intranasal infection (day 37 postimmunization). In addition to serum collection, tissues that were sampled included the lung-draining mediastinal lymph node (MLN), spleen, and priming-site-draining-PLN. Of the NP epitopes screened for immunogenicity, we were able to detect robust frequencies of peptide-specific cytokine-producing CD4 T cells at day 10 postimmunization (Fig. 1B). Additionally, there was no evidence of high precursor frequencies of peptide-specific CD4 T cells in unprimed mice (not detectable in IFA-immunized mice). Altogether, these experiments indicated that the NP-derived peptides identified by epitope mapping from infection with New Caledonia virus were intrinsically immunogenic as free synthetic peptides, allowing selective priming of CD4 T cells.
Peptide priming facilitates establishment of memory CD4 T cells that rapidly expand upon influenza virus challenge.
Next, the capacity of the NP peptide-priming regimen to persist and establish a memory CD4 T cell population was determined. Again, the PLN (Fig. 2B, open bar) was analyzed for the presence of NP peptide-specific memory CD4 T cells. Distal sites were also sampled, where we found that the spleen and MLN (Fig. 2A and C, open bars) both contained NP-specific cells. It was evident from the representation of peptide-specific CD4 T cells in the distal sites, spleen, and MLN that 4 to 5 weeks after subcutaneous peptide priming in the footpad, it was possible to generate memory CD4 T cells with the capacity to circulate through and populate the entire lymphoid compartment. The presence of peptide-specific CD4 T cells in the spleen and especially the MLN also served to provide a baseline for the frequency of peptide-reactive cells pre- and postinfection.
FIG 2.
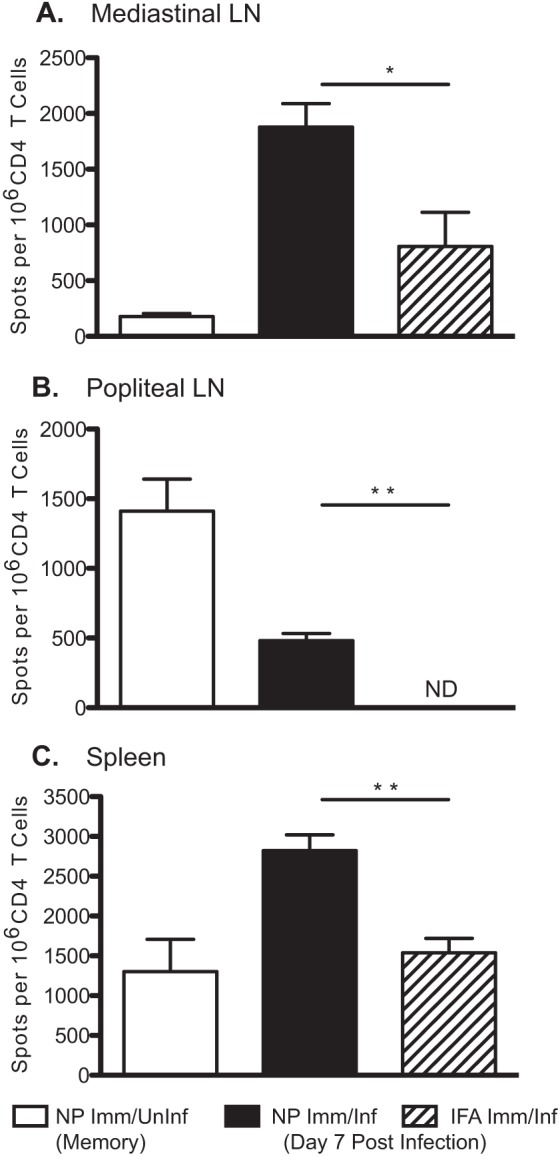
Influenza virus infection rapidly expands peptide-primed CD4 T cell memory. NP peptide- or IFA-immunized mice were challenged intranasally at 4 weeks postimmunization with 5 × 104 EID50 A/New Caledonia/20/99. On day 7 postinfection, CD4 T cells were isolated from MLN (A), PLN (B), and spleen (C) and tested in an IFN-γ ELISPOT assay for reactivity to the pool of NP peptides. Experimental groups included NP peptide-immunized and infected mice (Imm/Inf) and IFA-immunized and infected mice. Uninfected NP-peptide immunized mice (Imm/Uninf) were assayed as a control for memory at day 0 of infection (day 30 postimmunization). The data are presented as mean spots per million CD4 T cells from 3 to 5 mice per group and are representative of at least three experiments. ND, no detectable response for IFA-primed PLN. Error bars indicate standard deviations. *, P < 0.01; **, P < 0.005, Student's t test.
After establishing the presence of NP peptide-specific CD4 T cell memory, the ability of these cells to mobilize in response to an influenza virus infection challenge was evaluated. Tracking the magnitude of the NP peptide-specific CD4 T cell response following infection revealed that, compared to IFA control immunization, the establishment of CD4 memory through peptide priming augmented the early antigen-specific CD4 T cell response to intranasal infection. If the response seen in the NP-primed and infected group (Fig. 2, filled bars) is considered to be the boosted response, it is evident that NP-specific CD4 T cells are readily expanded by day 7 after infection. This is in contrast to the primary response (Fig. 2, hatched bars), where the frequency of influenza virus-reactive cells is much lower. The frequency of NP-specific CD4 T cells in both the MLN and spleen (Fig. 2A and C, respectively) of mice receiving the peptide-priming regimen 30 days prior to infection is approximately double the frequency of NP-specific CD4 T cells in control mice undergoing a primary response to infection. This result suggests that memory CD4 T cells residing in the MLN or recirculating memory cells recruited to the MLN rapidly respond to viral antigen and rapidly expand.
As expected in the PLN, there was no detectable expansion of influenza virus-specific CD4 T cells (Fig. 2B), consistent with a lack of influenza virus antigen in distal nodes. In the control group undergoing primary infection without prior CD4 T cell priming, there were no detectable NP-specific CD4 T cells. Interestingly, an apparent depletion of peptide-specific cells from this site occurred in the infected NP peptide-primed group, which may be an indication that central memory cells were recruited to sites of secondary antigen exposure, such as the MLN, the primary site of antigen access after intranasally inoculated respiratory infection (34–37).
The impact of peptide priming of memory CD4 T cells on B cell responses is evident through analyses of serum antibody responses to infection.
