

Abstract
Cowpox virus (CPXV) belongs to the genus Orthopoxvirus in the Poxviridae family. It infects a broad range of vertebrates and can cause zoonotic infections. CPXV has the largest genome among the orthopoxviruses and is therefore considered to have the most complete set of genes of all members of the genus. Since CPXV has also become a model for studying poxvirus genetics and pathogenesis, we created and characterized a complete set of single gene knockout bacterial artificial chromosome (BAC) clones of the CPXV strain Brighton Red. These mutants allow a systematic assessment of the contribution of single CPXV genes to the outcome of virus infection and replication, as well as to the virus host range. A full-length BAC clone of CPXV strain Brighton Red (pBRF) harboring the gene expressing the enhanced green fluorescent protein under the control of a viral late promoter was modified by introducing the mrfp1 gene encoding the monomeric red fluorescent protein driven by a synthetic early vaccinia virus promoter. Based on the modified BAC (pBRFseR), a library of targeted knockout mutants for each single viral open reading frame (ORF) was generated. Reconstitution of infectious virus was successful for 109 of the 183 mutant BAC clones, indicating that the deleted genes are not essential for virus replication. In contrast, 74 ORFs were identified as essential because no virus progeny was obtained upon transfection of the mutant BAC clones and in the presence of a helper virus. More than 70% of all late CPXV genes belonged to this latter group of essential genes.
INTRODUCTION
Cowpox virus (CPXV) belongs to the family Poxviridae and the genus Orthopoxvirus. While the prototype virus of the genus, the human pathogen variola virus, was declared eradicated in 1980, other members of the genus, including CPXV and monkeypox virus, still circulate within animal populations and cause zoonotic infections in Western Eurasia and Africa, respectively (1). CPXV is closely related to vaccinia virus (VACV) that was used as a heterotypic live virus vaccine against smallpox. Studies investigating the function of orthopoxvirus (OPV) genes have mainly been conducted using VACV (2–5), although most VACV strains are attenuated in many species. In addition, VACV lacks functional copies of several genes present in other OPVs due to loss or truncation of the respective ORFs compared to other species of the genus (6). Consequently, the function of a number of known or predicted OPV genes and their products remains unknown, and VACV seems not to be the ideal model for studying OPV gene function.
In contrast, CPXV has a number of advantages that make it a suitable model virus for studying OPV biology. With a size of ∼220 kbp, it has the largest genome of all OPVs, ∼30 kbp larger than that of VACV. In addition, CPXV possesses the most complete genome of all known OPVs in terms of number of genes (7, 8) and is considered to have the potential to evolve into new virus species with pathogenic potential comparable to that of more virulent viruses (9). CPXV can infect a broad range of domestic and wild animals, including bovines, elephants, primates, cats, and rodents, and can also cause zoonotic disease in humans (1). It has become a popular model for the study of poxvirus biology and pathogenesis, but comprehensive functional analysis of poxvirus genes requires targeted genome manipulations. Classical methods to modify poxvirus genomes relying on homologous recombination in vertebrate cells are laborious and time-consuming (10). Progeny viruses containing the desired modified genome are rare, because of low recombination efficiencies, which make necessary multiple rounds of plaque purification and sometimes even chemical selection or comprehensive screening protocols (10). Knockout mutants of genes important for virus replication might be difficult or almost impossible to purify because of their greatly impaired fitness. Moreover, mutant isolation might need multiple rounds of selection, even when complementing cell lines are used (11).
Bacterial artificial chromosome (BAC) technology is a powerful tool used to propagate and modify large genomic DNA fragments and has been used to clone an increasing number of different virus genomes, among them various poxviruses (12–16). Even after sequential introduction of six major deletions into a chorioallantois vaccinia virus Ankara BAC clone, no fortuitous mutations occurred (16). This underlines the suitability of the BAC technology for maintenance and modification of poxvirus genomes. Mutagenesis methods such as two-step markerless Red recombination (also referred to as “en passant” mutagenesis) allow efficient modification of BAC DNA in Escherichia coli ranging from single base pair mutation to the deletion or introduction of large pieces of DNA (17, 18). Red recombination only requires short homologous sequences (>28 bp) to insert foreign DNA into a target site, which makes the design and execution of mutagenesis relatively simple. For comparison, recombination in infected vertebrate cells requires homologous sequences of at least 200 bp in order to achive relatively high efficiencies (19).
In the present study, we constructed a knockout library of the CPXV strain Brighton Red (BR) genome, which was based on a full-length virus BAC clone termed pBRFseR. pBRFseR was constructed by introducing a red fluorescent marker (mRFP) into pBRF, a previously described full-length BAC clone (15). Thus, pBRFseR contains a mRFP driven by an early promoter and an enhanced green fluorescent protein (eGFP) under the control of a viral late promoter, both of which allowed us to monitor early and late gene expression of CPXV. We used Red recombination in E. coli to insert deletion cassettes containing stop codons, as well as a kanamycin resistance gene, which resulted in single-gene knockout mutants for each unique CPXV-BR open reading frame (ORF). After reconstitution of each of the mutant viruses, we identified different phenotypes and were able to categorize CPXV genes as essential for the transition to late gene expression, essential for the production of virus progeny despite the production of late viral proteins, or nonessential for viral replication in cell culture.
MATERIALS AND METHODS
Cell lines and viruses.
All cell lines were cultivated at 37°C under a 5% CO2 atmosphere. African green monkey cells Vero 76 (Collection of Cell Lines in Veterinary Medicine, Friedrich-Loeffler-Institut, Greifswald-Insel Riems, Germany) were maintained in Eagle's minimal essential medium with Earle's salts, 2.2 g of NaHCO3/liter, and stable l-glutamine (MEM; Biochrom, Berlin, Germany) supplemented with 5% fetal bovine serum (FBS; Biochrom), 65 μg of penicillin G/ml, and 100 μg of streptomycin (AppliChem GmbH, Darmstadt, Germany)/ml. Primary chicken embryo cells (CEC) were prepared from 11-day-old embryonated specific-pathogen-free eggs (VALO BioMedia GmbH, Osterholz-Scharmbeck, Germany) according to standard procedures and cultured in MEM containing 10% FBS (Biochrom) and antibiotics as described above. Recombinant and mutant CPXV were propagated on Vero cells, whereas fowlpox virus (FWPV; Nobilis-PD, strain WP; Intervet, Boxmeer, Netherlands [kindly provided by D. Lüschow, Freie Universität, Berlin, Germany]) was amplified on CEC.
Reconstitution of infectious virus from BAC DNA.
For virus reconstitution, 1 × 105 or 7 × 105 Vero cells seeded in one well of a 24- or 6-well plate, respectively, were transfected with ∼2 μg of purified BAC DNA using 1 to 4 μl of FuGENE HD transfection reagents (Promega, Mannheim, Germany) according to the manufacturer's instructions. Transfected cultures were infected with 20 to 500 PFU of FWPV at 2 h after transfection. Virus reconstitution was monitored using an Axiovert 100 fluorescence microscope (Carl Zeiss, Jena, Germany) by screening for fluorescent early and late gene expression markers. Images were taken from 48 to 240 h after transfection using an AxioCam MRm charge-coupled device camera (Zeiss). Image processing was performed with the AxioVision 4.8.2 software package (Zeiss). Elimination of residual helper virus was achieved by passaging the reconstituted viruses three times on Vero cells, which are nonpermissive for FWPV. Between individual passages, infected cells were lysed by freeze-thawing the cultures twice at −70°C. Confluent monolayers in 24- or 6-well plates were infected with 1 to 10 μl of freeze-thaw cell lysate from the previous passage. Monitoring of virus replication and imaging of single virus plaques was performed as described above. Mutant BAC clones, which could not be reconstituted on the first attempt, were used for repeated transfections on Vero cells with FWPV and on CEC using Shope fibroma virus (SFV; Merial, Lyon, France) as a helper virus.
