

Abstract
Highly pathogenic avian influenza H5N1 virus clades 2.3.4, 2.3.2, and 7 are the dominant cocirculating H5N1 viruses in poultry in China. However, humans appear to be clinically susceptible mostly to the 2.3.4 virus clade. Here, we demonstrated that A549 cells and human macrophages infected with clade 2.3.4 viruses produced significantly more viruses than those infected with the other two clades. Likewise, clade 2.3.4-infected macrophages caused the most severe cellular damage and strongest proinflammatory response.
TEXT
Highly pathogenic avian influenza (HPAI) H5N1 viruses have continued to evolve and diversify since their occurrence in China and other countries. Based on the hemagglutinin (HA) gene of the H5N1 viruses, 10 distinct phylogenetic clades (clades 0 to 9) have been identified (1, 2). HPAI H5N1 viruses have infected over 633 humans, and 377 of these cases were fatal according to the World Health Organization (3). These human H5N1 cases remains relatively uncommon, and only certain clades of H5N1 viruses are linked to human infections (4). In China, since 2005, epidemiological studies have confirmed that there are three dominant clades (2.3.2, 2.3.4, and 7) of H5N1 viruses cocirculating in poultry (5, 6). In principle, these three virus clades could be expected to confer similar rates of infection of humans. However, 83% (35/42) of confirmed human cases of H5N1 virus infection in China since 2005 were caused by clade 2.3.4 viruses (communications from Chinese Center for Disease Control and Prevention). At present, it is not known why humans appear to be most susceptible to the virus clade 2.3.4 and not to clade 2.3.2 or 7. To ascertain the human cytopathogenicity of clade 2.3.4, 2.3.2, and 7 HPAI H5N1 viruses, we compared their abilities to attach to human respiratory epithelial cells, replicate in human airway cells and macrophages, and induce host proinflammatory response in macrophages.
Three representative bird strains of H5N1 viruses of each clade, 2.3.4, 2.3.2, and 7, as well as a human strain of the 2.3.4 clade, A/Anhui/1/2005, were used in the comparative studies (Table 1). All the characterized avian H5N1 strains were isolated from apparently healthy birds in live poultry markets (5, 7, 8); the human H5N1 strain was isolated from a pregnant woman (9). Virus stocks were prepared in Madin-Darby canine kidney (MDCK) cells, and virus infectivity was based on 50% tissue culture infection dose (TCID50) assays on MDCK cells as previously described (8). The intravenous pathogenicity indexes of the 10 viruses (Table 1) were considerably greater than 1.2, indicating that they are highly pathogenic avian influenza viruses. All experiments with H5N1 influenza viruses were performed in a biosecurity level 3 containment approved by the Biosafety Management Committee of the State Key Laboratory of Pathogens and Bio-security.
TABLE 1.
H5N1 avian influenza viruses used in this study
| Virus strain | Abbreviation | Virus clade | IVPIa |
|---|---|---|---|
| A/Anhui/1/2005 | AH05 | 2.3.4 | 3.0 |
| A/tree sparrow/Jiangsu/1/2008 | JS08 | 2.3.4 | 3.0 |
| A/chicken/Jilin/Q023/2009 | JL09 | 2.3.4 | 2.92 |
| A/duck/Liaoning/Q1/2009 | LN09 | 2.3.4 | 3.0 |
| A/chicken/Huabei/0513/2007 | HB07 | 2.3.2 | 2.85 |
| A/chicken/Tianjin/QA22/2009 | TJ09 | 2.3.2 | 2.88 |
| A/chicken/Guangdong/QR11/2009 | GD09 | 2.3.2 | 2.90 |
| A/chicken/Sheny/0606/2008 | SY08 | 7 | 2.80 |
| A/chicken/Shanxi/QX2/2009 | SX09 | 7 | 2.85 |
| A/chicken/Ningxia/QH14/2009 | NX09 | 7 | 2.90 |
IVPI, intravenous pathogenicity index.
