

Abstract
Apolipoprotein L1 (APOL1) is a major component of the human innate immune response against African trypanosomes. Although the mechanism of the trypanolytic activity of circulating APOL1 has been recently clarified, the intracellular function(s) of APOL1 in human cells remains poorly defined. Like that of many genes linked to host immunity, APOL1 expression is induced by proinflammatory cytokines gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α). Additionally, IFN-γ-polarized macrophages that potently restrict HIV-1 replication express APOL1, which suggests that APOL1 may contribute to HIV-1 suppression. Here, we report that APOL1 inhibits HIV-1 replication by multiple mechanisms. We found that APOL1 protein targeted HIV-1 Gag for degradation by the endolysosomal pathway. Interestingly, we found that APOL1 stimulated both endocytosis and lysosomal biogenesis by promoting nuclear localization of transcription factor EB (TFEB) and expression of TFEB target genes. Moreover, we demonstrated that APOL1 depletes cellular viral accessory protein Vif, which counteracts the host restriction factor APOBEC3G, via a pathway involving degradation of Vif in lysosomes and by secretion of Vif in microvesicles. As a result of Vif depletion by APOL1, APOBEC3G was not degraded and reduced infectivity of progeny virions. In support of this model, we also showed that endogenous expression of APOL1 in differentiated U937 monocytic cells stimulated with IFN-γ resulted in a reduced production of virus particles. This finding supports the hypothesis that induction of APOL1 contributes to HIV-1 suppression in differentiated monocytes. Deciphering the precise mechanism of APOL1-mediated HIV-1 restriction may facilitate the design of unique therapeutics to target HIV-1 replication.
INTRODUCTION
Human apolipoprotein L1 (APOL1) is the product of a member of a family of six APOL genes grouped on chromosome 22 in the region encompassing bands q12.3 to q13.1 (1–4). Interestingly, the APOL1 gene is located in the vicinity of a cluster of restriction factor APOBEC3 genes (5) known to control the expression of endogenous retroelements and retroviruses (6). Among primates, only humans and gorillas express functional APOL1 (7), although different sets of APOL genes have been found in other primates (8). APOL1 is the only protein of the APOL family that is secreted into the bloodstream (9), where it associates with a fraction of high-density lipoprotein (HDL3) particles and protects against Trypanosoma brucei infections (7, 10, 11). The HDL3-APOL1 complex is endocytosed by the parasite and delivered to the lysosome. The acidic condition of the lysosome triggers conformational changes in APOL1 that allow its binding to the lysosomal membrane and formation of anion channels causing osmotic swelling that kills the parasite. In response, Trypanosoma brucei rhodesiense evades APOL1-dependent lysis by producing serum resistance-associated (SRA) protein that inactivates APOL1 (12). To escape inactivation by a parasite, APOL1 G1 and G2 variants emerged with mutations that prevent binding of SRA and inactivation of APOL1 (12). Unfortunately, APOL1 alleles that protect against T. brucei infections are highly associated with increased risk for the development of certain types of kidney diseases, including HIV-associated nephropathy (HIVAN), which almost exclusively affects people of African descent (13, 14). The intracellular function of APOL1 in mammalian cells is not well understood. As a member of the family of BH3-only proteins, APOL1 may interact with the family of Bcl2 proteins to help regulate their function in autophagy and apoptosis (15, 16). APOL1 is also upregulated by proinflammatory cytokines gamma interferon (IFN-γ) and tumor necrosis factor alpha (TNF-α) (3, 16). The fact that APOL1 levels are strongly increased in IFN-γ-polarized M1 macrophages that effectively restrict productive HIV-1 infection (17–19) prompted us to investigate whether APOL1 affects HIV-1 replication.
In this article, we report that APOL1 displays anti-HIV-1 activity in part by inhibition of HIV-1 transcription and by degradation of HIV-1 Gag in the endolysosomal compartment expanded through the activation of the transcription factor EB (TFEB). These events result in extensive degradation of HIV-1 Gag and viral accessory proteins that target host restriction factors. Specifically, APOL1-mediated depletion of Vif resulted in restoration of APOBEC3G (A3G) levels and decreased infectivity of progeny virions. We also demonstrate that IFN-γ stimulated expression of endogenous APOL1 in human primary macrophages. Expression of endogenous APOL1 in differentiated monocytic U937 cells reduced production of HIV-1 particles. These results delineate a unique mechanism by which APOL1 inhibits HIV-1 replication.
MATERIALS AND METHODS
Tissue culture and transfections.
293T and TZM-bl cells were propagated in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). SupT1, Jurkat, THP-1, and U937 cells were maintained in RPMI medium with 10% FBS; 293T cells were transfected using PolyFect (Qiagen) according to the manufacturer's recommendations. SilencerSelect small interfering RNAs (siRNAs; Life Technologies) were transfected using Lipofectamine RNAiMAX transfection reagent (Life Technologies). Transfections were performed in 6-well plates. Briefly, for siRNA experiments, 293T cells were seeded at 120,000 cells/well on day 1. On day 2, the cells were transfected with siRNA, and on day 3, transfections were repeated. On day 4, the cells were transfected with plasmid DNAs (total 2 μg of DNA/well) using PolyFect. Total DNA in transfections was balanced with pcDNA3.1. Twenty hours later (day 5), culture media were collected, filtered through 0.45-μm syringe filters, and ultracentrifuged to pellet the virus. Cell lysates were prepared using complete lysis-M buffer supplemented with protease inhibitors (Roche) and clarified by centrifugation. Cell lysates (30 to 50 μg) and pelleted virions were mixed with 2× Laemmli sample buffer, boiled, and separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE). Proteins transferred to nitrocellulose membranes were detected using SuperSignal West Dura chemiluminescence system (Pierce).
Preparation of human primary macrophages and differentiation into M1 macrophages.
Human primary monocytes were obtained from peripheral blood mononuclear cells (PBMCs) of healthy donors. Briefly, PBMCs were isolated using Ficoll-Paque PREMIUM (GE Healthcare), and monocytes were selected by plastic adherence. The cells were cultured in RPMI medium supplemented with 10% human AB serum (Lonza), 100 U/ml of penicillin-streptomycin, 2 mM glutamine, 1× nonessential amino acids, and 1 mM sodium pyruvate (Life Technologies) and then allowed to differentiate for 7 days. Macrophages were subsequently polarized into M1 cells by 18 h of stimulation with IFN-γ (100 ng/ml) (Cell Signaling). Macrophages were also stimulated with IFN-α (100 ng/ml) (Cell Signaling), as indicated below.
Generation of stable U937 cell lines expressing control and APOL1 shRNAs.
APOL1 short hairpin RNA (shRNA) and control shRNA lentiviral particles were purchased from Santa Cruz Biotechnology. U937 cells were incubated for 24 h with lentiviral particles in the presence of 5 μg/ml of Polybrene. After an additional 48 h, transduced cells expressing shRNA were selected with puromycin (1 μg/ml) for 7 days. Clones obtained by serial dilutions were expanded and incubated with IFN-γ (20 ng/ml) to stimulate APOL1 expression. After 24 h, the cells were tested by real-time PCR for the expression levels of APOL1 transcripts using APOL1-specific primers (OriGene) and by immunoblotting for the expression of APOL1 protein. Clones U937-control shRNA and U937-APOL1 shRNA were selected and used in our studies.
Virus preparation and infectivity assay.
Virus stocks for infection were prepared by transfecting 293T cells as described previously (20). HIV-1-green fluorescent protein (HIV-1-GFP) reporter virus was generated by transfecting 293T cells with the transfer vector pHR′-CMV-EGFP and expression vectors CMV-gag-pol and CMV-VSV-G (pLenti-P2A and pLenti-P2B, respectively; Applied Biological Materials). HIV-1 reporter and infectious viruses were titrated using a quantitative PCR (qPCR) lentivirus titration kit (Applied Biological Materials).
