

Abstract
Enterovirus 71 (EV71) is a major causative agent of hand, food, and mouth disease, which frequently occurs in young children. Since there are 11 subgenotypes (A, B1 to B5, and C1 to C5) within EV71, an EV71 vaccine capable of protecting against all of these subgenotypes is desirable. We report here the vaccine potential and protective mechanism of two chimeric virus-like particles (VLPs) presenting conserved neutralizing epitopes of EV71. We show that fusions of hepatitis B core antigen (HBc) with the SP55 or SP70 epitope of EV71, designated HBcSP55 and HBcSP70, respectively, can be rapidly generated and self-assembled into VLPs with the epitopes displayed on the surface. Immunization with the chimeric VLPs induced carrier- and epitope-specific antibody responses in mice. Anti-HBcSP55 and anti-HBcSP70 sera, but not anti-HBc sera, were able to neutralize in vitro multiple genotypes and strains of EV71. Importantly, passive immunization with anti-HBcSP55 or anti-HBcSP70 sera protected neonatal mice against lethal EV71 infections. Interestingly, anti-HBcSP70 sera could inhibit EV71 attachment to susceptible cells, whereas anti-HBcSP55 sera could not. However, both antisera were able to neutralize EV71 infection in vitro at the postattachment stage. The divergent mechanism of neutralization and protection conferred by anti-SP70 and anti-SP55 sera is in part attributed to their respective ability to bind authentic viral particles. Collectively, our study not only demonstrates that chimeric VLPs displaying the SP55 and SP70 epitopes are promising candidates for a broad-spectrum EV71 vaccine but also reveals distinct mechanisms of neutralization by the SP55- and SP70-targeted antibodies.
INTRODUCTION
Enterovirus 71 (EV71) is the major causative agent of hand, foot, and mouth disease, which is prevalent in the Asia-Pacific region. EV71 infection may result in severe neurological complications and even death (1–3). However, there is currently no available EV71 vaccine (4, 5).
EV71 is a member of the enterovirus genus of the Picornaviridae family. It possesses a single-stranded, positive-sense RNA genome, which is encapsidated within an icosahedral capsid consisting of 60 copies of each of VP1, VP2, VP3, and VP4 subunit proteins. Based on the VP1 sequence, EV71 is categorized into three genotypes (A, B, and C), which can be further divided into eleven subgenotypes (A, B1 to B5, and C1 to C5) (reviewed in references 1 and 2). Thus, an EV71 vaccine capable of protecting against all of these subgenotypes is desirable.
Increasing evidence has established that neutralizing antibodies play a key role in in vivo protection against lethal EV71 infection (reviewed in references 4 and 5). For example, Foo et al. showed that passive transfer of neutralizing antisera protects recipient mice against lethal EV71 challenge (6). Inactivated whole-virus and recombinant EV71 virus-like particles (VLPs) containing all capsid subunit proteins could elicit potent neutralizing antibodies (reviewed in references 4 and 5). Besides intact capsids, VP1 has been shown to be the main protein subunit capable of inducing neutralizing antibodies (7–9) and thus contain neutralizing epitopes. Indeed, two linear neutralizing epitopes, namely, SP70 and SP55, have been identified within VP1 (10). These two epitopes of different EV71 subgenotypes are highly conserved; in particular, the SP70 is identical among all subgenotypes (10), suggesting a potential for developing a peptide-based, universal EV71 vaccine. However, small synthetic peptides containing linear B-cell epitopes are usually poorly immunogenic (11, 12). Thus, an optimal delivery system is needed to maximize the immunogenic and protective potential of the SP55 and SP70 epitopes.
VLPs have proven to be an excellent platform for epitope presentation, owing to their ability to efficiently interact with antigen-presenting cells, to display heterologous epitopes at high density, and to provide T-cell help. Hepatitis B core antigen (HBc or HBcAg) expressed in recombinant systems can self-assemble into VLPs with T=3 or T=4 symmetry (13). HBc VLPs are extremely immunogenic and have been successfully used as a carrier system for presentation of foreign epitopes (13–15). For example, chimeric HBc particles expressing Plasmodium falciparum circumsporozoite (CS) protein epitopes have shown promising results in several clinical trials (16, 17).
In the present study, we evaluated the possibility of using HBc-based VLPs for delivery of SP55 and SP70 epitopes of EV71 to achieve enhanced immunogenicity and protection against EV71 infection in the murine model. Moreover, we discovered divergent mechanisms of neutralization by antibodies against the two EV71 epitopes.
MATERIALS AND METHODS
Cells and viruses.
RD and Vero cells were grown as described previously (18). EV71 strains used in the present study included EV71/BrCr, EV71/G081, EV71/G082, EV71/FY2, EV71/SH98, and a mouse-adapted virus termed EV71/MAV-VR. To prepare EV71/MAV-VR, the parental strain EV71/G082 was subjected to two rounds of in vivo adaptation in 1-day-old ICR mice, followed by in vitro recovery in RD cells. The resultant virus was then passaged once in Vero cells and once in RD cells, yielding the EV71/MAV-VR virus stock. CA16 strain SZ05 (GenBank accession no. EU262658) has been described previously (19). All viruses were propagated in RD cells as described previously (18, 19). The virus titers were determined by the Reed and Muench method based on a typical cytopathic effect (CPE) developing in the infected RD cells and expressed in 50% tissue culture infectious doses (TCID50).