Tropism of avian influenza virus for the human upper respiratory tract is regarded as an important determinant of avian-to-human transmission (10, 11). To investigate the tissue tropism of clade 2.3.4, 2.3.2, and 7 H5N1 viruses, we examined their binding patterns to human upper and lower respiratory tissue sections as previously described (12). Briefly, paraffin-embedded sections of human trachea and lung tissues, deparaffinized in xylene and rehydrated through an alcohol gradient, were incubated with virus suspensions (64 HA units in phosphate-buffered saline [PBS]), followed by incubation with a nucleoprotein (NP) antibody to detect attached viruses. As shown in Fig. 1, the pandemic H1N1/2009 virus, as a control, bound extensively to ciliated epithelial cells in the trachea and, to a lesser degree, alveolar lining. All three clades of H5N1 viruses attached extensively to alveolar cells, but there was little binding to the tracheal tract. Tissue sections pretreated with Arthrobacter ureafaciens sialidase abolished virus binding, indicating the function of sialic acid receptors in virus attachment. Thus, all three clades of H5N1 viruses appeared to possess similarly low binding affinity for the human upper respiratory epithelium but higher binding ability to the alveolar lining.
FIG 1.
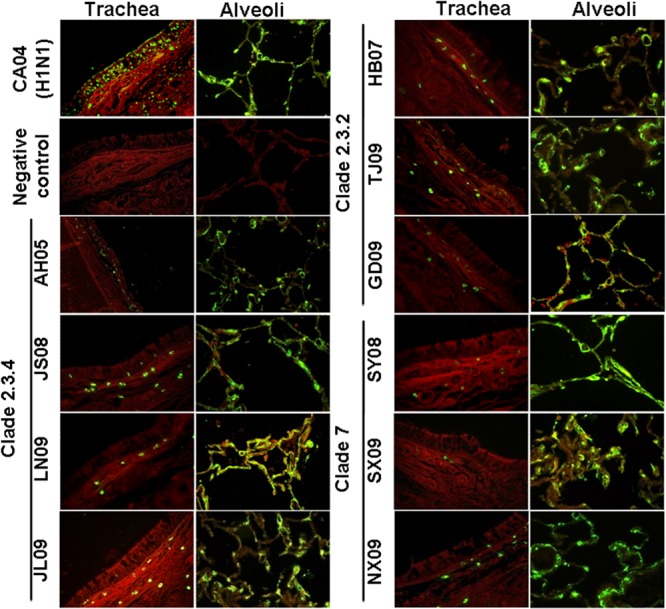
Binding of influenza viruses to human respiratory tissue sections. Human tracheal and alveolar tissue sections were incubated with clade 2.3.4 viruses (AH05, JS08, JL09, and LN09), clade 2.3.2 viruses (HB07, TJ09, and GD09), clade 7 viruses (SY08, SX09, and NX09), pandemic H1N1 (CA04) influenza virus, or negative control. Influenza virus nucleoprotein was subsequently localized by fluorescein isothiocyanate-labeled goat anti-mouse IgG.
Type II alveolar epithelial cells and macrophages are major cell targets of H5N1 virus in infected human patients (13, 14). To ascertain viable virus production of each virus clade, human A549 cells, a cell line widely used as an in vitro model for type II pulmonary epithelial cells (15), were infected with H5N1 viruses at a multiplicity of infection (MOI) of 0.001 and virus titers were analyzed over 72 h of infection. As evident in Fig. 2A, cells infected with viruses of clade 2.3.4 produced more infectious virus than did those infected with viruses of clade 2.3.2 or clade 7 from 12 h postinfection (hpi), reaching high titers at approximately 106.2 TCID50/ml at 72 h of infection. In contrast, clade 2.3.2 and clade 7 viruses showed peak titers at 104.5 and 103.5 TCID50/ml, respectively. To show that the differences in virus titers were not from differences in the early stage of infection, we determined viral protein expression levels by Western blotting with the cells infected by three clades of viruses at an MOI of 2 at 6 hpi. As shown in Fig. 2B, there were no differences in the expression levels of NP protein among the three virus clades. These results indicated that clade 2.3.4 viruses possessed the highest replication rate in human A549 epithelial cells.
FIG 2.
