

Abstract
Reverse transcription is an important early step in retrovirus replication and is a key point targeted by evolutionarily conserved host restriction factors (e.g., APOBEC3G, SamHD1). Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) is a major target of antiretroviral drugs, and concerns regarding drug resistance and off-target effects have led to continued efforts for identifying novel approaches to targeting HIV-1 RT. Several observations, including those obtained from monocyte-derived macrophages, have argued that ribonucleotides and their analogs can, intriguingly, impact reverse transcription. For example, we have previously demonstrated that 5-azacytidine has its greatest antiviral potency during reverse transcription by enhancement of G-to-C transversion mutations. In the study described here, we investigated a panel of ribonucleoside analogs for their ability to affect HIV-1 replication during the reverse transcription process. We discovered five ribonucleosides—8-azaadenosine, formycin A, 3-deazauridine, 5-fluorocytidine, and 2′-C-methylcytidine—that possess anti-HIV-1 activity, and one of these (i.e., 3-deazauridine) has a primary antiviral mechanism that involves increased HIV-1 mutational loads, while quantitative PCR analysis determined that the others resulted in premature chain termination. Taken together, our findings provide the first demonstration of a series of ribonucleoside analogs that can target HIV-1 reverse transcription with primary antiretroviral mechanisms that include premature termination of viral DNA synthesis or enhanced viral mutagenesis.
INTRODUCTION
Retroviruses are RNA viruses that replicate through a DNA intermediate (1, 2). Reverse transcription is an essential step in retroviral replication, and evolutionarily conserved host restriction factors (e.g., APOBEC3G, SamHD1) that target viral DNA synthesis have been discovered (3, 4). Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) represents a major target for the antiretroviral drugs used in antiretroviral therapy (ART). The continued concerns over the emergence of antiviral drug resistance and complications of ART due to off-target effects of the drugs have led to ongoing intense interest in the identification of novel approaches to interfering with this key step in the HIV-1 life cycle.
The backbone of ART remains the nucleoside reverse transcriptase inhibitors (NRTIs), which are included in virtually all ART regimens (5). NRTIs inhibit HIV-1 reverse transcription by acting as chain terminators, where incorporation of an NRTI into HIV-1 DNA results in chain termination. NRTIs typically lack a 3′-OH group, though noncanonical chain terminators exist as well. NRTI incorporation into viral DNA prevents the incorporation of the next nucleotide into the growing DNA strand. Nucleotide reverse transcriptase inhibitors (NtRTIs), such as tenofovir, are nucleotide analogs that are more readily converted to the active drug form. Many nonnucleoside reverse transcriptase inhibitors (NNRTIs) have been developed. NNRTIs are small molecules that generally interact allosterically with amino acid residues in the catalytic domain of HIV-1 RT and result in the inhibition of RT catalytic activity (6).
An intriguing and novel alternative therapeutic approach to targeting HIV-1 RT is by the use of ribonucleoside analogs. DNA and RNA polymerases are thought to have an evolutionary relationship to one another, which predicts that amino acid substitutions could control substrate selection. DNA polymerases differ from RNA polymerases in utilizing 2′-deoxynucleotides (i.e., dATP, dCTP, dGTP, and dTTP) rather than ribonucleotides (i.e., ATP, CTP, GTP, and UTP). Previous work with cellular DNA polymerases demonstrated that they selectively exclude UTP and that amino acid substitutions at two different positions within motif A of the DNA polymerase catalytic site can allow ribonucleotide incorporation and RNA synthesis (7). Observations from the Moloney murine leukemia virus (M-MLV) RT crystal structure predicted that deoxynucleotide selectivity and discrimination against ribonucleotides occurred through an unfavorable interaction between the aromatic ring of Phe-155 and the 2′-OH group of the incoming ribonucleoside triphosphate (NTP) (8). The F155V substitution was found to allow the incorporation of ribonucleotides and deoxyribonucleotides (9). Virus infectivity was lost, and minus-strand strong stop DNA could be synthesized, but complete minus-strand DNA synthesis failed, likely due to an inhibition of DNA synthesis by the incorporation of ribonucleotides (10). An aromatic ring at this amino acid residue was determined to be important for virus replication. In HIV-1 RT, Tyr-115 plays a role as a steric gate in preventing the incorporation of nucleotides with a 2′-hydroxyl group in a cation-independent manner, while its influence on fidelity was found to be modulated by Mg2+ or Mn2+ (11).
Several observations, including those obtained from monocyte-derived macrophages, have found that ribonucleotides and their analogs can impact reverse transcription (12–14). Quantitative tandem mass spectrometry was used to demonstrate that macrophages have deoxynucleotide triphosphate (dNTP) concentrations 22- to 320-fold lower than those observed in dividing target cells, along with a greater disparity between NTP and dNTP concentrations (14). A biochemical simulation of HIV-1 reverse transcription revealed that NTPs are efficiently incorporated into DNA in the macrophage but not in the T cell environment (13). The ribonucleoside analog 3′-deoxyadenosine was found to inhibit HIV-1 DNA synthesis more efficiently in human macrophages than in CD4+ T cells; the cell type differences were interpreted to be due to the relatively low dNTP concentrations compared to NTP concentrations in macrophages.
The ribonucleoside analog 5-azacytidine (5-AZC) exerts its anti-HIV-1 activity through incorporation into both viral RNA and DNA (15, 16). Interestingly, the most potent antiviral activity was attributed to incorporation of 5-AZC into viral DNA. However, it remains unclear whether 5-AZC is directly incorporated into viral DNA as a ribonucleoside triphosphate or if it is incorporated as a 2′-OH deoxynucleoside triphosphate subsequent to cell-mediated conversion. In either scenario, the mechanism of 5-AZC-mediated mutagenesis suggests that it is primarily incorporated into HIV-1 DNA during reverse transcription, leading to lethal mutagenesis, which was characterized by a significant increase in the mutational load due to G-to-C transversion mutations in the viral DNA (15).
Here we report on an investigation of a panel of ribonucleoside analogs for their ability to affect HIV-1 replication during the reverse transcription process. Our findings provide the first demonstration of a series of ribonucleoside analogs that can target HIV-1 reverse transcription with primary antiretroviral mechanisms that include premature termination of viral DNA synthesis or enhanced viral mutagenesis.
MATERIALS AND METHODS
Plasmids, cell lines, and chemical reagents.