Peptides, EV71 particles, and recombinant proteins.
The SP55 or SP70 peptides, representing the two linear neutralizing epitopes of EV71 identified previously (10), were synthesized by GL Biochem. Ltd. (Shanghai, China). Overlapping peptides, which span the entire amino acid sequence of VP1, have been described previously (18). Each peptide contains 15 amino acid residues. Cell culture-derived EV71 particles are present in two forms: the E-particle and the F-particle (20). E-particles are noninfectious, empty capsids consisting of VP1, VP3, and uncleaved VP0 proteins; F-particles are mature virions containing VP1, VP2, VP3, and VP4 proteins and an infectious viral genome (20). To prepare E- and F-particles of EV71, Vero cells were infected with EV71/G082 at a multiplicity of infection (MOI) of 0.1 and maintained in Dulbecco modified Eagle medium (DMEM) plus 2% fetal bovine serum (FBS) until all of the cells developed CPE. The culture was subjected to three freeze-thaw cycles and then clarified by centrifugation at 4,000 rpm (3,200 × g) for 10 min at 4°C. The supernatant was layered on a 20% sucrose cushion, followed by ultracentrifugation in Beckman SW28 Ti rotors at 27,000 rpm (131,000 × g) for 3 h at 4°C. The resulting pellet was resuspended with Tris-NaCl buffer and then clarified by centrifugation at 12,000 rpm (13,800 × g) for 10 min. Subsequently, the cleared virus suspension was layered onto a 15 to 55% sucrose gradient for ultracentrifugation with Beckman SW41 Ti rotor at 39,000 rpm (261,000 × g) and at 4°C for 3 h. Twenty-four fractions were taken from the top to the bottom and analyzed by Western blotting with an anti-EVVP0 polyclonal antibody (21). Based on the Western blot result, fractions 10 to 16 were identified to mainly contain E-particles possessing uncleaved VP0 capsid protein, and fractions 17 to 22 contain F-particles possessing VP2 and VP4, as described previously by Liu et al. (20). The fractions corresponding to E- and F-particles were pooled, respectively, dialyzed against phosphate-buffered saline (PBS), and then pelleted by ultracentrifugation through a 20% sucrose cushion. The pelleted E- and F-particles were resuspended in PBS and stored at −80°C until use. Recombinant EV71 VLP was prepared as described previously (18). Recombinant EV71 VP1 protein was described previously (21).
Construction of expression vectors.
The hepatitis B core antigen (HBc) gene within pICH-HBc (22) was released by NcoI/SacI digestion and then ligated into pIBT210 (23) to generate an intermediate plasmid pIBT-HBc. To make the HBcSP70 fusion, we obtained two complementary oligonucleotides (SP70-F, CTAGAGTACCCAACATTCGGAGAACATAAACAGGAGAAAGACCTTGAATATGGA; SP70-R, GATCTCCATATTCAAGGTCTTTCTCCTGTTTATGTTCTCCGAATGTTGGGTACT) designed to anneal and create XbaI and BglII overhangs at the two ends, respectively. The oligonucleotides were annealed and ligated into pIBT-HBc digested with XbaI and BglII, giving rise to an intermediate plasmid pIBT-HBcSP70. Similarly, the plasmid pIBT-HBcSP55 was made with synthetic oligonucleotides (SP55-F, 5′-CTAGAGCCAGATTCTAGGGAATCTCTTGCATGGCAAACTGCTACTAACCCTGGA-3′; SP55-R, 5′-GATCTCCAGGGTTAGTAGCAGTTTGCCATGCAAGAGATTCCCTAGAATCTGGCT-3′). The HBc, HBcSP70 and HBcSP55 fragments were then amplified from pIBT-HBc, pIBT-HBcSP70, and pIBT-HBcSP55, respectively, with the primers HBc-F-NcoI (5′-CTGCCATGGACATTGACCCTTACAAAG-3′) and HBc-R-XhoI (5′-GGCCTCGAGACATTGAGATTCCCTAGA-3′), digested with NcoI and XhoI, and then were ligated into pET28b to yield pET28-HBc, pET28-HBcSP70, and pET28-HBcSP55, respectively.
Expression and purification of fusion proteins.
The plasmids pET28-HBc, pET28-HBcSP70, and pET28-HBcSP55 were separately transformed into BL21 competent cells. A single transformed colony for each plasmid was inoculated into 5 ml of Luria-Bertani (LB) medium containing 50 μg of kanamycin/ml and grown overnight at 37°C with shaking. The overnight culture was inoculated 1:100 into fresh LB medium containing 50 μg of kanamycin/ml and grown at 37°C until the optical density at 600 nm (OD600) reached 0.6. Then, IPTG (isopropyl-β-d-thiogalactopyranoside) was added into the culture to a final concentration of 0.8 mM, and the culture was grown at 37°C for 3 h. The cells were harvested, resuspended in binding buffer (0.5 M NaCl, 20 mM Tris, 10 mM imidazole [pH 7.9]), and sonicated. After centrifugation, the resultant pellets containing inclusion bodies were resuspended in binding buffer containing 6 M urea and dissolved overnight. His-tagged proteins were purified under denaturing conditions using Ni-NTA His-Bind resins (Novagen, Darmstadt, Germany) according to the manufacturer's instructions and then eluted with the elution buffer (0.5 M NaCl, 20 mM Tris, 250 mM imidazole [pH 7.9]). For protein renaturation, the eluted samples were dialyzed serially against buffers (0.15 M NaCl, 20 mM Tris [pH 7.4]) with gradually decreased concentration (4, 3, 2, 1, and 0 M) of urea.
