

Abstract
The ORF49 tegument protein of varicella-zoster virus (VZV) is one of the core gene products that is conserved among herpesvirus family members. Although ORF49 is known to be a cell-tropic factor, its detailed functions remain elusive. ORF44 is another core gene product reported to be essential, although its characterization and detailed functional analysis have not been reported. These two core gene products form a complex in other herpesviruses beyond the host species and herpesvirus subfamilies. Here, we show that complex formation between ORF44 and ORF49 is conserved in VZV. We serendipitously found that binding is eliminated by an amino acid substitution at position 129 (phenylalanine 129), and four amino acids in the carboxyl-terminal half of the acidic cluster in ORF49 (i.e., aspartate-phenylalanine-aspartate-glutamate from positions 41 to 44 [41DFDE44]) were identified as its binding motif. Alanine substitutions in each domain rendered the ORF44F129A mutation lethal for VZV, similar to deletion of the entire ORF44. The phenotype of the ORF49-41AAAA44 mutation was comparable to that of the ORF49-defective virus, including small-plaque formation, impaired growth, and low infectious virus production. These results suggest that the interaction between ORF44 and ORF49 is essential for their role in VZV infection and that ORF49 is required for the efficient production of infectious progeny virus mediated by the conserved interaction between the two proteins.
INTRODUCTION
Varicella-zoster virus (VZV) is a member of the human alphaherpesvirus subfamily and the etiologic agent of two diseases: varicella is the result of primary infection with VZV, and herpes-zoster is caused by reactivation of the virus from the latent state (1). VZV shares many features, especially a tropism for epithelial and neural tissues, with other human alphaherpesvirus members, including herpes simplex viruses 1 and 2 (HSV-1 and -2, respectively), and with the nonhuman alphaherpesviruses. However, VZV spreads only via cell-to-cell infection in culture and is more akin to the betaherpesviruses (i.e., human herpesviruses 6 and 7) in its apparent T-cell-tropism (1).
The VZV genome is approximately 125 kb and contains at least 70 unique open reading frames (ORFs), and it is the smallest genome in terms of length and gene set among human herpesviruses (1–3). Of the 70 identified ORFs, 44 are core genes that are conserved among all human herpesvirus subfamilies (4). Recent genome-wide mutagenesis analysis showed that 34 ORFs among the core genes are essential for virus reconstitution in cell culture, whereas deletion of seven ORFs results in viral growth defects, and three ORFs are dispensable in cell culture or skin organ culture (5). Eight core genes encode tegument proteins, which are the structural components of the virion and are located between the nucleocapsid and the envelope.
VZV ORF49 encodes a nonessential tegument protein that functions as a cell-tropic factor in cell culture via an unknown mechanism (6). VZV ORF49 is the homolog of HSV-1 UL11 and human cytomegalovirus (HCMV) UL99, which are among the most extensively studied tegument protein-encoding genes. The UL11 and UL99 gene products, pUL11 and pp28, function in secondary envelopment (7–9), but they have different roles in the viral life cycle. HSV-1 UL11 is not essential for the viral life cycle; however, the UL11 deletion mutant forms small plaques, and the final titers are reduced to 80 to 95% of wild-type levels (10). In contrast, HCMV UL99 is an essential gene, and pp28-deficient mutants show extremely impaired growth in normal fibroblasts and produce no detectable infectious progeny (9). However, this mutant spreads from cell to cell via an unknown mechanism (11).
Several recent reports, beginning with one on HSV-1 UL16, which is a core gene within the intron of a conserved herpesvirus spliced gene, (12), showed that interactions between pUL11 and pUL16 homologs were conserved beyond the host species and herpesvirus subfamilies (13–15). HSV pUL16 localizes to the nucleus and the cytoplasm of infected cells and functions in virus entry and in nuclear and cytoplasmic egress (16–19); pUL16 homologs may function in secondary envelopment, as reviewed in reference 20. As described for UL11 homologs, whether UL16 homologs are required for the viral life cycle differs among viruses (13, 15, 21–25). In a genome-wide mutagenesis analysis, deletion of the entire gene region from the viral genome of VZV ORF44, the UL16 homolog, showed that it is an essential gene by loss-of-function analysis in the MeWo cell line (5), although this was not confirmed by a revertant virus generated for gain-of-function analysis.
The LI motif and the conserved acidic cluster of pUL11 are essential for its interaction with pUL16 of HSV-1, whereas the critical sequences in pUL16 have not been determined because it is highly sensitive to deletions. Its short N-terminal 75-amino-acid (aa) fragment was recently shown to include the pUL11 binding site, and its C-terminal region functions as the binding regulatory domain (26), although this has not been confirmed in the context of HSV-1-infection. HCMV pUL94 directs pp28 to the assembly compartment, where it plays a role in secondary envelopment. Amino acids 37 to 39, near the acidic cluster of pp28, and one of the conserved cysteine residues of pUL94 are involved in binding in the context of infection (24, 27). In VZV, potential ORF49 protein (ORF49p)-binding proteins, including the pUL16 homolog ORF44 protein (ORF44p), were identified by global screening using the yeast two-hybrid system (28, 29), although these interactions have not been confirmed, even by coexpression experiments in mammalian cells.
In our previous study on VZV ORF49 (6), ORF49p was identified as one of the cell-tropic factors for VZV lytic infection in cell culture. However, the precise function of ORF49 in cells in which the ORF49-defective virus showed impaired growth was not elucidated. To address this issue, we established a complete trans-complementation system for ORF49 and identified ORF44p as its binding partner in the context of infection. In the present study, we aimed to reveal the precise role of ORF49p by using this system and by analyzing the conserved mechanism of interaction between these proteins and its role in VZV infection.
MATERIALS AND METHODS
Cells and viruses.
The melanoma cell line MeWo was propagated in Dulbecco's modified Eagle's medium (DMEM) (Nissui Pharmaceutical, Ueno, Tokyo) supplemented with 8% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 0.6 mg/ml l-sodium glutamate, and 0.02 mg/ml gentamicin sulfate (Nacalai Tesque, Kyoto, Japan) (DMEM complete). MeWo cells stably expressing Cre recombinase, designated MeWo-Cre cells, were maintained in DMEM complete supplemented with 500 μg/ml G418 (Nacalai Tesque) (30). MeWoORF49 cells stably expressing ORF49 were generated as follows: MeWo cells were transfected with CAG/ORF49 (described below) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions, selected, and propagated in DMEM complete supplemented with 1.5 μg/ml puromycin (Invitrogen). Recombinant viruses derived from the parental VZV strain Oka (pOka), rpOka, rpOkaΔ44Rev, rpOkaORF44F129ARev, rpOkaORF44T128A, rpOkaORF44K130A, rpOkaORF49M1L, rpOkaORF49M1LRev, rpOkaORF49-41AAAA44, and rpOkaORF49-41AAAA44Rev were maintained in DMEM complete supplemented with 3% FBS.
Cell-free virus was prepared as described previously with slight modifications (6). At 48 h postinfection (hpi) by cell-to-cell spread, cells were harvested with a cell scraper (Iwaki, Tokyo, Japan), spun at 800 × g for 5 min at 4°C, and suspended in SGP buffer (phosphate-buffered saline [PBS] containing 0.1% l-sodium glutamate and 7% sucrose). The suspended cells were treated with an ultrasonic disruptor (UD-201; Tomy Seiko, Tokyo, Japan) at 1.5° for 30 s on ice and spun at 800 × g for 5 min at 4°C, and the supernatant was stored at −80°C until use. The purified viral particles were prepared as described in reference 6. Briefly, the cell-free virus solutions were subjected to Histodenz (Sigma-Aldrich) gradient purification (5 to 50% in PBS) by ultracentrifugation at 50,200 × g for 2 h at 4°C in a P40ST rotor (CP80WX; Hitachi Koki, Hitachinaka, Japan). Aliquots of the peak particle-containing fractions were subjected to ultracentrifugation at 52,600 × g for 2 h at 4°C in a P28S rotor (CP80WX; Hitachi Koki), and the pellets were stored at −80°C for further analyses.
Plasmids.