With clear evidence of peptide-specific CD4 T cell memory established, along with evidence for rapid CD4 T cell boosting, we next sought to determine whether the enhanced influenza virus-specific CD4 T cell response benefited the primary B cell response to infection. The CD4 T cells generated upon peptide priming would potentially have the capacity to provide B cell help for both the extrafollicular and germinal center responses (14, 15, 38–40). As before, mice were primed with the NP-derived peptides and then challenged 30 days later with intranasal infection of A/New Caledonia/20/99. The level of NP-specific antibody was tracked in serum from day 5 postinfection through day 10 using an ELISA (Fig. 3A).
FIG 3.
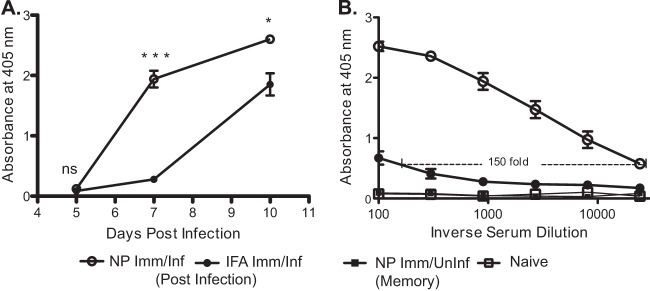
CD4 T cell help potentiates future serum antibody response to infection. Mice were primed and infected as illustrated in Fig. 1A. Sera were collected from the indicated cohorts of naive, NP peptide-immunized (infected or uninfected), and IFA-immunized and infected mice and tested for reactivity toward recombinant NP protein. NP protein-specific IgG was measured via ELISA. (A) Kinetics of NP-specific antibody accumulation in sera collected at three different time points: day 5, day 7, and day 10. Absorbance readings were made at a dilution 1:900, mean values from 2 to 5 individual mice are shown. (B) Serial dilutions (3-fold starting at 1:100) of sera from day 7 postinfection indicating fold increase in NP-specific IgG titers. The inverse dilution is shown. NP-specific IgG was ∼150-fold higher in NP peptide-primed and infected mice than in IFA-primed and infected mice. Data are shown as mean values from five individual mice, representative of three separate experiments. Error bars indicate standard deviations. *, P < 0.02; ***, P < 0.0001, Student's t test.
These assays revealed a distinct kinetic advantage in the serum antibody response to virus-derived NP protein in the NP peptide-primed group compared to the IFA-primed and infected cohort. This was evident on day 7 and as late as day 10 after infection. Quantification of antibody titers at day 7 (Fig. 3B) allows for an estimation of the relative levels of total NP-specific antibody in the two groups after infection. The NP peptide-primed and infected group had 150-fold greater NP-specific antibody than the control-primed and infected group. To eliminate the possibility of B cell reactivity to the original peptides used for priming, we also tracked serum antibody in NP peptide-primed but uninfected mice. As expected, the NP-specific antibody response developed only after infection, indicating that peptide priming did not result in detectable levels of NP protein-specific antibody (Fig. 3B), nor were any peptide-reactive antibodies identified after the peptide-priming regimen (data not shown).
Peptide priming of CD4 T cells supports the primary B cell response in the mediastinal lymph node.
The heightened levels of NP-specific serum IgG suggested that NP-specific CD4 T cells were able to potentiate the primary B cell response to influenza virus infection. To determine what contributions to the humoral response could be attributed to T cell-dependent B cell responses in the lung-draining lymph node, we isolated cells from the MLN of NP peptide-primed and infected or IFA-primed and infected mice and tested them in a B cell ELISPOT assay for reactivity to NP protein. In this way, we were able to enumerate the frequency of NP-specific antibody-secreting cells (ASC) in each lymph node. On day 7 postinfection (Fig. 4), only the mice that had been primed with the NP peptide pool had any detectable frequencies of NP-specific IgG-secreting B cells. This suggests that a memory CD4 T cell population established via selective epitope priming can support the early expansion and differentiation of antigen-specific naive B cells into antibody-secreting cells in response to infection, either through support of the extrafollicular antibody response or the germinal center reaction.
FIG 4.
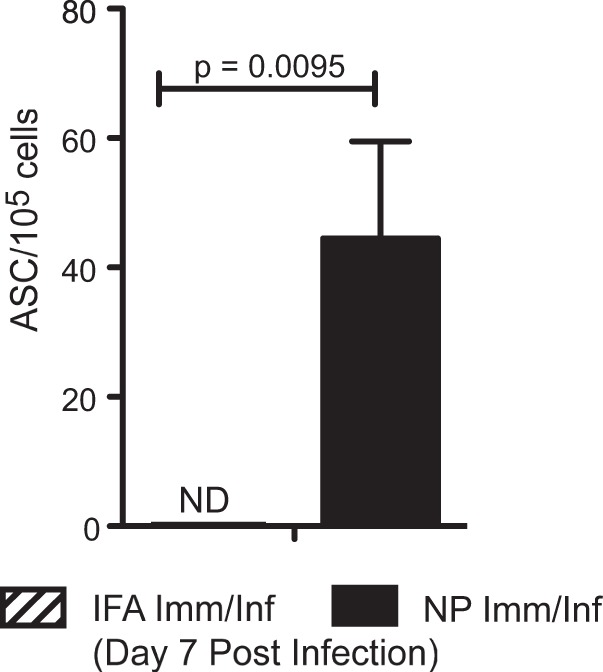
Accelerated NP protein-specific B cell response to infection in peptide-primed and infected animals. On day 7 postinfection, cells were isolated from the MLN of NP- peptide-immunized and infected mice and IFA-immunized and infected mice. Antibody-secreting cells were tested for reactivity toward recombinant NP protein from the A/New Caledonia/20/99 virus and detected by ELISPOT assay. IgG was measured with IgG-specific secondary antibody reagent. Data shown depict 5 mice per group, representative of three independent experiments. Error bars indicate standard deviations. ND, no detectable response for IFA-primed and infected mice.
Enhanced B cell responses to infection are evidenced by robust germinal center and follicular helper T cell populations.
After finding that both the serum antibody levels and ASC frequencies were augmented in mice with established influenza virus-specific CD4 memory, we evaluated whether the accelerated B cell response was associated with heightened frequency of Tfh or germinal center (GC) B cells. This issue was addressed through characterization of the various B cell and T cell populations in the MLN by flow cytometry at day 7 postinfection. As an indication of the progression of the adaptive humoral response to infection, the frequency of GC B cells (B220+ Fas+ GL-7+) (41–44) was quantified. In NP peptide-primed and infected mice, GC B cells were detected by day 7 (Fig. 5A), while in the primary response to infection (IFA-primed mice), these populations were essentially absent. The frequency of GC B cells was significantly higher in NP-primed and infected mice than in the IFA-primed group (Fig. 5C). This indicated that the primed CD4 memory population was able to facilitate the early expansion and differentiation of naive influenza virus-specific B cell populations.
