

Abstract
Hepatitis B virus (HBV) synthesizes its DNA genome through reverse transcription, which is catalyzed by viral polymerase (Pol). Previous studies suggested that the RNase H domain of hepadnaviral Pol may contribute to multiple steps of the viral genome replication, such as RNA encapsidation and viral DNA synthesis. However, specific residues of the RNase H domain that contribute to viral reverse transcription have not been determined. Therefore, we employed charged-to-alanine scanning mutagenesis to generate a set of single-substitution mutants of the RNase H domain and then analyzed their ability to support viral reverse transcription. Southern blot analysis showed that three mutants (R703A, D777A, and R781A mutants) yielded significantly reduced amounts of viral DNAs. However, none of these mutants were defective in RNA encapsidation. The data indicated that in the R703A and D777A mutants, minus-strand DNA synthesis was incomplete due to loss of catalytic activity of RNase H. In contrast, in the R781A mutant, the minus-strand DNA synthesis was near complete to some extent, while the plus-strand DNA synthesis (i.e., relaxed circular DNA) was severely impaired due to the defect in RNase H activity. Overall, our analysis revealed that three charged residues of the HBV Pol RNase H domain contribute to the catalysis of RNase H in removing the RNA template, but not in the RNA encapsidation.
INTRODUCTION
Hepatitis B virus (HBV), the prototypic member of the hepadnavirus family, is a major cause of liver disease worldwide, ranging from acute and chronic hepatitis to liver cirrhosis and hepatocellular carcinoma. Other members of the hepadnavirus family include woodchuck hepatitis virus (WHV) and duck hepatitis B virus (DHBV) (1). The DNA genome of hepadnaviruses is replicated through reverse transcription, which takes place within the viral capsid in the cytoplasm of infected cells (Fig. 1). Recognition of a stem-loop structure (an encapsidation signal designated ε near the 5′ end of the pregenomic RNA [pgRNA]) by viral polymerase (Pol) directs encapsidation of the pgRNA and Pol into a nascent capsid particle (2–4). Minus-strand DNA synthesis is initiated by protein priming using Pol as a primer and the bulge region of ε as a template (5, 6). Following synthesis of three or four nucleotides, the nascent minus-strand DNA switches templates to a position near the 3′ end of the pgRNA, i.e., the 3′ copy of direct repeat 1 (DR1*) (Fig. 1A). The minus-strand DNA synthesis then resumes, with the RNase H activity of the Pol degrading the pgRNA, proceeding to the 5′end of the pgRNA, and resulting in a full-length minus-strand DNA (Fig. 1B and C).
FIG 1.

Reverse transcription of the HBV genome. The pgRNA (dashed line) serves as a template for minus-strand DNA synthesis and contains 11-nt direct repeats (DR1 and DR2; open boxes) and epsilon stem-loop structures (ε). The oval represents HBV Pol protein. (A) The Pol protein initiates reverse transcription from the 5′ ε. The newly synthesized nascent minus-strand DNA (thick gray line) switches template to the 3′ copy of DR1. (B) Following the minus-strand template switch, the minus-strand DNA is extended, while RNase H activity of the Pol protein degrades the pgRNA. (C) A small RNA segment, which contains DR1, resulting from the RNase H activity of the Pol protein, is used as an RNA primer for plus-strand DNA synthesis. (D) The RNA primer then switches template from DR1 to DR2 and resumes the plus-strand DNA synthesis (thick black line). (E) To generate the mature RC DNA, growing plus-strand DNA switches templates from the 5′ end to the 3′ end of the minus-strand DNA. (F) Alternatively, the plus-strand DNA synthesis (i.e., in situ priming) initiates at DR1, resulting in DL DNA.
A short segment of RNA, the remnant of the pgRNA cleavage by RNase H activity, serves as an RNA primer for plus-strand DNA synthesis (7). Depending on whether or not the second template switch takes place during plus-strand DNA synthesis, one of two distinct double-strand DNA products—relaxed circular (RC) DNA or duplex linear (DL) DNA—is generated (Fig. 1E and F). The RC DNA is generated when the RNA primer translocates to DR2, termed primer translocation, near the 5′ end of the minus-strand DNA template (Fig. 1D). Following translocation, the plus-strand DNA synthesis initiated from DR2 proceeds to the 5′ end of the minus-strand template (8). For continuation of plus-strand DNA synthesis, an intramolecular template switch must occur (Fig. 1E). The third template switch, termed circularization, results in a relaxed circular conformation of the genome. In contrast, the DL DNA is generated when plus-strand DNA synthesis is initiated at DR1 without primer translocation, termed in situ priming (Fig. 1F).
In fact, reverse transcriptases (RTs) exhibit RNase H activity as well as RNA- and DNA-directed DNA polymerase activities (9). Unlike those of its retroviral counterpart, far less is known about the functional role of the RNase H domain in hepadnaviral Pol. Previous studies with DHBV suggested that the RNase H domain of DHBV Pol may contribute to multiple steps of viral genome replication, such as RNA encapsidation and minus-strand DNA synthesis (10, 11). The requirement of the RNase H domain in RNA packaging was demonstrated by analysis of deletion or substitution mutations in the DHBV RNase H domain. Regarding minus-strand DNA synthesis, substitution mutations of the putative catalytic residues in the RNase H domain affected removal of the RNA strand of RNA-DNA hybrids, synthesis of viral plus-strand DNA, and DNA polymerase activity (12).
The so-called “clustered charged-to-alanine (CA)” mutagenesis method has been successfully used to examine the contribution of charged amino acid residues to a specific function of the protein of interest (13). Specifically, the method is to replace two charged amino acids (i.e., arginine, lysine, aspartic acid, and glutamic acid) located within a sequence of, at most, five amino acids with alanine. Clusters of charged amino acids, typically located on the surface of proteins, likely participate in the molecular recognition process, while substitution with alanine would cause only a local alteration, if any, in an otherwise native polypeptide (14). Previously, we employed extensive CA mutagenesis to gain further insight into the contribution of the terminal protein (TP) domain of HBV Pol to viral reverse transcription, and we uncovered that a charged amino acid (i.e., Arg105) is critical for RNA encapsidation (15).