The pGEX/ORF44, pGEX/ORF44A, and pGEX/ORF44P plasmids were generated to express the full length (corresponding to aa 2 to 363), anterior half (aa 2 to 200), and posterior half (aa 181 to 363) of the ORF44 protein in Escherichia coli. The primer pairs ORF44ecoF4 (5′-ACCGAATTCGAATTACAACGCATATTTCCG-3′) and ORF44salR (5′-ACCGTCGACCTAGGTGGTTGTAGG-3′) for ORF44, ORF44ecoF4 and ORF44salR600 (5′-ACCGTCGACTAAATTAGGTTCCATAGCC-3′) for ORF44A, and ORF44ecoF541 (5′-ACCGAATTCGGAGTGTGGTGGTCAGACG-3′) and ORF44salR for ORF44P were used to amplify each indicated region of the ORF44 gene from the rpOka cDNA. The PCR products were inserted in frame into the pGEX6P-1 bacterial expression vector (GE Healthcare Bio-Sciences, Piscataway, NJ) via the EcoRI and SalI sites (underlined). The same procedure was used to construct pGEX/ORF61. The DNA fragment (positions 406 to 744) of ORF61 was cloned into pGEX6P-1 via the BamHI and SalI sites (underlined). The primer pair was ORF61bamF406 (5′-ACCGGATCCGGGCCCCTTCAATCGTCGG-3′) and ORF61salR744stop (5′-ACCGTCGACCTAGAATCTCGCGTTTCCCTC-3′). The eukaryotic ORF44 expression plasmid CAG/ORF44 or CAG/FLAGORF44, which was N-terminally tagged with FLAG (DYKDDDDK), was generated as follows: the entire ORF44 gene was amplified by PCR with the primers ORF44-25kpnF (5′-ACCGGTACCAATCCGCTAGACTG-3′) or ORF44FLAGkpnF4 (5′-ACCGGTACCGCCACCATGgactacaaagacgatgacgacaagGAATTACAACGCATATTTCCG-3′) and ORF44salR, and the PCR fragment was cut by KpnI and SalI (underlined) and cloned into pCAGGS-MCS-neo via the KpnI and XhoI sites. The FLAG tag coding sequence within the primer for ORF44FLAGkpnF4 is shown in lowercase letters. The ORF49 expression plasmid CAG/ORF49 was generated as follows: the entire ORF49 gene was amplified by PCR with the primers ORF49-24ecoF (5′-ACCGAATTCTTACATCAGCATTGCG −3′) and ORF49bamR (5′-ACCGGATCCTTAACATTTTGCGCATTTGG-3′), and the PCR fragment was cut by EcoRI and BamHI (underlined) and cloned into pCAGGS-MCS-puro via the EcoRI and BglII sites. The pCAGGS plasmid was kindly provided by Jun-ichi Miyazaki (Osaka University, Japan) (31). A Cre recombinase-expressing plasmid, pCX-Cre-neo, was previously generated (30) from pCX-Cre, which was a generous gift from Masaru Okabe (Osaka University, Japan).
Construction of mutant ORF44 and ORF49 expression plasmids.
The ORF44 mutant plasmids pGEX/ORF44F129A, CAG/ORF44T128A, CAG/ORF44F129A, CAG/ORF44K130A, CAG/FLAGORF44I121stop, CAG/FLAGORF44P136stop, and CAG/FLAGORF44F129AP136stop and the ORF49 mutant plasmid CAG/ORF49-41AAAA44 were generated with a QuikChange Lightning multisite-directed mutagenesis kit (Agilent Technologies, La Jolla, CA) according to the manufacturer's recommendations, using the primers listed in Table 1 based on pGEX/ORF44, CAG/ORF44, CAG/FLAGORF44, and CAG/ORF49, respectively. The ORF49 C-terminal deletion mutant plasmids CAG/ORF49N40, CAG/ORF49N44, and CAG/ORF49N48 were generated using the following primer pairs: ORF49-24ecoF (5′-ACCGAATTCTTACATCAGCATTGCG-3′) as the forward primer for all the deletion mutants and ORF49mycxhoR120 (5′-ACCCTCGAGcagatcctcttctgagatgagtttttgttcAAAGTCTTCAAAGAACTCTG-3′), ORF49mycxhoR132 (5′-ACCCTCGAGcagatcctcttctgagatgagtttttgttcCTCATCAAAGTCAAAGTCTT-3′), or ORF49mycxhoR144 (5′-ACCCTCGAGcagatcctcttctgagatgag tttttgttcCTCTGTTACATTCTCATCAAAGTC-3′) as the reverse primer for CAG/ORF49N40, CAG/ORF49N44, or CAG/ORF49N48. All of the reverse primers contained a c-myc tag, indicated by lowercase letters, and the PCR products were cloned into pCAGGS-MCS-puro via the EcoRI and XhoI sites (underlined).
TABLE 1.
Primers used for ORF44 or ORF49 mutations
| Primer | Sequencea | Amino acid substitution(s) |
|---|---|---|
| ORF44 361at-ta | 5′-TAT CCG GTT GAA AAC TAA GAC CAT GTT TTT GGA-3′ | Ile 121 to stop (TAA) |
| ORF44 382a-g | 5′-CAT GTT TTT GGA GCA GCG TTT AAG AAC CC-3′ | Thr 128 to Ala |
| ORF44 385tt-gc | 5′-TT TTT GGA GCA ACG GCT AAG AAC CCG ATC G-3′ | Phe 129 to Ala |
| ORF44 388aa-gc | 5′-TTT GGA GCA ACG TTT GCG AAC CCG ATC GCG-3′ | Lys 130 to Ala |
| ORF44 406ccc-taa | 5′-AAC CCG ATC GCG TAC TAA CTT CCA ACA TCT ATT-3′ | Pro 136 to stop (TAA) |
| ORF49 M1L | 5′-ATT GCG GTC ATT GCG TTG GGA CAA TCT TCA-3′ | Met 1 to Leu |
| ORF49 41AAAA44 | 5′-TTT GAA GAC TTT GCC GCT GCT GCG AAT GTA ACA GAG-3′ | Asp-Phe-Asp-Glu (41–44) to Ala-Ala-Ala-Ala |
Nucleotides that differ from those of the wild type are underlined.
Antibodies.
To produce a mouse monoclonal antibody (MAb) against ORF44, a glutathione S-transferase (GST)-ORF44A recombinant protein was expressed in E. coli BL21 transformed with pGEX/ORF44A, purified, and used to immunize mice; hybridoma clones producing the anti-ORF44 MAb were established as described previously (32). To produce polyclonal anti-ORF61 Abs, a GST-ORF61 fusion protein was purified from E. coli BL21 transformed with pGEX-ORF61 and used to immunize a rabbit (Sigma Genosys, Hokkaido, Japan). The anti-ORF61 Ab was purified with GST-conjugated normal human serum (NHS)-activated Sepharose and GST-ORF61-conjugated NHS-activated Sepharose. Rabbit polyclonal anti-ORF49 and anti-gB-C Abs were described previously (6). The mouse anti-glycoprotein E (anti-gE) (clone 9) Ab was described in reference 33. Mouse anti-glycoprotein H (gH) (VgIII-3) was obtained as described previously (33). Sheep anti-trans-Golgi network (anti-TGN46) Ab (AHP500G; AbD Serotec, Oxford, United Kingdom), anti-α-tubulin Ab (B-5-1-2; Sigma-Aldrich), and goat anti-GST Ab (GE Healthcare Bio-Sciences) were commercially available. Alexa Fluor 488-labeled donkey anti-mouse immunoglobulin G (IgG), Alexa Fluor 594-labeled donkey anti-rabbit IgG, and Alexa Fluor 647-labeled donkey anti-sheep IgG (Invitrogen) were used as secondary Abs, and Hoechst 33342 (Sigma-Aldrich) was used for nuclear staining in confocal microscopic analyses. ECL enhanced chemiluminescence anti-mouse or anti-rabbit IgG horseradish peroxidase-linked whole antibodies from donkey (GE Healthcare Bio-Sciences) were used as secondary Abs in immunoblotting.
Mutagenesis of viral genomes in E. coli.
The mutant bacterial artificial chromosomes (BACs) pOka-BACORF49M1L, containing a methionine-to-leucine substitution at residue Met-1, and pOka-BACORF49-41AAAA44, containing a tetra-alanine substitution at residues 41-Asp-Phe-Asp-Glu-44, were generated by recA-mediated allelic replacement in pOka-BAC-harboring DH10B transformed with pST76A-SR/pOkaORF49M1L and pST76A-SR/pOkaORF49-41AAAA44, respectively, which were derived from pST76A-SR/pOkaORF50 (including nucleotide positions 84361 to 89970 of pOka) (30), using the primers listed in Table 1 and a QuikChange Lightning multisite-directed mutagenesis kit. The revertant BACs pOka-BACORF49M1LRev and pOkaBACORF49-41AAAA44Rev were generated by recA-mediated allelic replacement using pST76A-SRORF50 transformed into DH10B cells harboring pOka-BACORF49M1L and pOka-BACORF49-41AAAA44, respectively.
To generate pOka-BACΔ44, in which the nucleotides (nt) 1 to 800 of the ORF44 gene were replaced with an FRT (flp recombinase recognition target) sequence, a linear fragment was amplified by PCR using the primer pair ORF44FRTfKMF0 (5′-TTAAACCCACAAGTACCCGGGCGGCAATCCGCTAGACTGTTTTTCTGCTCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCAGCAAGCGAACCGGAATTGC-3′) and ORF44FRTrKMR800 (5′-TCCCGCTGACCGGCCTTTCTCCACATACACGGAGCCCAACACACACAACCGAAGTTCCTATACTTTCTAGAGAATAGGAACTTCCTTTTTCAATTCAGAAGAACTC-3′) (FRT is underlined) using pCR2.1-TOPO as the template (Invitrogen). The amplified fragment was then transformed into DH10B harboring pOka-BAC (34) with pGETrec (a kind gift from Panayiotis A. Ioannou, The Murdoch Institute for Research into Birth Defects, Royal Children's Hospital, Melbourne, Australia) (35); recE/T recombination for pOka-BACΔ44KMr and excision of the kanamycin resistance gene from pOka-BACΔ44KMr by the flp/FRT system using pCP20 (a kind gift from Wilfried Wackernagel, Universität Oldenburg, Germany) (36), resulting in pOka-BACΔ44, were carried out as described previously (6).