SDS-PAGE and Western blot analysis.
Protein samples were subjected to SDS–12% PAGE and Western blotting as described previously (18) with the following modifications: the membranes were probed with a mouse anti-HBc monoclonal antibody (catalog no. ab8638; Abcam, Cambridge, MA) or a mouse anti-His monoclonal antibody (Abmart, Shanghai, China), followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma).
Sucrose gradient analysis.
Purified proteins were layered onto 10 to 50% sucrose gradients in PBS. After centrifugation in a Beckman SW60 Ti rotor at 39,000 rpm (205,000 × g) for 2.5 h at 4°C, 10 fractions were taken from top to bottom and subjected to enzyme-linked immunosorbent assay (ELISA) and Western blot analyses. For ELISA, a 96-well plate was coated with 1 μl of each fraction in 50 μl of PBS, followed by incubation at 4°C for 12 h; then and after each of the following steps, the plate was washed three times with PBST (PBS plus 0.05% Tween 20) buffer. Consecutively, the wells were blocked with 200 μl of 5% nonfat milk in PBST/well at 37°C for 1 h, incubated with 50 μl/well of the D5 monoclonal antibody (24) (12.5 μg/ml) in PBST plus 1% nonfat milk at 37°C for 2 h, and then incubated for 1 h with 50 μl of goat anti-mouse IgG-HRP (Sigma)/well. For color development, 50 μl of TMB mixture was added/well and reacted for 5 to 10 min; then, 50 μl of 1 N H3PO4 was added/well to stop the reaction. Absorbance was measured at 450 nm in a 96-well plate reader. Western blotting was performed as described above.
Electron microscopy.
Purified HBc, HBcSP55, and HBcSP70 were subjected to negative staining with 0.75% uranyl formate, and transmission electron microscopy was performed with a Tecnai G2 Spirit microscope operated at 120 kV. Images were recorded at ×100,000 microscope magnification on Gantan 2k×2k charge-coupled device camera.
Mouse immunization and serum antibody measurement.
Female BALB/c mice (6 to 8 weeks old, six mice per group) were used for immunogenicity studies. Antigens were adsorbed to Imject alum (Pierce, Rockford, IL) according to the manufacturer's instructions. Groups of mice were injected intraperitoneally with 10 μg of purified HBc, HBcSP55, and HBcSP70, respectively, at weeks 0, 2, and 4. A group of mice immunized with PBS plus Imject alum was included as control. Blood samples were collected 1 week before the first immunization and 2 weeks after each immunization. All mice were sacrificed at 2 weeks after the third immunization. The animal studies were approved by the Institutional Animal Care and Use Committee at the Institut Pasteur of Shanghai.
The specific IgG responses in serum samples were determined by ELISA as described previously (22, 25) with some modifications. Briefly, 96-well plate was coated with of 50 ng of HBc (PuZhen Biotech, Shanghai, China)/well or 200 ng of peptide diluted in PBS/well, followed by incubation at 4°C for 12 h. Then and after each of the following steps, the plate was washed three times with PBST buffer (PBS with 0.05% Tween 20). The wells were blocked with 5% nonfat milk for 1 h at 37°C, incubated with 50 μl of diluted sera/well for 2 h at 37°C, and then incubated with 50 μl of goat anti-mouse IgG-HRP (Sigma)/well for 1 h at 37°C. After color development, the absorbance was determined at 450 nm in a 96-well plate reader.
Neutralization assay.
The neutralization assay was conducted in a 96-well plate as described previously (18). Briefly, 50 μl of 2-fold serially diluted serum samples were mixed with equal volumes of 100 TCID50 of EV71 or CA16 (Table 1) in each well, followed by incubation at 37°C for 1 h. Then, 1.5 × 104 RD cells were added to each well of the 96-well plate, followed by incubation at 37°C with 5% CO2. After 72 h, the cells were observed under a microscope for the presence of cytopathic effects (CPE). The neutralization titers were read as the highest dilutions that fully protect the cells from CPE.
TABLE 1.
Neutralization capacity of the pooled antisera against different virusesa
| Pooled antisera | Neutralization titer for virus strain (subtype): |
|||||
|---|---|---|---|---|---|---|
| EV71/BrCr (A) | EV71/G082 (C4) | EV71/G081 (C4) | EV71/FY2 (C4) | EV71/MAV-VR | CA16/SZ05 (B1b) | |
| Anti-PBS | <8 | <8 | <8 | <8 | <8 | <8 |
| Anti-HBc | <8 | <8 | <8 | <8 | <8 | <8 |
| Anti-HBcSP55 | <8 | 32 | 16 | 16 | 8 | <8 |
| Anti-HBcSP70 | 128 | 64 | 64 | 32 | 64 | <8 |
The lowest serum dilution tested was 1:8.
Passive immunization and virus challenge.
