

Abstract
Last-generation nucleoside/nucleotide analogues are potent against hepatitis B virus (HBV) and have a high barrier to resistance. However, delayed responses have been observed in patients previously exposed to other drugs of the same class, long-term resistance is possible, and cure of infection cannot be achieved with these therapies, emphasizing the need for alternative therapeutic approaches. The HBV RNase H represents an interesting target because its enzyme activity is essential to the HBV life cycle. The goal of our study was to characterize the structure of the HBV RNase H by computing a 3-dimensional molecular model derived from E. coli RNase H and analyzing 2,326 sequences of all HBV genotypes available in public databases and 958,000 sequences generated by means of ultradeep pyrosequencing of sequences from a homogenous population of 73 treatment-naive patients infected with HBV genotype D. Our data revealed that (i) the putative 4th catalytic residue displays unexpected variability that could be explained by the overlap of the HBx gene and has no apparent impact on HBV replicative capacity and that (ii) the C-helix-containing basic protrusion, which is required to guide the RNA/DNA heteroduplex into the catalytic site, is highly conserved and bears unique structural properties that can be used to target HBV-specific RNase H inhibitors without cross-species activity. The model shows substantial differences from other known RNases H and paves the way for functional and structural studies as a prerequisite to the development of new inhibitors of the HBV cell cycle specifically targeting RNase H activity.
INTRODUCTION
Hepatitis B virus (HBV) infection is among the top 10 most frequent viral infections worldwide, with approximately 240 million chronic HBV carriers facing a dramatically high risk of life-threatening complications, such as liver cirrhosis or hepatocellular carcinoma (HCC) (1). HBV infection currently is the first cause of primary liver cancer worldwide, and mortality due to chronic hepatitis B exceeds 1 million per year. Prevention of these complications can be achieved if HBV replication is efficiently controlled in the long term. Two types of antiviral treatments can be used to achieve this goal: a finite treatment with injectable pegylated alpha interferon or lifelong oral administration of nucleoside and/or nucleotide analogues that target the HBV DNA polymerase (2).
The DNA-dependent and RNA-dependent HBV DNA polymerase is a multifunctional protein that consists of four domains, including a terminal protein (TP) primase, a reverse transcriptase (RT), an RNase H, and a variable spacer domain between the TP and RT domains (3). Nucleoside/nucleotide analogues are highly effective in controlling HBV replication by specifically inhibiting the enzymatic activity of the HBV RT (4). Treatment of chronic hepatitis B with RT inhibitors induces a rapid decrease of viremia. Long-term treatments with first-generation, low-barrier-to-resistance drugs, such as lamivudine or adefovir, have been associated with frequent viral breakthroughs due to the selection of drug-resistant HBV variants (5, 6). The current first-line HBV drugs, entecavir and tenofovir, are potent and have a high barrier to resistance (7). They ensure control of viral replication in the short term to midterm in the vast majority of treatment-naive patients. However, delayed responses have been observed in patients previously exposed to other drugs of the same class (8–10), development of resistance is possible in the long term, and cure of infection cannot be achieved with these therapies, emphasizing the need for other therapeutic approaches that target functions other than reverse transcription (11).
The HBV RNase H represents a potential therapeutic target, because its enzymatic activity is essential to the HBV life cycle. Indeed, the RNase H cleaves the RNA strand of RNA-DNA heteroduplexes formed during the viral genome replication cycle. Targeted drug discovery requires extensive knowledge of RNase H structure and variability. Attempts to experimentally determine the 3-dimensional (3-D) structure of the HBV DNA polymerase or its RNase H domain have been unsuccessful thus far. Experimentally solved RNase H structures from various organisms have been reported (12–21). The catalytic activity of type 1 RNases H has been shown to depend on a conserved tetrad of residues (DEDD), likely through a two-metal-ion-dependent mechanism (22–24). Another important functional feature of type 1 RNases H is the existence of a basic protrusion involved in substrate recognition (25). This protrusion sometimes (e.g., in Escherichia coli, Thermus thermophilus, murine Moloney leukemia virus [MMLV], xenotropic murine leukemia virus-related virus, prototype foamy virus, and Homo sapiens) but not always (e.g., in Bacillus halodurans, Sulfolobus tokodaii, human immunodeficiency virus type 1 [HIV-1], and HIV-2), includes an α-helix, called the C-helix, followed by a basic loop and the first residues of the D-helix.
The availability of numerous RNase H structures and their conservation across species support homology-based molecular modeling of the HBV RNase H. A molecular model of the HBV RNase H using the E. coli RNase H structure as the template has already been published (26). In the present work, we generated a new molecular model of HBV RNase H from the E. coli RNase H structure by using (i) sequences from different HBV genotypes generated by population sequencing and available in public databases and (ii) quasispecies sequences from a homogeneous population of 73 never-treated patients infected with HBV genotype D, generated by means of a highly sensitive method based on ultradeep pyrosequencing (UDPS).
MATERIALS AND METHODS
Molecular modeling.
A pairwise alignment of the HBV RNase H (UniProtKB Swiss-Prot accession number C7DSI5, residues 680 to 832; 153 residues) and the E. coli RNase H (PDB 1RDD) sequences was computed by means of the Clustal W 1.8 algorithm and manually refined in the C-terminal region of the sequences. The refinement was performed by combining data from (i) a structural alignment of MMLV (PDB 2HB5), E. coli (PDB 2RN2), HIV-1 (PDB 1HRH), and B. halodurans (PDB 1ZBF) RNases H published by Lim et al. (17), (ii) the pairwise sequence alignment of HBV and E. coli RNases H published by Potenza et al. (26), (iii) secondary structures derived from the 3-D structures by means of the DSSP algorithm (27), and (iv) predicted secondary structures obtained by combining PHD (28), DSC (29), and SOPMA (30) algorithms with the NPS@ server (31).