In this study, we employed CA mutagenesis to determine the role of charged residues of the HBV Pol RNase H domain. Of the 16 charged residues we examined, three single-substitution mutants yielded significantly reduced levels of viral DNA, suggesting a role for these charged residues in viral DNA synthesis. The data showed that two mutants (R703A and D777A mutants) failed to synthesize the viral DNA due to loss of the catalytic activity of the RNase H domain, whereas the R781A mutant normally synthesized single-stranded (SS) DNA and DL DNA but failed to synthesize RC DNA. The lack of RC DNA synthesis is perhaps due to a failure in generating the appropriate RNA primer essential for RC DNA synthesis due to a defect in RNase H activity. Overall, the minus-strand DNA synthesis appeared to be linked to RNase H activity in the R703A and D777A mutants but not in the R781A mutant.
MATERIALS AND METHODS
Cell culture and transfection.
HepG2 (ATCC; HB-8065) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum and antibiotic-antimycotic solution (Invitrogen) at 37°C in 5% CO2. Transfection via polyethylenimine (PEI) (linear; 25 kDa; Polysciences, Inc.) was performed as previously described (16), except for the following modifications. Cells were seeded at a confluence of 40 to 50%. The ratio of DNA to PEI was kept at 1:2 (μg/μg), and DNA and PEI were mixed in 150 mM NaCl at room temperature for 10 min. DNA-PEI complex solution was directly added to the medium. After 24 h, cells were fed with fresh medium and were harvested 4 days posttransfection.
Plasmid construction.
The nucleotide sequences of the HBV genome were numbered starting at the unique EcoRI site of the HBV ayw subtype (GenBank accession number V01460) (17). In this numbering system, nucleotide position 1 of HBV ayw subtype is the A of the EcoRI site (GAATTC). Details of HBV P-null replicon and pCMV-Pol constructs were previously described (15). Briefly, the HBV p-null construct, which expresses HBV pgRNA under the control of its own promoter and all the other proteins except for HBV polymerase, is made by introducing two mutations into the P open reading frame (ORF). The pCMV-Pol is an HBV polymerase expressing plasmid with three copies of the FLAG tag at its N terminus. All substitution mutants were generated by overlap extension PCR protocol, as previously described (18), and confirmed by DNA sequencing.
Isolation of viral DNAs.
Viral DNAs from cytoplasmic capsids were isolated from HepG2 cells after transfection (19). Cells were lysed in 0.5 ml of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 1% NP-40 for 10 min and were clarified by centrifugation to pellet debris and nuclei. For the detection of HBV Pol and its substitution mutants, 3% (vol/vol) fraction of lysates were taken after centrifugation and then analyzed by immunoblotting. The clarified cell lysates were adjusted to 10 mM MgCl2, 100 μg/ml of DNase I (Sigma), and 100 μg/ml of RNase A (Sigma) and incubated at 37°C to remove the transfected plasmid DNAs and unencapsidated HBV RNAs. After incubation for 3 h, the cell lysates were centrifuged, and cytoplasmic capsids were precipitated with polyethylene glycol (PEG 8000; USB). Precipitated capsids were resuspended in 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1 mM EDTA and digested with proteinase K (Sigma) to 240 μg/ml and SDS to 0.5% for 3 h. Viral DNAs were purified by phenol-chloroform extraction, precipitated with ethanol, and rehydrated for analysis of primer extension and Southern blotting. For primer extension analysis, viral DNAs were subsequently hydrolyzed by alkali treatment, neutralized, and precipitated with ethanol as described elsewhere (8). For Southern blot analysis, the alkaline hydrolysis procedure was omitted.
Southern blot analysis.
Viral DNAs from cytoplamic capsids of transfected HepG2 cells were analyzed by Southern blotting to measure viral replication intermediates, as described previously (20). In brief, viral DNAs isolated from a 35-mm plate were electrophoresed through a 1.3% agarose gel, transferred onto a nylon membrane, and subjected to UV cross-linking. The membrane was prehybridized and then hybridized with an [α-32P]dCTP-labeled HBV-specific probe for 12 h at 65°C. The membrane was washed and autoradiographed on an imaging plate. Images were obtained using a phosphorimager scanner (BAS-2500; Fujifilm). For Fig. 6C, viral DNAs isolated from a 100-mm plate were heat denatured at 95°C for 5 min and immediately cooled on ice before electrophoresis. The 1820-F primer (complementary to nucleotides [nt] 1820 to 1836), which binds to 3′ end of the minus-strand DNA, was 5′ end labeled with [γ-32P]ATP (21). The membrane blot was incubated with the full-length minus DNA-specific probe for 12 h at 42°C to facilitate annealing of the oligonucleotide probe to HBV DNAs on the membrane. For Fig. 7, viral DNAs were divided into two equal aliquots; one was left untreated, and the other was treated with Escherichia coli RNase H (New England BioLabs) at 37°C for 20 min before electrophoresis to determine whether pgRNA remains undegraded during the process of minus-strand elongation.
FIG 6.

Determination of defective steps in viral genome replication. (A) Diagram showing the oligonucleotide primer-binding sites. The name of each primer refers to the position of the first nucleotide. The letter F indicates that the primer has the same polarity as the plus-strand DNA, while the letter R indicates that the primer has the same polarity as the minus-strand DNA. (B and E) Primer extension analysis was performed to measure the amount of minus-strand DNA synthesis extended from DR1 with the 1665-F primer (B) and the amount of plus-strand DNA synthesis initiated from DR2 with the 1744-R primer (E). To normalize amplification efficiency between different samples, internal standard (I.S.) DNAs, which have the same primer-binding sites to each primer, were added to each DNA sample prior to the primer extension reaction. Replication intermediate (R.I.) bands are indicated by a closed arrowhead, while I.S. bands are indicated by an open arrowhead. A sequencing ladder is shown along with DR1 sequence, where the four nucleotides to which the nascent DNA anneals are highlighted in bold (B). A sequencing ladder is shown along with DR2 sequence (E). Representative data from five independent experiments are shown. (C) Southern blot analysis with heat-denatured DNA was performed to measure the amount of full-length minus-strand DNA. DNA samples were heat denatured before electrophoresis. The 1820-F primer was 5′ end labeled and used as a probe. Labeled bands comparable in size to DL DNA species are denoted as “A,” whereas those comparable to SS DNA species are denoted as “B.” The intensities of both A and B bands were combined to measure the level of full-length minus-strand DNA. Representative data from five independent experiments are shown. Two restriction fragments (3.2 kb and 2.2 kb) serve as size markers (open arrowhead). Bands denoted by asterisks likely represent spliced forms of DNA derived from spliced pgRNAs. (D and F) Histograms showing the efficiency of reverse transcription steps: minus-strand elongation (D) and primer translocation (F). The efficiency of each reverse transcription step was determined as described previously (21). Briefly, the efficiency of minus-strand elongation was calculated as the amount of full-length minus-strand DNA divided by the amount of minus-strand DNA synthesis extended from DR1 (D). Likewise, the efficiency of primer translocation was calculated as the level of plus-strand DNA initiated from DR2 divided by the level of full-length minus-strand DNA (F). Error bars and values above the error bars represent standard deviations and mean values, respectively. Statistical significance was determined using the Student t test. Differences were considered to be significant if the P value was <0.05. *, P < 0.05; **, P < 0.001. N/A, not applicable.