The protective efficacy of the experimental vaccines was evaluated by two in vivo assays. In one assay, four groups of neonatal ICR mice at <12 h of age were inoculated intraperitoneally (i.p.) with 100 μl of anti-PBS, anti-HBc, anti-HBcSP55, or anti-HBcSP70 serum/mouse, respectively, followed by i.p. inoculation with 3.55 × 106 TCID50 of EV71/MAV-VR at 24 h later. The challenged mice were monitored daily for survival and clinical score for 16 days. Clinical scores were graded as follows: 0, healthy; 1, reduced mobility; 2, limb weakness; 3, paralysis; and 4, death. In another assay, groups of adult mice were immunized with the experimental vaccines as described above and were then allowed to mate 1 day after the third immunization. The neonatal mice born to three immunized dams for each group were challenged at less than 24 h after birth by i.p. injection with 2.22 × 106 TCID50 of EV71/MAV-VR. The challenged mice were monitored daily for survival and clinical score for 17 days as described above.
Attachment inhibition assay.
For attachment inhibition assay, 3.8 × 105 TCID50 of EV71/G082 were mixed with different amounts (10, 2, and 0.4 μl) of the anti-HBc, anti-HBcSP55, or anti-HBcSP70 sera in a 200-μl final volume, respectively, and incubated at 37°C for 1 h. The serum-virus mixtures were added to 1.5 × 105 RD cells preseeded 1 day ahead in a 24-well plate and incubated at 4°C for 30 min. The cells were washed three times with DMEM containing 2% FBS to remove unbound virus. RNA was extracted by using TRIzol reagents (Invitrogen) and then reverse transcribed using oligo(dT) primers and Moloney murine leukemia virus reverse transcriptase (Promega) to produce cDNA. The resultant first-strand cDNAs were used as a template for quantitative real-time PCR (qRT-PCR) using the SYBR Premix Ex Taq kit (TaKaRa, Dalian, China) and Applied Biosystems 7900HT real-time PCR system. The primers used for EV71 amplification were EV71-RT-F (5′-AGTTGTGCAAGGATGCTAGT-3′) and EV71-RT-R (5′-CTCGTCACTAGCATTTGATG-3′). GAPDH mRNA was also measured, serving as an internal control, with the primers GAPDH-RT-F (5′-GAAGGTGAAGGTCGGAGTC-3′) and GAPDH-RT-R (5′-GAAGATGGTGATGGGATTTC-3′). The cycling conditions consisted of a denaturation step at 95°C for 30 s and 40 cycles of 95°C for 5 s and 60°C for 30 s. Data analysis was performed using the 2−ΔΔCT method. The CT values of all samples were first normalized with GADPH and then compared to the control (treated with virus only). The data are shown as percentages of the EV71 virus genome copy number of the treatment groups in relation to that of the virus-only control.
Postattachment neutralization assay.
For the postattachment assay, 50 μl of virus suspension containing 100 TCID50 of EV71/G082 was added to 3 × 104 RD cells in a 96-well plate, followed by incubation at 4°C for 1 h to allow virus to attach to cells. The plate was carefully washed for three times. Then, 50 μl of serially diluted sera were added to the wells and incubated at 37°C for 1 h. After three washes, 100 μl of DMEM plus 2% FBS was added to the plate/well, followed by incubation at 37°C with 5% CO2. After 72 h, the cells were observed for CPE. Neutralization effects were determined by measurement of cell viability using a MTT-based method as described previously (18).
Statistics.
Statistical significance was determined by the Student two-tailed t test using GraphPad Prism version 4.
RESULTS
Expression and assembly of chimeric VLPs displaying EV71 epitopes.
The region between amino acids 77 to 83 of the HBc molecule constitutes the major immunodominant region (MIR) and is located at the tips of the α-helical hairpins that form protruding spikes (26). Therefore, we designed fusion proteins with the MIR of HBc replaced by the SP55 (PDSRESLAWQTATNP) or the SP70 (YPTFGEHKQEKDLEY) epitope of EV71, so that they can be exposed on the surfaces of the resulting chimeric HBc particles for optimal immunogenicity. Accordingly, two constructs, designated pET28-HBcSP55 and pET28-HBcSP70, respectively, were generated (Fig. 1A). A C-terminal 6×His tag was also fused at the C terminus of the proteins to facilitate detection and purification.
FIG 1.
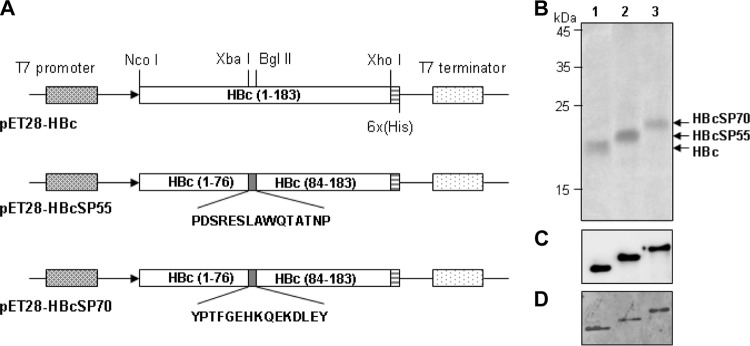
Expression of HBc fusion proteins in E. coli. (A) Schematic representation of the expression cassettes of the constructs used in the present study. (B to D) SDS-PAGE and Western blot analyses of the E. coli-expressed HBc fusion proteins. Protein samples were subjected to SDS–12% PAGE, stained with Coomassie blue (B), or blotted onto polyvinylidene difluoride membranes for Western blotting with monoclonal anti-HBc (C) or anti-His (D) antibody. Lane 1, HBc; lane 2; HBcSP55; lane 3, HBcSP70.