Generation of plasmid pACAA.
For the construction of plasmid pACAA, the vector pACYC177 (New England BioLabs, Frankfurt, Germany) was amplified via inverse PCR using the primers pAAfw and pAArv (see Table S1 in the supplemental material [http://www.vetmed.fu-berlin.de/en/einrichtungen/institute/we05/cowpox]). This resulted in the deletion of 205 bp from the plasmid and insertion of a bacterial promoter and the kanamycin resistance gene (aphAI), together with a 9-bp sequence (GCCGCGTGA) that codes for two alanines and a stop codon, while also containing a PaeI restriction site. After digestion of the PCR product with DpnI to eliminate template vector, the DNA was cleaved with PaeI and religated resulting in the vector pACAA. This plasmid was used as a template for the generation of the knockout library.
Generation of dual marker CPXV-BR BAC clone pBRFseR.
To generate transfer vector pEP-MVA-dVI-PK1L-mRFP (18), the gene encoding monomeric red fluorescence protein 1 (mrfp1) (20) was excised from plasmid pEP-ExpRFP1-in by using BamHI and SacI. The linear 763-bp fragment was inserted into the respective restriction sites in pEP-MVA-dVI-PK1L (Dai LianPan and Ingo Drexler, unpublished data).
Insertion of the PK1L-mRFP1 expression cassette into pBRF (15) was performed by two-step en passant Red recombination as described previously (17, 18) (Fig. 1). Briefly, the PK1L-mRFP1-aphAI fragment was amplified from pEP-MVA-dVI-PK1L-mRFP by using the primers mRFPfw and mRFPrv (see Table S1 in the supplemental material) and electroporated into E. coli strain GS1783 (17) harboring the pBRF BAC clone. Insertion of the PK1L-mRFP1 expression cassette into pBRF resulted in pBRFeR. In a second en passant mutagenesis procedure, the K1L promoter of the PK1L-mRFP1 cassette in pBRFeR was replaced by the previously published synthetic early promoter (Psyn7.5) (21). For this purpose, the aphAI-I-SceI fragment from plasmid pEP-kanS was amplified with the primers syn7.5fw and syn7.5rv (see Table S1 in the supplemental material) and used for en passant recombination exactly as described earlier (18). All BAC clones described above were verified by restriction fragment length polymorphism (RFLP) analysis and sequencing of the insertion site (data not shown).
FIG 1.

Insertion of a fluorescent marker for viral early gene expression into the mini-F region of pBRF. The PK1L-mRFP1-aphAI fragment was amplified from transfer vector pEP-MVA-dVI-PK1L-mRFP using the primers mRFPfw and mRFPrv. The cassette was integrated downstream of the existing late gene expression marker (eGFP) by en passant mutagenesis, resulting in pBRFeR. In a second en passant mutagenesis, the K1L promoter was exchanged for a previously published synthetic early promoter (Psyn7.5), resulting in pBRFseR. Dashed lines indicate recombination events between homologous sequences.
BAC mutagenesis.
Knockout BAC clones were generated by inserting PCR-derived marker cassettes into selected loci by one-step Red recombination (17) (Fig. 2). All knockout BAC clones are listed in Table 1. Knockout BAC clones were named according to the respective ORF deleted. For example, the CPXV010 deletion mutant was named pBRFseR_d10. PCR primers were designed to amplify the aphAI cassette from recombinant plasmid pACAA. Besides the marker cassette, PCR fragments contained at each end 40 bp of sequences that were homologous to the target locus in the CPXV BR sequence. The resulting PCR products were inserted into pBRFseR by conventional Red recombination, ultimately resulting in interruption of all predicted ORFs in CPXV (Table 1 and see Table S2 in the supplemental material [http://www.vetmed.fu-berlin.de/en/einrichtungen/institute/we05/cowpox]). Recombination was performed in E. coli strain GS1783 by electroporation of PCR products into GS1783 harboring pBRFseR. Bacteria were spread on LB agar plates containing 35 μg of chloramphenicol/ml and 35 μg of kanamycin/ml (Roth, Karlsruhe, Germany) to select for clones containing the insertion cassette.
FIG 2.
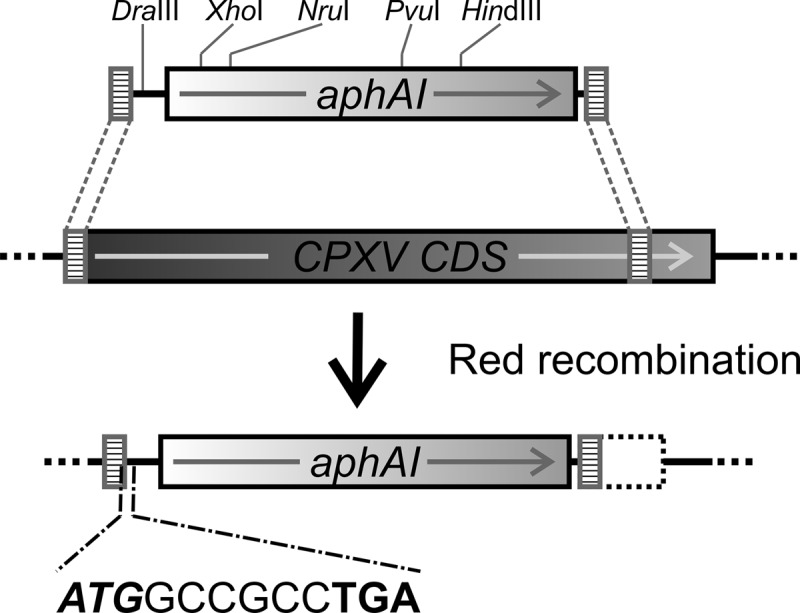
Schematic illustration of the generation of CPXV knockout mutants using Red recombination in E. coli. PCR fragments containing a kanamycin resistance gene aphAI and a sequence coding for two alanines (GCCGCG), followed by a stop codon (TGA) were amplified from plasmid pACAA. Homologous flanking sequences (40 bp) were added through 5′ overhangs of each PCR primer. PCR products were inserted into the target genes of the CPXV sequence by Red recombination. Restriction sites in PCR products for DraIII, HindIII, NruI, PvuI, and XhoI are indicated.
TABLE 1.