For Western blot analysis of viral proteins, filtered (0.45-μm filter) supernatants were concentrated by ultracentrifugation at 100,000 × g for 1 h. Infectivity assays were performed using TZM-bl indicator cells (21).
Expression vectors, siRNAs, antibodies, and immunoblotting.
HIV-1proviral constructs were obtained through the NIH AIDS Research and Reference Reagent program, Division of AIDS, NIAID, NIH. 1GA plasmid expressing Gag myristoylation mutant NL4-3 (22) was obtained from E. Freed (NCI, Frederick, MD). Plasmids expressing vif-negative NL4-3, NL-A1, and A3G-HA and rabbit anti-Vpu antibodies were obtained from K. Strebel (NIAID, NIH, Bethesda, MD). pNL-A1 is a subgenomic expression vector that expresses HIV-1 proteins except for Gag and Pol (23). APOL1 expression vector was purchased from OriGene. APOL1 constructs tagged at the C terminus with myc, GFP, or Ds-Red were generated in our laboratory. SilencerSelect predesigned siRNAs against Eps15, Rab7, Stx7, VAMP7, and Stx17 were purchased from Life Technologies and were used according to the manufacturer's protocol. The following antibodies were used: anti-Eps15, anti-Rab7, anti-lysosome-associated membrane protein 1 (anti-LAMP1), and anti-IRF-1 (Cell Signaling); anti-Stx7 (R&D Systems); anti-VAMP7 (ProSci); anti-Stx17 (Abgent), anti-APOL1, antiactin, and antitubulin (Sigma-Aldrich); anti-Alix, anti-CD81, anti-CD63, and anti-APOBEC3A (Santa Cruz Biotechnology); anti-Samhd1 (Abcam); and anti-Tsg101 (GeneTex). TFEB-GFP was purchased from Addgene. Antibodies against Vif (2221), Nef (2949), and Gag p17 and p24 were obtained through the NIH AIDS Research and Reference Reagent program, Division of AIDS, NIAID, NIH. Western blot analysis was performed as described previously (20).
Confocal microscopy.
293T cells were transfected on a 6-well plate, and 5 h after transfection, the cells were trypsinized and seeded in 2-well chamber slides (Lab-Tek II) precoated with collagen. The cells were transfected with plasmids expressing HIV-1 89.6 and APOL1-myc (see Fig. 1C) or TFEB-GFP and APOL1-dsRed (see Fig. 6). One microgram of HIV-1 89.6 and TFEB-GFP and 0.5 μg of APOL1 vector DNA were used for transfection (adjusted to 2 μg with pcDNA3). Lysosome staining was performed before cell fixation by incubation with 100 nM LysoTracker red DND-99 (Invitrogen) for 30 min at 37°C. For staining of intracellular Gag and APOL1-myc, cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. After washing, the cells were permeabilized with 0.3% Triton X-100 in fluorescence-activated cell sorting (FACS) buffer (5% normal goat serum and 1% bovine serum albumin [BSA] in phosphate-buffered saline [PBS]) for 5 min and blocked in FACS buffer for 30 min at room temperature. Gag and APOL1-myc were stained with primary rabbit anti-p17 and mouse anti-myc, respectively, for 1 h at room temperature, washed, and incubated with goat anti-rabbit Alexa Fluor 488 (for Gag staining) and goat anti-mouse Alexa Fluor 647 (for APOL1-myc staining) for 1 h at room temperature, washed, and mounted with SlowFade with 4′,6-diamidino-2-phenylindole DAPI (Invitrogen). The cells were observed under a laser scanning confocal microscope (Nikon TE2000) and images processed using Adobe Photoshop.
FIG 1.

APOL1 inhibits HIV-1production and infectivity. (A) 293T cells were seeded in 6-well plates and transfected with the indicated combination HIV-1 proviral construct DNA (1 μg), A3G (1 μg), and APOL1 (1 μg) expression vectors. Twenty hours after transfection, the whole-cell lysates (50 μg/lane) and pelleted viruses were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by immunoblotting for HIV-1 protein expression. (B) APOL1 inhibits intracellular accumulation of HIV-1 Vpu. 293T cells were cotransfected with the indicated HIV-1 proviral DNA and APOL1 or pcDNA3.1 vectors. The whole-cell lysates were analyzed by immunoblotting for the expression of Vpu, APOL1, and actin. (C) APOL1 depletes cellular HIV-1 Gag and stimulates accumulation of lysosomes. 293T cells were transfected with 1 μg of HIV-1 89.6 and 0.5 μg of pcDNA3.1 (control) or 0.5 μg of APOL1-myc vector (APOL1). Five hours later, the cells were trypsinized and a fraction of transfected cells was transferred into 2-well chamber dishes. Twenty hours after transfection, the cells were incubated for 30 min with LysoTracker red (100 nM), followed by fixation, permeabilization, and staining with rabbit anti-p17 Gag and goat anti-rabbit Alexa 488. For APOL1 labeling, the permeabilized cells were stained with mouse anti-myc and goat anti-mouse Alexa 647 antibodies. The cells were mounted with SlowFade with DAPI to stain nuclei (blue) and subjected to confocal microscopy. Number of lysosomes/cell (red fluorescent puncta) were determined and are presented as means ± SDs from 10 control cells and from 10 cells expressing APOL1 (pink). (D) APOL1 reduces HIV-1 infectivity. 293T cells were transfected with NL4-3 or JR-CSF alone or in combination with A3G and APOL1 (1 μg of DNA each). Cell supernatants containing released viruses were filtered, and equal amounts of HIV-1 (normalized by p24 ELISA) were added to TZM-bl indicator cells in triplicate. The relative infectivity of viruses was determined by a luciferase assay. Infectivity of the virus produced by cells transfected with HIV-1 proviral DNA only was set as 1.0 (control). Error bars indicate SDs.
FIG 6.

APOL1 promotes nuclear localization of TFEB and stimulates expression of lysosomal genes. (A) 293T cells were transfected with TFEB-GFP (1 μg) and APOL1-DsRed (APOL1) (1 μg) or pcDNA3 (control) (1 μg), and 5 h after transfection, a fraction of transfected cells was transferred into 2-well chamber dishes. Twenty-four hours after transfection, the cells were fixed, mounted in SlowFade with DAPI to stain nuclei (blue), and subjected to confocal microscopy. (B) Quantification of the subcellular distribution of TFEB-GFP in the absence (control) and presence of APOL1-DsRed (APOL1). The percentages of cells with cytoplasmic (C) or nuclear (N) localization of TFEB-GFP or both (C + N) is shown. ***, P < 0.001 for comparison of differences between nuclear TFEB-GFP ([N] + [C + N]) in cells transfected with TFEB-GFP (bar 1) and TFEB-GFP with APOL1-DsRed (bar 3). Nuclear localization of TFEB in the presence of 100 μM chloroquine (CLQ) was used as a positive control (36). (C) Real-time PCR analysis of TFEB target gene expression. 293T cells were transfected in triplicate with indicated increasing amounts of APOL1, total RNA was isolated, and synthesized cDNA was subjected to qPCR to detect expression of endogenous LAMP1, LAMP2, CD63, and TFEB RNAs (64). Gene expression was normalized to GAPDH mRNA expression. Error bars indicate SDs.
Endocytosis assays and FACS analysis. (i) Fluorescent dextran uptake.
Eighteen hours after transfection, 293T cells were incubated in complete medium (DMEM–10% FBS) with 250 μg/ml of lysine-fixable 10-kDa Oregon Green 488 dextran (Life Technologies) for 4 h at 37°C, followed by 2 h in dextran-free complete medium. After washing with PBS, live cells were imaged on a fluorescence microscope using standard filter sets. The cells were subsequently trypsinized, washed, and analyzed on a FACSCalibur (Becton, Dickinson). Data analysis was performed by using CellQuest Pro software (Becton, Dickinson).
(ii) DQ-Green BSA uptake.