The vectors were transferred into E. coli strain BL21 for expression of the target protein. HBc, HBcSP55, and HBcSP70 fusion proteins were highly expressed as inclusion bodies (data not shown). These proteins were first purified under denaturing conditions with Ni-NTA columns to near homogeneity and subsequently renatured. SDS-PAGE analyses show that the inferred molecular masses of HBcSP55 and HBcSP70 are slightly higher than that of unmodified HBc (Fig. 1B). As expected, all three proteins reacted with anti-HBc (Fig. 1C) and anti-His monoclonal antibodies (Fig. 1D) in Western blot assays.
Purified HBc, HBcSP55, and HBcSP70 proteins were subjected to sucrose gradient analyses for the evaluation of VLP assembly. Western blot analyses of the gradient fractions with anti-HBc monoclonal antibody showed that the strongest signals were detected in the fraction 7 for all three proteins (Fig. 2A), indicating that they are present in particulate form. Furthermore, ELISA analysis of the HBcSP70 fractions with a mouse monoclonal antibody D5 (24), which recognizes the SP70 epitope, reveals a profile of reactivity with the peak at the fraction 7 (Fig. 2B), suggesting that the SP70 is exposed on VLP surface for efficient antibody binding. As expected, no specific reactivity with the D5 monoclonal antibody was detected for the HBc or HBcSP55 fractions (Fig. 2B).
FIG 2.
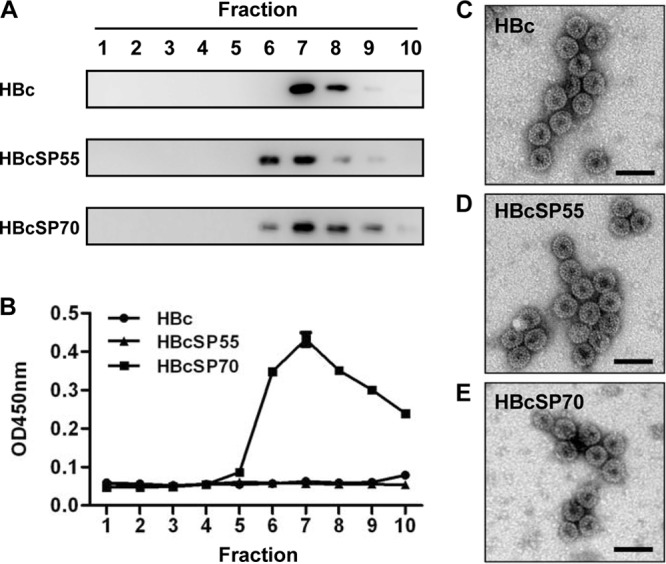
Assembly of chimeric VLPs. Purified proteins were layered onto 10 to 50% sucrose gradients and subjected to centrifugation as described in Materials and Methods. Ten fractions were taken from top to bottom and assayed. (A) Western blot analyses of HBc, HBcSP55, and HBcSP70 factions with HBc-specific monoclonal antibody. (B) ELISA analysis of the sucrose gradient fractions with a mouse monoclonal antibody D5 recognizing the SP70 epitope. The data are means of triplicate samples for each fraction. (C) Electron microscopy of HBc VLPs. (D) Electron microscopy of HBcSP55 VLPs. (E) Electron microscopy of HBcSP70 VLPs. Bar, 50 nm.
To obtain direct evidence of VLP formation, HBc, HBcSP55, and HBcSP70 preparations were subjected to negative staining electron microscopy. Empty particles with a diameter of ∼30 nm were observed for all three proteins (Fig. 2C to E). These data conclude that HBcSP55 and HBcSP70 fusion proteins self-assemble into chimeric VLPs presenting SP55 and SP70, respectively.
Chimeric VLPs elicit EV71-neutralizing antibodies in mice.
To evaluate the immunogenicity of chimeric VLPs, groups of six mice were immunized i.p. with HBc, HBcSP55, or HBcSP70, respectively. A control group of mice were injected i.p. with PBS. Mouse sera were collected 2 weeks after the last immunization and then analyzed for specific antibody response by ELISA at 1:100 dilution. As expected, anti-HBc antibodies were detected in mouse sera immunized with HBc, HBcSP55, or HBcSP70 but not in the PBS control group (Fig. 3A). SP55-specific antibodies were detected only in the HBcSP55-immunized mouse sera (Fig. 3B). Similarly, only the HBcSP70-immunized mice developed antibodies binding to the SP70 peptide (Fig. 3C).
FIG 3.

Chimeric VLPs elicited neutralizing antibody responses in mice. Groups of six mice were immunized i.p. at weeks 0, 2, and 4 with PBS, HBc, HBcSP55, or HBcSP70 as described in Materials and Methods. At week 6, sera were collected from each mouse. The antisera were diluted 1:100 with PBS and used in an ELISA to determine the HBc-specific antibody response (A), the SP55-specific antibody response (B), and the SP70-specific antibody response (C). (D) Antisera were serially diluted, starting from 1:8, to determine neutralizing titers against EV71/G082. Each symbol represents an individual mouse, and the line indicates the geometric mean value of the group.