FIG 7.

Three single mutants have a defect in RNase H activity. Cells were cotransfected with an HBV P-null replicon construct along with WT Pol or an individual single substitution mutant as indicated. Approximately 1/10 (WT) or 3/10 (each mutant) of the capsid-associated DNA isolated from a 60-mm plate was analyzed by Southern blotting. DNA samples were either left untreated or subjected to E. coli RNase H treatment at 37°C for 20 min. Longer RNase H treatment and higher RNase H concentrations had no effect on migration. After treatment, each sample was immediately separated on a 1.2% agarose gel (14 by 12 cm). Asterisks to the right of labeled bands indicate newly appearing DNA forms following exogenous RNase H treatment. The intensities of bands (*) shifted upon the RNase H treatment were reduced to 90% (D689A), 93% (E718A), 92% (D737A), 95% (R703A), 93% (D777A), and 90% (R781A). Size markers (0.7 kb to 3.2 kb) were generated by restriction enzyme digestion of full-length HBV genome (open arrowhead). The results shown in lanes 15 and 16 are short exposures of lanes 13 and 14.
Primer extension analysis.
Viral DNAs isolated from a 60-mm plate by the above-mentioned methods were divided into five equal aliquots. One aliquot of the sample was used together with the 1665-F primer (complementary to nt 1665 to 1689) to determine the amount of minus-strand DNA initiated at the 3′ copy of DR1 and extended. Two other aliquots of the sample were used together with the 1774-R primer (complementary to nt 1744 to 1722) to measure the level of plus-strand DNA initiated at DR2 before the circularization step. The other two aliquots of the sample were used together with the 1952-R primer (complementary to nt 1952 to 1930) to determine the amount of plus-strand DNA initiated at DR2 and circularized. All primers were 5′ end labeled using [γ-32P]ATP and polynucleotide kinase (New England BioLabs). Primer extension reaction was performed with Deep VentR (exo-) DNA polymerase (New England BioLabs) (20), and 100 to 200 pg of internal standard DNAs was added to each sample prior to the extension. The thermal cycler setting used for the extension with all three primers was 20 cycles of 95°C for 30 s, 53°C for 40 s, and 72°C for 30 s. Primer extension products were then denatured at 95°C for 5 min prior to electrophoresis through 6% polyacrylamide-7 M urea gels. The gel was dried, and autoradiography was conducted as mentioned for Southern blot analysis. The sizes of extended products were 165 nt for the 1665-F primer, 142 nt for the 1744-R primer, and 350 nt for the 1952-R primer. A sequencing ladder was generated using Sequenase, version 2.0, DNA sequencing kit (Affymetrix).
Western blot analysis.
Protein samples obtained from the above-mentioned cell lysis procedure were resolved by SDS-10% polyacrylamide gel electrophoresis (PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). After being blocked, the membrane was incubated with a mouse anti-FLAG M2 antibody (diluted 1:5,000; Sigma) to detect FLAG-tagged HBV polymerases. β-Actin was detected using a rabbit anti-actin antibody (diluted 1:5,000; Sigma). Proteins were visualized by an ECL detection system (AbFrontier, Republic of Korea). Images were quantified using LAS-4000 (Fujifilm).
RNA extraction and RPA.
RNAs from whole cells and cytoplasmic capsids were isolated 3 days after transfection using acid guanidinium thiocyanate-phenol-chloroform extraction (22, 23). Approximately twice more RNA of the capsid fraction (C), compared with total cytoplasmic fraction (T), was subjected to RNase protection analysis (RPA), according to the manufacturer's procedure (Ambion). Specifically, a riboprobe used to detect HBV-specific RNAs was generated by the in vitro transcription of the core region (complementary to nt 1903 to 2139). Each sample of RNA was hybridized with the [α-32P]UTP-labeled probe for 16 h at 42°C. RNase digestion was carried out with a mixture of RNase A and RNase T1 for 50 min at 37°C. The digested products were separated on 6% acrylamide-7 M urea gels. Images were obtained using a phosphorimager (BAS-2500; Fujifilm).
RESULTS
Some CA mutants in the HBV RNase H domain fail to synthesize viral DNA.
To explore the role of the RNase H domain in reverse transcription, we introduced CA mutagenesis in the RNase H domain of HBV Pol as described in Materials and Methods. Six CA mutants were first generated as shown in Fig. 2. Three single point mutants harboring an alanine substitution in the putative catalytic site (D689, E718, and D737) were also generated in parallel. We examined the ability of each mutant to support viral DNA synthesis via complementation procedure, as previously described (24). HepG2 cells were transfected with an individual CA mutant along with a HBV P-null replicon construct harboring a frameshift mutation in the P ORF that otherwise yields mature viral DNAs. Southern blot analysis of cytoplasmic core DNA showed that the wild-type (WT) Pol yielded three species of viral replication intermediates: RC, DL, and SS DNAs (Fig. 3A, lane 1). HBV Pol derived from two mutants—CA2 and CA6—supported WT levels of viral genome replication, suggesting that the amino acid residues mutated in CA2 and CA6 were not essential for viral genome replication (Fig. 3A, lanes 3 and 7). In contrast, HBV Pol derived from another two CA mutants—CA1 and CA4—yielded a barely detectable amount of viral DNAs, suggesting a role for the residues mutated in CA1 and CA4 in viral genome replication (Fig. 3A, lanes 2 and 5). Intriguingly, the remaining two mutants—CA3 and CA5—led to synthesis of reduced amounts of DL and SS DNAs but failed to produce a detectable level of RC DNA (Fig. 3A, lanes 4 and 6). Comparable expression of HBV Pol protein was measured by Western blot analysis (Fig. 3B), confirming that the barely detectable levels of viral DNAs in the CA1 and CA4 mutants were not due to a lack of Pol protein expression. A similar observation was made in Huh7 cells (data not shown). As expected, significantly reduced amount of viral DNAs were detectable in the putative catalytic-site mutants (Fig. 3C). Overall, the eight charged residues altered in the four CA mutants (CA1, CA3, CA4, and CA5) were subjected to further analysis.