Conditions for the DQ-Green BSA uptake assay were identical to those described for dextran uptake. Since DQ-Green BSA acquires fluorescence only after dequenching through proteolytic enzyme cleavage, this assay visualizes lysosomes with proteolytic activity. To establish the size of lysosomal fraction positive for DQ-Green BSA, 100 nM LysoTracker red DND-99 (Life Technologies) was added to the cell culture media 30 min before the end of a 2-h chase in DQ-Green BSA-free media. After washing with PBS, the cells were imaged on a fluorescence microscope and monitored by trypsinization, washing, and FACS analysis as described above.
Lysosomal-pool analysis.
Lysosomal-pool analysis was conducted by incubating 293T cells with 100 nM LysoTracker red DND-99 for 30 min at 37°C and then washing them with PBS. Cells were then trypsinized, washed with PBS, and analyzed by FACS. Increasing pools of lysosomes in cells expressing APOL1 correlated with the increased expression of lysosomal marker LAMP1, as determined by immunoblotting with rabbit anti-LAMP1 antibody. Protein band intensities were quantitated by densitometry (Bio-Rad).
Preparation and characterization of microvesicles.
293T cells were grown in complete medium supplemented with 10% FBS depleted of endogenous exosomes (also called microvesicles) as described previously (20). Microvesicles were collected by filtering the supernatant through a 0.45-μm polyvinylidene difluoride (PVDF) filter, diluted with PBS, and ultracentrifuged at 100,000 × g for 1 h. The pelleted microvesicles were lysed in SDS-PAGE loading buffer and analyzed by immunoblotting for exosome-specific markers (20).
Quantification of HIV-1 Gag RNA.
Total cellular RNA was extracted using an RNeasy kit (Qiagen), and the cDNA was prepared using an iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed on CFX96 (Bio-Rad) using conditions and HIV-1 p24 primers as described previously (24). HIV-1 Gag RNA levels were normalized against GAPDH mRNA.
HIV-1 Gag pulse-chase analysis.
293T cells were transfected with 1GA NL4-3 and APOL1 or pcDNA3 constructs (HIV-1DNA/APOL1 DNA ratio, 2:1) and subjected to [35S]Met-Cys pulse-chase analysis as described previously (25). Gag myristoylation mutant 1GA NL4-3 did not released virions, and thus, all HIV-1 Gag accumulated intracellularly (22). HIV-1 Gag was immunoprecipitated from cell lysates collected at various chase times using rabbit anti-p17 antibody (NIH AIDS Reagent Program) and protein A/G-Plus (Santa Cruz Biotechnology). Complexes were resolved by 10% SDS-PAGE, and gels were soaked for 90 min in En3Hance (Perkin-Elmer), dried, and exposed to X-ray film. Intensities of the Gag bands were quantified using Bio-Rad ChemiDoc Imager and Quantity One software. Gag half-life values (t1/2) in cells cotransfected with APOL1 or pcDNA3 were calculated using regression analysis.
Statistical analysis.
All experiments were repeated at least twice, with similar results. Unless otherwise indicated, the variation in one experiment was expressed by calculating standard deviation (SD) from the triplicates and the result presented as the mean value ± the SD.
RESULTS
APOL1 inhibits HIV-1 replication.
Since APOL1 expression is strongly upregulated in human primary macrophages by IFN-γ (18, 26), we investigated the role of APOL1 in antiviral activity. To test the anti-HIV activity of APOL1, we analyzed the production of T cell-tropic (NL4-3 and NL4-3 ΔVif), macrophage-tropic (AD8 and JR-CSF), and dually tropic (89.6) HIV-1 in 293T cells cotransfected with an APOL1 expression vector. As determined by densitometric analysis, APOL1 reduced the steady-state levels of HIV-1 Gag p55 and p24 by over 90% in cells transfected with all tested HIV-1 constructs (Fig. 1A). Reduction of the levels of pelletable virions (measured by p24 enzyme-linked immunosorbent assay [ELISA]) ranged approximately from 60 to 70% for NL4-3 and AD8 to more than 90% for HIV-1 JR-CSF and HIV-1 89.6.
In addition to an APOL1-mediated reduction of the steady-state levels of Gag, the levels of Vif and Nef were reduced by 70 to 90% (Fig. 1A). Similarly, APOL1 reduced the steady-state levels of Vpu, with about 60% reduction of AD8 Vpu and over 95% reduction of Vpu expressed by NL4-3, JR-CSF, and 89.6 (Fig. 1B). These results suggest that APOL1 may reduce HIV-1 infectivity by decreasing levels of viral factors essential for optimal virus infectivity. Thus, the observed reduction of cellular Vif could potentially lead to increased incorporation of A3G into budding virions and reduce virus infectivity. To test this possibility, virus constructs were cotransfected with APOL1 and/or A3G (Fig. 1A). In the absence of APOL1, A3G was notably depleted from cells, with the exception of cells expressing vif-negative NL4-3 (NL4-3ΔVif). However, coexpression of APOL1 and Vif-positive HIV-1 constructs prevented depletion of A3G, resulting in A3G levels similar to that observed in cells expressing NL4-3 ΔVif. Thus, blocking of A3G depletion by APOL1 in cells expressing Vif-positive HIV-1 may decrease the infectivity of progeny virions. Detection of elevated levels of A3G in pelleted virions produced in the presence of APOL1 (Fig. 1A) supports this hypothesis.
Confocal microscopy confirmed that in 293T cells transfected with APOL1 DNA, the 89.6 Gag signal was nearly undetectable (Fig. 1C). Interestingly, expression of APOL1 coincided with an increased signal from LysoTracker red (Fig. 1C), suggesting that APOL1 increases the lysosome pool in transfected cells. Indeed, quantification of LysoTracker-labeled lysosomes showed about a 2.2-fold increase in number of lysosomes in 293 T cells expressing APOL1, compared to control cells transfected with pcDNA3 (Fig. 1C).
APOL1 reduces HIV-1 infectivity.
Increased levels of A3G detected in cells expressing Vif-positive HIV-1 and APOL1 was accompanied by accumulation of A3G in pelleted virions, suggesting that virions would be less infectious. Thus, to determine the effects of APOL1 expression on HIV-1 infectivity, virus supernatants were normalized by p24 and tested using TZM-bl indicator cells (Fig. 1D). A3G reduced infectivity of wild-type NL4-3 by 67% and JR-CSF by 85%. The lower infectivity of JR-CSF may be related to higher levels of A3G detected in pelleted JR-CSF virions (Fig. 1A). The values for the inhibitory effect of APOL1 on the infectivity of wild-type NL4-3 and JR-CSF were 72 and 77%, respectively. Further, in the presence of A3G and APOL1, the infectivity of NL4-3 virus was decreased to about 9% of control NL4-3 values, while JR-CSF was essentially noninfectious. Although the precise mechanism by which APOL1 reduces HIV-1 infectivity is presently unknown, we speculate that depletion of Vif and Nef by APOL1 (Fig. 1) contributes to reduced virus infectivity.
APOL1 inhibits HIV-1 RNA expression.
The observed reduction of HIV-1 protein expression in the presence of APOL1 may, at least in part, result from inhibition of HIV-1 transcription. Thus, to assess an effect of APOL1 on the levels of intracellular HIV-1Gag RNA, 293T cells were transfected with 1GA NL4-3 defective in viral particle assembly and release (22). This approach simplifies the analysis of HIV-1 RNA, since the cells release no extracellular viral RNA. Real-time PCR analysis showed reduction of intracellular HIV-1 Gag RNA accumulation in an APOL1 dose-dependent manner (Fig. 2A), suggesting that APOL1 inhibits HIV-1 transcription. No detectable changes were observed in the expression of cellular actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA (data not shown), and only a moderate reduction of EGFP expressed from a cotransfected CMV-EGFP vector was observed, indicating that the observed inhibition of HIV-1 Gag expression does not result from nonspecific global suppression of protein synthesis by APOL1 (Fig. 2B). In fact, we observed accumulation of LAMP1 protein (see Fig. 5D) and the expression of several lysosome-associated mRNAs (see Fig. 6C) by APOL1.