CPXV-BR ORFs, mutant BACs, and reconstitution
| CPXV-BR gene | VACV-COP homolog(s) | Function of VACV homologsa | Mutant BAC | Reconstitution | Conservationb |
|---|---|---|---|---|---|
| CPXV001 | Unknown | ||||
| CPXV002 | Unknown | ||||
| CPXV003 | C23L | Chemokine binding protein | |||
| CPXV004 | Unknown | ||||
| CPXV005 | C22L | Tumor necrosis factor receptor | |||
| CPXV006 | C19L | Ankyrin | |||
| CPXV007 | Unknown | ||||
| CPXV008 | C17L | Ankyrin | |||
| CPXV009 | C16L | Unknown | |||
| CPXV010 | N2L | Alpha-amanitin sensitivity protein | pBRFseR d10 | Yes | No |
| CPXV011 | Ankyrin | ||||
| CPXV012 | Unknown | pBRFseR d12 | Yes | No | |
| CPXV013 | Kelch like | pBRFseR d13 | Yes | No | |
| CPXV014f | C22L, B28R | Tumor necrosis factor receptor | pBRFseR d14 | Yes | No |
| CPXV015f | Unknown | pBRFseR d15 | Yes | No | |
| CPXV016 | Ankyrin | ||||
| CPXV017 | Ankyrin | ||||
| CPXV018 | Unknown | pBRFseR d18 | Yes | No | |
| CPXV019 | Ankyrin | ||||
| CPXV020 | Unknown | pBRFseR d20 | Yes | No | |
| CPXV021 | C11R | Epidermal growth factor | pBRFseR d21 | Yes | No |
| CPXV022 | C10L, C4L | Interleukin-1 receptor antagonist | pBRFseR d22 | Yes | No |
| CPXV023 | Ubiquitin ligase/host defense modulator protein | pBRFseR d23 | Yes | No | |
| CPXV024 | Interleukin-18 binding protein | pBRFseR d24 | Yes | No | |
| CPXV025 | Ankyrin host range protein | ||||
| CPXV026 | Unknown | pBRFseR d26 | Yes | No | |
| CPXV027 | C9L | Ankyrin | |||
| CPXV028 | C8L | Unknown | pBRFseR d28 | Yes | No |
| CPXV029 | C7L | Host range virulence factor | pBRFseR d29 | Yes | No |
| CPXV030 | C6L | Unknown | pBRFseR d30 | Yes | No |
| CPXV031f | C5L | Unknown | pBRFseR d31 | Yes | No |
| CPXV032 | C5L | Unknown | pBRFseR d32 | Yes | No |
| CPXV033 | C10L, C4L | Interleukin-1 receptor antagonist | pBRFseR d33 | Yes | No |
| CPXV034 | C3L | Complement binding protein (secreted) | pBRFseR d34 | Yes | No |
| CPXV035 | C2L | Kelch-like protein | pBRFseR d35 | Yes | No |
| CPXV036 | C1L | Unknown | pBRFseR d36 | Yes | No |
| CPXV037 | N1L | Virokine/NF-κB inhibitor | pBRFseR d37 | Yes | No |
| CPXV038 | N2L | Alpha amanitin sensitivity protein | pBRFseR d38 | Yes | No |
| CPXV039 | M1L | Ankyrin | |||
| CPXV040 | M2L | NF-κB inhibitor | pBRFseR d40 | Yes | No |
| CPXV041 | K1L | Ankyrin and NF-κB inhibitor | |||
| CPXV042 | C12L, K2L, B13R, B14R | “Serpin 1,2,3” | pBRFseR d42 | Yes | No |
| CPXV043 | K3L | Interferon resistance and eIF2 alpha-like PKR inhibitor | pBRFseR d43 | Yes | No |
| CPXV044 | K4L | Nicking/joining enzyme | pBRFseR d44 | Yes | No |
| CPXV045 | K5L, K6L | Monoglyceride lipase (putative) | pBRFseR d45 | Yes | No |
| CPXV046 | K7R | Unknown | pBRFseR d46+47 | Yes | No |
| CPXV047 | Pseudo?c | Unknown | |||
| CPXV048 | F1L | Apoptosis inhibitor (associated with mitochondria) | pBRFseR d48 | Yes | No |
| CPXV049 | F2L | dUTPase | pBRFseR d49 | Yes | No |
| CPXV050 | F3L | Kelch-like protein | pBRFseR d50 | Yes | No |
| CPXV051 | F4L | Ribonucleotide reductase small subunit | pBRFseR d51 | Yes | No |
| CPXV051A | Pseudo? | Unknown | |||
| CPXV052 | F5L | Membrane protein (36 kDa) | pBRFseR d52 | Yes | No |
| CPXV053 | F6L | Unknown | pBRFseR d53 | Yes | No |
| CPXV054 | F7L | Unknown | pBRFseR d54 | Yes | No |
| CPXV055 | F8L | Cytoplasmic protein | pBRFseR d55 | Yes | No |
| CPXV056 | F9L | Disulfide bond formation pathway protein | pBRFseR d56 | No | P |
| CPXV057 | F10L | Serine/threonine kinase | pBRFseR d57+58 | No | P |
| CPXV058 | F_ORF_D Pseudo? | Unknown | |||
| CPXV059 | F11L | Unknown | pBRFseR d59 | Yes | No |
| CPXV060 | F12L | IEV-associated protein | pBRFseR d60 | Yes | C |
| CPXV061 | F13L | EEV phospholipase | pBRFseR d61 | Yes | C |
| CPXV062 | F14L | Unknown | pBRFseR d62 | Yes | No |
| CPXV063 | Unknown | pBRFseR d63 | Yes | No | |
| CPXV064 | F15L | Unknown | pBRFseR d64 | Yes | C |
| CPXV065 | F16L | Unknown | pBRFseR d65 | Yes | No |
| CPXV066 | F17R | DNA binding phosphoprotein | pBRFseR d66 | No | C |
| CPXV067 | E1L | Poly(A) polymerase large subunit | pBRFseR d67 | No | P |
| CPXV068 | E2L | Unknown | pBRFseR d68 | Yes | C |
| CPXV069 | E3L | Interferon resistance and PKR inhibitor | pBRFseR d69 | Yes | No |
| CPXV070 | E4L | RNA polymerase 30 subunit | pBRFseR d70 | No | C |
| CPXV071 | E5R | Virosome component protein | pBRFseR d71 | Yes | No |
| CPXV072 | E6R | required for the formation of immature virions | pBRFseR d72 | No | P |
| CPXV073 | E7R | EEV myristylated soluble protein | pBRFseR d73 | Yes | No |
| CPXV074 | E8R | Endoplasmic reticulum localized membrane protein | pBRFseR d74 | Yes | C |
| CPXV075 | E9L | DNA polymerase | pBRFseR d75 | No | P |
| CPXV076 | E10R | Disulfide bond formation pathway protein | pBRFseR d76 | No | P |
| CPXV077 | E11L | Virion core protein | pBRFseR d77 | No | No |
| CPXV078 | O1L | Unknown | pBRFseR d78 | Yes | No |
| CPXV078A | Pseudo? | Unknown | |||
| CPXV079 | O2L | Glutaredoxin 1 | pBRFseR d79 | Yes | No |
| CPXV080 | I1L | DNA binding protein | pBRFseR d80 | No | C |
| CPXV081 | I2L | Unknown | pBRFseR d81 | No | C |
| CPXV082 | I3L | DNA binding phosphoprotein | pBRFseR d82 | No | C |
| CPXV083 | I4L | Ribonucleotide reductase large subunit | pBRFseR d83 | Yes | No |
| CPXV084 | I5L | IMV protein | pBRFseR d84 | Yes | C |
| CPXV085 | I6L | Telomere binding protein | pBRFseR d85 | Yes | C |
| CPXV086 | I7L | Virion core protease | pBRFseR d86 | No | P |
| CPXV087 | I8R | RNA helicase/NPH-II | pBRFseR d87 | No | P |
| CPXV088 | G1L | Metalloprotease (predicted) | pBRFseR d88 | No | P |
| CPXV089 | G3L | virus fusion complex | pBRFseR d89 | No | C |
| CPXV090 | G2R | Viral late transcription factor | pBRFseR d90 | No | C |
| CPXV091 | G4L | Glutaredoxin 2 | pBRFseR d91 | No | C |