To construct pOka-BACΔ44Rev, the revertant BAC genome against pOka-BACΔ44, recA-mediated mutagenesis was performed as described previously (30), using the shuttle plasmid pST76A-SR/pOkaORF44. For pST76A-SR/pOkaORF44, a 3.2-kbp fragment of viral DNA corresponding to nt 79230 to 82453 of pOka was amplified from the pOka-BAC genome using the primer pair ORF43F1201 (5′-ACCGCTGCGTGTATAAATGCCCGGGTTGAC-3′) and ORF42/45F (5′-ACCATGTCATTGATAATGTTTGG-3′), cloned into pCR2.1-TOPO, sequenced, cut with EcoRI, blunted, and cloned into the plasmid pST76A-SR (kindly provided by Ulrich H. Koszinowski, Max von Pettenkofer Institut, Ludwig-Maximilians-Universität München, Munich, Germany) (37), which was cut with KpnI and blunted.
The shuttle plasmids for the ORF44 point mutant BACs, pST76A-SRORF44T128A (with an alanine substitution for threonine at residue Thr-128), pST76A-SR/pOkaORF44F129A (with an alanine substitution for phenylalanine at residue Phe-129), and pST76A-SR/pOkaORF44K130A (with an alanine substitution for lysine at residue Lys-130), were generated from pST76A-SR/pOkaORF44 with a QuikChange Lightning multisite-directed mutagenesis kit using the primers listed in Table 1. Each mutated shuttle plasmid was transformed into DH10B harboring pOka-BACΔ44KMr, and recA-mediated allelic replacement was performed as described elsewhere (30) to generate pOka-BACORF44T128A, pOka-BACORF44F129A, and pOka-BACORF44K130A. The revertant BAC for the ORF44F129A mutant BAC, pOka-BACORF44F129ARev, was generated by recA-mediated allelic replacement using pST76A-SRORF44 transformed into pOka-BACORF44F129A-harboring DH10B.
All of the purified BACs were digested with BamHI or EcoRI to ensure that the expected DNA fragments were present, and the whole region for allelic replacement was sequenced to ensure that unexpected deletions or substitutions were not present.
Reconstitution of recombinant viruses and excision of the BAC cassette.
MeWo cells or MeWoORF49 cells for rpOkaORF49M1L and rpOkaORF49-41AAAA44 seeded in one well of a 12-well plate were transfected with 3 μg of BAC DNA using Lipofectamine 2000 or X-treamGene HP (Roche Applied Science, Basel, Switzerland). After typical cytopathic effects (CPE) were seen in cells expressing green fluorescent protein (GFP), cell-free virus was prepared as described above and used to infect MeWo-Cre cells or pCX-Cre-neo-transfected MeWoORF49 cells for rpOkaORF49M1L and rpOkaORF49-41AAAA44 to excise the BAC cassette.
Immunoblotting, immunoprecipitation, and immunofluorescence.
Immunoblotting, immunoprecipitation, and immunofluorescence were performed as described previously (30, 32) with slight modifications. The proteins in immunoblots were visualized by Chemi-Lumi One Super (Nacalai Tesque) in combination with LAS4000mini (GE Healthcare Bio-Sciences). Radioimmunoprecipitation assay (RIPA) lysis buffer (0.01 M Tris-HCl [pH 7.4], 0.15 M NaCl, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 1 mM EDTA) supplemented with protease inhibitor cocktail (Sigma-Aldrich) was used for cell lysis, and the supernatants obtained by ultracentrifugation at 216,900 × g for 1 h at 4°C in a P50A3 rotor (CP80WX; Hitachi Koki) and precleared with protein G Sepharose 4 Fast Flow (GE Healthcare Bio-Sciences) were used for immunoblotting and immunoprecipitation. Immunofluorescence images were captured and analyzed by an FV1000D confocal microscope (Olympus, Tokyo, Japan).
In vitro binding assay.
To analyze the interaction of GST-ORF44 with ORF49, GST-ORF44, GST-ORF44F129A, or GST-ORF44P recombinant protein was expressed in and purified using BugBuster master mix (Merck Millipore, Darmstadt, Germany) from E. coli BL21 transformed with pGEX/ORF44, pGEX/ORF44F129A, or pGEX/ORF44P as described above. ORF49p was expressed in MeWo cells by transfection of CAG/ORF49 using Lipofectamine 2000 and solubilized as described above. GST-ORF44 recombinant protein was bound to glutathione Sepharose 4B (GE Healthcare Bio-Sciences) overnight at 4°C, washed with PBS three times, pelleted, and reacted with soluble ORF49p overnight at 4°C. The bead–GST-ORF44 recombinant protein–ORF49p complex was washed with RIPA lysis buffer three times, pelleted, suspended in SDS-PAGE sample buffer, boiled, and subjected to SDS-PAGE and immunoblotting as described above.
Protein identification by MS.
MeWo cells were infected with pOka by cell-to-cell infection and lysed with RIPA lysis buffer as described above. The cell lysates from pOka- or mock-infected MeWo cells were precleared with protein G Sepharose and subjected to immunoprecipitation with anti-ORF49 Ab cross-linked protein G Sepharose. The immunoprecipitates were separated by SDS-PAGE and stained with a SilverQuest silver staining kit (Invitrogen). Protein bands were excised from the gel and digested with trypsin (sequencing grade; Promega, Madison, WI) according to published procedures (38). Nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) analyses were performed on an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with a nano-electrospray ionization (nano-ESI) source (AMR, Tokyo, Japan) and coupled to a Paradigm MG4 pump (Michrom Bioresources, Auburn, CA) and autosampler (HTC PAL; CTC Analytics, Zwingen, Switzerland). A spray voltage of 1,800 V was applied. The peptide mixture was separated on a Magic C18 AQ column (100 μm by 150 mm, 3.0-μm particle size, 300 Å; Michrom Bioresources) with a flow rate of 500 nl/min. The linear gradient was as follows: 5% to 45% B in 30 min, 45% to 95% B in 0.1 min, 95% B for 2 min, and finally 5% B (solvent A = 0.1% formic acid in 2% acetonitrile, and B = 0.1% formic acid in 90% acetonitrile). Intact peptides were detected in the Orbitrap at a resolution of 60,000. For the LC-MS/MS analysis, six precursor ions were selected for subsequent MS/MS scans in a data-dependent acquisition mode following each full scan (m/z, 350 to 1,500). A lock mass function was used for the LTQ-Orbitrap to obtain constant mass accuracy during the gradient analysis. Peptides and proteins were identified by automated database searches using Proteome Discoverer v.1.1 (Thermo Fisher Scientific, Waltham, MA) against human entries or all entries of the Swiss-Prot protein database (version 3.26) with a precursor mass tolerance of 10 parts per million (ppm), a fragment ion mass tolerance of 0.8 Da, and strict trypsin specificity, allowing for up to two missed cleavages. Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation was allowed as a variable modification.
Plaque size and infectious-center assays for growth kinetics.
To analyze the growth kinetics of the recombinant viruses, infectious-center assays were performed as described previously (6) with slight modifications. Briefly, 5 × 105 MeWo or MeWoORF49 cells were seeded on one well of a 12-well plate and inoculated with 50 PFU of cell-free virus per well. For the plaque size measurement, the infected cells were cultured for 7 days. For the infectious-center assay, infected cells were harvested at 24-h intervals and then titrated on newly prepared cells. The cells were fixed in 30% methanol and 70% acetone and stained with an anti-gE MAb (clone 9) and secondary ECL anti-mouse IgG horseradish peroxidase-linked whole antibody (GE Healthcare Bio-Sciences). The stain was developed with 3,3′,5,5′-tetramethylbenzidine-H (TMB-H) peroxidase substrate (Moss, Inc., Pasadena, MD). Images of the plaques were captured and traced, and the number of plaques was counted, or the plaque area was measured using ImageJ (http://rsbweb.nih.gov/ij/).
RESULTS
ORF49 functions in the efficient production of infectious progeny virus.
To examine the mechanism of action of ORF49, we performed loss-of-function and gain-of-function analyses by generating an ORF49-defective virus, rpOkaORF49M1L, and its revertant virus, rpOkaORF49M1LRev, from the pOka-BACORF49M1L and pOka-BACORF49M1LRev genomes, respectively (Fig. 1A and C). In addition, the MeWoORF49 cell line was established to express ORF49 constitutively in MeWo cells in which the previous ORF49-defective virus, rpOkaΔ49, specifically showed an impaired growth phenotype (6), and gain-of-function analysis was performed by ORF49 trans-complementation assay.
FIG 1.

Schematics showing the plasmids and recombinant BAC genomes for ORF44 and ORF49. (A) Location of the ORF44 and ORF49 genes in the unique long region (UL) segment of the genome of VZV strain pOka; terminal repeats (TR), unique short region (US), and internal repeats (IR) are indicated. (B) The wild type, deleted Δ44 region, and amino acid substitutions in ORF44p are shown. (C) The wild type, amino acid substitutions at the first methionine or the carboxyl-terminal half of the acidic cluster from amino acid positions 41 to 44, and the carboxyl-terminal truncations within ORF49p are shown. The acidic cluster is indicated as a black box. (B and C) Unexpressed regions of the mutant proteins are shown in gray outlined shapes. The names of relevant BACs, shuttle plasmids, and mammalian expression plasmids containing mutations are shown on the right.