FIG 5.

Accelerated primary B cell response characterized by day 7 germinal center B cell and Tfh cell populations. Single-cell suspensions prepared from the MLN of an NP peptide pool or IFA-immunized mice were stained and analyzed for the frequency of GC B cells (B220+ GL-7+ Fas+) (A) and Tfh cells (CXCR5++ PD-1++ CD4+ CD44hi) (B). Representative FACS plots are shown. (C and D) Plots depicting frequencies of GC B cells and Tfh cells show the mean value for 10 (IFA) to 15 (NP) individual mice per group, combined from two separate experiments. GC B cell frequency is indicated as a percentage of the B220+ cell population. Tfh cell frequency is indicated as percentage of the CD4+ CD44hi cell population. *, P < 0.05; **, P < 0.01, Student's t test. (E) Correlation of Tfh cell frequency and GC B cell frequency in NP-primed or IFA-primed mice. Pearson test was performed assuming Gaussian distribution, and R2 and P values are shown.
Further confirming that NP peptide priming led to an accelerated B cell response upon infection, we observed a Tfh cell population (CD4+ CD44hi CXCR5+ PD-1+, where CD44hi indicates high-level expression of CD44) in the previously primed group that was either absent altogether or much less expanded in unprimed but infected mice after 7 days (Fig. 5B and D). In mice that had not been boosted by infection, there were no detectable Tfh cells in the MLN, as expected (data not shown). Given the closely interdependent nature of Tfh cells and the GC reaction, we sought to determine if the heightened Tfh cell frequency could be directly correlated to an increased frequency in GC B cells. The frequency of Tfh cells in NP-primed and infected mice correlated positively with the frequency of GC B cells (Fig. 5E) while the Tfh populations in the control-primed and infected group had no such relationship to GC B cell frequency (IFA primed). The correlative relationship indicates that of the animals showing an increased frequency of Tfh cells, NP-primed mice are better able to support the GC reaction at this time than IFA-primed mice. In the IFA-primed group, there was no correlative relationship of Tfh cell frequency to GC reaction even if the frequency of Tfh cells approached the levels found in NP-primed mice during the early stages of the adaptive response to infection. There was significant mouse-to-mouse variability in both the germinal center and Tfh response among the sampled animals, but we suspect that this variability reflects a lack of synchrony during the infections or in the mobilization of pertinent cell populations. The correlative relationship shown for NP-primed mice, despite the range of variation, clearly indicates a more advanced state of the adaptive immune response that is not evident at this early time point for control-primed mice.
NP-specific CD4 T cells do not support interprotein-specific antibody responses.
With clear evidence for the ability of NP-specific CD4 T cells to support the early robust NP-specific B cell response, we next sought to determine the ability of such cells to facilitate antigen-specific responses to other influenza virus proteins. Essentially, could NP-specific CD4 T cells boost the HA-specific antibody response and vice versa? As described above for NP-derived peptide priming, a selection of immunogenic HA-derived CD4 T cell epitopes was used in a peptide-priming regimen to generate HA-specific CD4 T cell memory. As shown with NP peptide priming, an analogous frequency of HA-specific cells was enumerated soon after priming (day 10), as well as at day 30 (memory) and day 7 postinfection (data not shown). Additionally, a similar boost in the serum antibody response was also observed (Fig. 6A).
FIG 6.
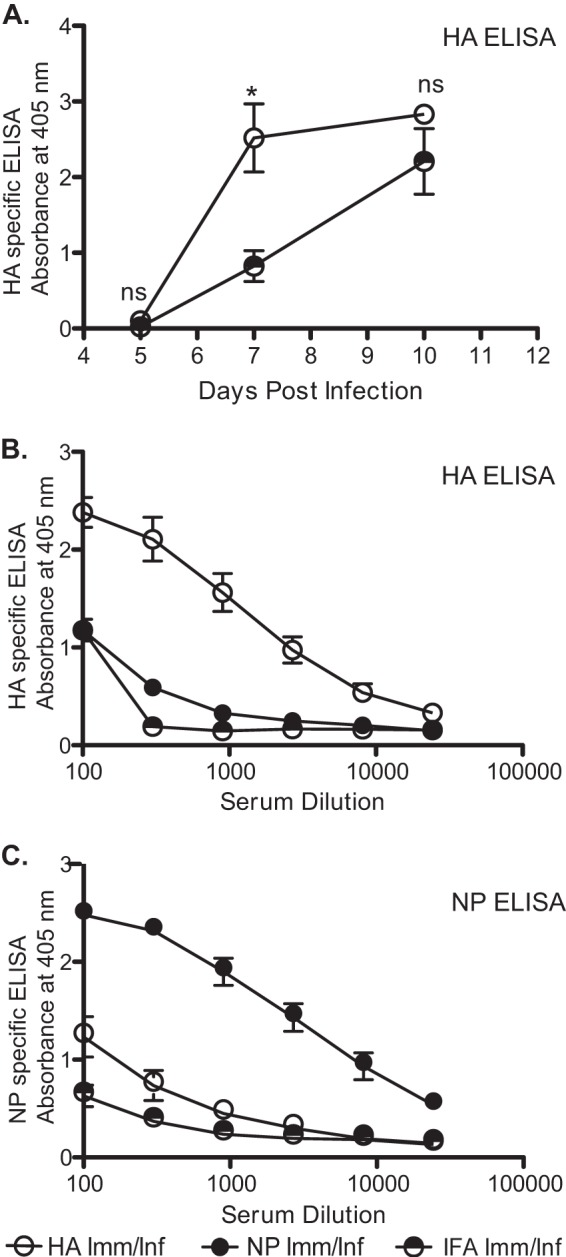
Provision of CD4 T cell help is selective for antibody responses of intramolecular specificity. Serum antibody responses to HA were determined in a manner similar to that for the ELISAs described in the legend of Fig. 3. Mice were HA peptide primed and then challenged with infection, with serum levels of HA-specific IgG evaluated on days 5, 7, and 10 postinfection. A serum dilution of 1:900 was used, and mean values from 2 to 5 individual mice, representative of three separate experiments, are shown (A). Peptide priming with influenza virus-derived epitopes boosts the serum antibody response of intramolecular-specific B cells for both HA-specific (B) and NP-specific (C) responses. Inverse 3-fold serum dilutions starting at 1:00 from primed and day 7 infected animals are shown. Data are shown as mean values from five individual mice, representative of three separate experiments. Error bars indicate standard deviations. ns, not significant; *, P < 0.05, Student's t test.