| CPXV092 | G5R | Unknown | pBRFseR d92 | No | P |
| CPXV093 | G5.5R | RNA polymerase 7 subunit | pBRFseR d93 | No | C |
| CPXV094 | G6R | Unknown | pBRFseR d94 | Yes | P |
| CPXV095 | G7L | Virion assembly protein | pBRFseR d95 | No | C |
| CPXV096 | Pseudo? | Unknown | |||
| CPXV097 | G8R | Viral late transcription factor 1 | pBRFseR d96+97 | No | C |
| CPXV098 | G9R | Entry fusion complex protein | pBRFseR d98 | No | P |
| CPXV099 | L1R | IMV myristylated membrane protein | pBRFseR d99 | No | P |
| CPXV100 | L2R | Crescent formation | pBRFseR d100 | No | C |
| CPXV101 | L3L | Internal virion protein | pBRFseR d101 | No | P |
| CPXV102 | L4R | Core package and transcription protein | pBRFseR d102 | No | P |
| CPXV103 | L5R | IMV entry and fusion protein | pBRFseR d103 | No | P |
| CPXV104 | J1R | Virion morphogenesis protein | pBRFseR d104 | No | C |
| CPXV105 | J2R | Thymidine kinase | |||
| CPXV106 | J3R | Poly(A) polymerase small subunit | pBRFseR d106 | No | P |
| CPXV107 | J4R | RNA polymerase 22 subunit | pBRFseR d107 | No | C |
| CPXV108 | J5L | Unknown membrane protein | pBRFseR d108 | Yes | P |
| CPXV109 | J6R | RNA polymerase 147 subunit | pBRFseR d109 | No | P |
| CPXV110 | H1L | Serine/Tyrosine phosphatase | pBRFseR d110 | Yes | C |
| CPXV111 | H2R | Entry and cell to cell fusion protein | pBRFseR d111 | No | P |
| CPXV112 | H3L | IMV heparin binding surface protein | pBRFseR d112 | Yes | P |
| CPXV113 | H4L | RNA polymerase associated protein RAP94 | pBRFseR d113 | No | P |
| CPXV114 | H5R | Viral late transcription factor 4 | pBRFseR d114 | No | C |
| CPXV115 | H6R | Topoisomerase type I | pBRFseR d115 | Yes | P |
| CPXV116 | Pseudo? | Unknown | |||
| CPXV117 | H7R | Crescent formation | pBRFseR d117+116 | No | C |
| CPXV118 | D1R | Large capping enzyme | pBRFseR d118 | No | P |
| CPXV119 | D2L | Virion core | pBRFseR d119+119A | No | C |
| CPXV119A | D_ORF_B | Unknown | |||
| CPXV120 | D3R | Virion core protein | pBRFseR d120 | No | C |
| CPXV121 | D4R | Uracil DNA glycosylase | pBRFseR d121 | No | P |
| CPXV122 | D5R | NTPase and DNA replication protein | pBRFseR d122 | No | P |
| CPXV123 | D6R | Viral early transcription factor small subunit | pBRFseR d123 | No | P |
| CPXV124 | D7R | RNA polymerase 18 subunit | pBRFseR d124 | No | P |
| CPXV125 | D8L | Carbonic anhydrase | pBRFseR d125 | Yes | No |
| CPXV126 | D9R | NTP-PPH containing mutT motif | pBRFseR d126 | Yes | C |
| CPXV127 | D10R | NPH-PPH RNA levels regulator containing mutT motif | pBRFseR d127 | Yes | P |
| CPXV128 | D11L | Helicase NPH-I | pBRFseR d128 | No | P |
| CPXV129 | D12L | Small capping enzyme | pBRFseR d129+130 | No | P |
| CPXV130 | Pseudo? | Unknown | |||
| CPXV131 | D13L | Virion coat protein rifampin resistance | pBRFseR d131 | No | P |
| CPXV132 | A1L | Viral late transcription factor 2 | pBRFseR d132 | No | P |
| CPXV133 | A2L | Viral late transcription factor 3 | pBRFseR d133 | No | P |
| CPXV134 | A2.5L | Thioredoxin like protein | pBRFseR d134 | No | C |
| CPXV135 | A3L | P4b precursor | pBRFseR d135 | No | P |
| CPXV136 | A4L | Core protein | pBRFseR d136 | Yes | C |
| CPXV137 | A5R | RNA polymerase 19 subunit | pBRFseR d137 | No | P |
| CPXV138 | A6L | Virion morphogenesis protein | pBRFseR d138 | No | C |
| CPXV139 | A7L | Viral early transcription factor large subunit | pBRFseR d139 | No | P |
| CPXV140 | A8R | Viral intermediate transcription factor 3 | pBRFseR d140 | No | C |
| CPXV141 | A9L | Membrane protein | pBRFseR d141 | No | P |
| CPXV142 | A10L | P4a precursor | pBRFseR d142 | No | P |
| CPXV143 | A11R | Membrane formation protein | pBRFseR d143 | No | P |
| CPXV144 | A12L | Structural protein | pBRFseR d144 | No | C |
| CPXV145 | A13L | Virion maturation protein | pBRFseR d145 | No | C |
| CPXV146 | A14L | IMV membrane protein (phosphorylated) | pBRFseR d146 | No | C |
| CPXV147 | A14.5L | IMV virulence factor (membrane protein) | pBRFseR d147 | Yes | C |
| CPXV148 | A15L | Unknown | pBRFseR d148 | No | C |
| CPXV149 | A16L | Entry and cell-to-cell fusion protein (myristilated) | pBRFseR d149 | No | P |
| CPXV150 | A17L | IMV membrane protein phosphorylated | pBRFseR d150 | No | C |
| CPXV151 | A18R | DNA helicase | pBRFseR d151 | No | P |
| CPXV152 | A19L | Unknown | pBRFseR d152+152A | No | C |
| CPXV152A | Pseudo? | Unknown | |||
| CPXV153 | A21L | Entry and cell-to-cell fusion protein | pBRFseR d153 | No | P |
| CPXV154 | A20R | DNA processivity factor | pBRFseR d154 | No | C |
| CPXV155 | A22R | Holliday junction resolvase | pBRFseR d155 | Yes | P |
| CPXV156 | A23R | Viral intermediate transcription factor 3 (45-kDa subunit) | pBRFseR d156 | No | P |
| CPXV157 | A24R | RNA polymerase 132 subunit | pBRFseR d157 | No | P |
| CPXV158 | A25L | A-type inclusion protein | pBRFseR d158 | Yes | No |
| CPXV159 | A26L | P4c precursor | pBRFseR d159 | Yes | No |
| CPXV160 | Pseudo? | Unknown | |||
| CPXV161 | A26L | P4c precursor | pBRFseR d161 | Yes | No |
| CPXV162 | A27L | Fusion protein | pBRFseR d162 | Yes | No |
| CPXV163 | A28L | IMV virus entry (membrane protein) | pBRFseR d163 | No | P |
| CPXV164 | A29L | RNA polymerase 35 subunit | pBRFseR d164 | No | P |
| CPXV165 | A30L | Virion morphogenesis protein | pBRFseR d165 | No | C |
| CPXV166 | A31R | Unknown | pBRFseR d166 | Yes | No |
| CPXV167 | A32L | DNA packaging and ATPase protein | pBRFseR d167 | No | P |
| CPXV168 | A33R | EEV glycoprotein | pBRFseR d168 | Yes | No |
| CPXV169 | A34R | EEV C-type lectin-like protein | pBRFseR d169 | Yes | C |
| CPXV170 | Pseudo? | Unknown | pBRFseR d170+171 | Yes | No |
| CPXV171 | A35R | Unknown | |||
| CPXV172 | A36R | IEV specific | pBRFseR d172 | Yes | No |
| CPXV173 | A37R | Unknown | pBRFseR d173 | Yes | No |
| CPXV174 | Unknown | pBRFseR d174 | Yes | No | |