FIG 2.
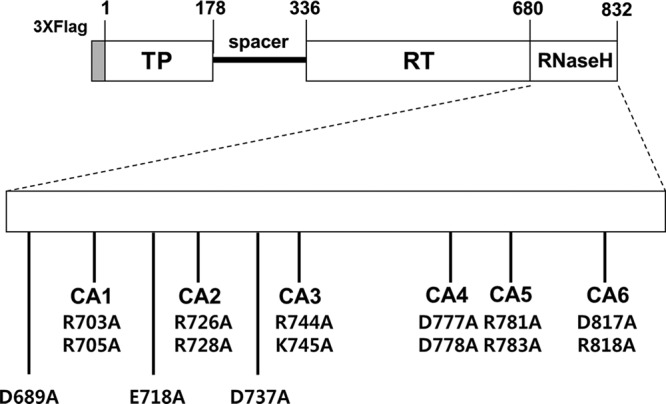
Schematic illustrating HBV Pol-expression plasmids used in this study. HBV Pol consists of four different subdomains: the terminal protein (TP), spacer, RT, and the RNase H domain. The positions of the clustered CA mutants in the HBV RNase H domain are indicated. The positions of alanine substitutions at the putative catalytic sites (D689, E718, and D737) are denoted as well. The amino acid number of each subdomain and three copies of the FLAG epitopes at its N terminus are labeled.
FIG 3.
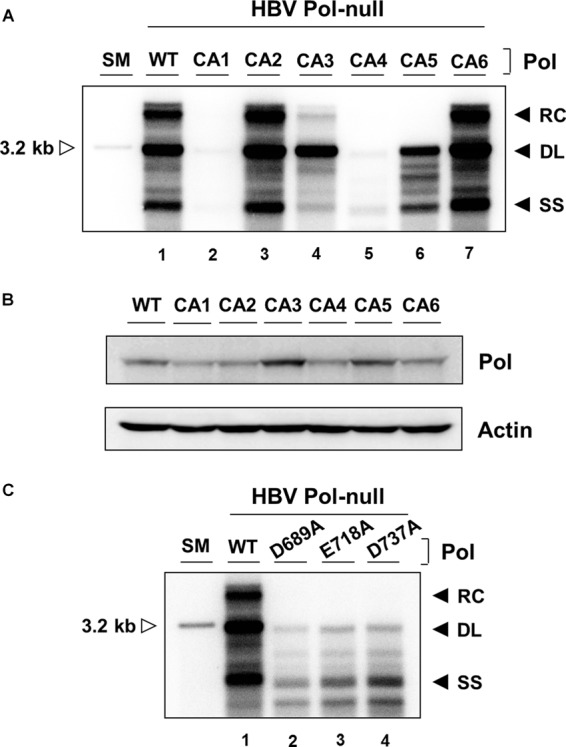
Analysis of CA mutants for the ability to support viral genome replication. (A) Cells were cotransfected with a HBV P-null replicon construct along with WT Pol or individual CA mutant constructs as indicated. Intracellular capsid-associated DNAs were isolated and analyzed by Southern blotting. The viral replication intermediates, RC, DL, and SS DNAs, are denoted. A restriction fragment representing one HBV genomic unit (3.2 kb) serves as a size marker (SM) (open arrowhead). The experiment was repeated four times, and representative data are shown. (B) Cytoplasmic lysates, equivalent to 3% input from the same cells transfected with the constructs indicated, were analyzed by immunoblotting. The levels of HBV Pol and β-actin were measured using anti-Flag and anti-actin antibodies, respectively. (C) Intracellular capsid-associated DNAs obtained from three catalytic-site mutants were isolated and analyzed similarly to the assay described above.
A single substitution reveals charged residues that are critical for viral DNA synthesis.
To determine the specific residue responsible for viral DNA synthesis of the two altered residues in the four CA mutants mentioned above, eight single-substitution mutants were generated. Each mutant DNA was transfected and then analyzed by Southern blotting as shown in Fig. 4A. In CA1, the R703A mutant was severely defective in viral DNA synthesis, while the R705A mutant was normal in viral DNA synthesis (Fig. 4A, lanes 2 and 3), suggesting that R703A fully accounts for the defect seen in CA1. Notably, a few bands migrating faster than DL or SS DNA were detectable in R703A mutant-transfected cells, mirroring those detected in the putative catalytic-site mutants (Fig. 3C). In CA3, the K745A mutant led to modestly reduced synthesis of the RC and SS DNAs, while the R744A mutant led to near-WT levels of viral DNA synthesis (Fig. 4A, lanes 4 and 5). Thus, the defect of the K745A mutant appears to fully account for the defect seen in CA3. Notably, the DL DNA synthesis proceeded normally, while the RC DNA synthesis was impaired in the K745A mutant. In CA4, the D777A mutant appeared severely defective in viral DNA synthesis, yielding a few fast-migrating bands (Fig. 4A, lane 6) similar in size and pattern to those seen with the putative catalytic-site mutants (Fig. 3C). In contrast, the D778A mutant led to modestly reduced synthesis of all three viral replication intermediates (Fig. 4A, lane 7). Therefore, the defect seen in CA4 seemed to be primarily attributable to the defect in the D777A mutant and, to a lesser extent, in the D778A mutants. In CA5, the R781A mutant led to significantly reduced synthesis of the RC DNA, while R783A led to near-WT-level synthesis of viral DNAs (Fig. 4A, lanes 8 and 9). Thus, R781A substitution appears to be fully responsible for the defect seen in CA5. Overall, the data show that five single-substitution mutants (R703A, K745A, D777A, D778A, and R781A mutants) were defective in yielding WT levels of viral DNAs (Fig. 4A). Comparable expression of HBV Pol protein was confirmed by Western blot analysis (Fig. 4B).