To determine if NP-specific or HA-specific memory CD4 T cells could boost B cell responses to nonhomologous proteins, serum from either priming group was tested for antibody reactivity to the alternative protein. Most strikingly in this set of experiments was our finding that CD4 T cell help for antibody responses was selective. Titration of serum antibody revealed that preexisting memory HA-specific CD4 T cells enhanced the antibody response to HA only, with little apparent help for the NP-specific antibody response. Conversely, NP-specific CD4 T cells were able to accelerate the B cell response to NP but not the HA protein (Fig. 6B and C). Thus, remarkably, we could find little evidence that HA-specific memory T cells were more proficient in providing help to NP-specific B cells than the primary, nonboosted CD4 T cells expanded in control IFA-primed mice or, similarly, that NP-specific memory T cells provided help to HA-specific B cells.
Collectively, our results indicate that during a primary response to live influenza virus infection, the number of CD4 T cells available to help the influenza virus-specific B cell response is a limiting factor in the kinetics and early magnitude of the primary B cell response to influenza virus challenge. Memory CD4 T cells in the draining lymph node, boosted in frequency by NP peptide priming, provide help in the form of follicular T helper cells for the early expansion of germinal center B cells. Furthermore, the selectivity of CD4 T cell help for B cells appeared to be dependent on the intramolecular rather than intermolecular protein specificity of both cell types.
DISCUSSION
In this study, we have demonstrated the impact of a simple influenza virus peptide-priming regimen to assess the potential limitations of CD4 T cells during a primary response to influenza virus infection. NP-specific CD4 T cell memory, elicited by peptide priming, can be reactivated and rapidly expand upon infection with influenza virus, resulting in an advantage in proliferation and peak numbers of CD4 T cells in the lung-draining lymph node compared to the primary response. As evidence for the helper function of this expanded T cell population, we show that NP-specific serum antibody titers are elevated by day 7 postinfection compared to levels with primary infection. At these early time points, there are also readily detectable populations of ASC and GC B cells in the MLN at a time when there is still live virus in the lung. As there is an accompanying increase in Tfh cell frequency in the primed and infected animals, it is likely that the established CD4 T cell memory is contributing directly to this population (23, 25, 26, 45), especially since there is a positive correlative relationship to GC B cell frequency. All of these studies have been successfully recapitulated in alternate mouse strains [B10.S (I-As) and A/J (I-AkEk)] involving different background genes (data not shown), major histocompatibility complex (MHC) molecules, and peptide epitopes to rule out the possibility of our observations being a strain-specific phenomenon. Finally, our results indicate that CD4 T cell help for antigen-specific B cell responses is likely predicated upon a shared intramolecular specificity; NP-specific CD4 T cells did not provide an effective boost to the HA antibody response and vice versa.
Our results demonstrate that the primary B cell response has the potential to progress more rapidly than what occurs during an unmanipulated primary B cell response to live influenza virus infection. Consistent with previous studies, this suggests that B cells acquire antigen, move to the T cell-B cell border in a CCR7-dependent manner, and wait upon the arrival of T cell signals to advance further toward ASC or the GC reaction (20, 46, 47). Otherwise, the number of influenza virus-specific CD4 T cells observed in the primary response to infection would be entirely sufficient to drive a robust B cell response at the advanced pace that was observed after peptide priming. Thus, CD4 T cell help for antibody responses can be limiting in the response to infection.
This is somewhat surprising as our laboratory has identified the magnitude and diversity of CD4 T cells in the primary response (27, 29, 30). These studies have revealed that in this strain of mice, within a week of infection, upwards of 30 epitopes can be recognized, totaling as many as 15,000 influenza virus-specific CD4 T cells per lymph node, leading to a broad repertoire and high frequency of influenza virus-specific helper cells. Previous studies suggest that increased frequencies of antigen-specific CD4 T cells could facilitate a more robust extrafollicular antibody response but without an accompanying boost in germinal center phenotype B cells (48), contrary to what our results have demonstrated. This apparent difference may be related to the phenotype of the precursor T cells at the time of infection as this previous work utilized T cell receptor (TCR)-transgenic naive and adoptively transferred cells, while the present study highlighted the contributions made by peptide-primed memory CD4 T cells drawn from the endogenous T cell repertoire.
Memory T cells are known to be more sensitive to antigen stimulation due to the presence of clustered T cell receptors on the cell surface and to the reduced dependence on costimulation (49–52). Accordingly, the memory CD4 T cells evident in the MLN on day 0 of infection proliferated rapidly to high frequencies by day 7, whereas CD4 T cells elicited in the primary response to infection were barely detectable in the draining lymph node. The provision of more rapid help for B cells could simply be the result of a larger pool of antigen-specific cells than with the primary response rather than any advantage related to differentiation stage. An increased frequency of antigen-specific CD4 T cells directly influences the probability of encountering an antigen-bearing, antigen-presenting cell early in infection. There was evidence of a local antigen-specific population in the MLN and of recruitment of NP-specific CD4 T cells from peripheral nodes such as the PLN upon infection as we observed an apparent decrease in the frequency of these cells in the PLN early after infection compared to the frequency in NP-primed mice not challenged with virus. Furthermore, the CXCR5+ phenotype of circulating memory CD4 T cells has been closely associated with central memory and may in fact be an indication of Tfh cell memory (25, 53, 54). Considering the early expansion of Tfh cells observed following immunization and infection, it would appear that peptide priming is capable of generating central memory that can later be recalled and stimulated for further differentiation into Tfh cells. The potentially elevated expression of CXCR5 by central memory T cells may make this population uniquely poised for future B cell help upon infection. It should be noted, though, that the relationship of Bcl6 and CXCR5 expression and their mutual induction for determination of a Tfh program are yet unclear (54, 55). Preexpression of this chemokine receptor in memory cells may bypass the early differentiation stages necessary for naive T cells to eventually gain follicular helper function and allow for more rapid migration to the interfollicular zone where initial B cell interactions take place (56).