The molecular model of the HBV RNase H was computed by means of Geno3D software (32). The modeling process builds models by following a protocol similar to the one used for structure resolution by nuclear magnetic resonance. Geno3D initiates the molecular modeling process by measuring spatial restraints (dihedral angles and interatomic distances) from the template structure for similar residues according to the query-template alignment. The restraints are then used in the distance geometry, simulated annealing, molecular dynamics, and energy minimization algorithms as implemented in the CNS software (version 1.2) (33).
RNase H variability in sequences generated by population sequencing retrieved from the HBVdb database.
HBV RNase H sequences were retrieved from release 1.0 of the HBVdb database (http://hbvdb.ibcp.fr) (34). All sequences were combined into one data set, called all, while the notD data set excluded the genotype D sequences. The genotype D data set, called Dpop, included only genotype D sequences. Dpop was created for direct comparison with the Dudps sequence data set, which was generated by ultradeep pyrosequencing (UDPS) from a cohort of genotype D-infected treatment-naive patients. The residue repertoire, residue frequencies, and normalized Shannon entropy (NSE) values were computed for these four data sets. The resulting data were used to identify RNase H positions showing variability according to an NSE cutoff of 0.1.
RNase H variability in quasispecies sequences generated by ultradeep pyrosequencing.
A representative subset of 80 patients (52 males and 28 females with a mean age [± standard deviation] of 52 ± 13 years) was randomly selected from a cohort of 579 treatment-naive hepatitis B e antigen (HBeAg)-negative patients chronically infected with HBV genotype D for UDPS sequence analysis of the RNase H domain. HBV DNA was extracted from 200 to 500 μl of serum with the QIAamp DNA blood kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's instructions. A 697-bp fragment encompassing the full-length RNase H domain of HBV DNA polymerase was PCR amplified with primers RNase-H-F (nucleotide positions 1031 to 1058, 5′-CACAATGTGGATATCCTGCCTTAATGC-3′) and RNase-H-R (nucleotide positions 1702 to 1728, 5′-CCTACTTCAAAGACTGTGTGTTTAAG-3′) in the presence of AmpliTaqGold (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. The following amplification program was used: initial denaturation at 95°C for 10 min; 40 PCR cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 40 s; and final elongation at 72°C for 7 min. PCR products were then purified using Amicon-Ultra columns (Millipore, Billerica, MA). A second, nested PCR round was performed with internal primers UDPS3719-F (nucleotide positions 1134 to 1154, 5′-CATGAACCTTTACCCCGTTG-3′) and UDPS3719-R (nucleotide positions 1661 to 1681, 5′-GACTCTTGGACTCCCAGCAA-3′), modified to introduce a GS FLX bead adaptor and a specific identity tag (multiplex identifier [MID]). HBV DNA could be amplified from the serum of 73 of the 80 patients studied. Amplicons containing the bead adaptor and MID were then pyrosequenced as previously described (35). Briefly, the amplicons were purified in Nucleofast 96 PCR plates (Clontech, Moutain View, CA), quantified with the Quant-iT PicoGreen double-stranded DNA (dsDNA) kit (Invitrogen, Carlsbad, CA), and emulsion-PCR amplified by means of an emPCR (emulsion-based clonal amplification PCR) kit (454 Life Sciences, Roche Diagnostics Corp., Branford, CT). The beads were counted with a Beckman Coulter Z1 particle counter (Beckman Coulter, Brea, CA) and deposited in a GS FLX titanium PicoTiterPlate (454 Life Sciences). The pyrosequencing reaction was performed in both directions with the GS FLX titanium sequencing kit on an FLX genome sequencer (454 Life Sciences).
Our in-house software package, Pyropack, was used to analyze UDPS data (35–38). Briefly, the sequences were ascribed to each patient by means of their identity tags and PyroClass. Then, the sequences were quality filtered and aligned, and substitutions were analyzed statistically by PyroMute to eliminate low-quality data and errors due to the procedure and keep only relevant sequences for final analysis. The consensus amino acid sequence was built and used as a reference to map genetic variability using PyroDyn.
Mapping of variable positions on the HBV RNase H molecular model.
The variable positions identified in the HBVdb and UDPS-generated sequences were mapped on the HBV RNase H molecular model by means of PyMOL software (0.99rc6, 2006; DeLano Scientific LLC [http://www.pymol.org]).
RESULTS
Molecular modeling of HBV RNase H.
Our manually refined pairwise sequence alignment used to compute the molecular model is shown in Fig. 1. It displayed 14.6% identity and 39.3% similarity, with a gap content of 27.0%. These values and secondary structure predictions indicated that HBV and E. coli RNases H are remote homologues and implied low-resolution molecular models. The first 60 residues of the HBV sequence (residues 680 to 739) were reliably aligned with the E. coli ones, with conserved and similar residues around the first 3 active site residues. The remaining residues were more difficult to align, and HBV D817 was aligned with the 4th catalytic residue of E. coli RNase H.
FIG 1.