None of the assayed viral proteins, including glycoprotein H (gH), ORF61 protein (ORF61p), ORF44p, and ORF49p, were detected in MeWo cells (Fig. 2A, lane 1). In MeWoORF49 cells, ORF49p was the only protein detected among the tested viral proteins (Fig. 2A, lane 2), and its expression level was comparable to that of ORF49p in rpOka-infected MeWo cells (Fig. 2A, lane 3). In rpOkaORF49M1L-infected cells, ORF49p was not detected in the infected MeWo cells (Fig. 2A, lane 4) or in the viral particles (Fig. 2C, lane 2), although gH and ORF61p were clearly expressed in infected cells (Fig. 2A, lane 4), as was gH in the viral particles (Fig. 2C, lane 2). The rpOkaORF49M1LRev line expressed all of the viral proteins tested (Fig. 2A, lane 5), similar to rpOka-infected MeWo cells (Fig. 2A, lane 3).
FIG 2.

Expression and interaction of viral proteins during ORF49 mutant virus infection. (A) Proteins expressed in mock-infected MeWo cells (lane 1), mock-infected MeWoORF49 cells (lane 2), rpOka-infected MeWo cells (lane 3), rpOkaORF49M1L-infected MeWo cells (lane 4), and rpOkaORF49M1LRev-infected MeWo cells (lane 5) were visualized with Abs against gH, ORF61p, α-tubulin, ORF44p, and ORF49p. (B) The interaction between ORF44p and ORF49p was analyzed in rpOkaORF49M1L-infected cells (lane 1) and rpOkaORF49M1LRev-infected cells (lane 2). Immunoprecipitates obtained with an anti-ORF49 Ab from each type of virus-infected cells were electrophoretically separated and visualized using anti-ORF44 and anti-ORF49 Abs. (C) The viral proteins incorporated into virions from rpOka-infected MeWo cells (lane 1), rpOkaORF49M1L-infected MeWo cells (lane 2), and rpOkaORF49M1L-infected MeWoORF49 cells (lane 3) were visualized using Abs against gH, ORF61p, ORF44p, and ORF49p. (D) Proteins expressed in rpOkaORF49-41AAAA44-infected MeWo cells (lane 1) and rpOkaORF49-41AAAA44Rev-infected MeWo cells (lane 2) were visualized using Abs against gH, ORF61p, α-tubulin, ORF44p, and ORF49p. (E) The interaction between ORF44p and ORF49p was analyzed in rpOkaORF49-41AAAA44-infected cells (lane 1) and rpOkaORF49-41AAAA44Rev-infected cells (lane 2). Immunoprecipitates obtained using an anti-ORF49 Ab from each type of virus-infected cells were electrophoretically separated and visualized with anti-ORF44 and anti-ORF49 Abs. (F) The viral proteins incorporated into virions from rpOkaORF49-41AAAA44-infected MeWo cells (lane 1), and rpOkaORF49-41AAAA44-infected MeWoORF49 cells (lane 2) were visualized with Abs against gH, ORF61p, ORF44p, and ORF49p.
To confirm the ORF49-defective phenotype and perform gain-of function analysis, plaque formation was analyzed on MeWo and MeWoORF49 cells (Fig. 3A). Plaque sizes were similar between rpOka-infected MeWo and MeWoORF49 cells (Fig. 3A, lanes 1 and 2), indicating that the exogenous expression of ORF49 had neither positive nor negative effects on normal VZV infection. Similar to our previous results using rpOkaΔ49, rpOkaORF49M1L formed significantly smaller plaques on MeWo cells (Fig. 3A, lane 3) than on MeWoORF49 cells, and this reduction was recovered in the revertant virus infection in MeWo cells (Fig. 3A, lane 5) and completely rescued by the exogenous expression of ORF49 in MeWoORF49 cells (Fig. 3A, lane 4). Consistent with the plaque formation assay results, rpOkaORF49M1L propagated on MeWo cells showed slower growth than rpOka or rpOkaORF49M1LRev on MeWo or MeWoORF49 cells in an infectious-center assay, and the growth impairment of rpOkaORF49M1L was completely rescued by exogenous ORF49p in MeWoORF49 cells (Fig. 3B).
FIG 3.

Growth properties of the ORF49M1L mutant virus in MeWo and MeWoORF49 cells. (A) Comparison of plaque sizes among recombinant viruses. MeWo cells or MeWoORF49 cells were infected with rpOka, rpOkaORF49M1L, or rpOkaORF49M1LRev (50 PFU/well) and cultured for 7 days. The infected cells were then stained with an anti-gE Ab, and the plaques were traced and measured by ImageJ software. Plaque size is shown with the standard error of the mean. Statistical significance was determined by Student's t test. (B) Growth kinetics of recombinant viruses in MeWo and MeWoORF49 cells. MeWo or MeWoORF49 cells were infected with rpOka, rpOkaORF49M1L, or rpOkaORF49M1LRev (50 PFU/well), harvested at the indicated times, serially diluted, added to newly prepared MeWo cells, and cultured for 5 days. The plaques were stained with an anti-gE Ab and counted. Each point represents the mean titer for two wells of one experiment. The experiments were performed twice independently. Statistical significance was determined by Student's t test.
During the preparation of cell-free viruses, another strikingly different phenotype of the ORF49 defect was observed. As summarized in Table 2, the titer of cell-free virus or plaque size formed by the cell-free virus infection of rpOka was almost the same whether the virus was propagated on MeWo or MeWoORF49 cells or titrated on MeWo or MeWoORF49 cells, again indicating that the exogenous ORF49p had no effect on normal VZV infection. When rpOkaORF49M1L was propagated on MeWo cells, the titer of cell-free virus was 3 to 5% of that observed with propagation of MeWoORF49 cells, and the plaque size depended on the kind of cells used for titration but not on the kind used for propagation. In addition, the rpOkaORF49M1L particles isolated from MeWoORF49 cells contained abundant ORF49p (Fig. 2C, lane 3) and produced almost the same titer of cell-free virus as the parental virus but formed significantly smaller plaques on MeWo cells (Table 2). This gain-of-function analysis performed using the ORF49 trans-complementation system suggested that the incoming ORF49p from the viral particles into the cells was not functional in any step and revealed that de novo ORF49p functioned in the production of infectious progeny viruses required for efficient propagation.
TABLE 2.
Comparison of cell-free virus titer and plaque formation
| Virus | Cells for: |
Titer (PFU/ml)a | Mean (SE) plaque size, mm2b | |
|---|---|---|---|---|
| Propagation | Titration | |||
| rpOka | MeWo | MeWo | 2.3 × 103 | 0.232 (0.00891) |
| MeWo | MeWoORF49 | 4.1 × 103 | 0.225 (0.00854) | |
| MeWoORF49 | MeWo | 4.0 × 103 | 0.232 (0.00911) | |
| MeWoORF49 | MeWoORF49 | 1.3 × 103 | 0.206 (0.00828) | |
| rpOkaORF49M1L | MeWo | MeWo | 1.5 × 102 | 0.161 (0.00601) |
| MeWo | MeWoORF49 | 1.7 × 102 | 0.235 (0.01319) | |
| MeWoORF49 | MeWo | 4.0 × 103 | 0.147 (0.00534) | |
| MeWoORF49 | MeWoORF49 | 6.3 × 103 | 0.228 (0.01474) | |
| rpOkaORF49-41AAAA44 | MeWo | MeWo | 1.2 × 102 | 0.141 (0.00726) |
| MeWo | MeWoORF49 | 1.3 × 102 | 0.212 (0.00669) | |
| MeWoORF49 | MeWo | 2.1 × 103 | 0.149 (0.00796) | |
| MeWoORF49 | MeWoORF49 | 4.7 × 103 | 0.227 (0.00740) | |
Titers of cell free viruses are shown from one experiment performed in duplicate.
Plaque sizes are shown as means (standard errors [SE]) from one experiment performed in duplicate.
Furthermore, to examine the role of ORF49 in the production of infectious progeny viruses in detail, we redesigned our study on ORF49 to investigate its function by analyzing its binding partners.
Identification of the ORF44 protein as the binding partner for ORF49.
ORF49p was immunoprecipitated from pOka- or mock-infected MeWo cells using an anti-ORF49 antibody (Ab), and the coimmunoprecipitating proteins were separated in a denaturing gel and visualized by silver staining (Fig. 4A). An approximately 36-kDa band was coimmunoprecipitated with the 13-kDa band corresponding to the ORF49p in pOka-infected MeWo cell lysates (Fig. 4A, lane 1). This 36-kDa band was identified as the ORF44 protein (ORF44p) of VZV by LC-MS/MS analysis (24.3% coverage of 363 amino acids).
FIG 4.