| CPXV175 | A38L | CD47-like protein | pBRFseR d175 | Yes | No |
| CPXV176 | A39R | Semaphorin | pBRFseR d176 | Yes | No |
| CPXV177 | A40R | Lectin homolog | pBRFseR d177 | Yes | No |
| CPXV178 | A41L | Secreted virulence factor | pBRFseR d178 | Yes | No |
| CPXV179 | A42R | Profilin homolog | pBRFseR d179 | Yes | No |
| CPXV180 | A43R | Membrane glycoprotein class I | pBRFseR d180 | Yes | No |
| CPXV181 | Unknown | pBRFseR d181 | Yes | No | |
| CPXV182 | A44L | Hydroxysteroid dehydrogenase | pBRFseR d182 | Yes | No |
| CPXV183 | A45R | Superoxide dismutase-like protein | pBRFseR d183 | Yes | No |
| CPXV184 | A46R | Interleukin-1 signaling inhibitor | pBRFseR d184 | Yes | No |
| CPXV185 | A47L | Unknown | pBRFseR d185 | Yes | No |
| CPXV186 | A48R | Thymidylate kinase | pBRFseR d186 | Yes | No |
| CPXV187 | A49R | Phosphotransferase anion transport protein (putative) | pBRFseR d187 | Yes | No |
| CPXV188 | A50R | DNA ligase | pBRFseR d188 | Yes | No |
| CPXV189 | A51R | Unknown | pBRFseR d189 | Yes | No |
| CPXV190 | A52R | Intracellular TLR and interleukin-1 signaling inhibitor | pBRFseR d190 | Yes | No |
| CPXV191 | A53R, A_ORF_T | Tumor necrosis factor receptor (CrmC) | pBRFseR d191 | Yes | No |
| CPXV192 | Pseudo? | Unknown | |||
| CPXV193 | A55R | Kelch-like protein | pBRFseR d193 | Yes | No |
| CPXV194 | A56R | Hemagglutinin | pBRFseR d194 | Yes | No |
| CPXV195 | A57R | Guanylate kinase | pBRFseR d195 | No | No |
| CPXV196 | B1R | Serine/threonine kinase | pBRFseR d196 | Yes | No |
| CPXV197 | B2R; B3R | Schlafen | pBRFseR d197 | Yes | No |
| CPXV198 | B4R | Ankyrin | |||
| CPXV199 | B5R | EEV complement control protein | pBRFseR d199 | Yes | No |
| CPXV200 | B6R | Unknown | pBRFseR d200 | Yes | No |
| CPXV201 | B7R | Virulence factor (endoplasmic reticulum associated) | pBRFseR d201 | Yes | No |
| CPXV202 | B8R | Interferon gamma receptor | pBRFseR d202 | Yes | No |
| CPXV203 | B9R | Virulence factor | pBRFseR d203 | Yes | No |
| CPXV204 | B10R | Kelch-like protein | pBRFseR d204 | Yes | No |
| CPXV205 | B11R | Unknown | pBRFseR d205 | Yes | No |
| CPXV206 | B12R | Serine/threonine kinase | pBRFseR d206 | Yes | No |
| CPXV207 | C12L, K2L, B13R, B14R | “Serpin 1,2,3” | |||
| CPXV208 | C16L, B15R, B22R | Unknown | pBRFseR d208 | Yes | No |
| CPXV209 | B16R | Interleukin-1β receptor | pBRFseR d209 | Yes | No |
| CPXV210 | B17L | Unknown | pBRFseR d210 | Yes | No |
| CPXV211 | B18R | Ankyrin | |||
| CPXV212 | B19R | Alpha/beta interferon receptor | pBRFseR d212 | Yes | No |
| CPXV213 | B20R | Ankyrin | |||
| CPXV214 | Pseudo? | Unknown | |||
| CPXV215 | Kelch-like protein | pBRFseR d214+215 | Yes | No | |
| CPXV216 | C12L, K2L, B13R, B14R | Unknown | pBRFseR d216 (ATG) | Yes | No |
| CPXV217 | C12L, K2L, B13R, B14R | “Serpin 1,2,3” | pBRFseR d217 | Yes | No |
| CPXV218 | C14L, C13L | Unknown | pBRFseR d218 | Yes | No |
| CPXV219 | Surface glycoprotein | pBRFseR d219 | Yes | No | |
| CPXV220 | C21L, C20L, C19L, B25R, B26R, B27R | Ankyrin | |||
| CPXV221 | CrmD | Tumor necrosis factor receptor (CrmD) | pBRFseR d221 | Yes | No |
Information was obtained from the PBRC.
C, gene families conserved in chordopoxviruses; P, gene families conserved in poxviruses (32).
Pseudo?, pseudogene (according to PBRC).
Knockout BAC verification.
For each of the knockout mutants, bacterial colonies resistant to chloramphenicol and kanamycin were selected and BAC DNA was extracted by alkaline lysis (22). For RFLP analysis, BAC DNA was cleaved with selected restriction enzymes and separated by 0.8% agarose gel electrophoresis for 16 h at 75 V in 1× TAE buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA [pH 8.4]). Each individual mutant BAC DNA was tested for the correct RFLP with at least two different restriction enzymes (see Table S3 in the supplemental material [http://www.vetmed.fu-berlin.de/en/einrichtungen/institute/we05/cowpox]). To confirm the in-frame insertion of the premature stop codon, PCR primers covering the original start and new stop codon of the respective target ORFs were designed for all mutants (see Table S3 in the supplemental material). PCR products overlapping with the modified loci were checked by agarose gel electrophoresis, purified using the GF-1 AmbiClean (PCR & Gel) nucleic acid extraction kit (Vivantis Technologies, Subang Jaya, Malaysia) and sequenced to verify the correct insertion of the marker cassette.
Bioinformatics analysis.
Searches for homologous poxvirus sequences, as well as prediction of gene expression kinetics and function of encoded proteins, were performed using the NCBI BLAST and the VectorNTI 9.1 software package (Invitrogen, Darmstadt, Germany) and were based on OPV sequences available at the Poxvirus Bioinformatics Resource Center (PBRC; http://www.poxvirus.org) and GenBank (23). Predicted incorporation of CPXV proteins into the virion was based on information for homologous proteins of VACV (7, 24–26).
RESULTS AND DISCUSSION
Generation of the dual marker CPXV-BR BAC clone pBRFseR.
We previously cloned the CPXV-BR genome as a BAC by inserting the mini-F harboring the egfp gene under the control of a viral late promoter into the locus of the nonessential thymidine kinase (TK, CPXV105) (15). In order to be able to monitor early and late viral gene expression in infected cells, we introduced the mrfp1 gene under the control of a synthetic early promoter, based on the 7.5-kDa VACV promoter, into the recombinant parental clone pBRF. Since early gene promoters tend to be weak and homology to existing sequences in the BAC might have led to genetic instability, we chose a synthetic early promoter, optimized for strong expression (21). The early marker expression cassette was inserted into the mini-F region of the BAC by two-step en passant Red mutagenesis.