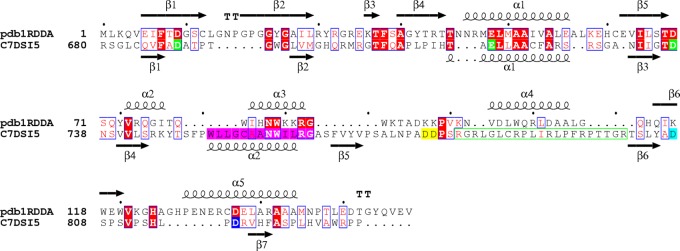
Pairwise sequence alignment of E. coli and HBV RNases H. The E. coli and HBV sequence are designated by pdb1RDDA and C7DSI5, respectively. Identical residues are shown in red boxes. Similar residues are in red letters. The first three catalytic residues of the HBV RNase H (D689, E718, and D737) are in green. The four D residue candidates for the 4th catalytic residue are in yellow (D777 and D778), cyan (D807), and blue (D817). The HBV RNase H putative C-helix is highlighted in magenta. The dots indicate gaps. Protein secondary structures deduced from the 3-D structure (E. coli) or predicted (HBV) are indicated above and below each sequence, respectively, with the numbering of each secondary structure element (e.g., α1 and β1). The figure was generated with the ESPript server (50).
A set of 10 models was computed with 10,675 distances and 314 dihedral restraints involving 108 residues. The agreement between the models was good, with a mean root mean square deviation (RMSD) of 1.54 Å over 81 Cα atoms. The stereochemistry of each model was checked by means of the PROCHECK software (39). Among the set of 10 computed models, model 8 was selected for further analysis according to energy value, stereochemistry, restraint violations, and the RMSD to the template. A superimposition of the template structure and selected model is presented in Fig. 2. Its RMSD with the PDB 1RDD template structure was 1.49 Å over 81 Cα atoms. Although this value indicated good agreement between the model and the template structure, there were local differences in close proximity to the putative C-helix (α2 predicted helix at residues 751 to 762). However, the accuracy of the molecular model in this region was poor, due to the lack of restraints as a consequence of weak identity and the presence of insertions in the HBV RNase H sequence before and after the putative C-helix.
FIG 2.
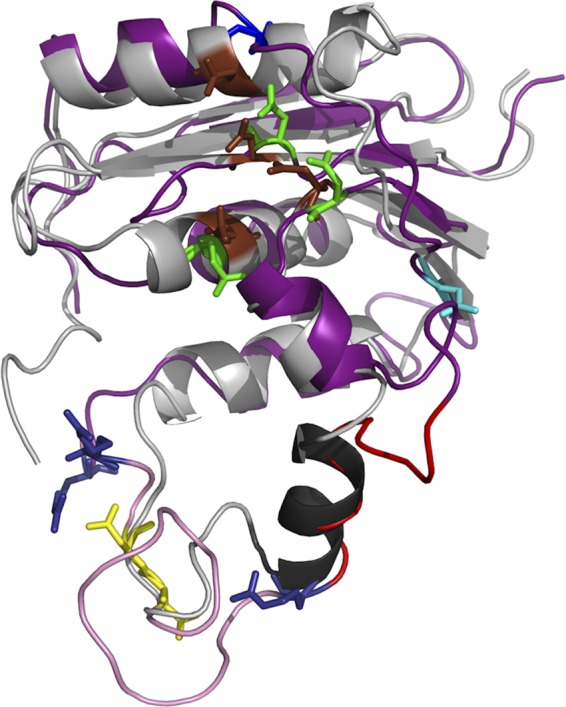
Cartoon representation of the superimposition of the E. coli RNase H structure and the HBV RNase H model. The E. coli experimental structure is gray, with the C-helix in black and catalytic residues as brown sticks. The HBV RNase H model is deep purple. The first three catalytic residues and the four D residue candidates for the 4th catalytic residue are shown in sticks and colored as in Fig. 1, i.e., green (D689, E718, and D737), yellow (D777 and D778), cyan (D807), and blue (D817; top of the model). The putative basic protrusion is shown in pink, with the C-helix in red and arginine residues in deep blue (bottom of the model).
Overall variability of HBV RNase H.
A total number of 2,326 HBV RNase H sequences were retrieved from release 1.0 of the HBVdb database (http://hbvdb.ibcp.fr) (34). They comprised 676 genotype A, 121 genotype B, 963 genotype C, 193 genotype D, 223 genotype E, 97 genotype F, 29 genotype G, and 24 genotype H sequences. These sequences were combined into the three data sets all, notD, and Dpop.
A total number of approximately 958,000 UDPS sequences was generated, with an average of 13,130 ± 3,220 sequences per patient. Their average length was 302 bp (range, 221 to 369 bp), and 63.8% of the full set of sequences was considered of excellent quality (PHRED score, >30). After eliminating unreliable or too-short sequences by means of PyroMute, 910,000 sequences remained available for subsequent analysis and constituted the 4th data set, Dudps.
Figure 3 shows plots of NSE per sequence position, based on the analysis of the 4 sequence data sets all, notD, Dpop, and Dudps. Overall, 81.7% of the positions (125/153) had an NSE of <0.1. Among the 28 remaining positions with an NSE of ≥0.1 listed in Table 1, 10 had NSEs of between 0.201 and 0.463. These results indicate that the HBV RNase H sequence is conserved among HBV isolates and quasispecies variants. In addition, the Dudps data set, generated by means of UDPS, a technology capable of identifying minor viral populations within complex quasispecies, identified a stretch of slightly variable positions (positions 741 to 753) with NSEs of between 0.101 and 0.205. Within this stretch, position 749 (NSE of 0.151) was also identified as variable in Dpop, whereas position 752 was more conserved (NSE of <0.1 for the 4 data sets).
FIG 3.

Distribution of normalized Shannon entropy (NSE) values along the RNase H sequence in the four sequence data sets studied. The histograms for the three sequence data sets from the public database are gray (all data set; all sequences), black (notD data set; all sequences of genotypes other than D), and cyan (Dpop data set; sequences of genotype D only). The histogram for the data set of genotype D sequences generated by means of ultradeep sequencing (Dudps data set) is blue. The HBV RNase H sequence positions are numbered on the x axis. The y axis reports the NSE values. The four data sets are shown along the z axis.
TABLE 1.