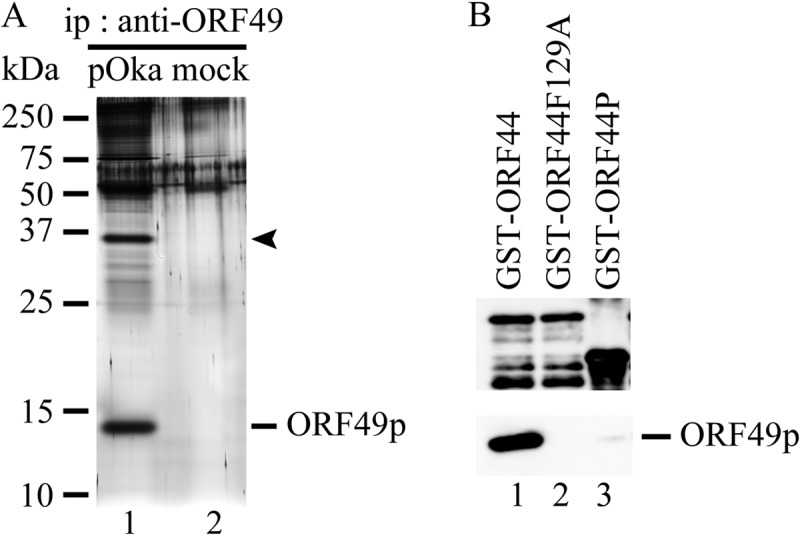
Identification of ORF44p as the binding partner of ORF49p by proteomic analysis and their in vitro binding assay. (A) pOka-infected MeWo cells expanded by cell-to-cell spread with full CPE at 2 to 3 days postinfection were lysed with RIPA buffer, and the binding molecules were coimmunoprecipitated with ORF49p using an anti-ORF49 Ab (lane 1). Mock-infected MeWo cells were used as a negative control (lane 2). The immunoprecipitates (ip) were electrophoretically separated and visualized by silver staining. (B) ORF49p expressed in and purified from MeWo cells was incubated with purified GST-ORF44 (lane 1), GST-ORF44F129A (lane 2), or GST-ORF44P (lane 3). Bound proteins were electrophoretically separated and visualized by anti-GST Abs (upper panel) and anti-ORF49 Abs (lower panel).
ORF44p was specifically detected as a 36-kDa band in all recombinant VZV-infected MeWo cells, including rpOkaORF49M1L, by immunoblotting with an anti-ORF44 Ab (Fig. 2A, lanes 3, 4, and 5). ORF44p was coimmunoprecipitated with ORF49p only in cells infected with the wild-type virus, rpOka, or rpOkaORF49M1LRev (data not shown) (Fig. 2B, lane 2) but not in the absence of ORF49p, as seen in rpOkaORF49M1L infection (Fig. 2B, lane 1). Thus, the interaction of ORF44p and ORF49p was specific and conserved in VZV.
ORF44 is essential for viral growth in cell culture.
Because ORF49 is completely dispensable for viral reconstitution and propagation in MRC-5 cells, and the interaction between ORF44p and ORF49p was confirmed, we predicted that the ORF44 deletion mutant would be viable at least in MRC-5 cells, despite the fact that loss-of-function analysis showed that it is essential in MeWo cells (5). The ORF44 deletion mutant virus could not be reconstituted from pOka-BACΔ44 (Fig. 1B) in either MeWo cells or MRC-5 cells; however, the revertant virus of pOka-BACΔ44 reconstituted from the pOka-BACΔ44Rev genome (Fig. 1B) showed almost the same plaque size and growth as the parental virus, rpOka, in MeWo cells (data not shown). These findings confirmed that ORF44 is essential for VZV growth in cell culture even in MRC-5 cells.
ORF44p binds to and depends on ORF49p for its accumulation on the TGN in coexpressing cells and infection.
When ORF44p was expressed alone by CAG/ORF44 transfection, it was dispersed throughout the cytoplasm and did not localize to the TGN (Fig. 5A). When ORF49p was expressed alone, it was predominantly localized to the juxtanuclear region with TGN46 (Fig. 5B), as reported previously (6). In cells coexpressing ORF44 and ORF49, ORF44p accumulated on the TGN with ORF49p (Fig. 5C), suggesting that the complex formation between ORF44p and ORF49p required no other viral factors and that it functioned in the accumulation of the ORF44p on the TGN. The expression of and interaction between ORF44p and ORF49p were confirmed by immunoblotting with the corresponding Abs and immunoprecipitation with anti-ORF49 Ab followed by immunoblotting with each Ab (Fig. 6A, lane 1, and B, lane 1, respectively). In rpOkaORF49M1LRev-infected MeWo cells, in spite of the broadly diffuse pattern seen for ORF44p, it appeared to accumulate on the TGN with ORF49p (Fig. 7B), as was seen in coexpressing cells (Fig. 5C), and this pattern was also observed in cells infected with rpOka (data not shown). In rpOkaORF49M1L-infected cells, ORF44p was dispersed as in cells expressing ORF44 alone and was not accumulated on the TGN (Fig. 7A), again indicating that the accumulation of ORF44p on the TGN depended on ORF49p and required no other viral factors.
FIG 5.

Localization of ORF44p and ORF49p in transiently transfected MeWo cells. MeWo cells were transfected with CAG/ORF44 (A) or CAG/ORF49 (B) or cotransfected with CAG/ORF44 and CAG/ORF49 (C). Cells were fixed at 48 h posttransfection and triple labeled for ORF44p (green), ORF49p (red), and TGN46 (blue). Nuclei were stained with Hoechst 33342 (cyan). Scale bars, 5 μm.
FIG 6.

Expression and interaction of ORF44p and ORF49p in cotransfected cells. MeWo cells were cotransfected with CAG/ORF49 (lanes 1 to 4 and 7 to 10) or CAG/ORF49-41AAAA44 (lanes 5 and 6) and CAG/ORF44 (lanes 1 and 5), CAG/ORF44T128A (lane 2), CAG/ORF44F129A (lanes 3 and 6), CAG/ORF44K130A (lane 4), CAG/FLAGORF44 (lane 7), CAG/FLAGORF44I121stop (lane 8), CAG/FLAGORF44P136stop (lane 9), or CAG/FLAGORF44F129AP136stop (lane 10) (A and B). Protein expression was visualized with anti-ORF44p and anti-ORF49p antibodies (A), and proteins immunoprecipitated by anti-ORF49 Ab from cotransfected cells were electrophoretically separated and visualized using anti-ORF44 and anti-ORF49 Abs (B).
FIG 7.
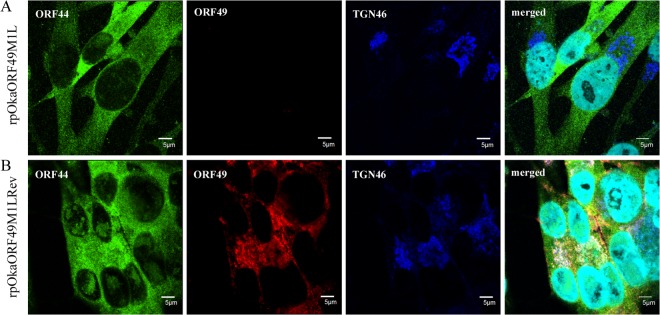
Localization of ORF44p and ORF49p in rpOkaORF49M1L-infected MeWo cells. rpOkaORF49M1L-infected (A) and rpOkaORF49M1LRev-infected (B) MeWo cells were fixed at 48 hpi and triple labeled for ORF44p (green), ORF49p (red), and TGN46 (blue). Nuclei were stained with Hoechst 33342 (cyan). Scale bars, 5 μm.
The phenylalanine at amino acid position 129 of ORF44p functions in the conserved interaction and is essential for VZV infection.
In the process of ORF44 cloning, one mutant showed a T-to-C substitution at nt 385, which led to a phenylalanine-to-serine transition at aa 129 (F129S). In cells coexpressing ORF44F129Sp and ORF49p, ORF44F129Sp did not accumulate on the TGN and was not coimmunoprecipitated with ORF49p (data not shown).
To examine whether 129F functions specifically in the conserved interaction in VZV, alanine scanning was performed around 129F (Fig. 1B). ORF44T128Ap, ORF44F129Ap, and ORF44K130Ap showed similar distributions, and none of these mutants localized to the TGN when expressed alone (data not shown), as observed in cells expressing ORF44p alone (Fig. 5A). Coexpression of ORF44T128Ap and ORF44K130Ap with ORF49p (Fig. 8A and C, respectively) resulted in their colocalization with ORF49p at the TGN, as observed in cells coexpressing ORF44p and ORF49p (Fig. 5C). In contrast, ORF44F129Ap was dispersed throughout the cytoplasm and failed to accumulate on the TGN, even when coexpressed with ORF49p (Fig. 8B). The levels of expression of ORF44T128Ap, ORF44F129Ap, and ORF44K130Ap were almost equal (Fig. 6A, lanes 2, 3, and 4, respectively), and ORF44T128Ap and ORF44K130Ap were coimmunoprecipitated with ORF49p (Fig. 6B, lanes 2 and 4, respectively), whereas ORF44F129Ap was not coimmunoprecipitated with ORF49p (Fig. 6B, lane 3).
FIG 8.

Localization and accumulation of ORF44 mutant proteins in MeWo cells expressing ORF49p. MeWo cells were cotransfected with CAG/ORF49 and CAG/ORF44T128A (A), CAG/ORF44F129A (B), CAG/ORF44K130A (C), CAG/FLAGORF44I121stop (D), CAG/FLAGORF44P136stop (E), or CAG/FLAGORF44F129AP136stop (F). Cells were fixed at 48 h posttransfection and triple labeled for ORF44 (green), ORF49 (red), and TGN46 (blue). Nuclei were stained with Hoechst 33342 (cyan). Scale bars, 5 μm.