With advances in epitope discovery, there is an increased interest in peptide-based vaccines and multieptiope synthetic vaccines (57, 58), and a peptide-priming regimen presents a unique opportunity to arm the memory pool of CD4 T cells without influencing the specificity of the B cell immune repertoire. In the case of influenza virus vaccination, repeated exposures to protein antigens from vaccine and infection may increase the levels of circulating antibodies that can limit the response to a novel exposure by eliminating the antigen source before a robust germinal center B cell response can be generated (59, 60). It is thought that preexisting antibody leads to more rapid clearance of antigen, thus blunting the adaptive immune response (61). Peptide priming circumvents this problem by providing monovalent, unstructured ligands, leading to poor B cell-stimulatory conditions. This allows for the full expansion of the CD4 T cell immune repertoire, which can greatly facilitate future antibody responses. The potential for original antigenic sin (62, 63) can also be avoided through use of a peptide-priming immunization strategy. By generating no B cell memory, the breadth of future responses will not be restricted by a preexisting B cell repertoire. This type of advantage would be directly applicable to situations where responses to vaccination are already suboptimal, as in the elderly or immunocompromised (64–68). Through provision of a selective boost to the CD4 T cell repertoire, intrinsic antibody interference and shifts in the specificity of the memory B cell repertoire can be bypassed.
The mechanisms that underlie the striking selectivity in help for antibody responses is unknown but most likely relates to the form of antigen available to viral antigen-specific B cells after infection in the draining lymph node, where the B cell response is initiated. After natural infection, many cells in the lung become infected (69), and though dendritic cells (DC) can be infected and synthesize virus proteins and can migrate to the draining lymph node (34, 35, 70, 71), there is limited evidence that these DC are able to release intact virions in the lymph node (72, 73). Thus, the form of virus protein available for B cell receptor-mediated uptake in the draining lymph node may consist primarily of membrane fragments and proteins released from dying infected cells that either passively drain to the LN or are actively carried by migratory APC. In contrast to uptake of intact virions by HA-specific B cells that presumably would lead to endosomal release of all structural proteins and subsequent MHC class II-restricted presentation to CD4 T cells, if viral proteins and membrane fragments are the main form of influenza virus antigens available in the lymph node, HA-specific B cells will internalize and display only peptides from the influenza viral proteins that are cointernalized with HA. They thus will recruit help only from the CD4 T cells specific for these immunoglobulin-internalized antigens. If HA proteins released from virally infected cells are aggregated with other membrane proteins such as neuraminidase (NA) and the matrix proteins as part of large lipid rafts (74), then it is possible that HA-specific B cells could present peptides from these proteins and thus recruit help from CD4 T cells of these specificities. In contrast to HA, the NP protein is likely released from dead and dying cells in the lung after infection and either drains directly to the lymph node or is carried by migratory APC. In either case, the soluble NP protein will be taken up by NP-specific B cells independently of membrane proteins such as HA. Accordingly, NP- and HA-specific B cells will display only peptides derived from the proteins that they are specific for or with which they are cointernalized and can only recruit help from the CD4 T cells specific for these peptides.
There have been previous studies closely examining the role of linked specificities in the provision of help for antibody responses to viral infection. Such a linkage was found in the context of vaccinia virus, with several potential mechanisms thoroughly discussed by the authors (75). From this study, it was surmised that the size of the virus particle (360 nm) in relation to the size of B cell endocytic vesicles (50 to 150 nm) could be a primary determinant of specificity linkage. Considering the size of influenza virus virions (80 nm) (75) and evidence for deposition and availability of intact viral particles in the lymph node (76), the potential for intermolecular help was feasible for influenza virus, through either B cell receptor-mediated uptake and processing of whole virions or an intrastructural mechanism as described above for proteins associated in a membrane system.
Early work explored the possibility of intermolecular help for influenza virus. These studies were limited by the use of single T cell clones transferred into nude mice in conjunction with infection (77, 78) or the use of an intraperitoneal model of immunization, with virus particles chemically denuded of HA protein followed by boost with HA-intact virions (79). It was evident from this and other work that, in a simplified system, a single specific clonal T cell population could provide help for an unlinked protein antibody response to infection or that repeated immunization with virus particles made intermolecular help possible. The experimental system we have outlined embodies the physiological context arising from natural influenza virus infection within which CD4 T cells and B cells interact, allowing for elucidation of the helper role that antigen-specific memory CD4 T cells are likely playing during infection.
We have demonstrated that CD4 T cell help is limiting for the primary antibody response to influenza virus infection and that it may indeed be quite selective, depending on B cell specificity. Whether the provision of help by peptide-primed CD4 memory leads to more rapid clearance and protection is unclear. There are many effector functions of CD4 T cells that facilitate protection from influenza virus infection, including potentiation of the early innate response and direct cytotoxicity (6, 24, 38, 80, 81; reviewed in references 82–85). We expect that the latter two functions of CD4 T cells may be carried out by cells reactive to peptides derived from many viral proteins, including HA, NA, NP, and M1, because these proteins are synthesized or available in many cells within the lung. Understanding whether CD4 T cells of different antigen specificities vary in their provision of effector or helper functions will be of great interest in future studies. The potential value of enhancing CD4 T cells reactive with key shared influenza virus-derived epitopes from multiple proteins is particularly important with the ongoing threats of pandemic influenza involving completely novel avian strains. For these and other newly emerging influenza viruses, there will be little or no preexisting protective antibody in most human populations and limited shared T cell epitopes among the variant HA and NA proteins. Under these conditions, an expanded influenza virus-specific CD4 T cell memory compartment, generated by targeted vaccination, may allow subjects to both more rapidly mobilize B cells for protective antibody upon infection and potentiate more vigorous responses to novel influenza virus vaccines, which have been shown to be poorly immunogenic (86–89).
ACKNOWLEDGMENTS
This work is supported by research grants from the NIAID Centers of Excellence for Influenza Research and Surveillance, the New York Influenza Center of Excellence, and the National Institutes of Health, including grants HHSN27220201200005C, HHSN266200700008C, R01AI51542, and 5T32AI007285.
We have no conflicting financial interests.