The neutralization activity of the antisera was first determined with a microneutralization assay using EV71/G082, a strain belonging to C4 genotype. As shown in Fig. 3D, sera from the PBS control group did not show any neutralization effect at 1:8 dilution (the lowest dilution tested). For the HBc group, one antiserum sample exhibited neutralization at 1:8 dilution most likely due to nonspecific inhibition; however, the geometric mean titer of the group was <8 and considered to be negative in neutralization. In contrast, HBcSP55- and HBcSP70-immunized sera exhibited neutralization, with titers ranging from 32 to 128.
Pooled antisera for each group were further tested for neutralization capacity against a panel of EV71 and CA16 strains. As shown in Table 1, the control sera (PBS and HBc groups) did not exhibit significant neutralization activity; the HBcSP55 immunized sera could neutralize the EV71 clinical strains of C4 genotype and a mouse-adapted EV71 strain (designated EV71/MAV-VR) but not the BrCr strain belonging to A genotype; the anti-HBcSP70 sera could neutralize all EV71 strains (Table 1). None of the antisera had neutralization effects on the CA16 strain.
Kinetics of antibody responses after VLP immunization.
In another immunization experiment to determine the kinetics of antibody production, sera were collected from the immunized mice at 2 weeks after each dose and analyzed at 1:100 dilution by ELISA. The first-dose antiserum for all of the groups did not show significant binding reactivity with SP55 or SP70 peptides. After the second dose, the anti-SP55 antibody response was positively detected only in the HBcSP55-immunized sera (Fig. 4). Although the post-second-dose HBcSP55 antisera were 2/6 positive for anti-SP55 (OD450 readings that were 0.1 above that of the control sera were considered positive) when analyzed at the 1:100 dilution, all of them (6/6) were found to be positive at 1:25 (data not shown). The anti-SP55 responses were greatly elevated after the third dose (Fig. 4B). The presence of anti-SP70 antibodies was readily detectable (6/6) after the second dose, and the titers significantly increased after the third dose (Fig. 4C). We also tested the neutralization capacity of the antisera pooled for each group. No significant neutralizing activity was detected for the PBS and HBc antisera regardless of the time point, whereas the neutralizing titers of HBcSP55 and HBcSP70 antisera increased after the third dose, from 16 to 32 and from 128 to 256, respectively.
FIG 4.

Kinetics of antigen-specific antibody responses. Groups of six mice were immunized i.p. at weeks 0, 2, and 4 with PBS, HBc, HBcSP55, or HBcSP70 as described in Materials and Methods. Sera were collected from each mouse 2 weeks after the second and third doses and analyzed by ELISA to measure the HBc-specific antibody response (A), the SP55-specific antibody response (B), and the SP70-specific antibody response (C). All sera were tested at a 1:100 dilution. Each symbol represents an individual mouse, and the line indicates the geometric mean value of the group.
Anti-HBcSP55 and anti-HBcSP70 sera protect mice against lethal challenge.
The in vivo protective efficacy of the neutralizing antisera was evaluated in neonatal mouse models of EV71 infection. In one experiment, groups of 1-day-old mice were passively transferred with different antisera and challenged 1 day later with a mouse-adapted EV71 strain, designated EV71/MAV-VR. After virus challenge, the mice were monitored for clinical symptom and survival for a period of 16 days. As shown in Fig. 5A and B, the mice injected with anti-PBS or anti-HBc sera showed significantly higher clinical scores and mortality than the one receiving anti-HBcSP55 sera, whereas all of the anti-HBcSP70 sera treated mice survived, only showing transient and minor symptoms at 7 and 8 dpi. These results demonstrate that passive transfer of anti-HBcSP55 and anti-HBcSP70 sera can provide in vivo protection against lethal EV71 infection and that the neutralizing antibody titer correlates with protective efficacy.
FIG 5.

Chimeric VLP-immunized sera passively protected mice against lethal EV71 challenge. (A and B) In one experiment, groups of 1-day-old mice were i.p. injected with 100 μl of pooled anti-PBS, anti-HBc, anti-HBcSP55, or anti-HBcSP70 mouse serum/mouse and, after 24 h, i.p. inoculated with 3.55 × 106 TCID50 of EV71/MAV-VR. The mice were subsequently observed on a daily base for survival (A) and clinical score (B) for 16 days. Clinical scores were graded as follows: 0, healthy; 1, reduced mobility; 2, limb weakness; 3, paralysis; and 4, death. (C and D) In another experiment, female BALB/c mice of 6 weeks old were i.p. immunized at weeks 0, 2, and 4 with PBS, HBc, HBcSP55, or HBcSP70, respectively. After the third immunization, the mice were allowed to mate. Neonatal mice born to the immunized dams within 24 h of birth were i.p. challenged with 2.22 × 106 TCID50 of EV71/MAV-VR. The challenged mice were subsequently monitored for survival (C) and clinical scores (D) for a period of 17 days, as described above.