FIG 2.

APOL1 inhibits HIV-1 Gag RNA expression. (A) 293T cells were transfected in triplicate with 1GA NL4-3 (22) in the absence or presence of the indicated amounts of APOL1 DNA expression vector. After 24 h, the total cellular RNA was extracted and cDNA synthesized. The expression of HIV-1 Gag RNA was analyzed by real-time qPCR analysis using HIV-1 p24 primer sets (24). The HIV-1 Gag RNA levels were normalized against GAPDH mRNA. (B) 293T cells were transfected with HIV-1 proviral construct DNA (1 μg), pEGFP-N1 (1 μg), and the indicated amounts of APOL1 expression vector. Twenty hours after transfection, the whole-cell lysates (40 μg per lane) and pelleted viruses were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by immunoblotting for the expression of Gag, APOL1, enhanced GFP (EGFP), and actin. Intensities of Gag and EGFP signals were obtained by densitometry. The expression of total Gag and EGFP was normalized against the expression of actin and presented as Gag/actin and EGFP/actin ratios. Error bars indicate SDs.
FIG 5.

APOL1 stimulates endocytosis and increases lysosome accumulation. (A) 293T cells in 6-well plates were transfected with pcDNA3.1 (control) (1 μg) or APOL1 expression vector (APOL1) (1 μg). Transfection efficiency in 293T cells was determined by transfecting the APOL1-GFP construct (1 μg) and counting GFP-positive cells by fluorescence microscopy. We observed more than 90% GFP-positive cells (data not shown). Sixteen hours after transfection, the cells were incubated with Oregon Green 488 dextran (250 μg/ml) for 4 h at 37°C, followed by 2 h in dextran-free complete medium. After a washing with PBS, live cells were imaged by fluorescence microscopy. To quantitate the uptake of fluorescently labeled dextran by cells, the cells were trypsinized, washed, and analyzed by FACS. A representative experiment is shown. (B) 293T cells transfected as for panel A were incubated with DQ-Green BSA (250 μg/ml) for 2 h at 37°C followed by 1 h in DQ-Green BSA-free complete medium. After a washing, the cells were incubated for 30 min in medium containing LysoTracker red (100 nM) and then washed, and images were obtained by fluorescence microscopy. (C) The uptake of DQ-Green BSA into lysosomes and the mass of LysoTracker red-positive lysosomes were quantitated by FACS as described for panel B, with the exception that DQ-Green BSA was used at 25 μg/ml. (D) 293T cells were transfected with increasing amounts of APOL1 vector (transfected DNA was balanced with pcDNA3.1), and after 18 h, the cell lysates were analyzed by immunoblotting for the expression of LAMP1, APOL1, and actin. Intensities of protein signals were obtained from densitometric scanning. The expression of LAMP1 was normalized against the expression of actin and presented as the LAMP1/actin ratio. In cells transfected with pcDNA3.1 only, the LAMP1/actin ratio was used as a reference.
APOL1 reduces stability of HIV-1 Gag protein.
Inhibition of HIV-1 transcription by APOL1 does not rule out the possibility that APOL1 also affects the stability of HIV-1 Gag, especially when the levels of APOL1 only moderately inhibit HIV-1 transcription. To determine whether APOL1 stimulates HIV-1 Gag degradation, we performed a pulse-chase stability assay (Fig. 3) as described previously (25). In this experiment, 293T cells were transfected with 1GA NL4-3 and APOL1expression vector at concentrations that inhibit HIV-1 Gag RNA expression by about 60% (Fig. 2A). The half-life of HIV-1 Gag proteins (p55 and p24) in control cells transfected with pcDNA3 was 3.6 h (Fig. 3B), similar to that in previous reports. In contrast, Gag was rapidly degraded in the presence of APOL1, with an estimated half-life of about 1.1 h. In addition, about a 40% decrease in total Gag synthesis was observed during the 30-min [35S]methionine-cysteine pulse period in cells transfected with APOL1 (Fig. 3A), consistent with Gag expression also being affected at the transcriptional level. Together, these results suggest that in addition to inhibition of HIV-1 Gag expression at the transcriptional level (Fig. 2A), APOL1 targets Gag for degradation.
FIG 3.

APOL1 reduces stability of HIV-1 Gag. (A) 293T cells were transfected with 1GA NL4-3 and APOL1 expression vector at a ratio of 2:1. Control cells were transfected with 1GA NL4-3 and pcDNA3. Eighteen hours after transfection, cells were labeled for 30 min with [35S]methionine-cysteine (25). Cell lysates were collected at the indicated chase times, and HIV-1 Gag was immunoprecipitated using rabbit anti-p17 antibody and protein A/G-Plus. Complexes were resolved by 10% SDS-PAGE and gels exposed to X-ray film. (B) Intensities of the Gag p55 and p24 bands were quantified using Bio-Rad ChemiDoc Imager and Quantity One software. Total Gag half-life values were normalized to chase time zero values. Gag half-life values in cells cotransfected with APOL1 or pcDNA3 were calculated using regression analysis.
APOL1 depletes Vif by secretion in microvesicles and lysosomal degradation.
Depletion of Vif by APOL1 prevented A3G degradation and thus could lead to increased encapsidation of A3G and reduced infectivity of HIV-1 virions. To examine the mechanism by which APOL1 depletes Vif from cells, we used the subgenomic HIV-1 vector pNL-A1, which expresses Vif (23). The cellular accumulation of Vif was reduced over 95% in the presence of APOL1 (Fig. 4A), which could in part result from decreased HIV-1 transcription (Fig. 2). However, in cells expressing APOL1 and treated with the calcium chelator BAPTA-AM [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)], which sequesters calcium and inhibits fusion events in the endolysosomal and exocytic pathways (27), the level of intracellular Vif was upregulated and reached about 40% of the Vif level detected in the absence of APOL1 (Fig. 4A), suggesting that the reduction of cellular Vif is only partly due to inhibition of HIV-1 transcription by APOL1. However, inhibition of lysosomal degradation by cell-permeative lysosomal protease inhibitors E64d, an inhibitor of cathepsins B, H, and L, and pepstatin A, an inhibitor of cathepsins D and E (28) (Fig. 4A), was less effective than BAPTA-AM treatment and resulted in a moderate increase in intracellular Vif level in cells expressing APOL1. These results led us to hypothesize that only a fraction of Vif was degraded in lysosomes, while a majority of intracellular Vif could be released from cells using an exocytic pathway inhibited by BAPTA-AM. To investigate this possibility, we isolated microvesicles released into culture media by cells transfected with Vif in the absence or presence of APOL1. We observed that although Vif accumulated intracellularly to high levels in the absence of APOL1, it was not detectably released in microvesicles. However, in the presence of APOL1, a significant fraction of Vif was released in association with Alix and CD81-positive microvesicles (Fig. 4A, “Microvesicles”). Treatment with BAPTA-AM totally inhibited APOL1-mediated Vif release and resulted in intracellular accumulation of Vif. Treatment with E64d and pepstatin A was less efficient than treatment with BAPTA-AM in recovery of Vif in the presence of APOL1, but in contrast to BAPTA-AM, lysosomal inhibitors moderately stimulated release of Vif in microvesicles. To investigate further the contribution of the endolysosomal pathway in Vif depletion, we exploited the role that Rab7A plays in promoting fusion of late endosomes with lysosomes (29). Depletion of Rab7A with siRNA resulted in a 35% increase of the steady-state levels of Vif in the absence of APOL1 and partial recovery of Vif in the presence of APOL1 (Fig. 4B, “Cell”); this suggests that Vif is partly degraded by a Rab7A-dependent pathway. A partial recovery of Vif in the presence of APOL1 in Rab7A-depleted cells prompted us to examine if Vif was released in microvesicles. Indeed, in the presence of APOL1, Vif was secreted and cosedimented with microvesicle-specific markers Alix and Tsg101 (Fig. 4B, “Microvesicles”). We have also detected APOL1 released in association with these extracellular vesicles. Furthermore, APOL1 stimulated a 3- to 4-fold secretion of Vif by Rab7A knockdown cells, compared to Vif levels released by control cells expressing Rab7A (Fig. 4B). Interestingly, depletion of Rab7A resulted in Vif secretion even in the absence of APOL1, but at an approximately 3-fold-lower level. Taken together, these observations suggest that depletion of Rab7A, which suppresses fusion events between endosomes and lysosomes, may redirect endosomes to the plasma membrane for fusion and the release of Vif into the extracellular space.