The results for F129S and F129A suggest that the binding site for ORF49p might reside in an N-terminal domain, as was recently reported for the pUL16 of HSV-1 (26). Therefore, stop codons were inserted at positions to either side of codon 129F (i.e., at I121stop and P136stop). In addition, a construct containing both the F129A and P136stop mutations was made (Fig. 1B). All three mutants were expressed at their predicted size (Fig. 6A, lanes 8, 9, and 10), and only the ORF44P136stop protein was coimmunoprecipitated with ORF49p (Fig. 6B, lane 9), whereas the ORF44I121stop and ORF44F129AP136stop proteins were not (Fig. 6B, lanes 8 and 10, respectively). Consistent with this, only the ORF44P136stop protein accumulated on the TGN with ORF49p (Fig. 8E), and the other two mutants did not (Fig. 8D and F), despite the diffused cytoplasmic localization of all mutants if expressed alone (data not shown). These results show that the binding site for ORF49p resides in the first third of ORF44p and that 129F plays a critical role in binding, either directly or indirectly. Furthermore, in GST-pulldown assays using GST-ORF44 (corresponding to aa 2 to 363), GST-ORF44F129A (aa 2 to 363 with F129A mutation), and GST-ORF44P (aa 180 to 363), only GST-ORF44 pulled down ORF49p expressed in and purified from MeWo cells (Fig. 4B), suggesting that there may not be a binding site within the C-terminal half of ORF44p; however, other C-terminal constructs have not been tested in this or other assays.
To analyze the impact of the 129F mutation in the context of infection, pOka-BACORF44T128A, pOka-BACORF44F129A, pOka-BACORF44K130A, and the revertant BAC for pOka-BACORF44F129A, pOka-BACORF44F129ARev, were generated (Fig. 1B). With the exception of the ORF44F129A mutant, the reconstitution of each virus with a mutation around 129F and of the revertant virus for the F129A mutant was successful, and all of the reconstituted viruses showed similar growth to rpOka (data not shown), suggesting that 129F of ORF44p may play a central role in the function of ORF44p in VZV infection, which occurs through its interaction with ORF49p.
The carboxyl-terminal half of the acidic cluster of ORF49p is required for the conserved interaction with ORF44p.
To map the binding domain of ORF49p for ORF44p using the accumulation of ORF44p as an indicator of the interaction, we generated a series of carboxyl-terminal-truncated mutants of ORF49p (Fig. 1C). The coexpression of ORF44p and ORF49N48p or ORF49N44p resulted in the accumulation of ORF44p at the juxtanuclear region with TGN46 and the mutant ORF49p (Fig. 9A and B). In contrast, ORF44p never colocalized with ORF49N40p (Fig. 9C) and was dispersed in the cytoplasm, as when it was expressed alone (Fig. 5A). These results suggested that the ORF44p-binding domain in ORF49p may be located between the aspartate at aa 41 and glutamate at aa 44, which is in the carboxyl-terminal half of the conserved acidic cluster (Fig. 1C) in the ORF49 homologs.
FIG 9.

Localization and accumulation of ORF44p in MeWo cells expressing ORF49 mutant proteins. MeWo cells were cotransfected with CAG/ORF44 and CAG/ORF49N48 (A), CAG/ORF49N44 (B), CAG/ORF49N40 (C), or CAG/ORF49-41AAAA44 (E), or transfected with CAG/ORF49-41AAAA44 alone (D). Cells were fixed at 48 h posttransfection and triple labeled for ORF44 (green), ORF49 (red), and TGN46 (blue). Nuclei were stained with Hoechst 33342 (cyan). Scale bars, 5 μm.
To confirm the specificity of the ORF44p and ORF49p interaction while avoiding (although not excluding) nonspecific effects on the function of ORF49p resulting from the destruction of the ORF49p backbone, the four residues (41DFDE44) identified as the candidate ORF44p-binding motif were replaced by alanine, resulting in ORF49-41AAAA44p (Fig. 1C). As shown in Fig. 9D, ORF49-41AAAA44p targeted the TGN, similar to ORF49p (Fig. 5B), but it was unable to accumulate ORF44p on the TGN (Fig. 9E). Furthermore, ORF49-41AAAA44p did not form a complex with ORF44p or ORF44F129Ap (Fig. 6B, lane 5 or 6) despite the efficient coexpression of all proteins (Fig. 6A, lanes 5 and 6).
The carboxyl-terminal half of the acidic cluster of ORF49p plays a central role in the function of ORF49p during infection.
rpOkaORF49-41AAAA44 showed almost the same phenotype as rpOkaORF49M1L, including the loss of the interaction with ORF44p (Fig. 2E, lane 1), the dispersed localization of ORF44p without accumulation on the TGN (Fig. 10A), impaired growth as assessed by plaque size and infectious-center assays (Fig. 11A and B, respectively), and reduced production of infectious progeny virus (to 3 to 10% of the wild-type level [Table 2]), excluding the apparent ORF49-41AAAA44p expression (Fig. 2D, lane 1, and Fig. 10A). These defects were completely rescued by revertant virus infection (Fig. 2D and E, lanes 2, Fig. 10B, and Fig. 11A and B) or by exogenous ORF49p in MeWoORF49 cells (Fig. 11A and B and Table 2). The expression of ORF49-41AAAA44p was detected as a faint and faster-migrating band than that of ORF49p in the revertant virus infection, but an equal amount of ORF44p was detected in both viruses with gH and ORF61p (Fig. 2D, lanes 1 and 2). Furthermore, whereas the interaction between ORF49-41AAAA44p and ORF44p was not detected at all (Fig. 2E, lane 1), ORF44p was incorporated into the rpOkaORF49-41AAAA44 particles in the absence or presence of exogenous ORF49p (Fig. 2F, lane 1 or 2), which was also the case in rpOkaORF49M1L infection (Fig. 2D and F).
FIG 10.
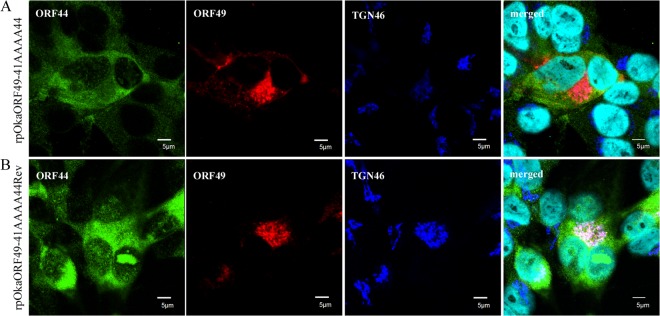
Localization of ORF44p and ORF49p in rpOkaORF49-41AAAA44-infected MeWo cells. rpOkaORF49-41AAAA44-infected MeWo cells (A) and rpOkaORF49-41AAAA44Rev-infected MeWo cells (B) were fixed at 48 hpi and triple labeled for ORF44p (green), ORF49p (red), and TGN46 (blue). Nuclei were stained with Hoechst 33342 (cyan). Scale bars, 5 μm.
FIG 11.

Growth properties of ORF49-41AAAA44 mutant virus in MeWo and MeWoORF49 cells. (A) Comparison of plaque sizes among recombinant viruses. MeWo cells or MeWoORF49 cells were infected with rpOka, rpOkaORF49-41AAAA44, or rpOkaORF49-41AAAA44Rev (50 PFU/well) and cultured for 7 days. Infected cells were stained with an anti-gE Ab, and the plaques were traced and measured by ImageJ software. Plaque size is shown with the standard error of the mean. Statistical significance was determined by Student's t test. (B) Growth kinetics of recombinant viruses on MeWo cells and MeWoORF49 cells. MeWo cells or MeWoORF49 cells were infected with rpOka, rpOkaORF49-41AAAA44, or rpOkaORF49-41AAAA44Rev (50 PFU/well), harvested at the indicated times, serially diluted, added to newly prepared MeWo cells, and cultured for 5 days. The plaques were stained with an anti-gE Ab and counted. Each point represents the mean titer for two wells of one experiment. Two experiments were performed independently. Statistical significance was determined by Student's t test.
Taken together, our results indicated that ORF49p functions in the efficient production of progeny viruses required for VZV infection through its interaction with the essential protein ORF44p.
DISCUSSION
In VZV, ORF49 encodes a nonessential tegument protein that is one of the cell-tropic factors in cell culture (6). In human fetal lung fibroblast MRC-5 cells, the growth of ORF49-defective virus is identical to that of its parental virus, whereas in the human melanoma MeWo cell line, it shows reduced growth; however, the cell tropism of VZV for these two most permissive cell lines has not been studied. In the previous study, we showed that it is a cell-tropic factor, although the step(s) at which ORF49 functions, including the entry, host gene modulation, viral gene expression, viral particle assembly, or egress, remained unclear. Nevertheless, we showed that it may play an important role in the production of a complete virion (6).
VZV is highly cell associated in vitro, producing extremely small amounts of infectious virus even when isolated intracellularly (in a particle-to-PFU ratio that varies from approximately 40,000 to 1,000,000) (39, 40), and the particles detected by electron microscopy (EM) analysis appear to be degraded, even in cells infected by the wild-type virus (41–43). The low infectivity and the presence of few or no infectious particles in the cell culture supernatant in VZV have made it difficult to construct a trans-complementation system. In such a system, the target viral gene is expressed on permissive cells on which the target gene-deleted virus is only capable of efficient replication similar to the wild-type virus infection on the parental cells. The infection of parental cells by the target gene-deleted virus isolated from cells expressing the target gene enables analysis of its function. This method is useful to confirm that the deletion phenotype is not caused by undesired mutations, which can also be determined by generating the revertant virus to repair the mutated gene within the viral genome. In addition, it can also be used to identify the target gene function, which cannot be determined by simply generating the mutant and revertant viruses and has been used widely in the mutagenic analysis of other herpesviruses, especially in the analysis of structural proteins. However, to the best of our knowledge, this system has been used successfully to analyze gene function in only one report (44) and to confirm that the deletion phenotype is independent of undesired mutation in two reports (45, 46) in VZV research.