Most-variable positions in HBV RNase H according to a normalized Shannon entropy threshold of 0.1 for the four sequence datasets used in the study
| Position | Residue | NSE value in: |
|||
|---|---|---|---|---|---|
| All | NotD | Dpop | Dudps | ||
| 681 | S | 0.217 | 0.22 | 0.097 | 0.031 |
| 698 | V | 0.08 | 0.003 | 0.121 | 0.092 |
| 699 | M | 0.136 | 0.078 | 0.095 | 0.066 |
| 702 | Q | 0.184 | 0.191 | 0.017 | 0.015 |
| 709 | Q | 0.156 | 0.041 | 0.463 | 0.449 |
| 732 | N | 0.22 | 0.182 | 0.01 | 0.086 |
| 733 | I | 0.111 | 0.051 | 0.115 | 0.094 |
| 734 | I | 0.076 | 0.022 | 0.203 | 0.214 |
| 741 | V | 0.004 | 0.005 | 0 | 0.104 |
| 742 | L | 0.001 | 0 | 0.01 | 0.101 |
| 743 | S | 0.004 | 0.004 | 0 | 0.103 |
| 744 | R | 0.001 | 0.001 | 0 | 0.111 |
| 745 | K | 0.008 | 0.008 | 0 | 0.125 |
| 747 | T | 0.004 | 0.004 | 0 | 0.113 |
| 748 | S | 0.005 | 0.005 | 0 | 0.126 |
| 749 | F | 0.03 | 0.006 | 0.151 | 0.205 |
| 750 | P | 0.001 | 0.001 | 0 | 0.115 |
| 751 | W | 0.001 | 0.001 | 0 | 0.123 |
| 753 | L | 0.006 | 0.006 | 0.01 | 0.109 |
| 756 | A | 0.185 | 0.189 | 0.041 | 0.056 |
| 786 | L | 0.132 | 0.137 | 0.056 | 0.303 |
| 787 | C | 0.25 | 0.216 | 0.313 | 0.345 |
| 792 | R | 0.233 | 0.238 | 0.076 | 0.012 |
| 795 | F | 0.109 | 0.115 | 0.024 | 0.043 |
| 796 | R | 0.225 | 0.232 | 0.036 | 0.042 |
| 807 | D | 0.18 | 0.143 | 0.041 | 0.085 |
| 817 | D | 0.257 | 0.265 | 0.034 | 0.234 |
| 830 | R | 0.145 | 0.146 | 0.109 | 0.124 |
Conservation of the catalytic tetrad.
Type 1 RNase H enzymes typically display four conserved residues that constitute the DEDD catalytic tetrad (24). In the present analysis, the first three catalytic residues of the tetrad (D689, E718, and D737) were found to be conserved across the four data sets analyzed (all, notD, Dpop, and Dudps). However, the third catalytic residue (D737) was less conserved in the Dudps data set, with 12 patients harboring a D737A substitution in variants that represented 15% to 50% of their viral quasispecies.
Four positions bearing a D residue (777, 778, 807, and 817) could be considered candidates for the 4th catalytic residue. However, none of them fully fulfilled the expected criteria, including a high level of conservation and a short distance from the 3 other catalytic residues. Although D777 was highly conserved across HBV genotypes, and mutating it was reported to dramatically affect the enzyme activity (40), this residue was too far from the three other catalytic residues (>25 Å) in our molecular model. Similarly, residue D778 was highly conserved but too far from the other catalytic residues.
Potenza et al. (26) suggested that D807 could be the 4th catalytic residue of HBV RNase H. Nevertheless, in our analysis, D807 was not conserved across all genotypes. As shown in Fig. 4A, V was the most frequent residue at position 807 in HBV genotypes A, C, E, and G (all and notD data sets). For genotype F, A and D residues were found at nearly equal frequencies. Seventy of the 73 patients infected with HBV genotype D analyzed by UDPS (Dudps data set) had a D as the most frequent residue at position 807, whereas the remaining three patients had an aliphatic substitution (Fig. 4A, patients P1 and P2, who each had a D807V substitution, and patient P3, with D807A) in their major viral population.
FIG 4.

Distribution of amino acids observed at positions 807 and 817 of the HBV RNase H. For both panels, the letters A (dark gray), D (black), G (gray), and V (light gray) on the right side of the bars indicate the residues observed at the given position for sequences retrieved from HBVdb (genotypes A to H, indicated by letters A to H under the bars) and UDPS-generated sequences from 73 patients (Pall under the bars), as well as 3 individual patients (P1 to P3). The height of each bar indicates the observed residue frequency multiplied by 100. (A) Distribution for position 807. Two patients (P1 and P2) showed a D807V substitution, and one patient (P3) a D807A substitution. (B) Distribution for position 817.
In our alignment, HBV RNase H D817 was aligned with the 4th catalytic residue of the E. coli RNase H sequence. As shown in Fig. 4B, D817 was well conserved across all HBV genotypes except genotypes A and H (all, notD, and Dpop data sets). Among the 73 patients tested by UDPS (Dudps data set), 51 had a dominant D817 viral population, 20 had a D817V substitution, and 2 had a D817A substitution. Moreover, one patient with a D817V substitution also had the D807A aliphatic substitution. The HBV DNA level was not substantially lower in this patient than in the other patients (3.5 versus 4.3 ± 1.8 log IU/ml, respectively).
Figure 5 shows a superimposition of residues D689, D737, D817, and E718 in our model with the corresponding residues in the E. coli structure (RMSD of 3.23 Å on 33 atoms). The image is compatible with D817 acting as the 4th catalytic residue. Importantly, these 4 putative catalytic residues were found at almost identical distances from each other in the 10 models generated during this study (data not shown).
FIG 5.