The HIV-1 vector NL4-3 MIG was used for single-cycle susceptibility and mutation frequency assays (17). The vector was pseudotyped with the vesicular stomatitis G protein (VSV-G; pHCMV_VSV-G; a kind gift from J. Burns, University of California, San Diego). The CEM-GFP and U373-MAGICXCR4 cell lines were obtained from the AIDS Reagent Program (provided by M. Emerman and J. Corbeil, respectively). Cell lines were maintained in RPMI and Dulbecco modified Eagle medium (DMEM) supplemented with 10% HyClone FetalClone III (FC3; Thermo Scientific), respectively. The HEK293T cell line was obtained from ATCC and maintained in DMEM with 10% FC3. Zidovudine (AZT) was obtained from the AIDS Reagent Program. The ribonucleoside analogs 5-flurocytidine, 5-azacytidine, and 2′-C-methylcytidine were obtained from Carbosynth Limited (Berkshire, United Kingdom). The ribonucleoside analogs 8-azaadenosine and formycin A were obtained from Berry and Associates, Inc. (Dexter, MI), and 3-deazauridine, cytidine, adenosine, and thymidine were obtained from Sigma-Aldrich (St. Louis, MO), Carbosynth Ltd. (Bershire, United Kingdom), and Berry and Associates (Dexter, MI). All ribonucleoside analogs were stored as solutions (1 M) in dimethyl sulfoxide (DMSO) at −20°C. Linear polyethylenimine (PEI; molecular weight [MW], 25,000) was obtained from Polysciences, Inc. (Warrington, PA).
Transfections, infections, and treatment of target cells with ribonucleoside analogs.
The HIV-1 vector was produced by PEI transfection of HEK293T cells. Briefly, 2 × 106 HEK293T cells in 5 ml medium were transfected with 20 μg of pNL4-3 MIG and 2.5 μg pHCMV_VSV-G. Medium was removed after 5 h, and 9 ml fresh medium was added. After 16 h, the viral supernatant was pooled, passed through a 0.2-μm-pore-size filter, and stored at −80°C. The virus titer on U373-MAGICXCR4 target cells was established so that 20 to 30% of the target cells were infected. To assess antiviral activity during viral infection, each ribonucleoside analog was added to target cells 2 h prior to addition of viral supernatant. Specifically, ribonucleoside analogs were added at a 1:1,000 dilution to 3 × 104 U373-MAGICXCR4 cells in a total volume of 500 μl.
Flow cytometry and mutation frequency analysis.
U373-MAGICXCR4 target cells were prepared for flow cytometry by washing in phosphate-buffered saline (PBS) and a final suspension in 2% FC3-PBS. Cells were analyzed for mCherry and green fluorescent protein (GFP) expression on a BD LSR II flow cytometer (BD Biosciences, San Jose, CA). Cells were first gated on the basis of forward scatter and side scatter, and a minimum of 10,000 gated cells were analyzed per sample. GFP was excited with a blue 488-nm laser, and emission was detected using 505LP and 525/50-nm filters. The mCherry reporter protein was excited with a 561-nm laser, and emission was detected using 595LP and 610/20-nm filters. No fluorescent compensation was necessary with this particular combination of fluorophores, lasers, and filters (data not shown). Control experiments were performed by transfection of mCherry or GFP into HEK293T cells and analysis of transfected cells (either kept separately or mixed together) by flow cytometry. Flow cytometry data were examined with FlowJo (v.7.6.1) software (Ashland, OR). Infectivity was determined by determining the percentage of mCherry-positive (mCherry+)/GFP-positive (GFP+) cells. The mutation frequency was calculated by dividing the number of cells in single-positive populations (mCherry+ only and GFP+ only) by the total number of infected cells. Infectivity and mutation frequency data were calculated relative to those for the nontreated vehicle-control cells.
Proviral DNA sequencing analysis.
PCR amplification was performed on total genomic DNA isolated from infected target cells treated with ribonucleoside analogs using a ZymoBead genomic DNA kit (Zymo Research, Irvine, CA). Reactions were performed using Taq from PCR Platinum Supermix at a 20-μl total volume. Total genomic template DNA (1 to 5 μl) was used in each reaction mixture. Oligonucleotides were designed to amplify the 5′ region of the reverse transcriptase-coding sequence within the pol gene. The positive-strand primer NL4-3_2550 (i.e., 5′-CCC ATT AGT CCT ATT GAG AC-3′) and the negative-strand primer NL4-3_3648 (i.e., 5′-GTT TCA CAT CAT TAG TGT GGG-3′) were used to amplify the 1.1-kb amplicons. Amplicons were resolved on a 0.8% TAE (Tris-acetate-EDTA) DNA gel, purified using a Promega Wizard SV gel and PCR cleanup kit (Madison, WI), and cloned into the pGEM-T Easy vector (Promega, Madison, WI) for DNA sequencing analysis. DNA sequences were aligned and compared to the HIV-1 subtype B NL4-3 sequence using the Seqman program of the Lasergene software package (DNASTAR, Madison, WI).
Cellular toxicity analysis.
HEK293T and U373-MAGICXCR4 cells were plated at a density of 7,500 cells per well in a 96-well flat-bottom plate. After 24 h, to allow attachment and growth, the cells were incubated with each ribonucleoside analog for 24 h. Cells were then washed and incubated with CellTiter-Glo solution (Promega, Madison, WI) according to the manufacturer's instructions.
Indirect competition assay.
An indirect competition assay was conducted as described above for the assessment of the antiviral activity of ribonucleoside analogs with minor protocol modifications. In particular, the natural ribonucleoside corresponding to the ribonucleoside analog was added simultaneously during treatment of cells.
Time-of-addition assay.
A time-of-addition assay was conducted as described above for the assessment of the antiviral activity of ribonucleoside analogs, except that each ribonucleoside analog was added at sequential times postinfection. The intervals ranged from 0.5 to 24 h after infection in parallel (i.e., time zero). The effective concentration that inhibited between 75% (EC75) and 90% (EC90) of viral growth for each ribonucleoside analog was used in order to observe a reversal of nearly complete inhibition of viral replication.
qPCR of late RT products.