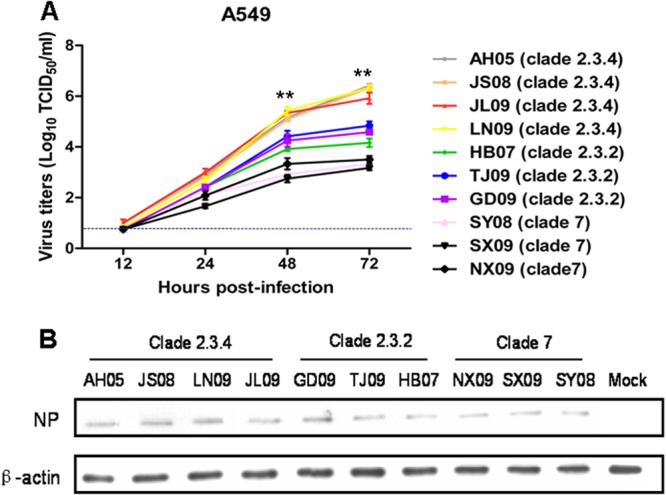
Viral growth properties in A549 cells. (A) Viable virus output from A549 cells infected with clade 2.3.4 (AH05, JS08, JL09, and LN09), clade 2.3.2 (HB07, TJ09, and GD09), and clade 7 (SY08, SX09, and NX09) viruses at an MOI of 0.001. At 12, 24, 48, and 72 h postinfection, virus titers in the supernatants were determined by TCID50 assays on MDCK cells. The values are expressed as means ± standard deviations (n = 3). **, P < 0.01 between clade 2.3.4 virus-infected cells and clade 2.3.2 virus-infected cells. The dashed line represents the detection limit of the TCID50 assay. (B) A549 cells were infected with clade 2.3.4 (AH05, JS08, JL09, and LN09), clade 2.3.2 (HB07, TJ09, and GD09), and clade 7 (SY08, SX09, and NX09) viruses at an MOI of 2. At 6 h of infection, the viral NP expression levels were determined by Western blotting. β-Actin was used as a loading control.
Macrophages in the lung play an important role in innate and adaptive immune responses to viruses and other intracellular pathogens (16, 17). Due to the difficulty in sourcing human alveolar macrophages, human monocyte-derived macrophages (MDMs) were used in the study of macrophage response to influenza virus infection (18–20). We prepared human MDMs from healthy donors as previously described (19). To determine the infection ability of clade 2.3.4, 2.3.2, and 7 HPAI H5N1 virus in macrophages, we infected macrophages with the representative virus strains at an MOI of 0.1 and examined the percentage of infected cells by the detection of viral NP using immunofluorescence at 6 or 24 hpi. As shown in Fig. 3A, the number of JS08 or AH05 (clade 2.3.4)-infected cells (>95%) was significantly higher (P < 0.05) than that of cells infected with HB07 (clade 2.3.2) (63% ± 6%) or SY08 (clade 7) (25% ± 3%) at 6 hpi. By 24 h of infection, similar proportions (>95%) of macrophages were infected by JS08, AH05 (clade 2.3.4), and HB07 (clade 2.3.2), which in turn were significantly higher (P < 0.05) than SY08 (clade 7) (47% ± 8%) virus-infected cells. Similar results were obtained with two additional virus strains of each clade (data not shown). To ensure that the H5N1 viruses at the calculated titers were infected at comparable doses, we also infected MDCK cells with the representative virus strains at an MOI of 0.1 and examined the percentage of infected cells by the detection of viral NP using immunofluorescence. As shown in Fig. 3B, MDCK cells were equally susceptible to infections by viruses from the three clades, with over 95% of cells expressing virus protein at 6 or 24 hpi. The results indicated that clade 2.3.4 viruses proliferated better in macrophages than did the other two virus clades.
FIG 3.