FIG 4.
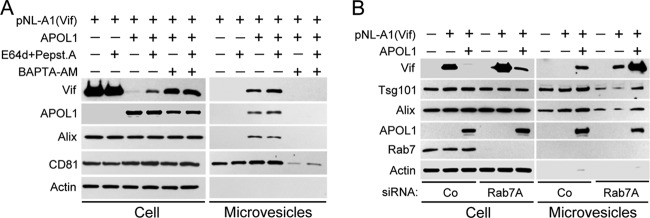
APOL1 depletes intracellular Vif by lysosomal degradation and stimulating its secretion in microvesicles. (A) 293T cells were transfected with Vif-expressing pNL-A1 vector alone or in combination with APOL1 expression vector. Five hours after transfection, the cells were treated with E64d (10 μg/ml) and pepstatin A (10 μg/ml). After 24 h, whole-cell lysates and pelleted microvesicles were separated by 10% SDS-PAGE and analyzed by Western blotting for the expression of Vif, APOL1, and microvesicle markers Alix and CD81. Secretion of microvesicles was inhibited by incubation of transfected cells with the calcium chelator BAPTA-AM (10 μM) for 16 h. (B) Depletion of Rab7A with siRNA inhibited lysosomal degradation of Vif and potentiated APOL1-mediated release of Vif in microvesicles. 293T cells were transfected twice with Rab7A siRNA, followed by transfection with pNL-A1 alone or with APOL1 vector. After 24 h, whole-cell lysates and pelleted microvesicles were separated by 10% SDS-PAGE and analyzed by Western blotting for the expression of Vif, APOL1, and Rab7 and microvesicle markers Alix and Tsg101. Co, control.
APOL1 stimulates endocytosis.
In a majority of cells, HIV-1 Gag was shown to assemble at the plasma membrane (30–33). Since APOL1 decreased steady-state levels of HIV-1 Gag (Fig. 1), we investigated whether APOL1 inhibited virus particle formation by rerouting Gag from the plasma membrane into the lysosomal compartment. We considered the possibility that APOL1 depletes Gag from the plasma membrane assembly sites by stimulating endocytosis. Thus, to investigate whether APOL1 stimulates endocytosis, we monitored internalization of Oregon Green 488 dextran, a marker of fluid-phase endocytosis (34). Fluorescence microscopy showed a significant increase in accumulation of fluorescent dextran in the endosomal-lysosomal compartment in the presence of APOL1 (Fig. 5A). FACS analysis confirmed a 3.9-fold increase in the uptake of fluorescent dextran in cells expressing APOL1 (Fig. 5A). To confirm this observation, we also measured the uptake of DQ-Green BSA, which labels enzymatically active lysosomes since self-quenched DQ-Green BSA fluoresces only when partly degraded in a lysosomes (35). In the presence of APOL1, the number of fluorescent puncta corresponding to DQ-Green BSA in lysosomes was increased 3.6-fold over control cells, as determined by FACS analysis (Fig. 5B and C). Moreover, in APOL1-expressing cells, about 84% of lysosomes were labeled with DQ-Green BSA during a 5-h incubation period; in contrast, only 35% of lysosomes colocalized with DQ-Green BSA in control cells (Fig. 5B). FACS analysis showed that in the presence of APOL1, the number of LysoTracker-labeled lysosomes increased about 3.2-fold over control cells (Fig. 5C). Stimulation of lysosome accumulation in the presence of APOL1 was indirectly confirmed by Western blotting, in which we observed an APOL1 concentration-dependent accumulation of lysosome-associated membrane protein 1 (LAMP1) (Fig. 5D).
APOL1 stimulates lysosomal biogenesis by promoting nuclear localization of TFEB and expression of TFEB target genes.
To investigate the mechanism of the APOL1-mediated expansion of the lysosomal compartment, we examined whether APOL1 stimulates lysosomal biogenesis. Most lysosomal genes exhibit coordinated transcriptional activation by TFEB, which translocates from the cytoplasm to the nucleus when activated (36–38). Using 293T cells transiently transfected with TFEB-GFP, we showed that approximately 75% of TFEB-GFP localized to the cytoplasm in the absence of APOL1 (Fig. 6B). We observed a dramatic difference when cells were transiently transfected with APOL1 and TFEB-GFP. Less than 10% of TFEB-GFP remained in the cytoplasm, and the majority of TFEB-GFP translocated into the nucleus (Fig. 6A and B). Real-time PCR analysis confirmed increased expression of TFEB target genes encoding lysosomal proteins LAMP1, LAMP2, and CD63 and a moderate expression of TFEB (Fig. 6C). Together, these results indicate that APOL1 stimulates lysosomal biogenesis, at least in part, via activation of TFEB and induction of the lysosomal transcriptional program.
A functional endocytic pathway is necessary for inhibition of HIV-1 replication by APOL1.
We hypothesized that APOL1-mediated stimulation of endocytosis inhibits HIV-1 particle production by diverting Gag from the plasma membrane assembly sites to the expanded pool of lysosomes. To test this hypothesis, we knocked down several genes that play critical roles in endocytosis by transfecting 293T cells with specific siRNAs. The cells were subsequently cotransfected with a doubly tropic HIV-1 (89.6) and APOL1 expression constructs, and the levels of intracellular Gag and released virions were analyzed by immunoblotting (Fig. 7). First, we evaluated the effect of depletion of Eps15, which inhibits clathrin-mediated endocytosis (39). In the absence of APOL1, depletion of Eps15 led to a modest (25 to 30%) increase in intracellular and virion-associated Gag (Fig. 7A). Similarly, depletion of Rab7 and syntaxin7 (Stx7), which localize to late endosomes (29, 40, 41), as well as VAM7, present on lysosomes (42), moderately increased virus production (Fig. 7B to D). In contrast, depletion of syntaxin 17 (Stx17), an autophagosomal SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) essential for fusion of autophagosomes with endosomes and lysosomes (43), had no effect on intracellular Gag and virus production (Fig. 7E). This observation suggests that in the absence of APOL1, Gag is not detectably targeted for degradation in lysosomes through an autophagosomal pathway. Together, these data indicate that in 293T cells a small fraction of synthesized Gag is endocytosed and degraded in lysosomes. However, in cells expressing APOL1, knockdown of Eps15, Stx7, Rab7, and VAMP7 stimulated virus production from 7.1-fold (Eps15 siRNA) to 11.4-fold (Stx7 siRNA), compared to virus produced by cells transfected with control siRNA and APOL1. In contrast, knockdown of Stx17 did not restore virus production (Fig. 7E), supporting the concept that macroautophagy does not play a significant role in APOL1-mediated degradation of Gag in 293T cells. Taken together, these results indicate that genes that play a crucial role in the endocytic pathway are necessary for APOL1-mediated suppression of HIV-1 replication.
FIG 7.