FIG 4.
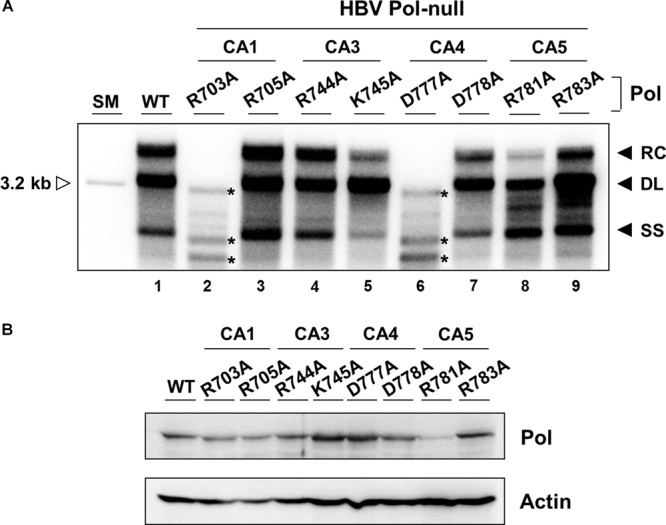
Three charged residues in the RNase H domain are critical for RC DNA synthesis. (A) Cells were cotransfected with an HBV P-null replicon construct along with WT Pol or an individual single substitution mutant as indicated. Southern blot analysis was performed to detect the three replication intermediates: RC, DL, and SS DNAs. Asterisks to the right of the labeled bands denote replication intermediates that appear in only two individual mutants (R703A and D777A). The experiment was repeated six times, and representative data are shown. (B) The expression of HBV Pols was evaluated by immunoblotting from cytoplasmic lysates (input: 3%) obtained from the same cells as transfected for panel A. β-Actin served as a loading control.
None of the charged residues are essential for RNA encapsidation.
To determine the step at which the above-mentioned single-site mutants are defective, we next examined RNA encapsidation, a step prior to viral reverse transcription. Cells were transfected, and then viral pgRNA was extracted from both capsids (C) and total cells (T) and analyzed by RNase protection analysis (RPA) as detailed in Materials and Methods. As shown in Fig. 5, none of the mutants appeared defective in RNA encapsidation, indicating that none of these charged residues are essential for RNA encapsidation. This finding led us to examine the viral reverse transcription.
FIG 5.
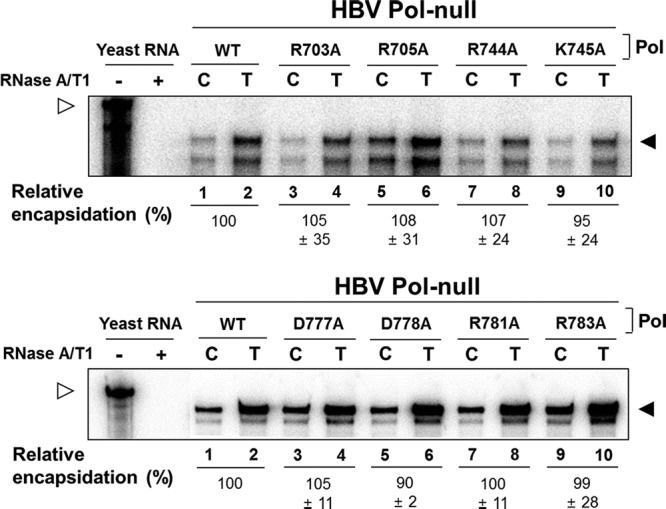
None of the charged residues are indispensable for RNA encapsidation. Cells were cotransfected with an HBV P-null replicon construct along with WT Pol or an individual single-substitution mutant as indicated. RNA from core particles (C) and total cytoplasmic fractions (T) was extracted and analyzed by RPA, as described in Materials and Methods. The protected fragment of pgRNA is indicated with a closed arrowhead, while the riboprobe is indicated with an open arrowhead. Yeast RNA served as a negative control. The encapsidation efficiency was estimated by comparing the amount of pgRNA detected in the C fraction to that detected in the T fraction (mean ± SD). Representative data from at least five independent experiments are shown.
Some mutants have a defect in minus-strand elongation.
To further narrow down the defect of the above-described five mutants in viral DNA synthesis, we next investigated whether the minus-strand DNA was normally synthesized. Primer extension analysis was performed as previously described (21), and the strategy is shown in Fig. 6A. We carried out primer extension analysis using the 1665-F primer to measure the amount of minus-strand DNA initiated at the 3′ copy of DR1 (Fig. 6A). The extended products from the minus-strand DNA initiated at DR1 were detected in cells transfected with a WT Pol (Fig. 6B, lane 1). In contrast, modestly reduced levels of minus-strand DNAs that were initiated at DR1 were detected in all five mutants (Fig. 6B, lanes 2 to 6). One interpretation of these results is that none of these charged residues are critically important for minus-strand DNA synthesis at DR1.
Though the above-mentioned mutants appeared not to be defective in initiation of minus-strand DNA, they nonetheless failed to accumulate RC DNA to WT levels. Thus, we next examined whether or not minus-strand DNA synthesis was completed by probing the very end of the minus-strand DNA with the 1820-F oligomer (Fig. 6A) following heat denaturation of viral DNAs, as previously described (21). Unexpectedly, both DL and SS DNAs were detectable in WT-transfected cells, even though the viral DNAs were fully heat denatured (Fig. 6C, lane 1). It appeared that the heat-denatured DNA became rapidly renatured following denaturation or during gel electrophoresis. Regardless of the DNA type (DL or SS), it is important to note that the oligonucleotide probe would detect the very end of the minus-strand DNA. Southern blot analysis with the oligonucleotide probe showed that the K745A, D778A, and R781A mutants had significantly reduced levels of DNA (Fig. 6C, lanes 3, 5, and 6). Intriguingly, two discrete bands (denoted by asterisks) were detected in the WT as well as the R781 mutant (Fig. 6C, lanes 1 and 6), in which one DNA species migrated faster than DL DNA, and another DNA species migrated faster than SS DNA. We speculate that these DNA species were derived from the spliced pgRNA (25). In contrast, no band was detected in either R703A or D777A mutant-transfected cells, indicating that the minus-strand DNA synthesis was not completed (Fig. 6C, lanes 2 and 4).