In another experiment, adult female BALB/c mice were immunized with the VLP vaccines and then allowed to mate. One-day-old neonatal mice born to the immunized dams were challenged with EV71/MAV-VR and subsequently monitored for clinical symptoms and survival on a daily basis for 17 days. Mice born to dams immunized with HBc VLPs or PBS gradually developed symptoms, including reduced mobility, limb weakness, and paralysis, and then died, with a final mortality rate of >70% at the end of the 17-day observation period (Fig. 5C and D). In contrast, 15 of 16 (93.8%) mice in HBcSP55 group survived, whereas all mice in the HBcSP70 group were protected from severe disease and death, only showing transient and minor symptoms at 8 and 9 dpi (Fig. 5C and D). These data indicate that maternal neutralizing antibodies from HBcSP55 or HBcSP70 VLP-immunized dams were able to protect newborn mice against lethal EV71 challenge.
Anti-HBcSP70, but not anti-HBcSP55, inhibits EV71 attachment to susceptible cells.
To explore the mechanism of neutralization by the immune antisera, we first examined whether the antisera could inhibit virus attachment to cell surface, which is the first step in virus entry. As shown in Fig. 6, anti-HBcSP55 sera treatment did not result in significant difference in total viral genome copy number compared to the anti-HBc sera treatment, which serves as a negative control in this experiment; in contrast, larger amounts (10 and 2 μl) of the anti-HBcSP70 sera led to significant decrease in cell surface-bound virus, whereas a smaller amount (0.4 μl) did not (Fig. 6). These data indicate that anti-HBcSP70, but not anti-HBcSP55 sera, inhibited EV71 binding to the cell surface in a dose-dependent manner.
FIG 6.
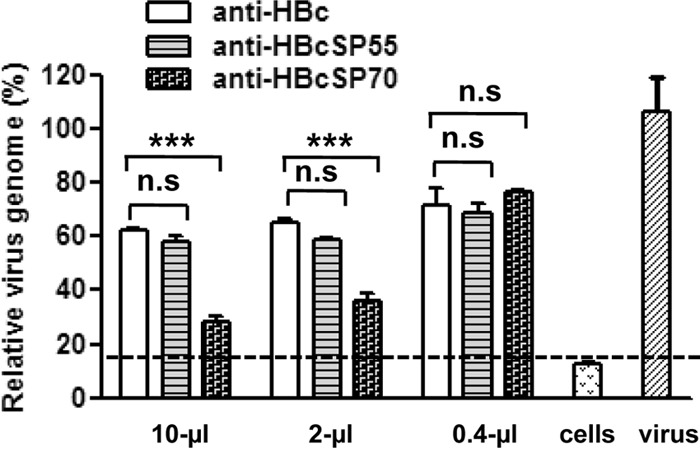
Inhibitory effect of the VLP-immunized sera on virus attachment. Different amounts of the VLP-immunized sera were incubated with 3.8 × 105 TCID50 of EV71 in a 200-μl final volume for 1 h at 37°C. The mixtures were added to RD cells and incubated for 1 h at 4°C for virus attachment. After incubation, unbound virus was carefully washed away. The total RNA of treated cells was then extracted and used for relative quantitation of genome copy of cell-bound EV71 by qRT-PCR as described in Materials and Methods. The y axis shows the ratio of relative EV71 genomic RNA level of antiserum-treated cells to that of the control (cells only infected with the virus). Means ± the standard errors of the mean (SEM) of triplicate wells are shown. The dotted line indicates the background level of the assay. The data are representative results of two independent experiments. Statistical significance was determined by the Student t test and is indicated as follows: n.s., P > 0.05; *, P < 0.05; **, P < 0.01; or ***, P < 0.001.
Binding property of anti-HBcSP55 and anti-HBcSP70.
To understand why anti-HBcSP70 but not anti-HBcSP55 could inhibit virus attachment, we evaluated the antisera for their capacity to bind different EV71 particles by ELISA. VP1 protein expressed in E. coli was used as a control in the assay. As shown in Fig. 7, anti-HBc produced only baseline levels of reactivity with all of the coating antigens; anti-HBcSP55 did not exhibit significant binding with E-particle, F-particle, or recombinant VLP but was able to react with recombinant VP1; in contrast, the anti-HBcSP70 reacted with all of the coating antigens tested, with higher readings for the three particle forms than for VP1. These data demonstrate that the anti-HBcSP55 could not bind normal EV71 virions, explaining its inability to inhibit virus attachment to cells.
FIG 7.
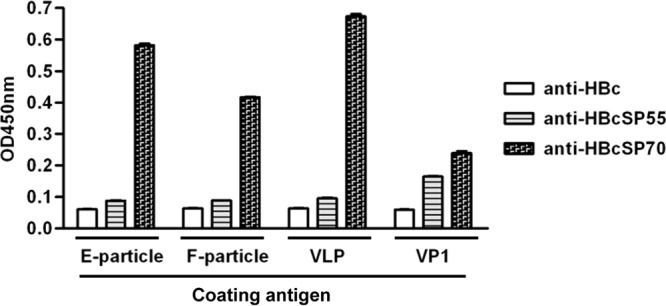
Ability of the VLP-immunized sera to bind different EV71 particles. The pooled antisera were diluted 1:100 and then used in an ELISA with EV71 E-particle, F-particle, VLP, or recombinant VP1 protein as the capture antigen, respectively. Means ± the SEM of the OD450 readings from triplicate wells are shown. The data are representative results of two independent experiments.