Inhibition of endocytosis by depletion of Eps15, Stx7, Rab7, or VAMP7 restores virus production in cells expressing APOL1. 293T cells were transfected on day 1 and day 2 with control siRNA (Co) or siRNAs that specifically target Eps15 (A), Stx7 (B), Rab7 (C), VAMP7 (D), or Stx17 (E). Twenty-four hours later, the cells were transfected with HIV-1 89.6 proviral DNA in the presence or absence of APOL1 vector. After an additional 24 h, pelleted virus from culture supernatants and corresponding cell lysates were collected and analyzed by immunoblotting for the expression of Gag, APOL1, and proteins targeted by siRNA. (E) Depletion of the autophagosomal marker Stx17 did not restore virus production by cells overexpressing APOL1.
APOL1 expression is inducible by IFN-γ in differentiated human monocytic U937 and THP-1 cells and in primary macrophages.
We next examined whether cells targeted by HIV-1 express endogenous APOL1. We found that APOL1 protein was not detectably expressed in unstimulated or PMA-stimulated SupT1 or Jurkat T cells (Fig. 8A). Moreover, treatment of PMA-stimulated T cells with IFN-γ, which was shown to induce APOL1 expression in human endothelial cells (16), did not stimulate accumulation of APOL1. Similarly, undifferentiated or PMA-differentiated monocytic THP-1 or U937 cells did not express APOL1. However, in contrast to T cells, treatment of PMA-differentiated THP-1 and U937 cells with IFN-γ stimulated expression of endogenous APOL1. To investigate whether human primary macrophages express APOL1, PBMC were isolated from the buffy coats of healthy blood donors and monocytes were isolated by plastic adherence and differentiated for 7 days into monocyte-derived macrophages (MDM) (17, 19, 44). MDM from the first donor were either left untreated or stimulated for 18 h with 2 different doses (25 or 50 ng/ml) of IFN-γ to polarize MDM into macrophages displaying the M1 phenotype (17, 19, 44). The expression of APOL1 was detected only in MDM stimulated with IFN-γ and not in resting MDM or PBMC (Fig. 8B, left side). As previously reported, expression of transcription factor IRF-1 was strongly stimulated in MDM treated with IFN-γ (45). To investigate the effect of IFN-α on APOL1 expression, MDM were stimulated with IFN-α or IFN-γ (Fig. 8B, right side). We also confirmed that untreated MDM do not express APOL1 (Fig. 8B). However, APOL1 was potently induced in MDM by IFN-γ. In comparison, treatment of MDM with IFN-α only marginally stimulated APOL1. However, IFN-α but not IFN-γ induced expression of Apobec3A (A3A), as recently reported (46). In contrast, the levels of Samhd1 in MDM did not change significantly upon treatment with IFN-α or IFN-γ, confirming recent observations (47). In conclusion, IFN-γ is a specific and potent stimulator of APOL1 expression in human primary macrophages.
FIG 8.

IFN-γ-stimulated expression of APOL1 in differentiated U937 monocytes contributes to inhibition of HIV-1 production. (A) APOL1 expression is inducible by IFN-γ in differentiated human monocytes (THP-1 and U937) but not in CD4+ T cells (SupT1 and Jurkat). The cells were stimulated for 24 h with PMA (50 ng/ml) or were left untreated (control). After a washing, PMA-treated cells were cultured in media alone (PMA) or were treated for another 24 h with IFN-γ (20 ng/ml) (PMA/IFN-γ). As an additional control, 293T cells were transfected with APOL1 vector (0.5 μg). Cell lysates (50 μg/lane) were analyzed by immunoblotting for the expression of APOL1 and actin. (B) (Left side) Seven-day differentiated MDM were either left untreated or stimulated for 18 h with 2 different doses (25 or 50 ng/ml) of IFN-γ. Cell lysate proteins (50 μg/lane) were resolved by 10% SDS-PAGE, and the levels of APOL1, IRF-1, and Samhd1 were analyzed by Western blotting. Expression of transcription factor IRF-1 served as a positive control for IFN-γ treatment (45). (Right side) Seven-day differentiated MDM were either left untreated (control) or stimulated for 18 h with IFN-α or IFN-γ at 100 ng/ml. Cell lysate proteins were resolved by 4 to 12% gradient SDS-PAGE, and the levels of APOL1, IRF-1, A3A, and Samhd1 were analyzed by Western blotting. (C) U937 clones expressing control shRNA (control) or APOL1 shRNA (APOL1) were stimulated for 24 h with PMA (50 ng/ml), followed by treatment with IFN-γ (20 ng/ml) as indicated. After 24 h, the cells were rinsed and exposed to HIV-1 89.6 (50 ng of p24 per 106 cells) pseudotyped with VSV-G. After 4 h of incubation, the cells were washed and cultivated for another 48 h, followed by collection of supernatants and cell lysates. Pelleted virus and lysates were analyzed by immunoblotting for the expression of HIV-1 Gag, APOL1, and actin. Virus production was determined by the expression of pelletable p24. Protein signals were obtained from densitometric scanning. Virus released from PMA-treated U937-CoshRNA cells (control) was used as a reference (100%).
Expression of endogenous APOL1 in PMA-differentiated U937 inhibits production of HIV-1.
To investigate whether endogenous APOL1 inhibits HIV-1 production in differentiated monocytes, we established U937 cell lines expressing APOL1 shRNA (U937-APOL1shRNA) or control shRNA (U937-CoshRNA). While APOL1 was induced by IFN-γ in PMA-differentiated U937-CoshRNA cells, expression of APOL1 in U937-APOL1shRNA cells stimulated with IFN-γ was reduced by approximately 84% compared to that in U937-CoshRNA cells (Fig. 8C). To examine whether endogenous APOL1 affected HIV-1 production, the cells were first stimulated with phorbol myristate acetate (PMA) and 24 h later were treated with IFN-γ to induce endogenous APOL1. Since IFN-γ treatment partly inhibits virus replication by downregulating HIV-1 entry receptors (48), U937 clones were infected with vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped HIV-1 NL4-3, and virus production was monitored by immunoblotting of pelletable Gag released into the cell supernatant. In the absence of IFN-γ, U937-CoshRNA and U937-APOL1shRNA cells produced almost identical amounts of Gag p24. Stimulation of U937-CoshRNA cells with IFN-γ resulted in a 53% reduction of virus production compared with that of unstimulated cells. In contrast, stimulation of U937-APOL1shRNA cells with IFN-γ reduced virus production by only 23% compared to that of unstimulated U937-APOL1shRNA cells. Under these conditions, we observed an approximately 6-fold reduction in APOL1 expression. Together, these results suggest that induction of APOL1 in U937 cells contributes to reduced virus production by the cells.
DISCUSSION
In this study, we demonstrated that APOL1 inhibits HIV-1 replication by reducing HIV-1 transcription and by targeting HIV-1 Gag for degradation. In addition, a pulse-chase stability assay showed that HIV-1 Gag became significantly less stable in the presence of suboptimal APOL1 levels that incompletely inhibited HIV-1 transcription. Consistent with this observation, we found that APOL1 stimulated endocytosis and significantly expanded the lysosomal compartment. Interestingly, the increased number of lysosomes in the presence of APOL1 may facilitate proteolytic degradation of endocytosed Gag and viral accessory proteins. Whether accumulation of lysosomes results solely from the stimulation of lysosome biogenesis or, additionally, from the inhibition of autophagosomal flux, which consumes a significant pool of lysosomes, is unclear. Here we show that APOL1 stimulates lysosomal biogenesis by promoting nuclear localization of TFEB, a master regulator of lysosomal biogenesis (36, 37), and expression of TFEB target genes coding for lysosomal proteins LAMP1, LAMP2, and CD63. Interestingly, expansion of the lysosomal compartment was recently shown in Trex1-deficient CD4+ T cells and macrophages, which coincided with enhanced nuclear translocation of TFEB (49, 50). This lysosomal expansion was hypothesized to be intimately involved in the antiviral phenotype of Trex1-deficient cells. Moreover, induction of Trex1 by IFN-γ in mouse macrophages (51) suggests that if Trex1 is similarly induced by IFN-γ in human macrophages, it may reduce lysosomal biogenesis induced by APOL1 and possibly modulate anti-HIV-1 effects of APOL1. Thus, it would be worth evaluating how these two factors intersect in macrophages to control HIV-1 replication and regulate innate immune responses to HIV-1.