Successful integration resulting in the dual marker pBRFseR BAC clone was checked by RFLP analysis and sequencing (data not shown). Upon reconstitution and serial virus passage, we obtained fully replicating virus (vBRFseR) expressing both the red and green fluorescent proteins (Fig. 3A). We performed time course experiments to test the kinetics of the expression of both fluorescent markers. Vero cells were infected with vBRFseR using a multiplicity of infection (MOI) of 1. The early red and the late green fluorescence signals were readily detectable at 6 and 12 h postinfection (p.i.), respectively.
FIG 3.

Detection of dual marker expression in infected cell culture. (A) Reconstitution of vBRFseR on Vero cells using FWPV as a helper virus. Expression of both mRFP and eGFP can be detected upon reconstitution. (B) Expression of mRFP and eGFP during infection. mRFP can be detected at 6 h p.i.; eGFP can be detected at 12 h p.i.; araC can block eGFP but not mRFP expression in infected cells, as evidenced by the expression of mRFP only. Scale bars, 200 μm.
Furthermore, addition of 50 μg of 1-β-d-arabinofuranosyl cytidine (araC)/ml to the cell culture did not affect expression of the early mRFP marker, while completely blocking expression of the late eGFP marker (Fig. 3B). This clearly showed that the newly introduced marker was indeed expressed in the early phase of viral replication, whereas the eGFP marker was only expressed in late stages of the replication cycle after successful replication of viral DNA (Fig. 3).
Targeted knockout of all unique CPXV-BR ORFs.
The overall aim of the present study was to generate single knockout mutants for each of the 216 unique CPXV ORFs representing ORF CPXV010 to ORF CPXV221. We did not include genes present in the terminal inverted repeats (TIR; ORFs CPXV001 to CPXV009 and CPXV222 to CPXV229). Thirteen of the 216 unique ORFs were determined to be pseudogenes (according to the PBRC). Knockout mutants of all 16 ankyrin repeat protein-encoding genes, including 12 unique genes and the 4 ORFs in the TIR regions of the genome, were generated previously (B. K. Tischer, unpublished data). The thymidine kinase encoding gene CPXV105 is interrupted by the mini-F replicon, and a BAC-based mutant of CPXV207 coding for CrmA was described previously (15, 27). The six predicted kelch-like protein encoding genes were deleted by en passant mutagenesis for another study (Tischer, unpublished; see Fig. S1 in the supplemental material [http://www.vetmed.fu-berlin.de/en/einrichtungen/institute/we05/cowpox]). In the case of gene CPXV216, which is short and in close proximity to gene CPXV217, the start codon was replaced by en passant mutagenesis to avoid an interruption of the promoter of CPXV217.
We generated the knockout mutants of the remaining 182 ORFs by inserting the engineered PCR-derived bacterial selection marker using classical Red recombination (see Materials and Methods). We ensured disruption of the targeted ORF by positioning the bacterial selection marker such that it would certainly interrupt viral gene expression of the respective CPXV ORF and replacing between 200 and 1,000 bp of each gene depending on the respective length. Further, the insertions were targeted to be immediately after the start codon and an in-frame stop codon was added to avoid the production of larger truncated proteins, which could still be functional. Since poxvirus genes usually contain multiple start codons, we analyzed every ORF for additional downstream start codons and deleted the first two in-frame AUG codons. For example, the vaccinia virus E3L ORF can encode two functional proteins, p19 and p25, by initiating translation at its first or second AUG codon (28), and this gene was inactivated according to the outlined principle.
The knockout strategy described above would lead to double deletions in the case of overlapping ORFs, which occur frequently in poxvirus genomes. In CPXV-BR, for example, CPXV036 and CPXV037 overlap head to tail, with the promoter of CPXV036 located in the coding sequence of CPXV037. Thus, deletion of CPXV037 may lead to the simultaneous abrogation of CPXV036 expression. In this and similar cases, we avoided interruption of the promoter of the downstream gene or damaging the integrity of the upstream gene by shifting the targeted position of the deletion cassette.
We confirmed successful deletion of each individual ORF by RFLP analysis. The inserted deletion cassette contained DraIII, HindIII, PvuI, NruI, and XhoI sites (Fig. 2). Therefore, cleavage with one of these restriction enzymes resulted in a change of the restriction pattern of the mutated BAC in comparison to the parental pBRFseR (see Fig. S2 in the supplemental material [http://www.vetmed.fu-berlin.de/en/einrichtungen/institute/we05/cowpox]), confirming the presence of the marker sequence. All mutants were checked with at least two different restriction enzymes (see Table S3 in the supplemental material). In all cases, the observed restriction pattern and the sequencing results were in complete agreement with the in silico predictions (data not shown).
Virus reconstitution.
We performed virus reconstitution by transfecting Vero cells with BAC DNA in the presence of FWPV as helper virus. The obtained virus clones were passaged three times on Vero cells to remove FWPV helper virus. Of the 183 single ORF deletion mutants, 109 knockout viruses could be reconstituted, whereas reconstitution was unsuccessful for 74 knockout viruses (Table 1).
In order to minimize host range limitations of the reconstitution system, we used two independent systems to generate BAC-derived CPXV mutants. Mutants that did not reconstitute on Vero cells in two independent experiments were also tested on CEC and SFV as a helper virus to confirm the result (see Materials and Methods). We termed genes “essential” if virus reconstitution failed twice with each of the two systems. Since it is well known that for VACV the CPXV069 homolog E3L confers a host range phenotype on Vero cells (29, 30), we used BHK21 cells and SFV as the helper virus for reconstitution. Similar host range limitations might be true for other CPXV mutants that could not be reconstituted using the Vero and CEC systems and will be addressed in future studies.
Different phenotypes upon virus reconstitution.
Signals emitted by mRFP and eGFP indicate early and late viral gene expression, respectively. Monitoring mRFP and eGFP fluorescence upon virus reconstitution or infection provided information about the involvement of individual CPXV genes in virus replication. According to the observed fluorescence signals, three different mutant virus phenotypes were observed: (i) no plaque formation, (ii) formation of small, red fluorescence-only plaques with single double-fluorescent cells in the center, and (iii) formation of wild-type-like plaques expressing both fluorescence markers (Fig. 4).
FIG 4.

Different phenotypes upon reconstitution. (A) No plaque formation (vBRFseR-d195); (B) formation of small, “red fluorescence-only” plaques with single, double-fluorescent cells in the center (vBRFseR-d121); (C) formation of wild-type-like plaques that exhibit mRFP and eGFP fluorescence (vBRFseR-d158). Scale bars, 200 μm.
The red plaques expressed only mRFP but not eGFP except for a single cell in the center of the plaque (Fig. 4). The most likely explanation is that the FWPV helper virus was able to compensate for the deleted CPXV-BR gene in transfected cells. Due to complementation, the viral replication cycle could be completed in the central cell of the plaque, leading to the production of infectious viral particles. However, in neighboring cells that were infected subsequently with the mutant CPXV but not with helper FWPV, the replication cycle was aborted before the transition to late gene expression, as reflected by the lack of the green fluorescent marker. This hypothesis is supported by the fact that virus progeny was lost by repeated passage of progeny virus, which diluted and ultimately eliminated the helper virus.
Essential genes of CPXV.