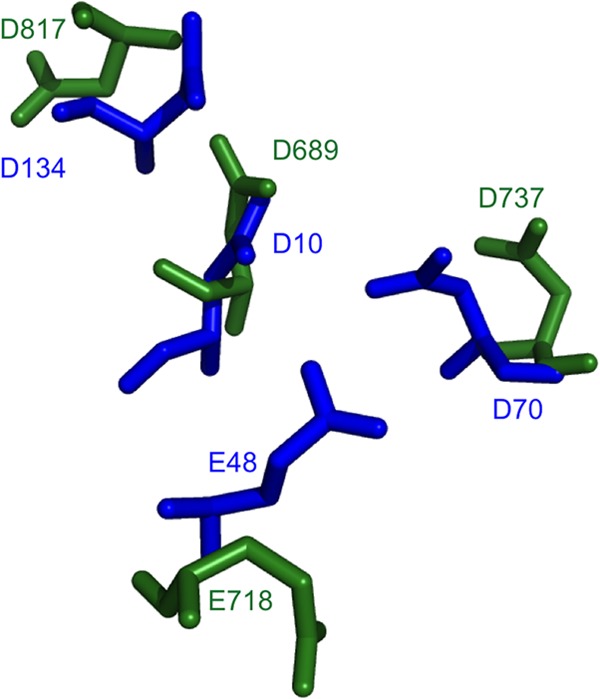
Superimposition of residues D689, D737, D817, and E718 in our model with the corresponding residues in the E. coli structure. HBV and E. coli RNase H residues are shown by blue and green sticks, respectively.
Basic protrusion and C-helix of HBV RNase H.
As shown in Fig. 1 and 2, our sequence alignment and secondary structure predictions identified a basic protrusion in HBV RNase H, between residues 751 and 783 (751-WLLGCAANWILRGASFVYVPSALNPADDPSRGR-783). This protrusion contained a predicted C-helix (helix α2, residues 751-WLLGCAANWILR-762). The NWXXRG residue motif was conserved in both HBV (758-NWILRG-763) and E. coli (84-NWKKRG-89) C-helices. Moreover, the basic protrusion was highly conserved across the 4 data sets (except for positions 751 and 753, which were found to be more variable in the Dudps data set, with NSEs of 0.123 and 0.109, respectively). Strikingly, the HBV C-helix contained only one basic residue (R762) that could be involved in substrate recognition. Finally, compared to E. coli, the HBV RNase H basic protrusion was longer due to two insertions, before and after the C-helix, respectively.
HBV RNase H primer grip.
The primer grip of RNases H is a group of residues that establishes contact with the bases of the RNA-DNA heteroduplex substrate (23). In particular, an aromatic residue (Y or F) within the B-helix plays a key role in substrate binding. According to our alignment, the residue in the HBV sequence that was equivalent to E. coli Y73 was the aliphatic V740, a residue unlikely to play a role in nucleic acid substrate binding. Instead, Y746 appeared to be the most appealing candidate for substrate binding, due to its conservation in the four data sets and its localization 12 positions before the C-helix. The F749 and W751 residues could also be considered candidates for substrate binding, but the NSEs were higher at these positions in the Dudps data set. According to secondary structure predictions, a minor structural difference appeared to exist between the basic protrusions of the HBV RNase H and other enzymes: indeed, a β-strand was predicted instead of the B-helix. This strand could be part of a larger β-sheet that could involve a second predicted β-strand (766-FVYV-769) in this region of the HBV RNase H.
Mapping of variable positions on the RNase H molecular model.
Positions with an NSE of ≥0.1 were mapped onto the molecular model of HBV RNase H. As shown in Fig. 6, variable positions were mainly located outside the molecular surface in contact with the nucleic acid substrate, except for the putative 4th catalytic residue at position 817. The positions identified as variable by UDPS only were principally located between the basic protrusion and the catalytic site.
FIG 6.

Mapping of the HBV RNase H variable positions on the molecular model. The model is drawn in cartoon mode with the surface represented. The first three catalytic residues and the four D residue candidates for the 4th catalytic residue are colored as in Fig. 1 and 2, i.e., green (D689, E718, and D737), yellow (D777 and D778), cyan (D807), and blue (D817; top of the model), respectively. The putative basic protrusion is shown in pink, with the C-helix in red and arginine residues in deep blue (bottom of the model), as in Fig. 2. Variable positions are in orange or in brown if identified by UDPS only.
DISCUSSION
New HBV drug development should now focus on targets other than the HBV reverse transcriptase. Because of its enzymatic activity and its important role in the HBV life cycle, the RNase H domain of the viral polymerase represents an attractive candidate for therapeutic inhibition. To better characterize its key structural features, we computed a molecular model and extensively analyzed sequences available in public databases (essentially generated by population sequencing) and sequences obtained by UDPS. We identified residues likely to be involved in the primer grip and suggested that residue D817 could act as the 4th catalytic residue of the enzyme. Our results also supported the existence of a basic protrusion spanning a C-helix, another argument supporting the conclusion that HBV RNase H is a type 1 RNase H. Our molecular model must, however, be considered with caution because its resolution is low as a result of the relatively weak identity between HBV and E. coli sequences. This is particularly true at the C-terminal end of the sequence, probably as a result of the overlapping reading frame with the HBx protein that induces additional constraints on the RNase H codons. However, this model, based on the analysis of numerous sequences from different HBV genotypes and quasispecies variants, is of the utmost value to understand the structural organization of the HBV RNase H in the frame of type 1 RNases H. It may also be useful to design in vitro functional experiments; however, such models remain limited and their relevance to the natural infection is debated.