Remarkably, Trex1-deficient cells have reduced mammalian target of rapamycin (mTOR) activity. Suppression of mTOR-dependent phosphorylation of TFEB promotes its activation and nuclear translocation. This suggests that APOL1 expression could also inhibit mTOR activity. Consistent with this hypothesis, APOL1 has been implicated in autophagy, another process that is activated by suppression of mTOR activity (15, 16). Interestingly, mTOR has been shown to also control NF-κB activity (52), hence providing a potential explanation for the APOL1 inhibition of HIV-1 transcription.
Downregulation of key components of the endocytic pathway that mediate fusion between late endosomes (Stx7 and Rab7) and lysosomes (VAMP7) moderately increased virus production and steady-state accumulation of Gag in cells, indicating that endocytosis does not significantly influence virus production in 293T cells (30, 33). However, the expression of APOL1 in cells in which endocytic flux was perturbed by downregulation of Eps15, Rab7, Stx7, or VAMP7 significantly restored virus production, suggesting that a functional endocytic pathway is necessary for APOL1-mediated inhibition of HIV-1 replication.
In addition to potent inhibition of virus production, APOL1 also decreased virus infectivity. We observed that APOL1 downregulated viral accessory proteins Vif, Nef, and Vpu, which counteract host restriction factors. Thus, we propose that APOL1 reduces HIV-1 infectivity by depleting viral accessory proteins and preventing degradation of host restriction factors. For example, the cellular cytidine deaminase A3G potently inhibits replication of vif-deficient HIV-1 in human cells (53). However, unrestricted replication of wild-type HIV-1 in A3G-positive cells is attributed to viral accessory protein Vif, which eliminates A3G either by proteasome-mediated degradation (54–57) or by degradation-independent mechanisms (58). Here, we show that APOL1 prevents degradation of A3G in cells expressing wild-type HIV-1 partly by inhibiting HIV-1 gene expression as well as by targeting Vif for lysosomal degradation and secretion in microvesicles. Thus, depletion of Vif by APOL1 restores intracellular A3G levels sufficiently to reduce infectivity of wild-type progeny viruses, possibly by enhancing incorporation of A3G into virions.
To investigate whether APOL1 affects HIV-1 replication in cells that are natural targets for HIV-1, we analyzed the expression of APOL1 in T cells and monocytic cells. We have found that only differentiated monocytic THP-1 and U937 cells expressed detectable levels of APOL1 protein when stimulated with IFN-γ. This finding is especially intriguing since IFN-γ-polarized M1 macrophages inhibit HIV-1 replication (17, 59) and also express APOL1 transcripts and proteins (18, 26). We have confirmed these results and showed that only primary macrophages stimulated with IFN-γ expressed APOL1. In contrast, treatment with IFN-α only marginally stimulated APOL1. Although expression of restriction factor Samhd1 in macrophages (60, 61) was not significantly affected by treatment with IFN-γ or IFN-α in macrophages as reported previously (47), antiviral activity of Samhd1 can be regulated posttranscriptionally by phosphorylation (62). Thus, we cannot rule out the possibility that APOL1 acts together with Samhd1 or other factors to restrict HIV-1 in IFN-γ-polarized macrophages. These observations prompted us to examine whether endogenous APOL1 expressed in differentiated and IFN-γ-stimulated U937 cells contributes to anti-HIV-1 activity. However, interferon-stimulated macrophages express a multitude of restriction factors and microRNA (miRNAs) that affect HIV-1 replication (63), and thus, it may be challenging to elucidate the contribution of APOL1 under such conditions. Nevertheless, we have established U937 cell lines that stably express APOL1 shRNA. While IFN-γ stimulation of U937 cells expressing control shRNA reduced HIV-1 production by approximately 53%, cells expressing APOL1 shRNA were more permissive and reduced virus production only by 23%. Importantly, in the absence of IFN-γ, the two cell lines showed almost identical levels of permissiveness to HIV-1, indicating that the observed difference in HIV-1 production is due to differential expression of APOL1 between cell lines. Thus, analysis of the effect of APOL1 on HIV-1 replication in human primary macrophages is warranted.
Based on the evidence presented in this work, we propose that APOL1 inhibits virus replication partly by reducing HIV-1 transcription and stimulating endocytosis targeting HIV-1 Gag for degradation in an expanding lysosomal compartment. A schematic of this model is shown in Fig. 9. APOL1-mediated degradation and secretion of viral accessory protein Vif in microvesicles produce virions with decreased infectivity. The combined effects of these pathways significantly limit virus replication. Further understanding of the mechanism by which APOL1 cooperates with other restriction factors in macrophages to limit HIV-1 infection will have implications for the design of novel therapeutic approaches against HIV-1.
FIG 9.

Proposed model for APOL1-mediated inhibition of HIV-1 expression. (Left side) In the absence of APOL1, HIV-1 Gag is mainly targeted to the plasma membrane, where virus assembly takes place. Expression of HIV-1 Vif depletes cells of A3G and preserves infectivity of the virus progeny. (Right side) In the presence of APOL1, transcription of HIV-1 is partly inhibited, which leads to decreased synthesis of Gag protein. Gag protein is targeted for degradation in the lysosomal compartment, which expands due to stimulation of lysosome biogenesis induced by nuclear translocation of TFEB in cells expressing APOL1. Here, Gag is also targeted to the plasma membrane, but increased endocytosis redirects Gag to endosomes that efficiently fuse with an increased pool of lysosomes. Infectivity of produced virions is partly reduced by APOL1-dependent suppression of HIV-1 transcription and reduction of cellular Vif levels by degradation in lysosomes and secretion in microvesicles (Exo). As a result, host restriction factor A3G is not degraded but encapsidated and reduces HIV-1 virion infectivity. LYS, lysosomes; LE/MVB, late endosomes or multivesicular bodies; EE, early endosomes.
ACKNOWLEDGMENTS
This work was supported by NIH grants R21 DK089980 (W.P.), R21 DK094735 (W.P.), 8 U54 MD007593 (NIMHD), and G12MD007586 (NIMHD) and by Vanderbilt Institute for Clinical and Translational Research Scholar Award 5 KL2 RR024977 (H.E.T.).
We thank K. Strebel (NIAID, NIH), E. Freed (NCI, NIH), C. Aiken (Vanderbilt University), F. Diaz-Griffero (Albert Einstein College of Medicine), and K. Skorecki (Technion, Haifa, Israel) for reagents and helpful discussions.
The authors declare no conflict of interests.