Wild-type-like formation of plaques by mutant viruses indicated that the gene product of the deleted ORF was dispensable for virus replication on Vero cells. This was the case for 109 of the 183 (60%) mutant viruses that we generated (Table 1 and Fig. 5). All six kelch-like protein single deletion mutants, as well as the mutant virus lacking all six genes, were also successfully reconstituted. During continuous passage on Vero cells, some mutant viruses exhibiting red and green fluorescent plaques lost their ability to produce infectious progeny. This suggested that the respective ORFs are essential for virus replication on these cells. The “red fluorescence-only” mutants (12/183) lost the ability to form plaques by repeated passage, and the respective mutants were consequently grouped with the mutants, for which no plaque formation was detectable from the start (62/183). Hence, the respective ORFs were considered essential for CPXV replication on Vero cells, and a total of 40% of the generated mutant viruses were grouped in this category.
FIG 5.

Overview of the results from reconstitution experiments of mutant BAC clones on Vero cells and CEC. The numbers of genes that are not essential for virus replication are highlighted in green. Deletion of “red” ORFs (“red fluorescence only”; red underline) resulted in virus progeny with a blocked transition to late gene expression. Gene numbers highlighted in black are essential, as characterized by the absence of production of any virus progeny upon three attempts at reconstitution. Gray numbers indicate putative ORFs, which were not deleted for generation of the library, since this group contains pseudogenes, small ORFs that completely overlap with other ORFs or genes within the TIR. The color of the ORFs indicates gene expression kinetics according to their homologues in VACV, as indicated on the right.
Based on data from our mutant library, as well as information from the PBRC and previous studies performed on VACV (2, 4), a genome map was created to provide an overview about CPXV genes and their transcription kinetics (early, intermediate, and late) (Fig. 5). In accordance with previous studies, we found that 71% of all late genes were essential for virus replication. The products of all essential late genes are predicted to be incorporated into mature virions (24–26, 31). We also confirmed that most essential genes were located in the central portion of the genome, whereas most nonessential genes clustered toward the genomic termini. All except for 3 of the 74 genes determined to be essential were located in the central part of the CPXV-BR genome, where the central part accounts for 50% of the genome (Fig. 6). More than 50% (7/12) of the ORFs whose deletion resulted in the “red fluorescence-only” phenotype code for proteins that are involved in RNA transcription and are also predicted as structural components of the mature virion. Consistent with their essential nature, all 12 “red fluorescence-only” ORFs were located in the central part of the genome.
FIG 6.

Distribution of essential and nonessential genes in the CPXV genome. (A) The flanking 50% of the CPXV genome consists almost exclusively of nonessential genes. (B) Most essential genes, as well as all genes of the “red fluorescence-only” phenotype, are located in the central 50% of the genome. However, the central part does also contain nonessential genes (∼40% of the center genes are essential).
A previous extensive comparison of 21 completely sequenced poxvirus genomes revealed that 49 gene families are conserved among poxviruses and that 41 additional families are conserved in the Chordopoxvirinae (32). Based on this high degree of conservation, the authors of one study predicted these gene families to be essential for poxvirus replication (30), and some of the genes were already found to be essential by independent studies (Table 2). However, an exhaustive investigation to confirm this analysis of genes essential for poxvirus replication was lacking. We found that most of the conserved genes were also essential as derived from our data set, with the exception of 17 genes (Table 2), most of which are conserved in the Chordopoxvirinae but not all Poxviridae. Among these 17 genes, 10 have already been described to be nonessential in in vitro experiments (for references, see Table 2). For example, the VACV type I topoisomerase gene (H6R) is conserved among all poxviruses but is not essential for virus replication in vitro (33). However, the mutant virus exhibited reduced infectivity, which could be ascribed mainly to lower early transcription rather than to direct effects on the processing of viral DNA. Similarly, the VACV homologues D9R and D10R, which are encoding proteins likely involved in decapping host cell mRNAs, are not essential for virus replication if knocked out separately (34). A double deletion mutant for both ORFs failed, however, to reconstitute, possibly indicating mutually compensation of the two genes, which is most likely also the case in our knockout library. Clearly, further experiments are needed to confirm this hypothesis. Interestingly, we found CPXV077 and CPXV195 (homologs of VACV E11L and A57L, respectively) to be essential for virus replication in vitro, even though both are not conserved in poxviruses or chordopoxviruses.
TABLE 2.
Literature summary of nonessential conserved CPXV genes
| CPXV-BR gene | VACV-COP homolog | Family name | Conservationa | Essential according to this study | Essential according to other publications | Reference |
|---|---|---|---|---|---|---|
| CPXV060 | F12L | Actin tail, microtubule | P | No | No | 34 |
| CPXV061 | F13L | Phospholipase extracellular enveloped virion | P | No | No | 35 |
| CPXV064 | F15L | Unknown | P | No | ||
| CPXV068 | E2L | Unknown | P | No | No | 36 |
| CPXV074 | E8R | Endoplasmic reticulum-localized MP | P | No | ||
| CPXV084 | I5L | Unknown VP13 | P | No | No | 37 |
| CPXV085 | I6L | Unknown | P | No | ||
| CPXV108 | J5L | Late MP, essential | C | No | Yes | 38 |
| CPXV110 | H1L | Tyrosine-serine phosphatase | P | No | Yes | 39 |
| CPXV112 | H3L | Intracellular mature virus morphogenesis viral protein (VP55) | C | No | No | 40 |
| CPXV115 | H6R | Topoisomerase type I | C | No | No | 32 |
| CPXV126 | D9R | mutT motif, nucleoside triphosphate pyrophosphohydrolase | P | No | No | 33 |
| CPXV127 | D10R | Nucleophosphohydrolase-pyrophosphohydrolase downregulator | C | No | No | 33 |
| CPXV136 | A4L | Core protein | P | No | Yes | 41 |
| CPXV147 | A14.5L | IMV MP, virulence factor | P | No | No | 42 |
| CPXV155 | A22R | Holliday junction resolvase | C | No | Yes | 43 |
| CPXV169 | A34R | Extracellular enveloped virion glycoprotein | P | No | No | 44 |
According to a previously published study (28). C, gene families conserved in chordopoxviruses; P, gene families conserved in poxviruses.
In conclusion, we have created the first complete targeted BAC knockout library of a large DNA virus. Reconstitution of mutant clones has yielded novel insight into the importance of single viral genes for viral replication. With the insertion of fluorescent markers for early and late viral gene expression, the library at hand can be used for high-throughput screens to identify genes involved in various processes of the virus replication cycle, including features of host range, virulence factors, and gene products involved in immunomodulation.
ACKNOWLEDGMENTS
We thank the Nationale Forschungsplattform für Zoonosen and the German Federal Ministry of Education and Research (BMBF 01KI1102) for funding this project.