To calculate the efficiency of the minus-strand elongation step, the level of full-length minus-strand DNA probed with 1820-F oligomer was divided by the level of minus-strand DNA synthesis from the 5′ end, which was first normalized to the level of its internal standard (Fig. 6D). Quantitation revealed that the K745A mutant exhibited near-WT elongation efficiency, whereas the D778A and R781A mutants exhibited modestly reduced elongation efficiency.
The R781A mutant has a defect in RNA primer translocation.
It remains possible that the other steps of viral replication, such as primer translocation during the plus-strand DNA synthesis, could be affected by certain mutants. To determine a step at which these mutants are defective, primer extension analysis with the 1744-R primer was carried out to measure the amount of plus-strand DNA synthesis initiated at DR2 (Fig. 6A), as previously performed (26). As shown in Fig. 6E, the analysis showed that plus-strand DNAs that initiated at DR2 were reduced significantly in the R703A and D777A mutants and to a lesser extent in the R781A mutant (Fig. 6E, lanes 2, 4, and 6), but they were only modestly reduced in the K745A and D778A mutants (Fig. 6E, lanes 3 and 5). Quantitation was done to examine the extent to which primer translocation proceeds; the level of plus-strand DNA synthesis initiated at DR2 was divided by the level of full-length, minus-strand DNA (Fig. 6F). The data showed that the K745A and D778A mutants did not have a defect in primer translocation. Taken together, our results indicate that the reductions in viral replication levels in the K745A and the D778A mutants are largely attributable to a defect in the minus-strand synthesis (i.e., minus-strand initiation and elongation). Intriguingly, the R781A mutant had a significant decrease in primer translocation. Thus, our results indicate that the R781A mutant has a defect in at least three different steps of viral genome replication: (i) minus-strand initiation, (ii) minus-strand elongation, and (iii) RNA primer translocation during plus-strand DNA synthesis.
To exclude the possibility that steps following primer translocation could be impaired by certain mutations, we also performed primer extension analysis using 1952-R primers, which can measure the amount of elongating plus-strand DNA that had undergone the third template switch (Fig. 6A and data not shown). The level of plus-strand DNA that continued to the circularization step divided by the level of plus-strand DNA that initiated synthesis from DR2 was then calculated. It was found that three mutants (K745A, D778A, and R781A mutants) that we considered display no defect in circularization (data not shown). Collectively, the R703A, D777A, and R781A mutants exhibited a phenotype that is consistent with a defect in RNA primer translocation or a prior step.
Three mutants have a defect in RNase H activity.
As mentioned above, a few bands migrating faster than DL or SS DNA were detectable in R703A and D777A mutant- transfected cells (Fig. 4A, lanes 2 and 6). Several discrete viral nucleic acid species detected by the catalytic triad mutants of the RNase H domain of HBV or DHBV were found to be RNA-DNA duplexes resulting from a loss of RNase H activity (12). Hence, it is not unreasonable to speculate that our three mutants (R703A, D777A, and R781A mutants) could have similarly lost their catalytic activity. To determine whether the RNase H activities of the three mutants were lost, the viral DNA replication intermediates extracted from transfected cells were treated with Escherichia coli RNase H before gel electrophoresis, as performed previously (27). This experiment is based upon the premise that the migration of viral RNA-DNA hybrid molecules would shift upon exogenous RNase H treatment, as the residual RNA is removed. As expected, the migration pattern of the characteristic fast-migrating bands of the three triad mutants (D689A, E718A, and D737A mutants) was changed upon exogenous RNase H treatment (Fig. 7, lanes 3 to 8). Likewise, in the R703A and D777A mutants, the migration pattern of the characteristic fast-migrating bands was changed in parallel (Fig. 7, lanes 9 to 12) Further, the observation that the migration patterns of the hybrid molecules of these mutants were indistinguishable from those of the catalytic triad mutants led us to speculate that these charged residues contribute to catalysis of the RNase H, either by constituting the catalytic residues or by being involved in metal ion coordination (27).
In contrast, unlike the R703A and D777A mutants, the R781A mutant led to a rather abundant accumulation of DL and SS DNA but a lack of RC DNA synthesis (Fig. 4A, lane 8, and Fig. 7, lane 13). Nonetheless, upon exogenous RNase H treatment, the shift of DL and SS DNAs was also observed, implicating the presence of residual RNA in two DNA species. Importantly, no such shift was observed in the WT, as well as the K745A and D778A mutants (Fig. 7, lane 2; data not shown). It is also notable that the DL and SS DNAs in the R781A mutant migrated a bit slower than those of the WT, consistent with the presence of residual RNA (Fig. 7, lane 1 versus 13). The residual RNA that caused the shift upon exogenous RNase H treatment is attributable to the reduced catalytic activity of the RNase H derived from the R781A mutant. Overall, we conclude that the catalytic activities of the RNase H domain were affected in all three mutants but in subtle, distinct ways: in the R703A and D777A mutants, DNA synthesis is coupled to RNase H activity, whereas in the R781A mutant, DNA synthesis is uncoupled from RNase H activity (see Fig. 8).
FIG 8.

Diagram of hypothetical RNA-DNA hybrid molecules generated by three single-substitution mutants. (A) Two mutants (R703A and D777A mutants) failed to synthesize full-length minus-strand DNA. As a result, hybrid molecules consisting of short-length minus-strand DNAs and undigested viral RNA fragments are detected. These two mutants manifested a phenotype that is indistinguishable from those of catalytic triad mutants (D689A, E718A, and D737A mutants). Our interpretation is that the RT activity is coupled to the RNase H activity in these mutants. (B) In contrast to the mutants in panel A, the full-length minus-strand DNA is synthesized in the R781A mutant. Hybrid molecules consisting of the full-length minus-strand DNA and some undigested residual viral RNAs may be generated. Moreover, the plus-strand DNA synthesis (i.e., in situ priming) proceeds to some extent. Our interpretation is that the RT activity is uncoupled to the RNase H activity in the R781A mutant.