U373-MAGICXCR4 cells were plated into a 12-well plate at a density of 60,000 cells per well. Twenty-four hours after the cells were plated, the EC50 of each ribonucleoside analog was added to the medium for 2 h prior to infection with the HIV-1 vector. AZT treatment was done using the EC75. The cell culture supernatants containing vector virus were treated with DNase I (2.5 U/ml) for 2 h at 37°C. As a control to detect possible plasmid DNA carryover, an aliquot of DNase I-treated vector virus was heat inactivated for 30 min at 95°C. At 18 h postinfection, cells were harvested and 10% of them were replated for viral infectivity analysis after 48 h, while the remaining 90% were collected for downstream lysis and quantitative PCR (qPCR) analysis. As an additional precaution to remove plasmid DNA, cells were washed six times with 1 ml PBS. Total cellular DNA was purified using a ZymoBead genomic DNA kit and eluted into 35 μl. qPCR mixtures contained 1 μl of purified cellular DNA as the template, along with 500 nM forward and reverse primers, in 20-μl-total-volume reaction mixtures. Power SYBR green PCR master mix (Applied Biosystems) was used to provide the dNTPs, polymerase (AmpliTaq Gold DNA polymerase), fluorescent readout (SYBR green I dye), and buffer components. Primers for the 18S rRNA reference gene were used to normalize sample-to-sample variation. The primers for 18S rRNA were forward primer 5′-GTA ACC CGT TGA ACC CCA TT-3′ and reverse primer 5′-CCA TCC AAT CGG TAG TAG GG-3′. The primers for the 143-bp late reverse transcription product were forward primer 5′-TGT GTG CCC GTC TGT TGT GT-3′ and reverse primer 5′-GAG TCC TGC GTC GAG AGA TC-3′. A 10-fold-serial-dilution standard curve of a known quantity of plasmid HIV-1 DNA was used to quantitate late reverse transcription products, while a similar standard curve for 18S rRNA quantity was performed using pCR-18S plasmid DNA. Cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The amplification efficiencies of each reaction were interpreted only if they were within a 10% range of 100%. Gene expression was analyzed using the ΔΔCT threshold cycle (CT) method using Bio-Rad software.
Statistical analyses.
All statistical analyses and graphical representations were done using GraphPad Prism (v.5.0) software (GraphPad Software, La Jolla, CA). The concentration-response curves were determined by fitting the data points to a nonlinear regression. From these curves, the EC50 and 95% confidence intervals were determined.
RESULTS
Anti-HIV-1 activity of ribonucleoside analogs in the absence of cytotoxicity.
In order to investigate the potential anti-HIV-1 activity and ability to alter the mutation frequency of ribonucleoside analogs, we used a recently described single-cycle assay that we developed for the rapid analysis of HIV-1 infectivity and mutation frequency (17). Briefly, an HIV-1 vector (pNL4-3 MIG) containing a gene cassette carrying the mCherry gene, an internal ribosomal entry site (IRES) from encephalomyocarditis virus, and the green fluorescent protein (GFP) gene was used for analysis of virus infectivity and mutation frequency. In order to make vector virus, pNL4-3 MIG was transiently transfected into HEK293T cells along with a VSV-G envelope expression plasmid (Fig. 1). Vector virus was harvested and used to infect permissive target cells (U373-MAGICXCR4). For experiments in which the anti-HIV-1 activity of ribonucleoside analogs was being assessed, target cells were pretreated for 2 h prior to infection. At 48 h postinfection, cells were harvested and analyzed by flow cytometry to determine infectivity in the presence and absence of a compound(s) as well as determine the mutation frequency (Fig. 1).
FIG 1.
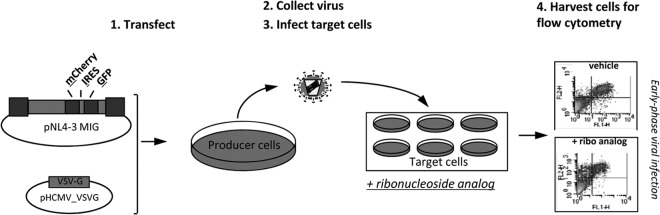
Single-cycle HIV-1 vector assay to determine the antiviral potency and mutation frequency of ribonucleoside analogs. The HIV-1 vector pNL4-3 MIG containing the dual-reporter mCherry and GFP cassette was cotransfected with the VSV-G envelope to produce pseudotyped vector virus. The titer in the cell-free viral supernatant was determined, and the supernatant was used to infect permissive U373-MAGICXCR4 target cells. Cells were collected and analyzed for fluorescent marker gene expression by flow cytometry to determine relative infectivity and the mutation frequency. Experimental drugs could be used to treat either the target cells (top right) or the virus-producing cells (bottom right) in order to investigate the antiretroviral targets in the early or late phase of HIV-1 replication.
The assay described in Fig. 1 was used to screen a small panel of ribonucleoside analogs (Table 1), which included 8-azaadenosine, formycin A, 3-deazauridine, 5-fluorocytidine, and 2′-C-methylcytidine. Figure 2 shows that each of these ribonucleoside analogs possesses potent anti-HIV-1 activity in the absence of cytotoxicity. As indicated in Table 1, 8-azaadenosine was found to have a selectivity index (SI; SI is equal to EC50/toxic concentration at which viability is reduced by 50% [TC50]) of 37, while the selectivity indices of formycin A, 3-deazauridine, 5-fluorocytidine, and 2′-C-methylcytidine were determined to be 39, >68, >40, and >8.8, respectively. These data demonstrate that new ribonucleosides that have potent anti-HIV-1 activity have been identified and that the antiviral activity is associated with the early phase of HIV-1 replication. Antiviral activity was also observed when virus-producing cells were treated with each of the 5 ribonucleoside analogs (see Fig. S1 in the supplemental material). The activities of each ribonucleoside analog were, in general, comparable when treating either target cells or virus-producing cells, though the average EC50s of 3-deazauridine and 5-fluorocytidine were slightly lower during the early phase of HIV-1 replication (i.e., reverse transcription; Table 1), and the average EC50s of 8-azaadenosine, formycin A, and 2′-C-methylcytidine were slightly lower when virus-producing cells were treated (i.e., during viral RNA synthesis; see Fig. S1 in the supplemental material).
TABLE 1.