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Vanhollebeke B, Pays E. 2006. The function of apolipoproteins L. Cell. Mol. Life Sci. 63:1937–1944. 10.1007/s00018-006-6091-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duchateau PN, Pullinger CR, Cho MH, Eng C, Kane JP. 2001. Apolipoprotein L gene family: tissue-specific expression, splicing, promoter regions; discovery of a new gene. J. Lipid Res. 42:620–630 [PubMed] [Google Scholar]
- 3.Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJ. 2002. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics 79:539–546. 10.1006/geno.2002.6729 [DOI] [PubMed] [Google Scholar]
- 4.Page NM, Butlin DJ, Lomthaisong K, Lowry PJ. 2001. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics 74:71–78. 10.1006/geno.2001.6534 [DOI] [PubMed] [Google Scholar]
- 5.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79:285–296. 10.1006/geno.2002.6718 [DOI] [PubMed] [Google Scholar]
- 6.Chiu YL, Greene WC. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26:317–353. 10.1146/annurev.immunol.26.021607.090350 [DOI] [PubMed] [Google Scholar]
- 7.Poelvoorde P, Vanhamme L, Van Den Abbeele J, Switzer WM, Pays E. 2004. Distribution of apolipoprotein L-I and trypanosome lytic activity among primate sera. Mol. Biochem. Parasitol. 134:155–157. 10.1016/j.molbiopara.2003.11.006 [DOI] [PubMed] [Google Scholar]
- 8.Smith EE, Malik HS. 2009. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 19:850–858. 10.1101/gr.085647.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O'Connor PM, Malloy MJ, Kane JP. 1997. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 272:25576–25582 [DOI] [PubMed] [Google Scholar]
- 10.Lugli EB, Pouliot M, Portela Mdel P, Loomis MR, Raper J. 2004. Characterization of primate trypanosome lytic factors. Mol. Biochem. Parasitol. 138:9–20. 10.1016/j.molbiopara.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 11.Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E. 2005. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 309:469–472. 10.1126/science.1114566 [DOI] [PubMed] [Google Scholar]
- 12.Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Paturiaux-Hanocq F, Andris F, Lins L, Pays E. 2009. C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS Pathog. 5:e1000685. 10.1371/journal.ppat.1000685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, Skorecki K. 2010. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum. Genet. 128:345–350. 10.1007/s00439-010-0861-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. 2010. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329:841–845. 10.1126/science.1193032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. 2008. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 283:21540–21549. 10.1074/jbc.M800214200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. 2008. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy 4:1079–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. 2009. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J. Immunol. 182:6237–6246. 10.4049/jimmunol.0803447 [DOI] [PubMed] [Google Scholar]
- 18.Beyer M, Mallmann MR, Xue J, Staratschek-Jox A, Vorholt D, Krebs W, Sommer D, Sander J, Mertens C, Nino-Castro A, Schmidt SV, Schultze JL. 2012. High-resolution transcriptome of human macrophages. PLoS One 7:e45466. 10.1371/journal.pone.0045466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cassetta L, Kajaste-Rudnitski A, Coradin T, Saba E, Chiara GD, Barbagallo M, Graziano F, Alfano M, Cassol E, Vicenzi E, Poli G. 26 April 2013. M1 polarization of human monocyte-derived macrophages restricts pre-and post-integration steps of HIV-1 replication. AIDS 10.1097/QAD.0b013e328361d059 [DOI] [PubMed] [Google Scholar]
- 20.Khatua AK, Taylor HE, Hildreth JE, Popik W. 2009. Exosomes packaging APOBEC3G confer human immunodeficiency virus resistance to recipient cells. J. Virol. 83:512–521. 10.1128/JVI.01658-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khatua AK, Taylor HE, Hildreth JE, Popik W. 2010. Non-productive HIV-1 infection of human glomerular and urinary podocytes. Virology 408:119–127. 10.1016/j.virol.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. 1994. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 68:5311–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strebel K, Daugherty D, Clouse K, Cohen D, Folks T, Martin MA. 1987. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 328:728–730. 10.1038/328728a0 [DOI] [PubMed] [Google Scholar]
- 24.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, David M. 2012. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491:125–128. 10.1038/nature11433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD, Pearson RB. 2012. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene 31:1949–1962. 10.1038/onc.2011.394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez FO, Gordon S, Locati M, Mantovani A. 2006. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177:7303–7311 [DOI] [PubMed] [Google Scholar]
- 27.Savina A, Furlan M, Vidal M, Colombo MI. 2003. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J. Biol. Chem. 278:20083–20090. 10.1074/jbc.M301642200 [DOI] [PubMed] [Google Scholar]
- 28.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. 2005. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1:84–91. 10.4161/auto.1.2.1697 [DOI] [PubMed] [Google Scholar]
- 29.Vanlandingham PA, Ceresa BP. 2009. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 284:12110–12124. 10.1074/jbc.M809277200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435. 10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balasubramaniam M, Freed EO. 2011. New insights into HIV assembly and trafficking. Physiology (Bethesda) 26:236–251. 10.1152/physiol.00051.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ono A. 2009. HIV-1 assembly at the plasma membrane: Gag trafficking and localization. Future Virol. 4:241–257. 10.2217/fvl.09.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finzi A, Orthwein A, Mercier J, Cohen EA. 2007. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J. Virol. 81:7476–7490. 10.1128/JVI.00308-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahreiss L, Menzies FM, Rubinsztein DC. 2008. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9:574–587. 10.1111/j.1600-0854.2008.00701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. 2008. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10:776–787. 10.1038/ncb1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. 2012. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31:1095–1108. 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. 2012. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5:ra42. 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. 2009. A gene network regulating lysosomal biogenesis and function. Science 325:473–477 [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, De Camilli P. 1998. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature 394:793–797. 10.1038/29555 [DOI] [PubMed] [Google Scholar]
- 40.Mullock BM, Smith CW, Ihrke G, Bright NA, Lindsay M, Parkinson EJ, Brooks DA, Parton RG, James DE, Luzio JP, Piper RC. 2000. Syntaxin 7 is localized to late endosome compartments, associates with Vamp 8, and is required for late endosome-lysosome fusion. Mol. Biol. Cell 11:3137–3153. 10.1091/mbc.11.9.3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura N, Yamamoto A, Wada Y, Futai M. 2000. Syntaxin 7 mediates endocytic trafficking to late endosomes. J. Biol. Chem. 275:6523–6529. 10.1074/jbc.275.9.6523 [DOI] [PubMed] [Google Scholar]
- 42.Pryor PR, Mullock BM, Bright NA, Lindsay MR, Gray SR, Richardson SC, Stewart A, James DE, Piper RC, Luzio JP. 2004. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO Rep. 5:590–595. 10.1038/sj.embor.7400150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itakura E, Kishi-Itakura C, Mizushima N. 2012. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151:1256–1269. 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 44.Gobeil LA, Lodge R, Tremblay MJ. 2012. Differential HIV-1 endocytosis and susceptibility to virus infection in human macrophages correlate with cell activation status. J. Virol. 86:10399–10407. 10.1128/JVI.01051-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lehtonen A, Matikainen S, Julkunen I. 1997. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J. Immunol. 159:794–803 [PubMed] [Google Scholar]
- 46.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. 2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38:4274–4284. 10.1093/nar/gkq174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goujon C, Schaller T, Galao RP, Amie SM, Kim B, Olivieri K, Neil SJ, Malim MH. 2013. Evidence for IFNalpha-induced, SAMHD1-independent inhibitors of early HIV-1 infection. Retrovirology 10:23. 10.1186/1742-4690-10-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faltynek CR, Finch LR, Miller P, Overton WR. 1989. Treatment with recombinant IFN-gamma decreases cell surface CD4 levels on peripheral blood monocytes and on myelomonocyte cell lines. J. Immunol. 142:500–508 [PubMed] [Google Scholar]
- 49.Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. 2013. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol. 14:61–71. 10.1038/ni.2475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11:1005–1013. 10.1038/ni.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serra M, Forcales SV, Pereira-Lopes S, Lloberas J, Celada A. 2011. Characterization of Trex1 induction by IFN-gamma in murine macrophages. J. Immunol. 186:2299–2308. 10.4049/jimmunol.1002364 [DOI] [PubMed] [Google Scholar]
- 52.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. 2008. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22:1490–1500. 10.1101/gad.1662308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 54.Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009–2013. 10.1016/j.cub.2003.10.034 [DOI] [PubMed] [Google Scholar]
- 55.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 56.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 279:7792–7798 [DOI] [PubMed] [Google Scholar]
- 57.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407. 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- 58.Opi S, Kao S, Goila-Gaur R, Khan MA, Miyagi E, Takeuchi H, Strebel K. 2007. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 81:8236–8246. 10.1128/JVI.02694-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cassol E, Cassetta L, Alfano M, Poli G. 2010. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 87:599–608. 10.1189/jlb.1009673 [DOI] [PubMed] [Google Scholar]
- 60.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. 10.1038/nature10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. 10.1038/nature10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13:441–451. 10.1016/j.chom.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cobos Jiménez V, Booiman T, de Taeye SW, van Dort KA, Rits MA, Hamann J, Kootstra NA. 2012. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]