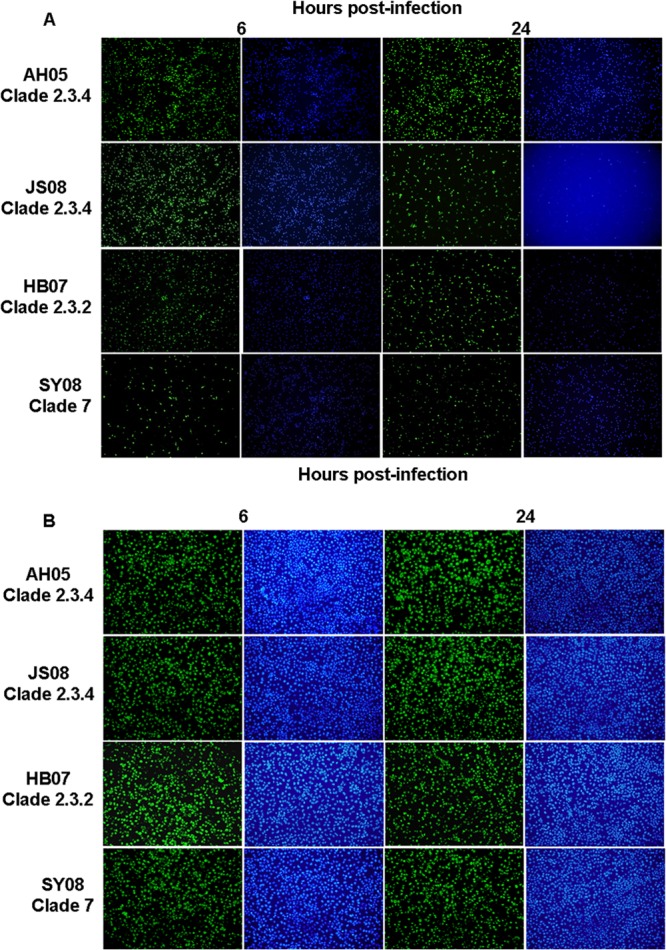
Immunofluorescence staining of macrophages (A) and MDCK cells (B) at 6 and 24 h postinfection with JS08 (clade 2.3.4), AH05 (clade 2.3.4), HB07 (clade 2.3.2), or SY08 (clade 7). Influenza virus nucleoprotein was detected with a fluorescein isothiocyanate-conjugated mouse antibody (left column at each time point). Nuclei were detected with 4′,6-diamidino-2-phenylindole (right column).
Culture supernatants of macrophages infected at an MOI of 0.001 with the three clades of viruses were titrated by TCID50 assays on MDCK cells. As shown in Fig. 4, there was no significant difference in virus titers between the clade 2.3.4 and 2.3.2 viruses at each time point. However, from 12 h of infection onward, clade 2.3.4 and 2.3.2 viruses produced more viable progeny viruses (P < 0.05) than did clade 7 viruses. At their peak, the mean outputs of clade 2.3.4 and 2.3.2 viruses were 59 times and 41 times higher than that of clade 7 viruses, respectively. Thus, although all of the three virus clades were able to replicate in macrophages, virus replication rates varied considerably between individual viruses.
FIG 4.
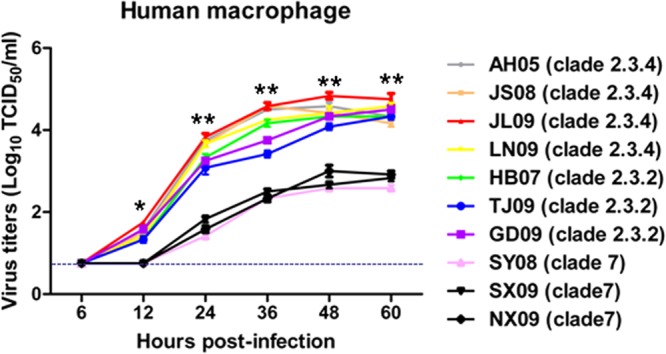
Viable virus output from macrophages infected with clade 2.3.4 (AH05, JS08, JL09, and LN09), clade 2.3.2 (HB07, TJ09, and GD09), or clade 7 (SY08, SX09, and NX09) virus at an MOI of 0.001. At 6, 12, 24, 36, 48, and 60 h postinfection, virus titers in the supernatants were determined by TCID50 assays on MDCK cells. The values are expressed as means ± standard deviations (SD) (n = 3). *, P < 0.05, and **, P < 0.01, between clade 2.3.4-virus infected cells and clade 7 virus-infected cells. The dashed line represents the detection limit of the TCID50 assay.
Morphological changes of macrophages infected at an MOI of 0.1 with the three clade viruses were examined (Fig. 5A). Clade 2.3.4 viruses produced the most severe cytopathic effects, exhibiting extensive damage and detachment of cells as early as 12 hpi, followed by clade 2.3.2 viruses and clade 7 viruses, which caused the least severe cellular disruption even at 48 hpi. We further examined the surface morphology of macrophages infected with the three clades of viruses at an MOI of 0.1 at 24 hpi by scanning electron microscopy. Clade 2.3.4 virus-infected macrophages showed severe morphological damage and extensive cellular disintegration, in sharp contrast to mock-infected cells. Clade 2.3.2 virus-infected cells displayed less-severe damage and the presence of numerous fine pseudopodia extending from each cell body. Clade 7 virus-infected macrophages were least morphologically affected, and there were no obvious changes observed compared with uninfected controls (Fig. 5B). These results demonstrated that clade 2.3.4 viruses were able to cause the most severe cytopathic damage in human macrophages.