Z.X. was supported by the Chinese Scholarship Council and the Dahlem Research School. D.Z. received a scholarship from the International Max Planck Research School and is a member of the Centre for Infection Biology and Immunology Research Training Group in Berlin, Germany.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Essbauer S, Pfeffer M, Meyer H. 2010. Zoonotic poxviruses. Vet. Microbiol. 140:229–236. 10.1016/j.vetmic.2009.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assarsson E, Greenbaum JA, Sundstrom M, Schaffer L, Hammond JA, Pasquetto V, Oseroff C, Hendrickson RC, Lefkowitz EJ, Tscharke DC, Sidney J, Grey HM, Head SR, Peters B, Sette A. 2008. Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. U. S. A. 105:2140–2145. 10.1073/pnas.0711573105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876. 10.1126/science.1183173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9908. 10.1128/JVI.05446-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sivan G, Martin SE, Myers TG, Buehler E, Szymczyk KH, Ormanoglu P, Moss B. 2013. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:3519–3524. 10.1073/pnas.1300708110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hendrickson RC, Wang C, Hatcher EL, Lefkowitz EJ. 2010. Orthopoxvirus genome evolution: the role of gene loss. Viruses 2:1933–1967. 10.3390/v2091933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubser C, Hue S, Kellam P, Smith GL. 2004. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 85:105–117. 10.1099/vir.0.19565-0 [DOI] [PubMed] [Google Scholar]
- 8.Shchelkunov SN, Safronov PF, Totmenin AV, Petrov NA, Ryazankina OI, Gutorov VV, Kotwal GJ. 1998. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology 243:432–460. 10.1006/viro.1998.9039 [DOI] [PubMed] [Google Scholar]
- 9.Shchelkunov SN. 2011. Emergence and reemergence of smallpox: the need for development of a new generation smallpox vaccine. Vaccine 29:D49–D53. 10.1016/j.vaccine.2011.05.037 [DOI] [PubMed] [Google Scholar]
- 10.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr. Protoc. Protein Sci. Chapter 5:Unit 5. 10.1002/0471142727.mb1617s43. [DOI] [PubMed] [Google Scholar]
- 11.Holzer GW, Falkner FG. 1997. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 71:4997–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tischer BK, Kaufer BB. 2012. Viral bacterial artificial chromosomes: generation, mutagenesis, and removal of mini-F sequences. J. Biomed. Biotechnol. 2012:472537. 10.1155/2012/472537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domi A, Moss B. 2002. Cloning the vaccinia virus genome as a bacterial artificial chromosome in Escherichia coli and recovery of infectious virus in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 99:12415–12420. 10.1073/pnas.192420599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cottingham MG, Andersen RF, Spencer AJ, Saurya S, Furze J, Hill AV, Gilbert SC. 2008. Recombination-mediated genetic engineering of a bacterial artificial chromosome clone of modified vaccinia virus Ankara (MVA). PLoS One 3:e1638. 10.1371/journal.pone.0001638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth SJ, Hoper D, Beer M, Feineis S, Tischer BK, Osterrieder N. 2011. Recovery of infectious virus from full-length cowpox virus (CPXV) DNA cloned as a bacterial artificial chromosome (BAC). Vet. Res. 42:3. 10.1186/1297-9716-42-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meisinger-Henschel C, Spath M, Lukassen S, Wolferstatter M, Kachelriess H, Baur K, Dirmeier U, Wagner M, Chaplin P, Suter M, Hausmann J. 2010. Introduction of the six major genomic deletions of modified vaccinia virus Ankara (MVA) into the parental vaccinia virus is not sufficient to reproduce an MVA-like phenotype in cell culture and in mice. J. Virol. 84:9907–9919. 10.1128/JVI.00756-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 18.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 19.Yao XD, Evans DH. 2001. Effects of DNA structure and homology length on vaccinia virus recombination. J. Virol. 75:6923–6932. 10.1128/JVI.75.15.6923-6932.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99:7877–7882. 10.1073/pnas.082243699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakrabarti S, Sisler JR, Moss B. 1997. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 23:1094–1097 [DOI] [PubMed] [Google Scholar]
- 22.Sambrook JJ, Russell DDW. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 23.Lefkowitz EJ, Upton C, Changayil SS, Buck C, Traktman P, Buller RM. 2005. Poxvirus Bioinformatics Resource Center: a comprehensive poxviridae informational and analytical resource. Nucleic Acids Res. 33:D311–D316. 10.1093/nar/gki110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127–2140. 10.1128/JVI.80.5.2127-2140.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Vliet K, Mohamed MR, Zhang L, Villa NY, Werden SJ, Liu J, McFadden G. 2009. Poxvirus proteomics and virus-host protein interactions. Microbiol. Mol. Biol. Rev. 73:730–749. 10.1128/MMBR.00026-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoder JD, Chen TS, Gagnier CR, Vemulapalli S, Maier CS, Hruby DE. 2006. Pox proteomics: mass spectrometry analysis and identification of vaccinia virion proteins. Virol. J. 3:10. 10.1186/1743-422X-3-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roth SJ, Klopfleisch R, Osterrieder N, Tischer BK. 2012. Cowpox virus serpin CrmA is necessary but not sufficient for the red pock phenotype on chicken chorioallantoic membranes. Virus Res. 163:254–261. 10.1016/j.virusres.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 28.Yuwen H, Cox JH, Yewdell JR, Bennink JR, Moss B. 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195:732–744. 10.1006/viro.1993.1424 [DOI] [PubMed] [Google Scholar]
- 29.Beattie E, Kauffman EB, Martinez H, Perkus ME, Jacobs BL, Paoletti E, Tartaglia J. 1996. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 12:89–94. 10.1007/BF00370005 [DOI] [PubMed] [Google Scholar]
- 30.Hornemann S, Harlin O, Staib C, Kisling S, Erfle V, Kaspers B, Hacker G, Sutter G. 2003. Replication of modified vaccinia virus Ankara in primary chicken embryo fibroblasts requires expression of the interferon resistance gene E3L. J. Virol. 77:8394–8407. 10.1128/JVI.77.15.8394-8407.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233–247. 10.1016/j.virol.2006.08.025 [DOI] [PubMed] [Google Scholar]
- 32.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590–7600. 10.1128/JVI.77.13.7590-7600.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Da Fonseca F, Moss B. 2003. Poxvirus DNA topoisomerase knockout mutant exhibits decreased infectivity associated with reduced early transcription. Proc. Natl. Acad. Sci. U. S. A. 100:11291–11296. 10.1073/pnas.1534874100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parrish S, Moss B. 2006. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J. Virol. 80:553–561. 10.1128/JVI.80.2.553-561.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang WH, Wilcock D, Smith GL. 2000. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 74:11654–11662. 10.1128/JVI.74.24.11654-11662.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blasco R, Moss B. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 65:5910–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domi A, Weisberg AS, Moss B. 2008. Vaccinia virus E2L null mutants exhibit a major reduction in extracellular virion formation and virus spread. J. Virol. 82:4215–4226. 10.1128/JVI.00037-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sood CL, Ward JM, Moss B. 2008. Vaccinia virus encodes I5, a small hydrophobic virion membrane protein that enhances replication and virulence in mice. J. Virol. 82:10071–10078. 10.1128/JVI.01355-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zajac P, Spehner D, Drillien R. 1995. The vaccinia virus J5L open reading frame encodes a polypeptide expressed late during infection and required for viral multiplication. Virus Res. 37:163–173. 10.1016/0168-1702(95)00025-L [DOI] [PubMed] [Google Scholar]
- 40.Liu K, Lemon B, Traktman P. 1995. The dual-specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J. Virol. 69:7823–7834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Fonseca FG, Wolffe EJ, Weisberg A, Moss B. 2000. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J. Virol. 74:7518–7528. 10.1128/JVI.74.16.7518-7528.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams O, Wolffe EJ, Weisberg AS, Merchlinsky M. 1999. Vaccinia virus WR gene A5L is required for morphogenesis of mature virions. J. Virol. 73:4590–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Betakova T, Wolffe EJ, Moss B. 2000. The vaccinia virus A14.5L gene encodes a hydrophobic 53-amino-acid virion membrane protein that enhances virulence in mice and is conserved among vertebrate poxviruses. J. Virol. 74:4085–4092. 10.1128/JVI.74.9.4085-4092.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia AD, Moss B. 2001. Repression of vaccinia virus Holliday junction resolvase inhibits processing of viral DNA into unit-length genomes. J. Virol. 75:6460–6471. 10.1128/JVI.75.14.6460-6471.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McIntosh AA, Smith GL. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]