The first three residues of the DEDD catalytic tetrad (D689, E718, and D737) were found to be conserved across genotypes and within the viral quasispecies analyzed by UDPS. In contrast, four aspartate residues could possibly be involved as the 4th member of the catalytic tetrad. Although our analysis indicated that D777 and D778 were conserved, they could not be aligned with the 4th catalytic residue of the E. coli RNase H without inserting numerous gaps in the HBV sequence, leaving more than 40 of its C-terminal residues unaligned. Concomitant with the submission of our article, a work by Tavis et al. (41) was published in which mutating D777 abolished RNase H activity, confirming previous results by Chang et al. (40). It cannot be excluded that the D777A substitution abolished the enzyme activity by disrupting the RNase H structure, at least locally, invalidating D777 as a catalytic residue. In spite of these reports, D777 and D778 were both unlikely to act as the 4th catalytic residue according to our analysis.
D807 and D817 were more reliably aligned with the 4th catalytic D residue of E. coli RNase H. However, these two positions showed some variability, as indicated by sequence analysis. UDPS analysis allowed us to characterize HBV variability at the individual patient level, taking into consideration intermediate and minor viral variants along with major ones. Analysis of the Dudps data set at position 807 showed D807V and D807A substitutions in the majority of quasispecies variants in 3 of 73 cases. Based on our analysis, D817 is more likely to be the 4th catalytic residue, as it is the most conserved residue across genotypes (except genotype A) and the distances between this residue and the other members of the catalytic tetrad are compatible with this function in the different models generated in this study and when comparing our model to the E. coli RNase H structure. However, UDPS analysis showed that 20 of the 73 genotype D-infected patients harbored a D817V substitution, whereas only 2 harbored a D817A substitution in viral variants from their quasispecies. One patient harbored a double D807A and D817V substitution (Fig. 4A, P3), ruling out a possible complementation between both D residues.
Conservation of the histidine at position 814 also supported the hypothesis that D817 could be the 4th catalytic residue. Indeed, the importance of a conserved histidine residue has been demonstrated for E. coli (H124), with a mutation to alanine inducing greatly reduced kcat and increased Km values (25, 42). The histidine residue could act as a proton shuttle in the catalysis mechanism (43). Interestingly, among the sequences with no RNase H activity studied by Tavis et al. (41), some displayed substitutions at positions 807, 814, and 817, among others. Overall, although it does not definitively prove it, our study strongly suggests that D817 is the 4th residue of the HBV RNase H catalytic tetrad.
An intriguing result of our study was the discovery of viral variants harboring substitutions in the catalytic tetrad of the enzyme, which could be highly prevalent within patient-circulating quasispecies (up to 100%). Such substitutions were observed at position D737 and at the putative 4th catalytic residues D807 and D817. The existence of such substitutions at positions 807 and 817 could be explained by the need to maintain a glycine residue in the overlapping HBx open reading frame. Indeed, taking into account that the 3rd nucleotide of HBx codons corresponds to the 2nd nucleotide of RNase H codons, changing the 3rd nucleotide of the glycine codon (in order to maintain a glycine residue in HBx) results in the observed D, A, V, or G residues in the HBV RNase H sequence. Whether the corresponding RNase H remains functional is unknown. Some studies suggested a possible one-metal-ion-dependent mechanism, making the 4th catalytic residue of the tetrad dispensable (14, 44, 45). However, more recent studies support a two-metal-ion-dependent mechanism in which RNase H is inactivated when the 4th catalytic residue is mutated (16, 18, 43, 46, 47). In our study, the patients with substitutions at positions D807 and/or D817 did not have substantially lower HBV DNA levels than the others. This finding is in keeping with the former hypothesis but could also be the result of transcomplementation by RNases H from other viral variants or from the infected host cell (48).
Our results support that the HBV RNase H contains a basic protrusion spanning a C-helix. However, the protrusion appears to have distinct features compared to similar protrusions from other organisms. Indeed, the HBV RNase H protrusion is longer than that of E. coli and contains fewer basic residues. In addition, the HBV RNase H basic protrusion appears to contain β-strands along with the C-helix. However, the folding of the protrusion could not be assessed with our model because of the weak identity and the presence of insertions/deletions relative to E. coli sequences. The differences we observed in the HBV RNase H protrusion could be involved in the “template commitment” mechanism specifically described in a native context for the avian HBV RNase H (49). Overall, these data indicate that the HBV RNase H basic protrusion has a specific signature that can be used as a target of future HBV-specific inhibitors. Its exact function in the HBV life cycle, however, remains elusive.
In conclusion, the HBV RNase H appears to be a type 1 RNase H with a shared conserved active site tetrad. Conservation of the catalytic site among type 1 RNases H suggests that inhibitory compounds that target other type 1 RNases H should be tested against HBV. This conclusion is supported by the recent identification of 21 candidate HBV RNase H inhibitors among antagonists of the HIV RNase H and integrase, 12 of which inhibited the HBV RNase H in enzyme assays and 1 of which inhibited HBV replication in cell-based assays (41). Altogether, these results and ours suggest that drug design efforts against these HIV enzymes can be used to guide anti-HBV RNase H drug discovery. Our results also suggest that the unique structural features of the HBV RNase H basic protrusion make it an attractive target for improving the specificity of HBV RNase H inhibitors. They could explain the recently reported differences in the anti-HBV inhibitory efficiencies of the different inhibitors of HIV RNase H and integrase (41), which may be related to the lack of a C-helix in the HIV-1 RNase H (12, 17). Overall, our results set the basis for further studies of HBV RNase H, including mutagenesis of key residues identified here in order to improve the enzyme solubility and activity, that will open the way to drug screening and optimization of specific inhibitors.