In the present study, we established a trans-complementation system for ORF49. The ORF49 trans-complementation system allowed identification of the precise function of ORF49, which could not be determined by generating its defective virus following EM analysis on MeWo cells. In the EM analysis, no significant differences between the wild-type and ORF49-defective viruses were detected with regard to the intracellular and cell surface viral particle counts or morphology (T. Sadaoka and Y. Mori, unpublished observation), possibly leading to their obvious difference in infectivity, which was reduced by 10-fold or higher in the defective virus. Additionally, in both viruses, the infected cells or viral particles isolated from the same quantity of infected cells contained almost the same amount of viral proteins. The trans-complementation system in combination with the results of other analyses described above indicated that ORF49 functions in the production of efficient infectious viruses. The results of EM analysis and immunoblotting suggested that the ORF49 defect did not cause the reduction of viral protein synthesis and viral particle assembly and egress. The cell-free virus titration and plaque formation analyses using the trans-complementation system showed that the ORF49p released from the virion into the cells during the entry step was not functional, but de novo ORF49p synthesized during lytic replication functioned in the production of efficient infectious virus required for cell-free and cell-to-cell viral transmission modes. However, how the deletion of ORF49 impaired infectivity remained unclear.
To gain further insight into the function of ORF49 during VZV infection, we confirmed ORF44p as its binding partner, as reported in other herpesviruses (12–15), and examined the conserved interaction between these proteins by analyzing their binding properties. We identified 129F in ORF44p as being essential for accumulation on the TGN through the interaction with ORF49p: whether it functions in the binding directly or indirectly is unknown. Simultaneously, 41DFDE44 of the carboxyl-terminal half of the acidic cluster within ORF49p was identified as the binding motif for ORF44p. Among these critical amino acids of ORF44p and ORF49p, each phenylalanine seems to function in the binding. As the phenylalanine is an aromatic and hydrophobic amino acid, it prefers to be buried in protein hydrophobic cores. However, 129F of ORF44p is surrounded by polar amino acid 128T and charged amino acid 130K and 42F of ORF49p by charged amino acids 41D, 43D, and 44E, and there is possibility that these two phenylalanines are exposed at the protein surface. The phenylalanine side chain is fairly nonreactive and is thus rarely directly involved in protein function, although it can play a role in substrate recognition. In particular, hydrophobic amino acids can be involved in binding/recognition of hydrophobic ligands, and the aromatic side chain can also be involved in interactions with other aromatic side chains via stacking interactions (47). In coexpression of ORF44 and ORF49, ORF49F42A mutation alone disrupted the interaction and failed to accumulate ORF44p on the TGN, as seen in ORF49-41AAAA44 mutation, while individual ORF49D41A, -D43A, or -E44A mutation had no effect on them (T. Sadaoka and Y. Mori, unpublished observation). On the other hand, ORF44F129A showed an impaired phenotype in terms of their interaction and its accumulation on the TGN, and neither ORF44T128A nor K130A mutation had any effect on them. However, in the context of infection, ORF49F42A alone could not abrogate the interaction and had no effect on virus growth (T. Sadaoka and Y. Mori, unpublished observation) different from that of ORF49-41AAAA44 mutation (discussed below), while only ORF44F129A was truly lethal for infectious virus production/reconstitution, and again ORF44T128A and ORF44K130A had no effect. These findings may prompt us to conclude that the core machinery of the binding is the noncovalent attractive force between two aromatic rings of phenylalanine and that the additional binding force via charged amino acids around 42F of ORF49p is required in the context of infection; however, there is another possibility—that the ORF44F129A mutation just disrupts the protein structure itself, leading to the loss of interaction. As mentioned above, phenylalanine prefers to be buried in protein hydrophobic cores, and the interaction among the aromatic residues is also important in the protein folding and the structural stabilization of protein (48, 49). Our results about the interaction property revealed that the binding domain within ORF44p is located at the first 136 residues and 129F is essentially involved in the binding, but could not be determined as the precise binding domain, and at the same time, there was no apparent degradation or lower expression of ORF44F129Ap in comparison with ORF44p in both prokaryotic expression and eukaryotic expression systems. To address these issues, further analyses by making an N-terminal truncation for refining the interaction domain and another 129F substitution with tyrosine, which differs only in that it contains a hydroxyl group in place of the ortho hydrogen on the benzene ring, for more preferable substitution to maintain structural stability will be helpful and ongoing.
Assessment of the function of ORF44 in the context of infection did not reveal new findings, with the exception of the F129A mutant, which showed the same phenotype as the deletion mutant. The ORF44 deletion and F129A mutation were lethal for progeny virus production/reconstitution in MeWo and MRC-5 cells; however, an effective trans-complementation system for ORF44 as for ORF49 was not successfully established, and at what step(s) in the lytic infection ORF44 essentially functions remained unclear. To find the nonessential but important functions of ORF44 in the context of infection, we turned back to analyzing the ORF49 function by generating ORF49-41AAAA44 virus, in which ORF49p specifically lost the interaction with ORF44p, following comparison of the phenotype between ORF49-defective virus and ORF49-41AAAA44 virus. The rpOkaORF49-41AAAA44 virus showed the same phenotype as the rpOkaORF49M1L virus, indicating that the function of ORF49p in the efficient production of progeny viruses was completely dependent on the interaction with ORF44p at 41DFDE44. These results suggest that ORF44p is fully functional only in the presence of ORF49p and vice versa and has essential functions during infection, which are independent of the interaction with ORF49p or redundantly supported by other viral factors in the absence of ORF49p.
In the absence of the interaction with ORF49p during infection, ORF44p was detected throughout the cytoplasm and rarely colocalized with the TGN (or with a reorganized organ containing TGN-derived membranes known to be induced by viral infection, although it has not been found in VZV infection), the recognized site of viral assembly; however, incorporation of ORF44p into viral particles was comparable to that observed in wild-type virus infection. These results indicate that ORF44p was not directly incorporated into the particles through the TGN via its interaction with ORF49p, at least in the absence of ORF49p. In HSV-1, the amount of pUL16 packaged into the viral particles was severely reduced in the absence of pUL11 (50), but there are some other interaction partners that potentially function in incorporating pUL16 into the viral particles (i.e., pUL21 and glycoprotein E) (51, 52). In VZV, by global screening using the yeast two-hybrid system, some candidates for ORF44p binding partner have been reported (28, 29), but in our observations, none of these viral proteins other than ORF49p could accumulate ORF44p on the TGN; one viral protein could alter the localization of ORF44p into the nucleus; however, whether it functions in the incorporation of ORF44p into the viral particles remained unclear (T. Sadaoka and Y. Mori, unpublished observation). Anyway, additional ORF44p binding partners active during either the wild-type virus or ORF49-defective virus infection remain to be identified so far, and the complexity of the herpesvirus protein-protein network requires a solid approach to elucidate the essential roles of ORF44 during viral infection further through the interactions with other viral proteins.
In summary, in the present study, we established a trans-complementation system for ORF49 and identified ORF44p as the binding partner for ORF49p. We showed that (i) ORF49p functions in the efficient production of infectious virus, (ii) no other viral factor is required for binding, (iii) residue 129F of ORF44p is critical not only for binding to ORF49p but also for progeny virus production/reconstitution, (iv) the carboxyl-terminal half of the acidic cluster (41DFDE44) of ORF49p is the binding motif for ORF44p, and (v) the efficient production of infectious progeny virus by ORF49p is dependent on its interaction with ORF44p. Further analyses of the role of ORF44 mediated by its interaction with ORF49 or other as yet unidentified viral proteins may shed light on the conserved infection mechanisms of the Herpesvirinae and those unique to VZV.
ACKNOWLEDGMENTS
We thank Eiko Moriishi (National Institute of Biomedical Innovation, Osaka, Japan) for technical assistance, Panayiotis A. Ioannou (Cell and Gene Therapy Research Group, The Murdoch Children's Research Institute, The University of Melbourne, Royal Children's Hospital, Melbourne, Australia) for providing pGETrec, Wilfried Wackernagel (Genetics, Department of Biology and Environmental Sciences, Universität Oldenburg, Germany) for pCP20, Jun-ichi Miyazaki (Division of Stem Cell Regulation Research, Osaka University Graduate School of Medicine, Japan) for pCAGGS, Masaru Okabe (Department of Experimental Genome Research, Genome Information Research Center, Osaka University, Japan) for pCX-Cre, and Ulrich H. Koszinowski (Max von Pettenkofer Institut fur Virologie, Ludwig-Maximilians-Universität München, Germany) for pHA2 and pST76A-SR.
This study was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas (21022031 to Y.M.) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, a Grant-in-Aid for Scientific Research (B) (20390138 to Y.M.), a Grant-in-Aid for Young Scientists (B) (20790363 and 22790432 to T.S.), a Grant-in-Aid for Scientific Research (C) (24590551 to T.S.) from the Japan Society for the Promotion of Science (JSPS), and a grant from the Uehara Memorial Foundation (to T.S.).