FIG 5.

Morphological changes of human macrophages caused by three clades of H5N1 viruses. (A) Phase-contrast microscopy of macrophages infected with JS08 (clade 2.3.4), HB07 (clade 2.3.2), SY08 (clade 7), or mock control at an MOI of 0.1 for 12, 24, 36, and 48 h. (B) Scanning electron micrograph of human macrophages infected with JS08 (clade 2.3.4), HB07 (clade 2.3.2), SY08 (clade 7), or mock control at an MOI of 0.1 for 24 h.
Since macrophages are a major source of cytokine and chemokine production during infection, we analyzed the expression of interleukin-1β (IL-1β), IL-6, IL-8, tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), and monocyte chemotactic protein 1 (MCP-1) in the culture supernatants of macrophages infected with the three clades of viruses at an MOI of 0.1 for 12 h (Fig. 6). Clade 2.3.4 viruses induced the highest expression of IL-1β, IL-6, IL-8, TNF-α, IFN-γ, and MCP-1 compared with the other two virus clades (P < 0.01). Interestingly, clade 7 viruses appeared to have little effect on the induction of cytokine or chemokine production in infected macrophages. These findings indicated that clade 2.3.4 was the most potent of the three clades to induce the production of cytokines and chemokines in human macrophages.
FIG 6.

Cytokine and chemokine production in human macrophages infected with the three H5N1 virus clades. At 12 h of infection at an MOI of 0.1, the supernatants of the infected cells were collected and the proteins of 6 cytokines (IL-1β, IL-6, IL-8, TNF-α, IFN-γ, and MCP-1) were determined by cytometric bead array assays. The values are expressed as means ± standard deviations (n = 3). **, P < 0.01 between clade 2.3.4 virus-infected cells and clade 2.3.2 virus-infected cells.
In summary, we found no apparent difference in binding affinity for the human respiratory tract among the three virus clades. However, clade 2.3.4 viruses were clearly the most prolific of the three clades in the de novo production of infectious virus from alveolar epithelial cells and macrophages. Furthermore, clade 2.3.4 viruses induced the earliest and most severe cytopathic damage and the strongest proinflammatory response in human macrophages. These findings were consistent with the clinical severity and frequency of human cases of H5N1 virus infection caused by clade 2.3.4 virus in China.
ACKNOWLEDGMENTS
This work was supported by the National Science Fund for Distinguished Young Scholars (31025029), the National Basic Research Program (973 Program, 2011CB504702) of China, the Chang Jiang Scholars Program, the Chinese Universities Scientific Fund, and the Biotechnology and Biological Sciences Research Council (UK) China Partnering Award.
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.Duan L, Bahl J, Smith GJ, Wang J, Vijaykrishna D, Zhang LJ, Zhang JX, Li KS, Fan XH, Cheung CL, Huang K, Poon LL, Shortridge KF, Webster RG, Peiris JS, Chen H, Guan Y. 2008. The development and genetic diversity of H5N1 influenza virus in China, 1996–2006. Virology 380:243–254. 10.1016/j.virol.2008.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO/OIE/FAO 2008. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 14:e1. 10.3201/eid1407.071681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO 2013. Cumulative number of confirmed human cases of avian influenza A/(H5N1) reported to WHO, 2003–2013. World Health Organization, Geneva, Switzerland: http://www.who.int/influenza/human_animal_interface/H5N1_cumulative_table_archives/en/ [Google Scholar]
- 4.Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z, Hayden FG, Nguyen DH, de Jong MD, Naghdaliyev A, Peiris JS, Shindo N, Soeroso S, Uyeki TM. 2008. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 358:261–273. 10.1056/NEJMra0707279 [DOI] [PubMed] [Google Scholar]