DISCUSSION
Unlike for related retroviruses, the precise role of the RNase H domain of hepadnaviral Pol during viral reverse transcription remains largely unexplored. Our results showed that of the 16 residues we altered, three charged residues (R703, D777, and R781) in the RNase H domain of HBV Pol are important for viral reverse transcription; mutants with changes at the former two were defective in minus-strand synthesis, while the latter was defective in both minus-strand synthesis and plus-strand DNA (i.e., RC DNA) synthesis. The results imply that the catalytic activity of the RNase H plays roles not only in removal of the RNA template but also in a subsequent step leading to RC DNA synthesis (perhaps by generating the RNA primer needed for the RC DNA synthesis).
Based on the defects in synthesis of viral DNA replication intermediates, the three mutants could be classified into two groups. The first group (R703A and D777A mutants) failed to accumulate viral DNA replication intermediates but resulted in a few fast-migrating species on a Southern blot (Fig. 4A, lanes 2 and 6). The failure by the R703A and D777A mutants to complete minus-strand DNA synthesis suggests that minus-strand DNA synthesis was halted during elongation (Fig. 8A). Moreover, the observation that a few bands migrating faster than DL or SS DNA were detected in R703A and D777A mutant-transfected cells (Fig. 4A, lanes 2 and 6), mirroring those detected in the putative catalytic-site mutants (Fig. 3C), led us to speculate that the R703A and D777A mutants are catalytically inactive. An earlier report on the DED (catalytic triad) mutants of DHBV showed that these viral DNA species are, in fact, hybrid molecules of RNA and minus-strand DNA resulting from incomplete synthesis of the minus-strand DNA (12). Indeed, the sensitivity to an exogenous RNase H treatment (Fig. 7, lanes 10 and 12) led us to conclude that R703A and D777A mutants have a defect in completion of minus-strand DNA synthesis due to loss of RNase H activity. It is notable that the mutant phenotype of these two residues (R703 and D777) is indistinguishable from that of the catalytic-site mutants. The implication is that these two residues (R703 and D777) may play a critical role in the catalytic process, similar to the DED residues. Relevantly, while the manuscript for this article was being prepared, Tavis and coworkers reported that the D777 residue is essential for the RNase H activity (27). Overall, it was speculated that the “DEDD” motif, rather than the DED motif, constitutes the active site of the HBV RNase H, as the case of HIV RNase H, although sequence similarity at the amino acid level is only modest (27).
In contrast, the R781A mutant first appeared to lead to normal accumulation of DL and SS DNAs but failed to synthesize RC DNA (Fig. 4, lane 8). For instance, the analysis to detect the very end of the minus-strand DNA showed that minus-strand DNA synthesis was nearly completed to some extent in the R781A mutant (Fig. 6C, lane 6, and D). To our surprise, however, two DNA species that comigrated with the DL and SS DNAs were significantly shifted upon exogenous RNase H treatment (Fig. 7, lanes 13 and 14), a finding that suggests the existence of some residual RNA. Presumably, this residual RNA was significantly larger than the short RNA primer, as no such shift was observed in the WT (Fig. 7, lane 2). The detection of a decent amount of residual RNA in viral DNA replication intermediates in the R781A mutant is not necessarily inconsistent with the interpretation that the minus-strand DNA was nearly completed. One possibility is that HBV Pol of the R781A mutant proceeds to synthesize the minus-strand DNA to the very end of the RNA template without complete removal of the RNA template (Fig. 8B). Specifically, this mutant RNase H with altered cleavage specificity may generate RNA primers with different lengths, leading to predominant in situ priming for DL DNA synthesis. This phenomenon implies that the RT and RNase H activities could be uncoupled under certain circumstances (12). Overall, the R781A mutant was defective in three steps during viral reverse transcription: (i) the initiation in minus-strand DNA synthesis (49% of the level of the WT), (ii) the elongation of minus-strand DNA synthesis (38% of the level of the WT), and (iii) the RNA primer translocation during plus-strand RC DNA synthesis (47% of the level of the WT).
In addition to above-mentioned three mutants (R703A, D777A, and R781A mutants), which were severely defective in RC DNA synthesis, it was notable that RC DNA synthesis was reduced, to a lesser extent, in K745A and D778A mutants (Fig. 4A, lanes 5 and 7). Inconsistent with an expectation from the reduced RC DNA, the data showed that the primer translocation for the RC DNA synthesis appeared not to be defective (Fig. 6F). Thus, a question is which step of viral reverse transcription is defective. In fact, our data indicated that the K745A and D778A mutants were modestly defective in the initiation of minus-strand DNA synthesis (Fig. 6B, lanes 3 and 5) and appeared to be somewhat defective in minus-strand elongation (Fig. 6D), although the reduction in the K745A mutant was not statistically significant. Overall, the reduction of the RC DNA in the K745A and D778A mutants is almost fully accounted for by the defects seen in minus-strand DNA synthesis. Nonetheless, the aforementioned conclusion cannot account for abundant accumulation of the DL DNA in the K745A mutant (Fig. 4A, lane 5). It could be due to the overestimation of the primer translocation efficiency shown in Fig. 6F, in which the value obtained from the primer extension analysis (Fig. 6E) was divided by the amount of the full-length minus-strand DNA shown in Fig. 6C. The latter could be underestimated as a consequence of using only one short-length probe (i.e., 1820-F; 17-mer). Therefore, it remains possible that the K745A mutant is indeed defective in RNA primer translocation during plus-strand RC DNA synthesis.
Overall, we found that five charged residues are essential for the efficient accumulation of RC DNA synthesis. Three single point mutants harboring an alanine substitution at R703, D777, and R781 were severely defective in RC DNA synthesis, while the remaining two single point mutants harboring an alanine substitution at K745 and D778 were modestly defective in RC DNA synthesis. An interpretation is that the former three residues are involved in the catalysis of RNase H activity, as evidenced by the presence of residual RNA in the DNA replication intermediates, while the latter two residues are not directly involved in the catalysis, as evidenced by the lack of residual RNA in the DNA replication intermediates (Fig. 7 and data not shown). Nonetheless, we cannot exclude the possibility that the defects were, to some extent, attributable to misfolding caused by substitution mutations.