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Moffat J, Ku CC, Zerboni L, Sommer M, Arvin A. 2007. VZV: pathogenesis and the disease consequences of primary infection, p 675–688 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 2.Baines JD, Pellett PE. 2007. Genetic comparison of human alphaherpesvirus genomes, p 61–69 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 3.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759–1816. 10.1099/0022-1317-67-9-1759 [DOI] [PubMed] [Google Scholar]
- 4.Davison AJ. 2007. Comparative analysis of the genomes, p 10–26 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 5.Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog; 6:e1000971. 10.1371/journal.ppat.1000971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadaoka T, Yoshii H, Imazawa T, Yamanishi K, Mori Y. 2007. Deletion in open reading frame 49 of varicella-zoster virus reduces virus growth in human malignant melanoma cells but not in human embryonic fibroblasts. J. Virol. 81:12654–12665. 10.1128/JVI.01183-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baines JD, Jacob RJ, Simmerman L, Roizman B. 1995. The herpes simplex virus 1 UL11 proteins are associated with cytoplasmic and nuclear membranes and with nuclear bodies of infected cells. J. Virol. 69:825–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Britt WJ, Jarvis M, Seo JY, Drummond D, Nelson J. 2004. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J. Virol. 78:539–543. 10.1128/JVI.78.1.539-543.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605. 10.1128/JVI.77.19.10594-10605.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacLean CA, Dolan A, Jamieson FE, McGeoch DJ. 1992. The myristylated virion proteins of herpes simplex virus type 1: investigation of their role in the virus life cycle. J. Gen. Virol. 73:539–547. 10.1099/0022-1317-73-3-539 [DOI] [PubMed] [Google Scholar]
- 11.Silva MC, Schroer J, Shenk T. 2005. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. U. S. A. 102:2081–2086. 10.1073/pnas.0409597102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417–11424. 10.1128/JVI.77.21.11417-11424.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo H, Wang L, Peng L, Zhou ZH, Deng H. 2009. Open reading frame 33 of a gammaherpesvirus encodes a tegument protein essential for virion morphogenesis and egress. J. Virol. 83:10582–10595. 10.1128/JVI.00497-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Cui Z, Zhang Z, Wei H, Zhou Y, Wang M, Zhang XE. 2009. The tegument protein UL94 of human cytomegalovirus as a binding partner for tegument protein pp28 identified by intracellular imaging. Virology 388:68–77. 10.1016/j.virol.2009.03.007 [DOI] [PubMed] [Google Scholar]
- 15.Maninger S, Bosse JB, Lemnitzer F, Pogoda M, Mohr CA, von Einem J, Walther P, Koszinowski UH, Ruzsics Z. 2011. M94 is essential for the secondary envelopment of murine cytomegalovirus. J. Virol. 85:9254–9267. 10.1128/JVI.00443-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meckes DG, Jr, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 81:13028–13036. 10.1128/JVI.01306-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nalwanga D, Rempel S, Roizman B, Baines JD. 1996. The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 226:236–242. 10.1006/viro.1996.0651 [DOI] [PubMed] [Google Scholar]
- 18.Oshima S, Daikoku T, Shibata S, Yamada H, Goshima F, Nishiyama Y. 1998. Characterization of the UL16 gene product of herpes simplex virus type 2. Arch. Virol. 143:863–880. 10.1007/s007050050338 [DOI] [PubMed] [Google Scholar]
- 19.Yeh PC, Meckes DG, Jr, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693–10700. 10.1128/JVI.01230-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 21.Baines JD, Roizman B. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65:938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228. 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klupp BG, Bottcher S, Granzow H, Kopp M, Mettenleiter TC. 2005. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 79:1510–1522. 10.1128/JVI.79.3.1510-1522.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J. Virol. 86:2523–2532. 10.1128/JVI.06548-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401. 10.1073/pnas.1635160100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chadha P, Han J, Starkey JL, Wills JW. 2012. Regulated interaction of tegument proteins UL16 and UL11 from herpes simplex virus. J. Virol. 86:11886–11898. 10.1128/JVI.01879-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips SL, Cygnar D, Thomas A, Bresnahan WA. 2012. Interaction between the human cytomegalovirus tegument proteins UL94 and UL99 is essential for virus replication. J. Virol. 86:9995–10005. 10.1128/JVI.01078-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stellberger T, Hauser R, Baiker A, Pothineni VR, Haas J, Uetz P. 2010. Improving the yeast two-hybrid system with permutated fusions proteins: the varicella zoster virus interactome. Proteome Sci. 8:8. 10.1186/1477-5956-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, Haas J. 2006. Herpesviral protein networks and their interaction with the human proteome. Science 311:239–242. 10.1126/science.1116804 [DOI] [PubMed] [Google Scholar]
- 30.Sadaoka T, Yanagi T, Yamanishi K, Mori Y. 2010. Characterization of the varicella-zoster virus ORF50 gene, which encodes glycoprotein M. J. Virol. 84:3488–3502. 10.1128/JVI.01838-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. 10.1016/0378-1119(91)90434-D [DOI] [PubMed] [Google Scholar]
- 32.Sadaoka T, Yamanishi K, Mori Y. 2006. Human herpesvirus 7 U47 gene products are glycoproteins expressed in virions and associate with glycoprotein H. J. Gen. Virol. 87:501–508. 10.1099/vir.0.81374-0 [DOI] [PubMed] [Google Scholar]
- 33.Okuno T, Yamanishi K, Shiraki K, Takahashi M. 1983. Synthesis and processing of glycoproteins of varicella-zoster virus (VZV) as studied with monoclonal antibodies to VZV antigens. Virology 129:357–368. 10.1016/0042-6822(83)90175-7 [DOI] [PubMed] [Google Scholar]
- 34.Nagaike K, Mori Y, Gomi Y, Yoshii H, Takahashi M, Wagner M, Koszinowski U, Yamanishi K. 2004. Cloning of the varicella-zoster virus genome as an infectious bacterial artificial chromosome in Escherichia coli. Vaccine 22:4069–4074. 10.1016/j.vaccine.2004.03.062 [DOI] [PubMed] [Google Scholar]
- 35.Narayanan K, Williamson R, Zhang Y, Stewart AF, Ioannou PA. 1999. Efficient and precise engineering of a 200 kb beta-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 6:442–447. 10.1038/sj.gt.3300901 [DOI] [PubMed] [Google Scholar]
- 36.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 37.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729. 10.1128/JVI.74.17.7720-7729.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858. 10.1021/ac950914h [DOI] [PubMed] [Google Scholar]
- 39.Carpenter JE, Henderson EP, Grose C. 2009. Enumeration of an extremely high particle-to-PFU ratio for varicella-zoster virus. J. Virol. 83:6917–6921. 10.1128/JVI.00081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiraki K, Takahashi M. 1982. Virus particles and glycoprotein excreted from cultured cells infected with varicella-zoster virus (VZV). J. Gen. Virol. 61:271–275. 10.1099/0022-1317-61-2-271 [DOI] [PubMed] [Google Scholar]
- 41.Gabel CA, Dubey L, Steinberg SP, Sherman D, Gershon MD, Gershon AA. 1989. Varicella-zoster virus glycoprotein oligosaccharides are phosphorylated during posttranslational maturation. J. Virol. 63:4264–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gershon AA, Sherman DL, Zhu Z, Gabel CA, Ambron RT, Gershon MD. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 68:6372–6390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harson R, Grose C. 1995. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J. Virol. 69:4994–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ali MA, Li Q, Fischer ER, Cohen JI. 2009. The insulin degrading enzyme binding domain of varicella-zoster virus (VZV) glycoprotein E is important for cell-to-cell spread and VZV infectivity, while a glycoprotein I binding domain is essential for infection. Virology 386:270–279. 10.1016/j.virol.2009.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 81:13200–13208. 10.1128/JVI.01148-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamagishi Y, Sadaoka T, Yoshii H, Somboonthum P, Imazawa T, Nagaike K, Ozono K, Yamanishi K, Mori Y. 2008. Varicella-zoster virus glycoprotein M homolog is glycosylated, is expressed on the viral envelope, and functions in virus cell-to-cell spread. J. Virol. 82:795–804. 10.1128/JVI.01722-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Betts MJ, Russell RB. 22 May 2003. Amino acid properties and consequences of substitutions. Chapter 14 In Barnes MR, Gray IC. (ed), Bioinformatics for geneticists. Wiley, Chichester, West Sussex, England. 10.1002/0470867302.ch14 [DOI] [Google Scholar]
- 48.Burley SK, Petsko GA. 1985. Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229:23–28. 10.1126/science.3892686 [DOI] [PubMed] [Google Scholar]
- 49.Eidenschink LA, Kier BL, Andersen NH. 2009. Determinants of fold stabilizing aromatic-aromatic interactions in short peptides. Adv. Exp. Med. Biol. 611:73–74. 10.1007/978-0-387-73657-0_32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meckes DG, Jr, Marsh JA, Wills JW. 2010. Complex mechanisms for the packaging of the UL16 tegument protein into herpes simplex virus. Virology 398:208–213. 10.1016/j.virol.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han J, Chadha P, Meckes DG, Jr, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein E of herpes simplex virus. J. Virol. 85:9437–9446. 10.1128/JVI.05207-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harper AL, Meckes DG, Jr, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 84:2963–2971. 10.1128/JVI.02015-09 [DOI] [PMC free article] [PubMed] [Google Scholar]