ACKNOWLEDGMENTS
J.H. received a doctoral fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche (contract 0012). C.R. received a doctoral fellowship from the National Agency for Research on AIDS and Viral Hepatitis (ANRS). This work was supported by the Finovi Foundation (contract 051274 2010-2011), the Fondation pour la Recherche Médicale (FRM), and the ANRS. Sequence data analyses and molecular modeling were performed on the PRABI computing platform (contract GIS-IBiSA 2009-2011).
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Wei Y, Neuveut C, Tiollais P, Buendia MA. 2010. Molecular biology of the hepatitis B virus and role of the X gene. Pathol. Biol. (Paris) 58:267–272. 10.1016/j.patbio.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 2.European Association for the Study of the Liver 2012. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J. Hepatol. 57:167–185. 10.1016/j.jhep.2012.02.010 [DOI] [PubMed] [Google Scholar]
- 3.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. 10.1016/0092-8674(92)90599-8 [DOI] [PubMed] [Google Scholar]
- 4.Locarnini S, Mason WS. 2006. Cellular and virological mechanisms of HBV drug resistance. J. Hepatol. 44:422–431. 10.1016/j.jhep.2005.11.036 [DOI] [PubMed] [Google Scholar]
- 5.Locarnini S. 2008. Primary resistance, multidrug resistance, and cross-resistance pathways in HBV as a consequence of treatment failure. Hepatol. Int. 2:147–151. 10.1007/s12072-008-9048-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–608.e1-2. 10.1053/j.gastro.2009.08.063 [DOI] [PubMed] [Google Scholar]
- 7.European Association for the Study of the Liver 2009. EASL clinical practice guidelines: management of chronic hepatitis B. J. Hepatol. 50:227–242. 10.1016/j.jhep.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 8.Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, Trépo C, Zoulim F. 2008. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J. Hepatol. 48:747–755. 10.1016/j.jhep.2008.01.027 [DOI] [PubMed] [Google Scholar]
- 9.van Bömmel F, de Man RA, Wedemeyer H, Deterding K, Petersen J, Buggisch P, Erhardt A, Hüppe D, Stein K, Trojan J, Sarrazin C, Böcher WO, Spengler U, Wasmuth HE, Reinders JG, Möller B, Rhode P, Feucht HH, Wiedenmann B, Berg T. 2010. Long-term efficacy of tenofovir monotherapy for hepatitis B virus-monoinfected patients after failure of nucleoside/nucleotide analogues. Hepatology 51:73–80. 10.1002/hep.23246 [DOI] [PubMed] [Google Scholar]
- 10.Patterson SJ, George J, Strasser SI, Lee AU, Sievert W, Nicoll AJ, Desmond PV, Roberts SK, Locarnini S, Bowden S, Angus PW. 2011. Tenofovir disoproxil fumarate rescue therapy following failure of both lamivudine and adefovir dipivoxil in chronic hepatitis B. Gut 60:247–254. 10.1136/gut.2010.223206 [DOI] [PubMed] [Google Scholar]
- 11.Zoulim F. 2012. Are novel combination therapies needed for chronic hepatitis B? Antiviral Res. 96:256–259. 10.1016/j.antiviral.2012.09.006 [DOI] [PubMed] [Google Scholar]
- 12.Davies JF, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. 1991. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252:88–95. 10.1126/science.1707186 [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa K, Okumura M, Katayanagi K, Kimura S, Kanaya S, Nakamura H, Morikawa K. 1993. Crystal structure of ribonuclease H from Thermus thermophilus HB8 refined at 2.8 A resolution. J. Mol. Biol. 230:529–542. 10.1006/jmbi.1993.1169 [DOI] [PubMed] [Google Scholar]
- 14.Katayanagi K, Okumura M, Morikawa K. 1993. Crystal structure of Escherichia coli RNase HI in complex with Mg2+ at 2.8 A resolution: proof for a single Mg(2+)-binding site. Proteins 17:337–346. 10.1002/prot.340170402 [DOI] [PubMed] [Google Scholar]
- 15.Ren J, Bird LE, Chamberlain PP, Stewart-Jones GB, Stuart DI, Stammers DK. 2002. Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. U. S. A. 99:14410–14415. 10.1073/pnas.222366699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. 2005. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121:1005–1016. 10.1016/j.cell.2005.04.024 [DOI] [PubMed] [Google Scholar]
- 17.Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP. 2006. Crystal structure of the Moloney murine leukemia virus RNase H domain. J. Virol. 80:8379–8389. 10.1128/JVI.00750-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. 2007. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell 28:264–276. 10.1016/j.molcel.2007.08.015 [DOI] [PubMed] [Google Scholar]
- 19.You DJ, Chon H, Koga Y, Takano K, Kanaya S. 2007. Crystal structure of type 1 ribonuclease H from hyperthermophilic archaeon Sulfolobus tokodaii: role of arginine 118 and C-terminal anchoring. Biochemistry 46:11494–11503. 10.1021/bi700830f [DOI] [PubMed] [Google Scholar]
- 20.Kim HY, Kwon HD, Jang TS, Lim J, Lee HS. 2012. Mathematical modeling of triphasic viral dynamics in patients with HBeAg-positive chronic hepatitis B showing response to 24-week clevudine therapy. PLoS One 7:e50377. 10.1371/journal.pone.0050377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leo B, Schweimer K, Rösch P, Hartl MJ, Wöhrl BM. 2012. The solution structure of the prototype foamy virus RNase H domain indicates an important role of the basic loop in substrate binding. Retrovirology 9:73. 10.1186/1742-4690-9-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerritelli SM, Crouch RJ. 2009. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 276:1494–1505. 10.1111/j.1742-4658.2009.06908.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champoux JJ, Schultz SJ. 2009. Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 276:1506–1516. 10.1111/j.1742-4658.2009.06909.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tadokoro T, Kanaya S. 2009. Ribonuclease H: molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 276:1482–1493. 10.1111/j.1742-4658.2009.06907.x [DOI] [PubMed] [Google Scholar]