Whether or not the RNase H domain contributes to encapsidation remains unclear. Earlier work reported that HBV Pol harboring a C-terminal truncation of the RNase H domain failed to package pgRNA, suggesting a role for the RNase H domain in encapsidation (28). Subsequently, a natural variant of DHBV encoding C711Y substitution in the RNase H domain was shown to be defective in RNA encapsidation (10, 12). It should be noted that all substitution mutants examined in these earlier studies involved a drastic amino acid alteration, e.g., C711Y (11). Thus, a defect of the mutants could simply be attributable to the particular amino acid chosen as the replacement. Furthermore, only limited genetic analysis of the HBV counterpart has been performed (11). In contrast, our data showed that none of the charged residues we examined are important for RNA encapsidation. Nonetheless, it remained possible that other charged residues could be essential for encapsidation. To address this issue, in addition to the aforementioned charged residues, eight other charged residues (R680A, R762A, R788A, R792A, R796A, R801A, D807A, and R830A), of which seven were arginine residues, were analyzed in parallel. The data showed that none of these charged residues are essential for viral genome replication or encapsidation (data not shown). Hence, it appeared that none of the charged residues encoded in the RNase H domain are essential for viral RNA encapsidation.
Current treatments for chronic hepatitis B largely rely on nucleoside analogs (NUCs) such as lamivudine, adefovir, entecavir, and tenofovir (29). Although viral suppression is achieved in up to 95% of patients treated with NUCs, hepatitis B surface antigen (HBsAg) loss, which is a major treatment endpoint that allows for treatment cessation, is achieved in less than 10% of patients after 5 years of NUC administration. It is expected that addition of one or more drugs with a novel target to the current regimen should markedly improve the response to therapy (30). In this regard, the RNase H activity of HBV Pol represents an attractive target for anti-HBV therapy (27, 31).
ACKNOWLEDGMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant 2009-0089134). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (NRF-2011-Fostering Core Leaders of the Future Basic Science Program).
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.Seeger C, Zoulim F, Mason W. 2007. Hepadnaviruses, p 2977–3029 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Hirsch RC, Loeb DD, Pollack JR, Ganem D. 1991. Cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 65:3309–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeong JK, Yoon GS, Ryu WS. 2000. Evidence that the 5′-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J. Virol. 74:5502–5508. 10.1128/JVI.74.12.5502-5508.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Junker-Niepmann M, Bartenschlager R, Schaller H. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loeb DD, Hirsch RC, Ganem D. 1991. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: implications for other reverse transcribing elements. EMBO J. 10:3533–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Staprans S, Loeb DD, Ganem D. 1991. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J. Virol. 65:1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res. 134:235–249. 10.1016/j.virusres.2007.12.024 [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Robinson WS, Marion PL. 1992. Naturally occurring point mutation in the C terminus of the polymerase gene prevents duck hepatitis B virus RNA packaging. J. Virol. 66:1282–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Marion PL. 1996. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. J. Virol. 70:6151–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seeger C, Leber EH, Wiens LK, Hu J. 1996. Mutagenesis of a hepatitis B virus reverse transcriptase yields temperature-sensitive virus. Virology 222:430–439. 10.1006/viro.1996.0440 [DOI] [PubMed] [Google Scholar]
- 14.Alber T. 1989. Mutational effects on protein stability. Annu. Rev. Biochem. 58:765–798. 10.1146/annurev.bi.58.070189.004001 [DOI] [PubMed] [Google Scholar]
- 15.Shin Y-C, Park S, Ryu W-S. 2011. A conserved arginine residue in the terminal protein domain of hepatitis B virus polymerase is critical for RNA pregenome encapsidation. J. Gen. Virol. 92:1809–1816. 10.1099/vir.0.031914-0 [DOI] [PubMed] [Google Scholar]
- 16.Kim S, Wang H, Ryu WS. 2010. Incorporation of eukaryotic translation initiation factor eIF4E into viral nucleocapsids via interaction with hepatitis B virus polymerase. J. Virol. 84:52–58. 10.1128/JVI.01232-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. 1979. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 281:646–650. 10.1038/281646a0 [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Lee H-J, Shin M-K, Ryu W-S. 2004. Versatile PCR-mediated insertion or deletion mutagenesis. Biotechniques 36:398–400 [DOI] [PubMed] [Google Scholar]
- 19.Tang H, McLachlan A. 2001. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc. Natl. Acad. Sci. U. S. A. 98:1841–1846. 10.1073/pnas.98.4.1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin M-K, Lee J, Ryu W-S. 2004. A novel cis-acting element facilitates minus-strand DNA synthesis during reverse transcription of the hepatitis B viruses genome. J. Virol. 78:6252–6262. 10.1128/JVI.78.12.6252-6262.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewellyn EB, Loeb DD. 2011. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 85:1298–1309. 10.1128/JVI.01957-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cha MY, Ryu DK, Jung HS, Chang HE, Ryu WS. 2009. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 90:978–986. 10.1099/vir.0.009928-0 [DOI] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat. Protoc. 1:581–585. 10.1038/nprot.2006.83 [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Lee J, Ryu WS. 2009. Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsidation. J. Virol. 83:8032–8040. 10.1128/JVI.00332-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abraham TM, Lewellyn EB, Haines KM, Loeb DD. 2008. Characterization of the contribution of spliced RNAs of hepatitis B virus to DNA synthesis in transfected cultures of Huh7 and HepG2 cells. Virology 379:30–37. 10.1016/j.virol.2008.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haines KM, Loeb DD. 2007. The sequence of the RNA primer and the DNA template influence the initiation of plus-strand DNA synthesis in hepatitis B virus. J. Mol. Biol. 370:471–480. 10.1016/j.jmb.2007.04.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. 2013. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 9:e1003125. 10.1371/journal.ppat.1003125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartenschlager R, Junker-Niepmann M, Schaller H. 1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 64:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon H, Lok AS. 2011. Hepatitis B therapy. Nat. Rev. Gastroenterol. Hepatol. 8:275–284. 10.1038/nrgastro.2011.33 [DOI] [PubMed] [Google Scholar]
- 30.Zoulim F. 2012. Are novel combination therapies needed for chronic hepatitis B? Antiviral Res. 96:256–259. 10.1016/j.antiviral.2012.09.006 [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Cheng X, Cao F, Huang A, Tavis JE. 2013. β-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 99:221–229. 10.1016/j.antiviral.2013.06.007 [DOI] [PubMed] [Google Scholar]