Footnotes
Published ahead of print 24 October 2013
REFERENCES
- 1.Nichol KL, Treanor JJ. 2006. Vaccines for seasonal and pandemic influenza. J. Infect. Dis. 194(Suppl 20):S111–S118. 10.1086/507544 [DOI] [PubMed] [Google Scholar]
- 2.Osterholm MT, Kelley NS, Sommer A, Belongia EA. 2012. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 12:36–44. 10.1016/S1473-3099(11)70295-X [DOI] [PubMed] [Google Scholar]
- 3.Jackson LA, Gaglani MJ, Keyserling HL, Balser J, Bouveret N, Fries L, Treanor JJ. 2010. Safety, efficacy, and immunogenicity of an inactivated influenza vaccine in healthy adults: a randomized, placebo-controlled trial over two influenza seasons. BMC Infect. Dis. 10:71. 10.1186/1471-2334-10-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Treanor JJ, Talbot HK, Ohmit SE, Coleman LA, Thompson MG, Cheng PY, Petrie JG, Lofthus G, Meece JK, Williams JV, Berman L, Breese Hall C, Monto AS, Griffin MR, Belongia E, Shay DK. 2012. Effectiveness of seasonal influenza vaccines in the United States during a season with circulation of all three vaccine strains. Clin. Infect. Dis. 55:951–959. 10.1093/cid/cis574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinfurter JT, Brunner K, Capuano SV, III, Li C, Broman KW, Kawaoka Y, Friedrich TC. 2011. Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLoS Pathog. 7:e1002381. 10.1371/journal.ppat.1002381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL. 2010. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J. Virol. 84:9217–9226. 10.1128/JVI.01069-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherle PA, Palladino G, Gerhard W. 1992. Mice can recover from pulmonary influenza virus infection in the absence of class I-restricted cytotoxic T cells. J. Immunol. 148:212–217 [PubMed] [Google Scholar]
- 8.Mozdzanowska K, Furchner M, Maiese K, Gerhard W. 1997. CD4+ T cells are ineffective in clearing a pulmonary infection with influenza type A virus in the absence of B cells. Virology 239:217–225. 10.1006/viro.1997.8882 [DOI] [PubMed] [Google Scholar]
- 9.Topham DJ, Doherty PC. 1998. Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J. Virol. 72:882–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, Randall TD. 2005. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J. Immunol. 175:5827–5838 http://www.jimmunol.org/content/175/9/5827 [DOI] [PubMed] [Google Scholar]
- 11.Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. 2002. Compromised influenza virus-specific CD8+-T-cell memory in CD4+-T-cell-deficient mice. J. Virol. 76:12388–12393. 10.1128/JVI.76.23.12388-12393.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riberdy JM, Christensen JP, Branum K, Doherty PC. 2000. Diminished primary and secondary influenza virus-specific CD8+ T-cell responses in CD4-depleted Ig−/− mice. J. Virol. 74:9762–9765. 10.1128/JVI.74.20.9762-9765.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eichelberger MC, Wang ML, Allan W, Webster RG, Doherty PC. 1991. Influenza virus RNA in the lung and lymphoid tissue of immunologically intact and CD4-depleted mice. J. Gen. Virol. 72:1695–1698. 10.1099/0022-1317-72-7-1695 [DOI] [PubMed] [Google Scholar]
- 14.Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, Forster R. 2000. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192:1545–1552. 10.1084/jem.192.11.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, Yu D, Fagarasan S, Tarlinton DM, Cunningham AF, Vinuesa CG. 2011. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 208:1377–1388. 10.1084/jem.20102065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. 2010. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 207:353–363. 10.1084/jem.20091738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, Nutt SL, Tarlinton DM. 2010. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 207:365–378. 10.1084/jem.20091777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sangster MY, Riberdy JM, Gonzalez M, Topham DJ, Baumgarth N, Doherty PC. 2003. An early CD4+ T cell-dependent immunoglobulin A response to influenza infection in the absence of key cognate T-B interactions. J. Exp. Med. 198:1011–1021. 10.1084/jem.20021745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. 2000. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192:1553–1562. 10.1084/jem.192.11.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, Cyster JG. 2002. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 416:94–99. 10.1038/416094a [DOI] [PubMed] [Google Scholar]
- 21.Nayak JL, Fitzgerald TF, Richards KA, Yang H, Treanor JJ, Sant AJ. 2013. CD4+ T-cell expansion predicts neutralizing antibody responses to monovalent, inactivated 2009 pandemic influenza A(H1N1) virus subtype H1N1 vaccine. J. Infect. Dis. 207:297–305. 10.1093/infdis/jis684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, Flano E, Mejias A, Albrecht RA, Blankenship D, Xu H, Pascual V, Banchereau J, Garcia-Sastre A, Palucka AK, Ramilo O, Ueno H. 2013. Induction of ICOS+ CXCR3+ CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med. 5:176ra132. 10.1126/scitranslmed.3005191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacLeod MK, David A, McKee AS, Crawford F, Kappler JW, Marrack P. 2011. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J. Immunol. 186:2889–2896. 10.4049/jimmunol.1002955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strutt TM, McKinstry KK, Kuang Y, Bradley LM, Swain SL. 2012. Memory CD4+ T-cell-mediated protection depends on secondary effectors that are distinct from and superior to primary effectors. Proc. Natl. Acad. Sci. U. S. A. 109:E2551–E2560. 10.1073/pnas.1205894109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, Sallusto F, Tangye SG, Mackay CR. 2011. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J. Immunol. 186:5556–5568. 10.4049/jimmunol.1002828 [DOI] [PubMed] [Google Scholar]
- 26.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh S, Sabzghabaei N, Lavecchio EM, Punaro M, Pascual V, Banchereau J, Ueno H. 2011. Human blood CXCR5+ CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34:108–121. 10.1016/j.immuni.2010.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richards KA, Chaves FA, Krafcik FR, Topham DJ, Lazarski CA, Sant AJ. 2007. Direct ex vivo analyses of HLA-DR1 transgenic mice reveal an exceptionally broad pattern of immunodominance in the primary HLA-DR1-restricted CD4 T-cell response to influenza virus hemagglutinin. J. Virol. 81:7608–7619. 10.1128/JVI.02834-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alam S, Sant AJ. 2011. Infection with seasonal influenza virus elicits CD4 T cells specific for genetically conserved epitopes that can be rapidly mobilized for protective immunity to pandemic H1N1 influenza virus. J. Virol. 85:13310–13321. 10.1128/JVI.05728-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards KA, Chaves FA, Sant AJ. 2009. Infection of HLA-DR1 transgenic mice with a human isolate of influenza a virus (H1N1) primes a diverse CD4 T-cell repertoire that includes CD4 T cells with heterosubtypic cross-reactivity to avian (H5N1) influenza virus. J. Virol. 