- 5.Jiang WM, Liu S, Chen J, Hou GY, Li JP, Cao YF, Zhuang QY, Li Y, Huang BX, Chen JM. 2010. Molecular epidemiological surveys of H5 subtype highly pathogenic avian influenza viruses in poultry in China during 2007–2009. J. Gen. Virol. 91:2491–2496. 10.1099/vir.0.023168-0 [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Shi J, Zhong G, Deng G, Tian G, Ge J, Zeng X, Song J, Zhao D, Liu L, Jiang Y, Guan Y, Bu Z, Chen H. 2010. Continued evolution of H5N1 influenza viruses in wild birds, domestic poultry, and humans in China from 2004 to 2009. J. Virol. 84:8389–8397. 10.1128/JVI.00413-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Q, Ma J, Kou Z, Pu J, Lei F, Li T, Liu J. 2010. Characterization of a highly pathogenic avian influenza H5N1 clade 2.3.4 virus isolated from a tree sparrow. Virus Res. 147:25–29. 10.1016/j.virusres.2009.09.014 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Sun Y, Sun H, Pu J, Bi Y, Shi Y, Lu X, Li J, Zhu Q, Gao GF, Yang H, Liu J. 2012. A single amino acid at the hemagglutinin cleavage site contributes to the pathogenicity and neurovirulence of H5N1 influenza virus in mice. J. Virol. 86:6924–6931. 10.1128/JVI.07142-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu Y, Yu H, Li D. 2006. Lethal avian influenza A (H5N1) infection in a pregnant woman in Anhui Province, China. N. Engl. J. Med. 354:1421–1422. 10.1056/NEJMc053524 [DOI] [PubMed] [Google Scholar]
- 10.Sorrell EM, Schrauwen EJ, Linster M, De Graaf M, Herfst S, Fouchier RA. 2011. Predicting ‘airborne’ influenza viruses: (trans-) mission impossible? Curr. Opin. Virol. 1:635–642. 10.1016/j.coviro.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Riel D, den Bakker MA, Leijten LM, Chutinimitkul S, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T. 2010. Seasonal and pandemic human influenza viruses attach better to human upper respiratory tract epithelium than avian influenza viruses. Am. J. Pathol. 176:1614–1618. 10.2353/ajpath.2010.090949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y. 2012. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486:420–428. 10.1038/nature10831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao R, Dong L, Dong J, Wen L, Zhang Y, Yu H, Feng Z, Chen M, Tan Y, Mo Z, Liu H, Fan Y, Li K, Li CK, Li D, Yang W, Shu Y. 2010. A systematic molecular pathology study of a laboratory confirmed H5N1 human case. PLoS One 5:e13315. 10.1371/journal.pone.0013315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau LT, Lu J, Gao Z, Zhang B, McNutt MA, Lu M, Anderson VM, Gong E, Yu AC, Lipkin WI. 2007. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137–1145. 10.1016/S0140-6736(07)61515-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieber M, Smith B, Szakal A, Nelson-Rees W, Todaro G. 1976. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer 17:62–70. 10.1002/ijc.2910170110 [DOI] [PubMed] [Google Scholar]
- 16.Kreijtz JH, Fouchier RA, Rimmelzwaan GF. 2011. Immune responses to influenza virus infection. Virus Res. 162:19–30. 10.1016/j.virusres.2011.09.022 [DOI] [PubMed] [Google Scholar]
- 17.Wijburg OL, DiNatale S, Vadolas J, van Rooijen N, Strugnell RA. 1997. Alveolar macrophages regulate the induction of primary cytotoxic T-lymphocyte responses during influenza virus infection. J. Virol. 71:9450–9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mok KP, Wong CH, Cheung CY, Chan MC, Lee SM, Nicholls JM, Guan Y, Peiris JS. 2009. Viral genetic determinants of H5N1 influenza viruses that contribute to cytokine dysregulation. J. Infect. Dis. 200:1104–1112. 10.1086/605606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakabe S, Iwatsuki-Horimoto K, Takano R, Nidom CA, Le M, Nagamura-Inoue T, Horimoto T, Yamashita N, Kawaoka Y. 2011. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J. Gen. Virol. 92:1428–1434. 10.1099/vir.0.030346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakabe S, Takano R, Nagamura-Inoue T, Yamashita N, Nidom CA, Iwatsuki-Horimoto K, Kawaoka Y. 2013. Differences in cytokine production in human macrophages and in virulence in mice are attributable to the acidic polymerase protein of highly pathogenic influenza A virus subtype H5N1. J. Infect. Dis. 207:262–271. 10.1093/infdis/jis523 [DOI] [PMC free article] [PubMed] [Google Scholar]