Structures and SIs of ribonucleoside analogs
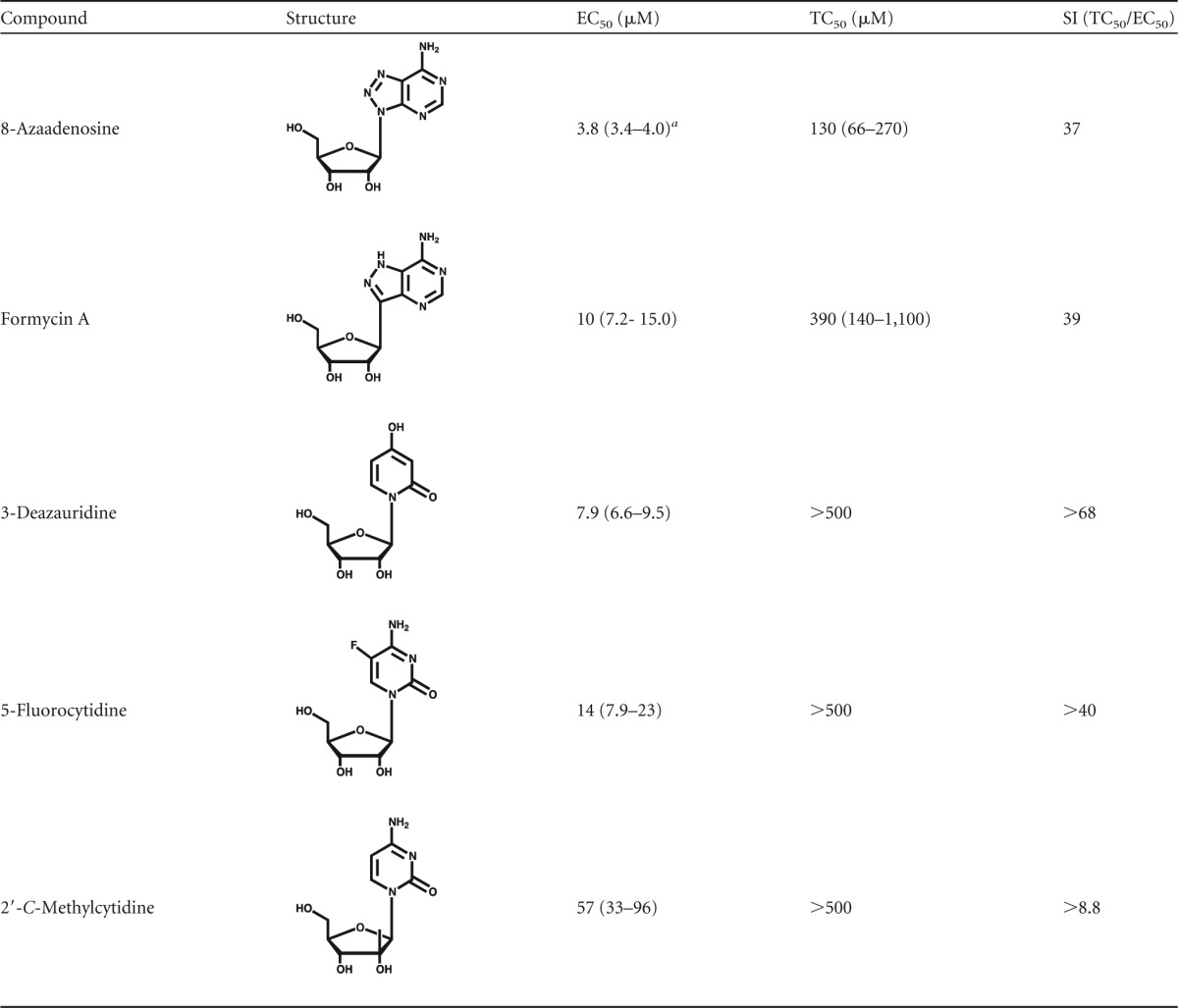
Values in parentheses indicate the 95% confidence intervals.
FIG 2.

Anti-HIV-1 activity of ribonucleoside analogs. Permissive target cells were pretreated with increasing concentrations of each ribonucleoside analog under investigation and then infected with equivalent amounts of the HIV-1 vector. At 48 h postinfection, cells were analyzed by flow cytometry to determine the percentage of infected cells, displayed relative to the amount of the vehicle control on a log-linear plot. Cell viability was determined by measuring the abundance of ATP after a 24-h exposure to each ribonucleoside analog. The EC50s and the TC50s were determined using a nonlinear regression equation for best fit.
Reverse transcription as the target of antiretroviral activity.
We next investigated the step in the viral life cycle that was the likely target of the antiretroviral activity for the ribonucleosides under investigation. To do this, permissive target cells were synchronously infected with the pNL4-3 MIG vector virus, and at various time intervals postinfection, each compound was added at its EC75 and EC90 (Fig. 3). An increase in viral infectivity was interpreted as being indicative of a phase during viral replication that the virus was no longer susceptible to inhibition. The nucleoside RT inhibitor control lamivudine (3TC) blocked replication, as predicted, during reverse transcription (at ∼2.5 h postinfection; Fig. 3). Both 3-deazauridine and 5-fluorocytidine were observed (Fig. 3A) to lose antiretroviral efficacy at about 3.5 h postinfection, which is indicative of the antiviral target being the reverse transcription phase of the viral life cycle. Analysis of 2′-C-methylcytidine, 8-azaadenosine, and formycin A showed that they possess anti-HIV-1 activity that was lost in a time frame (2 to 4 h; Fig. 3) which is indicative of activity against a particular antiviral target, including reverse transcription and perhaps a step after reverse transcription (e.g., viral RNA synthesis). Importantly, addition of ribonucleosides at 24 h postinfection (i.e., after the completion of viral replication in the single-cycle assay) had no effect on either viral replication or cellular toxicities.
FIG 3.

Time-of-addition assay defines reverse transcription to be the antiretroviral target of ribonucleoside analogs. Permissive target cells were synchronously infected with the HIV-1 vector (time zero). At sequential time intervals postinfection, each ribonucleoside analog was added at the EC75 and EC90 to monitor the increase in viral infectivity, which is indicative of a phase during viral replication that is no longer susceptible to inhibition. The nucleoside reverse transcriptase inhibitor 3TC, which is a chain terminator of reverse transcription, was used as a control (it blocked replication at ∼2.5 h postinfection). HIV-1 vector replication was blocked at ∼3.5 h for 3-deazauridine and between 2 and 4 h for 2′-C-methylcytidine, 8-azaadenosine, and formycin A. The data shown are representative of those from three independent experiments.
Lack of correlation between infectivity loss and reduced amounts of reverse transcription products in the presence of 3-deazauridine.
To determine whether the loss of viral infectivity correlates with a reduction in the amounts of reverse transcription products, reverse transcription product formation was analyzed by qPCR analysis. Targets cells were pretreated with each ribonucleoside and infected with the pNL4-3 MIG vector virus. At 18 h postinfection (i.e., after the completion of reverse transcription), 90% of the infected cells were harvested and analyzed by qPCR for relative the amount of late (U5-gag) viral DNA product formation. The remaining 10% of the infected cells were kept in culture until 72 h postinfection for analysis of marker gene expression by flow cytometry. Relative to the no-drug control, the AZT control revealed that the amount of viral DNA formation generally correlated with (and did not exceed) the relative amount of cells expressing marker genes (scored as infected cells) (Fig. 4). These data are predictive of a chain-terminating reverse transcriptase inhibitor. The 5-azacytiine control had a profile that is expected of a viral mutagen or possibly a delayed chain terminator, in that the relative amount of viral DNA formation did not correlate with or exceed the relative amount of cells expressing marker genes (scored as infected cells). To help exclude the possibility of carryover of plasmid DNA being detected in the qPCR assay, a heat-inactivated virus control was included to exclude the possibility that this was a contributor to the signals detected in the qPCR assay. The ribonucleosides under study (i.e., 8-azaadenosine, formycin A, 5-fluorocytidine, and 2′-C-methylcytidine) had profiles in which a loss of infectivity correlated with decreased RT product formation. This observation was consistent with the conclusion that they possess the ability to act as chain terminators of HIV-1 reverse transcription. However, the present data are not able to directly distinguish between antiretroviral activity based upon chain termination and an alternative form of inhibition of viral DNA synthesis.