83:6566–6577. 10.1128/JVI.00302-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nayak JL, Richards KA, Chaves FA, Sant AJ. 2010. Analyses of the specificity of CD4 T cells during the primary immune response to influenza virus reveals dramatic MHC-linked asymmetries in reactivity to individual viral proteins. Viral Immunol. 23:169–180. 10.1089/vim.2009.0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards KA, Chaves FA, Sant AJ. 2011. The memory phase of the CD4 T-cell response to influenza virus infection maintains its diverse antigen specificity. Immunology 133:246–256. 10.1111/j.1365-2567.2011.03435.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards KA, Chaves FA, Alam S, Sant AJ. 2012. Trivalent inactivated influenza vaccines induce broad immunological reactivity to both internal virion components and influenza surface proteins. Vaccine 31:219–225. 10.1016/j.vaccine.2012.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaves FA, Lee AH, Nayak JL, Richards KA, Sant AJ. 2012. The utility and limitations of current Web-available algorithms to predict peptides recognized by CD4 T cells in response to pathogen infection. J. Immunol. 188:4235–4248. 10.4049/jimmunol.1103640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ho AW, Prabhu N, Betts RJ, Ge MQ, Dai X, Hutchinson PE, Lew FC, Wong KL, Hanson BJ, Macary PA, Kemeny DM. 2011. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J. Immunol. 187:6011–6021. 10.4049/jimmunol.1100987 [DOI] [PubMed] [Google Scholar]
- 35.Moltedo B, Li W, Yount JS, Moran TM. 2011. Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog. 7:e1002345. 10.1371/journal.ppat.1002345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brimnes MK, Bonifaz L, Steinman RM, Moran TM. 2003. Influenza virus-induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic protein. J. Exp. Med. 198:133–144. 10.1084/jem.20030266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN. 2008. Clearance of influenza virus from the lung depends on migratory langerin+ CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 205:1621–1634. 10.1084/jem.20071365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DM, Dilzer AM, Meents DL, Swain SL. 2006. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J. Immunol. 177:2888–2898 http://www.jimmunol.org/content/177/5/2888.long [DOI] [PubMed] [Google Scholar]
- 39.Boyden AW, Legge KL, Waldschmidt TJ. 2012. Pulmonary infection with influenza A virus induces site-specific germinal center and T follicular helper cell responses. PLoS One 7:e40733. 10.1371/journal.pone.0040733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. 2010. T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol. Rev. 237:72–89. 10.1111/j.1600-065X.2010.00937.x [DOI] [PubMed] [Google Scholar]
- 41.Victora GD, Nussenzweig MC. 2012. Germinal centers. Annu. Rev. Immunol. 30:429–457. 10.1146/annurev-immunol-020711-075032 [DOI] [PubMed] [Google Scholar]
- 42.Oliver AM, Martin F, Kearney JF. 1997. Mouse CD38 is down-regulated on germinal center B cells and mature plasma cells. J. Immunol. 158:1108–1115 [PubMed] [Google Scholar]
- 43.Cervenak L, Magyar A, Boja R, Laszlo G. 2001. Differential expression of GL7 activation antigen on bone marrow B cell subpopulations and peripheral B cells. Immunol. Lett. 78:89–96. 10.1016/S0165-2478(01)00239-5 [DOI] [PubMed] [Google Scholar]
- 44.Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, Nussenzweig MC. 2011. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 208:1243–1252. 10.1084/jem.20102477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. 2011. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35:583–595. 10.1016/j.immuni.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okada T, Cyster JG. 2006. B cell migration and interactions in the early phase of antibody responses. Curr. Opin. Immunol. 18:278–285. 10.1016/j.coi.2006.02.005 [DOI] [PubMed] [Google Scholar]
- 47.Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, O'Garra A, Cahalan MD, Cyster JG. 2005. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 3:e150. 10.1371/journal.pbio.0030150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothaeusler K, Baumgarth N. 2010. B-cell fate decisions following influenza virus infection. Eur. J. Immunol. 40:366–377. 10.1002/eji.200939798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai W, Yu M, Huang MN, Okoye F, Keegan AD, Farber DL. 2011. Transcriptional control of rapid recall by memory CD4 T cells. J. Immunol. 187:133–140. 10.4049/jimmunol.1002742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar R, Ferez M, Swamy M, Arechaga I, Rejas MT, Valpuesta JM, Schamel WW, Alarcon B, van Santen HM. 2011. Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity 35:375–387. 10.1016/j.immuni.2011.08.010 [DOI] [PubMed] [Google Scholar]
- 51.London CA, Lodge MP, Abbas AK. 2000. Functional responses and costimulator dependence of memory CD4+ T cells. J. Immunol. 164:265–272 http://www.jimmunol.org/content/164/1/265.long [DOI] [PubMed] [Google Scholar]
- 52.Rogers PR, Dubey C, Swain SL. 2000. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J. Immunol. 164:2338–2346 http://www.jimmunol.org/content/164/5/2338.full [DOI] [PubMed] [Google Scholar]
- 53.Weber JP, Fuhrmann F, Hutloff A. 2012. T-follicular helper cells survive as long-term memory cells. Eur. J. Immunol. 42:1981–1988. 10.1002/eji.201242540 [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, Martin-Orozco N, Wang Y, Chang SH, Esplugues E, Flavell RA, Tian Q, Dong C. 2012. Bcl6 expression specifies the T follicular helper cell program in vivo. J. Exp. Med. 209:1841–1852, S1841–S1824. 10.1084/jem.20120219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34:932–946. 10.1016/j.immuni.2011.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM. 2011. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34:947–960. 10.1016/j.immuni.2011.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szurgot I, Szolajska E, Laurin D, Lambrecht B, Chaperot L, Schoehn G, Chroboczek J. 2013. Self-adjuvanting influenza candidate vaccine presenting epitopes for cell-mediated immunity on a proteinaceous multivalent nanoplatform. Vaccine 31:4338–4346. 10.1016/j.vaccine.2013.07.021 [DOI] [PubMed] [Google Scholar]
- 58.Cruz LJ, Rueda F, Tacken P, Albericio F, Torensma R, Figdor CG. 2012. Enhancing immunogenicity and cross-reactivity of HIV-1 antigens by in vivo targeting to dendritic cells. Nanomedicine 7:1591–1610. 10.2217/nnm.12.131 [DOI] [PubMed] [Google Scholar]
- 59.Gross PA, Sperber SJ, Donabedian A, Dran S, Morchel G, Cataruozolo P, Munk G. 1999. Paradoxical response to a novel influenza virus vaccine strain: the effect of prior immunization. Vaccine 17:2284–2289. 10.1016/S0264-410X(98)00478-2 [DOI] [PubMed] [Google Scholar]