- 25.Kanaya S, Katayanagi K, Morikawa K, Inoue H, Ohtsuka E, Ikehara M. 1991. Effect of mutagenesis at each of five histidine residues on enzymatic activity and stability of ribonuclease H from Escherichia coli. Eur. J. Biochem. 198:437–440. 10.1111/j.1432-1033.1991.tb16033.x [DOI] [PubMed] [Google Scholar]
- 26.Potenza N, Salvatore V, Raimondo D, Falanga D, Nobile V, Peterson DL, Russo A. 2007. Optimized expression from a synthetic gene of an untagged RNase H domain of human hepatitis B virus polymerase which is enzymatically active. Protein Expr. Purif. 55:93–99. 10.1016/j.pep.2007.04.005 [DOI] [PubMed] [Google Scholar]
- 27.Kabsch W, Sander C. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637. 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 28.Rost B, Sander C. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232:584–599. 10.1006/jmbi.1993.1413 [DOI] [PubMed] [Google Scholar]
- 29.King RD, Sternberg MJ. 1996. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 5:2298–2310. 10.1002/pro.5560051116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geourjon C, Deléage G. 1995. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 11:681–684 [DOI] [PubMed] [Google Scholar]
- 31.Combet C, Blanchet C, Geourjon C, Deléage G. 2000. NPS@: network protein sequence analysis. Trends Biochem. Sci. 25:147–150. 10.1016/S0968-0004(99)01540-6 [DOI] [PubMed] [Google Scholar]
- 32.Combet C, Jambon M, Deléage G, Geourjon C. 2002. Geno3D: automatic comparative molecular modelling of protein. Bioinformatics 18:213–214. 10.1093/bioinformatics/18.1.213 [DOI] [PubMed] [Google Scholar]
- 33.Brunger AT. 2007. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2:2728–2733. 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 34.Hayer J, Jadeau F, Deléage G, Kay A, Zoulim F, Combet C. 2013. HBVdb: a knowledge database for hepatitis B virus. Nucleic Acids Res. 41:D566–D570. 10.1093/nar/gks1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez C, Chevaliez S, Bensadoun P, Pawlotsky JM. 2013. Characterization of the dynamics of hepatitis B virus resistance to adefovir by ultra-deep pyrosequencing. Hepatology 58:890–901. 10.1002/hep.26383 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez C, Chevaliez S, Pawlotsky JM. September 2010. PyroClass. French patent IDDN.FR. 001.360005.001.S.P. 2010.000.31230
- 37.Rodriguez C, Chevaliez S, Pawlotsky JM. September 2010. PyroMute. French patent IDDN.FR. 001.120008.001.S.C. 2010.000.31230
- 38.Rodriguez C, Chevaliez S, Pawlotsky JM. September 2010. PyroDyn. French patent IDDN.FR. 001.370024.000.S.P. 2010.000.31230
- 39.Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R, Thornton JM. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8:477–486. 10.1007/BF00228148 [DOI] [PubMed] [Google Scholar]
- 40.Chang LJ, Hirsch RC, Ganem D, Varmus HE. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. 2013. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 9:e1003125. 10.1371/journal.ppat.1003125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oda Y, Yoshida M, Kanaya S. 1993. Role of histidine 124 in the catalytic function of ribonuclease HI from Escherichia coli. J. Biol. Chem. 268:88–92 [PubMed] [Google Scholar]
- 43.Ho MH, De Vivo M, Dal Peraro M, Klein ML. 2010. Understanding the effect of magnesium ion concentration on the catalytic activity of ribonuclease H through computation: does a third metal binding site modulate endonuclease catalysis? J. Am. Chem. Soc. 132:13702–13712. 10.1021/ja102933y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura H, Oda Y, Iwai S, Inoue H, Ohtsuka E, Kanaya S, Kimura S, Katsuda C, Katayanagi K, Morikawa K. 1991. How does RNase H recognize a DNA.RNA hybrid? Proc. Natl. Acad. Sci. U. S. A. 88:11535–11539. 10.1073/pnas.88.24.11535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lener D, Budihas SR, Le Grice SF. 2002. Mutating conserved residues in the ribonuclease H domain of Ty3 reverse transcriptase affects specialized cleavage events. J. Biol. Chem. 277:26486–26495. 10.1074/jbc.M200496200 [DOI] [PubMed] [Google Scholar]
- 46.Yang W, Hendrickson WA, Crouch RJ, Satow Y. 1990. Structure of ribonuclease H phased at 2 A resolution by MAD analysis of the selenomethionyl protein. Science 249:1398–1405. 10.1126/science.2169648 [DOI] [PubMed] [Google Scholar]
- 47.Kashiwagi T, Jeanteur D, Haruki M, Katayanagi K, Kanaya S, Morikawa K. 1996. Proposal for new catalytic roles for two invariant residues in Escherichia coli ribonuclease HI. Protein Eng. 9:857–867. 10.1093/protein/9.10.857 [DOI] [PubMed] [Google Scholar]
- 48.Campbell AG. 2001. Expression of Moloney murine leukemia virus RNase H rescues the growth defect of an Escherichia coli mutant. J. Virol. 75:6212–6217. 10.1128/JVI.75.13.6212-6217.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gong Y, Yao E, Tavis JE. 2001. Evidence that the RNAseH activity of the duck hepatitis B virus is unable to act on exogenous substrates. BMC Microbiol. 1:12. 10.1186/1471-2180-1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gouet P. 2003. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 31:3320–3323. 10.1093/nar/gkg556 [DOI] [PMC free article] [PubMed] [Google Scholar]