FIG 4.
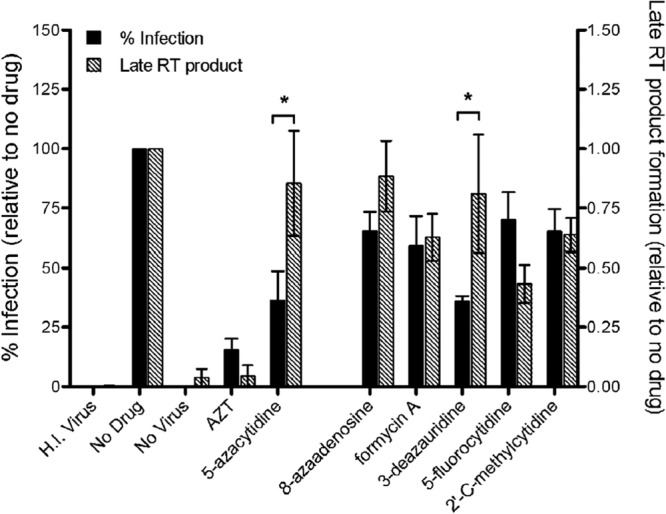
Loss of infectivity does not correlate with decreased viral DNA product formation when virus is exposed to 3-deazauridine. Targets cells were pretreated with each ribonucleoside analog at its EC50 (AZT was used at the EC75) and infected with the HIV-1 vector. At 18 h postinfection (i.e., after the completion of reverse transcription), cells were harvested and 90% of the total was used for qPCR analysis of the relative amount of late (U5-gag) viral DNA product formation. The remaining 10% of the cells were cultured until 72 h postinfection and then analyzed for marker gene expression by flow cytometry. The percentage of cells infected and the amount of viral DNA normalized to the amount of the cellular 18S rRNA gene were set relative to those for the no-drug viral controls. H.I. Virus, heat-inactivated virus. Data shown are from 4 independent experiments. *, P < 0.05.
Natural ribonucleosides antagonize the anti-HIV-1 activity of ribonucleoside analogs.
In order to further investigate the ability of ribonucleoside analogs to perturb reverse transcription, we pretreated target cells with a ribonucleoside analog alone or in combination with the corresponding natural ribonucleoside (e.g., 8-azaadenosine alone or 8-azaadenosine along with adenosine) prior to infection with pNL4-3 MIG vector virus. At 48 h postinfection, cells were collected and analyzed by flow cytometry. Figure 5 shows the results from cells treated with the natural purine adenosine and either 8-azaadenosine or formycin A in combination (Fig. 5A) or cells treated with the natural pyrimidine cytidine and either 5-fluorocytidine, 3-deazauridine, or 2′-C-methylcytidine in combination (Fig. 5B). As a control for antagonism by a natural nucleoside, AZT (a dideoxynucleoside chain terminator) was observed to be antagonized by the natural deoxynucleoside thymidine. Taken together, these results indicate that, in general, the antiviral activity of the ribonucleoside analogs was antagonized by competition with the parental natural ribonucleosides. This observation provides a further indication that ribonucleoside analogs can have an impact on HIV-1 infectivity and implies that ribonucleoside pools can also impact HIV-1 infectivity.
FIG 5.

The antiviral activity of ribonucleoside analogs is antagonized by competition with natural ribonucleosides. Target cells were pretreated either with a ribonucleoside analog alone or together with its corresponding natural ribonucleoside prior to infection with the HIV-1 vector. At 48 h postinfection, cells were harvested and analyzed by flow cytometry. (A) Results for cells treated with either 8-azaadenosine or formycin A alone or with 8-azaadenosine or formycin A in combination with adenosine and cells treated with AZT alone or with AZT in combination with thymidine; (B) results for cells treated with 5-flurocytidine, 3-deazauridine, or 2′-C-methylcytidine alone or with 5-flurocytidine, 3-deazauridine, or 2′-C-methylcytidine in combination with cytidine. Data shown are from 3 independent experiments.
Increased HIV-1 mutational load and altered mutation spectra due to 3-deazauridine.
To investigate whether the loss of HIV-1 infectivity could be associated with the incorporation of ribonucleoside analogs that result in noncanonical base pairing and subsequent mutation, the ability of the ribonucleoside analogs to alter the mutation frequency and/or mutation spectrum was analyzed. Target cells were pretreated at the EC75 of each ribonucleoside analog, and at 48 h postinfection, the cells were harvested and the total genomic DNA was purified. The 5′ region of the reverse transcriptase-coding sequence in the HIV-1 vector pol gene (amino acids 1 to 366) was PCR amplified and cloned for DNA sequencing analysis. Figure 6 shows a summary of the DNA sequencing results for experiments where cells were treated with each ribonucleoside analog, as well as a no-drug control and a viral mutagen control (i.e., 5-AZC). Compared to the no-drug control, 3-deazauridine led to a significant increase in mutation frequency (chi-square analysis, P = 0.0314), as did the 5-AZC control mutagen (chi-square analysis, P = 0.0012). The other ribonucleoside analogs (i.e., 8-azaadenosine, 5-fluorocytidine, formycin A, and 2′-C-methylcytidine) did not lead to a significant change in mutation frequency. Analysis of the mutation spectra confirmed the significant appearance of G-to-C mutations associated with the treatment of target cells with 5-AZC. The mutation spectra observed following 3-deazauridine treatment revealed the appearance of other mutation types (i.e., A to T, T to A, T to G, and C to G) that were not observed with the no-drug control (i.e., G to A, A to G, C to T, and T to C), but their frequencies were low and did not predominate over the mutation types observed in the absence of drug. The mutation spectra observed for the other ribonucleoside analogs were not significantly different from those observed with the no-drug control.
FIG 6.

The ribonucleoside 3-deazauridine causes altered mutation spectra and increased mutation frequencies in HIV-1. Targets cells were treated at the EC75 for each ribonucleoside analog, and at 48 h postinfection, cells were harvested and the total genomic DNA was purified. The HIV-1 pol gene (specifically, the 5′ region of the HIV-1 RT open reading frame, corresponding to amino acids 1 to 366) was amplified by PCR and cloned. (A to G) Tabulated summary of the mutation spectra as a percentage of the total mutations as well as the absolute values. The total number of clones sequenced, the total number of mutations scored, and the total number of bases sequenced are also indicated. Mutation frequency (freq.) was calculated on the basis of the ratio of the total numbers of mutations to the total number of bases sequenced. The fold difference in mutation frequency (Fold Δ mut. freq.) relative to that of the no-drug control was determined.