- 60.Sasaki S, He XS, Holmes TH, Dekker CL, Kemble GW, Arvin AM, Greenberg HB. 2008. Influence of prior influenza vaccination on antibody and B-cell responses. PLoS One 3:e2975. 10.1371/journal.pone.0002975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dormitzer PR, Galli G, Castellino F, Golding H, Khurana S, Del Giudice G, Rappuoli R. 2011. Influenza vaccine immunology. Immunol. Rev. 239:167–177. 10.1111/j.1600-065X.2010.00974.x [DOI] [PubMed] [Google Scholar]
- 62.Webster RG. 1966. Original antigenic sin in ferrets: the response to sequential infections with influenza viruses. J. Immunol. 97:177–183 [PubMed] [Google Scholar]
- 63.Kim JH, Skountzou I, Compans R, Jacob J. 2009. Original antigenic sin responses to influenza viruses. J. Immunol. 183:3294–3301. 10.4049/jimmunol.0900398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beck CR, McKenzie BC, Hashim AB, Harris RC, Nguyen-Van-Tam JS. 2012. Influenza vaccination for immunocompromised patients: systematic review and meta-analysis by etiology. J. Infect. Dis. 206:1250–1259. 10.1093/infdis/jis487 [DOI] [PubMed] [Google Scholar]
- 65.Ljungman P. 2012. Vaccination of immunocompromised patients. Clin. Microbiol. Infect. 18(Suppl 5):93–99. 10.1111/j.1469-0691.2012.03971.x [DOI] [PubMed] [Google Scholar]
- 66.de Lavallade H, Garland P, Sekine T, Hoschler K, Marin D, Stringaris K, Loucaides E, Howe K, Szydlo R, Kanfer E, Macdonald D, Kelleher P, Cooper N, Khoder A, Gabriel IH, Milojkovic D, Pavlu J, Goldman JM, Apperley JF, Rezvani K. 2011. Repeated vaccination is required to optimize seroprotection against H1N1 in the immunocompromised host. Haematologicaae 96:307–314. 10.3324/haematol.2010.032664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Opravil M, Fierz W, Matter L, Blaser J, Luthy R. 1991. Poor antibody response after tetanus and pneumococcal vaccination in immunocompromised, HIV-infected patients. Clin. Exp. Immunol. 84:185–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang I, Hong MS, Nolasco H, Park SH, Dan JM, Choi JY, Craft J. 2004. Age-associated change in the frequency of memory CD4+ T cells impairs long term CD4+ T cell responses to influenza vaccine. J. Immunol. 173:673–681 http://www.jimmunol.org/content/173/1/673.long [DOI] [PubMed] [Google Scholar]
- 69.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. U. S. A. 107:11531–11536. 10.1073/pnas.0914994107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.VanoOsten Anderson R, McGill J, Legge KL. 2010. Quantification of the frequency and multiplicity of infection of respiratory- and lymph node-resident dendritic cells during influenza virus infection. PLoS One 5:e12902. 10.1371/journal.pone.0012902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim TS, Braciale TJ. 2009. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One 4:e4204. 10.1371/journal.pone.0004204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bender A, Albert M, Reddy A, Feldman M, Sauter B, Kaplan G, Hellman W, Bhardwaj N. 1998. The distinctive features of influenza virus infection of dendritic cells. Immunobiology 198:552–567. 10.1016/S0171-2985(98)80078-8 [DOI] [PubMed] [Google Scholar]
- 73.Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, Murphy KM, Garcia-Sastre A, Merad M. 2012. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Invest. 122:4037–4047. 10.1172/JCI60659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veit M, Thaa B. 2011. Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011:370606. 10.1155/2011/370606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, Peters B, Rafii-El-Idrissi Benhnia M, Hoffmann J, Su HP, Singh K, Garboczi DN, Head S, Grey H, Felgner PL, Crotty S. 2008. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 28:847–858. 10.1016/j.immuni.2008.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, Pitcher LA, Degn SE, Kim YA, Cloninger MJ, Martinez-Pomares L, Gordon S, Turley SJ, Carroll MC. 2010. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 11:427–434. 10.1038/ni.1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scherle PA, Gerhard W. 1986. Functional analysis of influenza-specific helper T cell clones in vivo. T cells specific for internal viral proteins provide cognate help for B cell responses to hemagglutinin. J. Exp. Med. 164:1114–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scherle PA, Gerhard W. 1988. Differential ability of B cells specific for external vs. internal influenza virus proteins to respond to help from influenza virus-specific T-cell clones in vivo. Proc. Natl. Acad. Sci. U. S. A. 85:4446–4450. 10.1073/pnas.85.12.4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Russell SM, Liew FY. 1979. T cells primed by influenza virion internal components can cooperate in the antibody response to haemagglutinin. Nature 280:147–148. 10.1038/280147a0 [DOI] [PubMed] [Google Scholar]
- 80.Brown DM, Lee S, Garcia-Hernandez MDLL, Swain SL. 2012. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J. Virol. 86:6792–6803. 10.1128/JVI.07172-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, Swain SL. 2012. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Invest. 122:2847–2856. 10.1172/JCI63689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun J, Braciale TJ. 2013. Role of T cell immunity in recovery from influenza virus infection. Curr. Opin. Virol. 3:425–429. 10.1016/j.coviro.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boonnak K, Subbarao K. 2012. Memory CD4+ T cells: beyond “helper” functions. J. Clin. Invest. 122:2768–2770. 10.1172/JCI65208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kai McKinstry K, Dutton RW, Swain SL, Strutt TM. 2013. Memory CD4 T cell-mediated immunity against influenza A virus: more than a little helpful. Arch. Immunol. Ther. Exp. (Warsz.) 61:341–353. 10.1007/s00005-013-0236-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sant AJ, McMichael A. 2012. Revealing the role of CD4+ T cells in viral immunity. J. Exp. Med. 209:1391–1395. 10.1084/jem.20121517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manzoli L, Salanti G, De Vito C, Boccia A, Ioannidis JP, Villari P. 2009. Immunogenicity and adverse events of avian influenza A H5N1 vaccine in healthy adults: multiple-treatments meta-analysis. Lancet Infect. Dis. 9:482–492. 10.1016/S1473-3099(09)70153-7 [DOI] [PubMed] [Google Scholar]
- 87.Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. 2006. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N. Engl. J. Med. 354:1343–1351. 10.1056/NEJMoa055778 [DOI] [PubMed] [Google Scholar]
- 88.Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O'Brien D, Wolff M, Rabinovich G, Blackwelder W, Katz JM. 2001. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine 19:1732–1737. 10.1016/S0264-410X(00)00395-9 [DOI] [PubMed] [Google Scholar]
- 89.Nicholson KG, Colegate AE, Podda A, Stephenson I, Wood J, Ypma E, Zambon MC. 2001. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 357:1937–1943. 10.1016/S0140-6736(00)05066-2 [DOI] [PubMed] [Google Scholar]