DISCUSSION
We report here the discovery of ribonucleoside analogs that have antiretroviral activity against HIV-1. Given the importance of HIV-1 reverse transcriptase as an antiviral target, against the backdrop of continued concerns of antiviral drug resistance and off-target effects during the long-term management of HIV-1 infection, the identification of novel targets directed at HIV-1 RT is a rational means for keeping one step ahead of the rapid ability of HIV-1 to mutate and evolve. Recent observations indicate that ribonucleoside analogs represent a novel approach toward antiretroviral therapy. Our findings of several new ribonucleoside analogs (i.e., 8-azaadenosine, formycin A, 3-deazauridine, 5-fluorocytidine, and 2′-C-methylcytidine) with potent anti-HIV-1 activity in the absence of cell cytotoxicity help provide a basis for subsequent studies focused on the antiviral mechanism of action as well as the discovery of analogs for clinical translation.
All of the ribonucleoside analogs analyzed in this study possessed activity with a correlation between reduced infectivity and effects on HIV-1 reverse transcription. The selectivity indices (ranging from >68 for 3-deazauridine to >8.8 for 2′-C-methylcytidine) provide a clear indication that potent antiretroviral activity was observed among this panel of ribonucleoside analogs. These effects included the rebound of viral replication in time-of-addition experiments at time points beyond that of the time frame during which reverse transcription occurs, diminution of viral DNA synthesis due to drug treatment, antagonism from parental ribonucleosides, and in one instance (i.e., 3-azauridine) the elevation in virus mutation frequency. The ability of these ribonucleoside analogs to affect HIV-1 infectivity by their incorporation into viral RNA through RNA polymerase II was not as potent as that observed when permissive target cells were treated (Fig. 2; see Fig. S1 in the supplemental material). The kinetics observed in the time-of-addition experiments for 8-azaadenosine, formycin A, and 2′-C-methylcytidine may be a reflection of the combined antiretroviral activity at both the reverse transcription and viral RNA transcription steps of the HIV-1 life cycle.
There are no previous reports of antiviral activity associated with 8-azaadenosine. Formycin A derivatives have previously been noted to possess antiretroviral activity, though no detailed studies have been conducted (18, 19). Our study, to the best of our knowledge, is the first to demonstrate the antiretroviral activity of formycin A. Many previous studies identified the activity of 3-deazauridine against riboviruses (20–26) and retroviruses (23, 27). Interestingly, 3-deazauridine was previously observed to potentiate the anti-HIV-1 activity of 3TC and dideoxycytosine (28). The observed drug potentiation by 3-deazauridine, a CTP synthase inhibitor, was associated with a decrease of dCTP pool levels. The antiviral activity of 5-fluorocytidine against RNA and DNA viruses has been previously reported (29), as has the anti-HIV-1 activity of the reduced form of 5-fluorocytidine, 2′-deoxy-5-fluorocytidine (30), and that of the derivative 2′,3′-dideoxy-beta-l-5-fluorocytidine (30). Previous reports have demonstrated the antiviral activity of 2′-C-methylcytidine as a delayed chain terminator against RNA viruses, such as hepatitis C virus (HCV) and foot-and-mouth disease virus (FMDV) (31–33). Our observations of anti-HIV-1 activity associated with 2′-C-methylcytidine are the first to be reported.
Since 5-AZC is a ribonucleoside analog, it was originally thought that its antiviral activity would primarily be attributed to its incorporation into viral RNA and a subsequent increase in HIV-1 mutation frequency. In support of this, several previous studies have shown that 5-AZC can be incorporated into RNA (34–37). One study further demonstrated that 5-AZC is a weak competitive inhibitor, having a 20-fold lower affinity for RNA polymerase II than CTP (38). However, our previous work demonstrated that the most potent antiviral activity of 5-AZC is associated with its effect on the early phase of HIV-1 replication (i.e., reverse transcription), yet we were unable to distinguish which form of 5-AZC was ultimately used as a substrate for RT the triphosphorylated ribonucleoside or the 2′-OH-reduced 5-aza-2′-dCTP analogue (decitabine). In either event, while 5-AZC increased the HIV-1 mutation frequency in both the late and early phases of HIV-1 replication, it had the greatest effect on the early phase of replication (15). These data provide one line of evidence that 5-AZC exerts its antiviral activity on both phases of replication through an increase in mutation frequency. Although 5-AZC led to a modest increase in mutation frequency, similar increases in mutation rates have been shown to be sufficient to lethally mutagenize other RNA viruses (39–43). Lethal mutagenesis modeling studies suggest that small increases in viral mutation rates should lead to a disproportionately larger decrease in viral infectivity (44, 45).
Ribonucleoside analogs with mutagenic potential have also been explored for their antiviral activity against riboviruses (46–48). Ribonucleoside analogs have been used to block retroviral replication, but the mechanism of action is not clear. At least three models could explain the mechanism by which ribonucleoside analogs inhibit retrovirus replication. First, ribonucleoside analogs could be incorporated into HIV-1 RNA during transcription of the genome-length RNA (42, 49). Alternatively, the ribonucleoside analog could be incorporated into viral DNA by RT following its reduction to the 2′-deoxynucleotide form. A third possibility is a combination of the previous two antiviral mechanisms, which would account for incorporation into both DNA and RNA. Finally, emerging evidence suggests that ribonucleotides can be directly incorporated into HIV-1 DNA during reverse transcription (13, 14). This work uncovers a possible mechanism in cells with skewed NTP/dNTP ratios, such as macrophages, suggesting that RT is able to scavenge ribonucleotides when available deoxyribonucleotides are rate limiting. To date, a detailed understanding of how ribonucleosides, including those ribonucleosides with mutagenic potential, manifest an antiretroviral effect is not well established. Our observation of the anti-HIV-1 activity of 2′-C-methylcytidine provides one line of support for the direct incorporation of the ribonucleoside analog, as the 2′-C-methyl group likely prevents reduction to the deoxyribonucleoside form. Incorporation of a ribonucleoside analog not only could reduce viral DNA synthesis but also could increase the viral mutation rate (12).
Further studies to investigate the selection of resistance to antiviral ribonucleoside analogs are warranted by these studies. Given the evolutionary association between DNA and RNA polymerases, as well as the findings of previously published studies of single amino acid residues in RT influencing dNTP versus NTP incorporation, it is plausible that HIV-1 RT drug resistance mutations could have an impact on the discrimination between dNTPs and NTPs, which could further enhance the antiviral activity of a nucleoside analog.
A practical implication for antiretroviral studies of nucleoside analogues is that the synthesis of deoxyribonucleoside analogues is usually more complicated, expensive, and time-intensive than that of ribonucleoside analogues. Therefore, discovery of ribonucleoside analogues with anti-HIV-1 activities has practical advantages. In addition, commercially available ribonucleosides are commonly less expensive than the corresponding deoxyribonucleosides. It is also important to note that the base sugar bond of ribonucleoside analogs is typically more stable than that of deoxyribonucleoside analogs, which would enhance and accelerate the discovery of new nucleoside analogs with anti-HIV-1 activity.
In conclusion, we have discovered ribonucleoside analogs that have activity against HIV-1. HIV-1 RT is a major target of antiretroviral drugs, and concerns regarding drug resistance and off-target effects have led to continued efforts to identify novel approaches to targeting HIV-1 RT. Several previous observations, including those involving macrophages, have argued that ribonucleotides and their analogs can affect reverse transcription. Previous work with 5-AZC has emphasized the utility of ribonucleoside analogs as mutagens that have their greatest antiviral potency during reverse transcription by enhancement of G-to-C transversion mutations. Our observation of ribonucleosides that possess anti-HIV-1 activity by either premature chain termination or increased HIV-1 mutational loads provides the basis for further detailed analyses of both the antiviral mechanism of action and the potential for clinical translation.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by NIH grants R01 GM56615 and R21 AI96937. M.J.D. received support from NIH grant T32 DA007097.
Footnotes
Published ahead of print 23 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02444-13.
REFERENCES
- 1.Baltimore D. 1970. RNA-dependent DNA polymerase in virions of RNA tumor viruses. Nature 226:1209–1211. 10.1038/2261209a0 [DOI] [PubMed] [Google Scholar]
- 2.Temin HM, Mitzutani S. 1970. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226:1211–1213. 10.1038/2261211a0 [DOI] [PubMed] [Google Scholar]
- 3.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. 10.1038/nature10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 5.Arribas JR, Eron J. 2013. Advances in antiretroviral therapy. Curr. Opin. HIV AIDS 8:341–349. 10.1097/COH.0b013e328361fabd [DOI] [PubMed] [Google Scholar]
- 6.Jayaweera D, Dilanchian P. 2012. New therapeutic landscape of NNRTIs for treatment of HIV: a look at recent data. Expert Opin. Pharmacother. 13:2601–2612. 10.1517/14656566.2012.742506 [DOI] [PubMed] [Google Scholar]
- 7.Patel PH, Loeb LA. 2000. Multiple amino acid substitutions allow DNA polymerases to synthesize RNA. J. Biol. Chem. 275:40266–40272. 10.1074/jbc.M005757200 [DOI] [PubMed] [Google Scholar]
- 8.Georgiadis MM, Jessen SM, Ogata CM, Telesnitsky A, Goff SP, Hendrickson WA. 1995. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure 3:879–892. 10.1016/S0969-2126(01)00223-4 [DOI] [PubMed] [Google Scholar]
- 9.Gao G, Orlova M, Georgiadis MM, Hendrickson WA, Goff SP. 1997. Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. U. S. A. 94:407–411. 10.1073/pnas.94.2.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao G, Goff SP. 1998. Replication defect of Moloney murine leukemia virus with a mutant reverse transcriptase that can incorporate ribonucleotides and deoxyribonucleotides. J. Virol. 72:5905–5911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cases-Gonzalez CE, Gutierrez-Rivas M, Menendez-Arias L. 2000. Coupling ribose selection to fidelity of DNA synthesis. The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 275:19759–19767. 10.1074/jbc.M910361199 [DOI] [PubMed] [Google Scholar]
- 12.Daddacha W, Noble E, Nguyen LA, Kennedy EM, Kim B. 2013. Effect of ribonucleotides embedded in a DNA template on HIV-1 reverse transcription kinetics and fidelity. J. Biol. Chem. 288:12522–12532. 10.1074/jbc.M113.458398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy EM, Amie SM, Bambara RA, Kim B. 2012. Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J. Biol. Chem. 287:14280–14288. 10.1074/jbc.M112.348482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy EM, Gavegnano C, Nguyen L, Slater R, Lucas A, Fromentin E, Schinazi RF, Kim B. 2010. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J. Biol. Chem. 285:39380–39391. 10.1074/jbc.M110.178582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dapp MJ, Clouser CL, Patterson S, Mansky LM. 2009. 5-Azacytidine can induce lethal mutagenesis in human immunodeficiency virus type 1. J. Virol. 83:11950–11958. 10.1128/JVI.01406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dapp MJ, Patterson SE, Mansky LM. 2013. Back to the future: revisiting HIV-1 lethal mutagenesis. Trends Microbiol. 21:56–62. 10.1016/j.tim.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rawson JM, Heineman RH, Beach LB, Martin JL, Schnettler EK, Dapp MJ, Patterson SE, Mansky LM. 2013. 5,6-Dihydro-5-aza-2′-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorg. Med. Chem. 21:7222–7228. 10.1016/j.bmc.2013.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neidle S, Urpi L, Serafinowski P, Whitby D. 1989. The anti-conformation of 2′,3′-dideoxy nucleosides may be essential for anti-HIV activity: evidence from the crystal structure of 2′,3′-dideoxy formycin A. Biochem. Biophys. Res. Commun. 161:910–916. 10.1016/0006-291X(89)92685-5 [DOI] [PubMed] [Google Scholar]
- 19.Sarih L, Agoutin B, Lecoq O, Weill D, Jullien P, Heyman T. 1985. Inhibition of Rous sarcoma virus production by formycin. Virology 145:171–175. 10.1016/0042-6822(85)90212-0 [DOI] [PubMed] [Google Scholar]
- 20.Burns NJ, III, Barnett BB, Huffman JH, Dawson MI, Sidwell RW, De Clercq E, Kende M. 1988. A newly developed immunofluorescent assay for determining the Pichinde virus-inhibitory effects of selected nucleoside analogues. Antiviral Res. 10:89–98. 10.1016/0166-3542(88)90017-4 [DOI] [PubMed] [Google Scholar]
- 21.Kawana F, Shigeta S, Hosoya M, Suzuki H, De Clercq E. 1987. Inhibitory effects of antiviral compounds on respiratory syncytial virus replication in vitro. Antimicrob. Agents Chemother. 31:1225–1230. 10.1128/AAC.31.8.1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khare GP, Sidwell RW, Huffman JH, Tolman RL, Robins RK. 1972. Inhibition of RNA virus replication in vitro by 3-deazacytidine and 3-deazauridine. Proc. Soc. Exp. Biol. Med. 140:880–884. 10.3181/00379727-140-36571 [DOI] [PubMed] [Google Scholar]
- 23.Shannon WM, Arnett G, Schabel FM., Jr 1972. 3-Deazauridine: inhibition of ribonucleic acid virus-induced cytopathogenic effects in vitro. Antimicrob. Agents Chemother. 2:159–163. 10.1128/AAC.2.3.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smee DF, Sidwell RW, Clark SM, Barnett BB, Spendlove RS. 1981. Inhibition of bluetongue and Colorado tick fever orbiviruses by selected antiviral substances. Antimicrob. Agents Chemother. 20:533–538. 10.1128/AAC.20.4.533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smee DF, Sidwell RW, Clark SM, Barnett BB, Spendlove RS. 1982. Inhibition of rotaviruses by selected antiviral substances: mechanisms of viral inhibition and in vivo activity. Antimicrob. Agents Chemother. 21:66–73. 10.1128/AAC.21.1.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodman DR, Williams JC. 1977. Effects of 2-deoxy-d-glucose and 3-deazauridine individually and in combination on the replication of Japanese B encephalitis virus. Antimicrob. Agents Chemother. 11:475–481. 10.1128/AAC.11.3.475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shannon WM, Brockman RW, Westbrook L, Shaddix S, Schabel FM., Jr 1974. Inhibition of gross leukemia virus-induced plaque formation in XC cells by 3-deazauridine. J. Natl. Cancer Inst. 52:199–205 [DOI] [PubMed] [Google Scholar]
- 28.Gao WY, Johns DG, Mitsuya H. 2000. Potentiation of the anti-HIV activity of zalcitabine and lamivudine by a CTP synthase inhibitor, 3-deazauridine. Nucleosides Nucleotides Nucleic Acids 19:371–377. 10.1080/15257770008033015 [DOI] [PubMed] [Google Scholar]
- 29.Beres J, Bentrude WG, Kruppa G, McKernan PA, Robins RK. 1985. Synthesis and antitumor and antiviral activities of a series of 1-beta-d-ribofuranosyl-5-halocytosine (5-halocytidine) cyclic 3′,5′-monophosphates. J. Med. Chem. 28:418–422. 10.1021/jm00382a005 [DOI] [PubMed] [Google Scholar]
- 30.Lin TS, Guo JY, Schinazi RF, Chu CK, Xiang JN, Prusoff WH. 1988. Synthesis and antiviral activity of various 3′-azido analogues of pyrimidine deoxyribonucleosides against human immunodeficiency virus (HIV-1, HTLV-III/LAV). J. Med. Chem. 31:336–340. 10.1021/jm00397a011 [DOI] [PubMed] [Google Scholar]
- 31.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. 2005. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 48:5504–5508. 10.1021/jm0502788 [DOI] [PubMed] [Google Scholar]
- 32.Deval J, Powdrill MH, D'Abramo CM, Cellai L, Gotte M. 2007. Pyrophosphorolytic excision of nonobligate chain terminators by hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 51:2920–2928. 10.1128/AAC.00186-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goris N, De Palma A, Toussaint JF, Musch I, Neyts J, De Clercq K. 2007. 2′-C-Methylcytidine as a potent and selective inhibitor of the replication of foot-and-mouth disease virus. Antiviral Res. 73:161–168. 10.1016/j.antiviral.2006.09.007 [DOI] [PubMed] [Google Scholar]
- 34.Cihak A, Vesely J, Sorm F. 1965. Incorporation of 5-azacytidine into liver ribonucleic acids of leukemic mice sensitive and resistant to 5-azacytidine. Biochim. Biophys. Acta 108:516–518. 10.1016/0005-2787(65)90046-8 [DOI] [PubMed] [Google Scholar]
- 35.Doskocil J, Sorm F. 1967. The action of 5-azacytidine on bacteria infected with bacteriophage T4. Biochim. Biophys. Acta 145:780–791. 10.1016/0005-2787(67)90137-2 [DOI] [PubMed] [Google Scholar]
- 36.Li LH, Olin EJ, Buskirk HH, Reineke LM. 1970. Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 30:2760–2769 [PubMed] [Google Scholar]
- 37.Paces V, Doskocil J, Sorm F. 1968. Incorporation of 5-azacytidine into nucleic acids of Escherichia coli. Biochim. Biophys. Acta 161:352–360. 10.1016/0005-2787(68)90113-5 [DOI] [PubMed] [Google Scholar]
- 38.Lee TT, Momparler RL. 1977. Kinetic studies with 5-azacytidine-5′-triphosphate and DNA-dependent RNA polymerase. Biochem. Pharmacol. 26:403–406. 10.1016/0006-2952(77)90199-X [DOI] [PubMed] [Google Scholar]
- 39.Harris KS, Brabant W, Styrchak S, Gall A, Daifuku R. 2005. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Res. 67:1–9. 10.1016/j.antiviral.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 40.Holland JJ, Domingo E, De La Torre JC, Steinhauer DA. 1990. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J. Virol. 64:3960–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julias JG, Pathak VK. 1998. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 72:7941–7949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. U. S. A. 96:1492–1497. 10.1073/pnas.96.4.1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sierra S, Davila M, Lowenstein PR, Domingo E. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 74:8316–8323. 10.1128/JVI.74.18.8316-8323.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Domingo E. 2003. Quasispecies and the development of new antiviral strategies. Prog. Drug Res. 60:133–158. 10.1007/978-3-0348-8012-1_4 [DOI] [PubMed] [Google Scholar]
- 45.Eigen M. 2002. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. U. S. A. 99:13374–13376. 10.1073/pnas.212514799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuang RY, Suzuki S, Chuang TK, Miyagi T, Chuang LF, Doi RH. 2005. Opioids and the progression of simian AIDS. Front. Biosci. 10:1666–1677. 10.2741/1651 [DOI] [PubMed] [Google Scholar]
- 47.Cuevas JM, Gonzalez-Candelas F, Moya A, Sanjuan R. 2009. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 83:5760–5764. 10.1128/JVI.00201-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Graci JD, Too K, Smidansky ED, Edathil JP, Barr EW, Harki DA, Galarraga JE, Bollinger JM, Jr, Peterson BR, Loakes D, Brown DM, Cameron CE. 2008. Lethal mutagenesis of picornaviruses with N-6-modified purine nucleoside analogues. Antimicrob. Agents Chemother. 52:971–979. 10.1128/AAC.01056-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graci JD, Cameron CE. 2004. Challenges for the development of ribonucleoside analogues as inducers of error catastrophe. Antivir. Chem. Chemother. 15:1–13 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.