Because anti-HBcSP55 binds to recombinant VP1 protein and the SP55 synthetic peptide, we next determined the amino acid residues critical for this binding by evaluating reactivity of anti-HBcSP55 with overlapping synthetic peptides. Figure 8A shows that only peptide 33 exhibited significant reactivity with anti-HBcSP55 as did the SP55 peptide, suggesting that both the N terminus (-PDS-) and the C terminus (-WQTAT-) are required for efficient binding. Using the same approach, the SP70 epitope sequence essential for anti-HBcSP70 recognition was narrowed down to FGEHKQEKDL (Fig. 8B).
FIG 8.

Epitope mapping by peptide ELISA. Anti-HBcSP55 (A) and anti-HBcSP70 (B) sera were diluted 1:100 and then used in an ELISA with the indicated synthetic peptides as the capture antigen. Means ± the SEM of OD450 reading from triplicate wells are shown. The data are representative results of three independent experiments. The amino acid sequence for each synthetic peptide was shown. The amino acids identical to the SP55 or the SP70 epitopes were highlighted in red.
Both anti-HBcSP55 and anti-HBcSP70 could neutralize infection at the postattachment stage.
To determine whether the immune sera could inhibit virus entry at the postattachment stage, virus-bound cells were treated with each of the antisera for 1 h at 4°C, washed, and then incubated for 3 days at 37°C. The cells were observed for CPE and analyzed for viability by a MTT assay. As shown in Fig. 9, the anti-HBc-treated cells exhibited severe CPE regardless of the antiserum dilutions; in contrast, cells treated with either the anti-HBcSP55 or the anti-HBcSP70 sera diluted up to 1:16 appeared to grow normally, as did the uninfected cells, indicating that the neutralization activities of the anti-HBcSP55 and anti-HBcSP70 sera were preserved when virus was bound to the cells. Therefore, both anti-HBcSP55 and anti-HBcSP70 were able to inhibit infection at the postattachment step.
FIG 9.
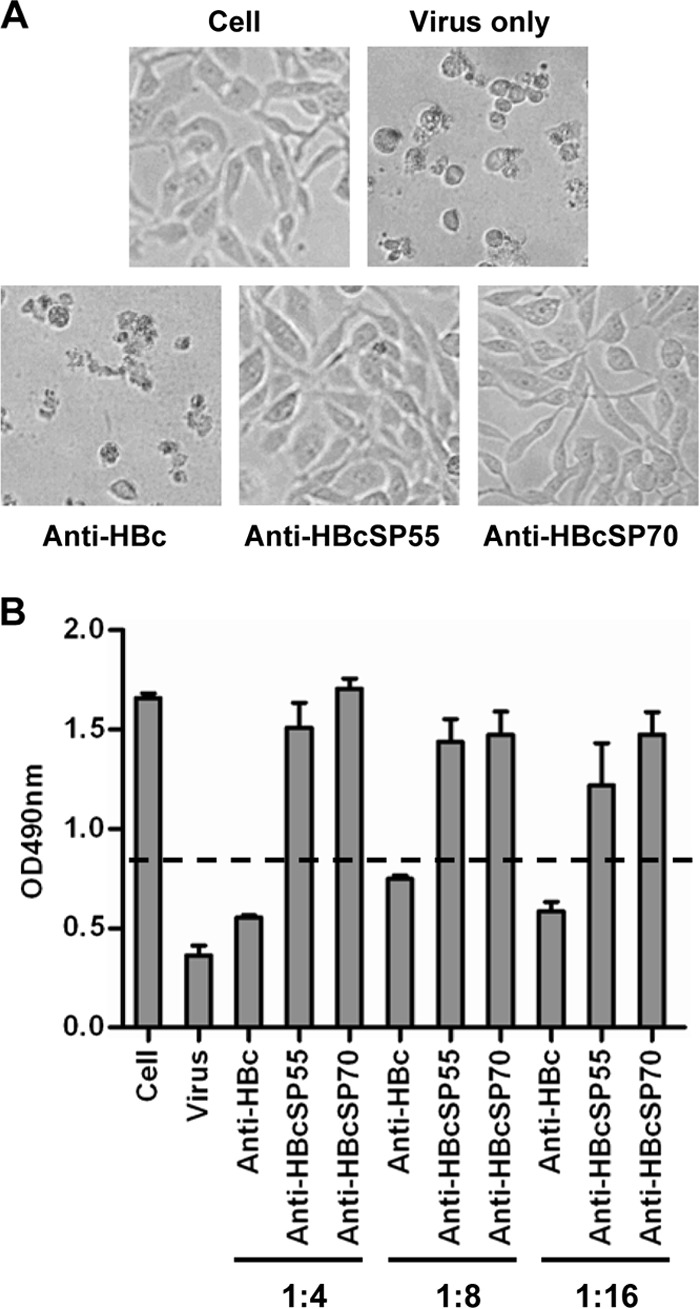
Postattachment neutralization by the VLP-immunized sera. EV71 (100 TCID50) was allowed to attach to RD cells for 1 h at 4°C. After washing away unbounded virus, the cells were then incubated for 1 h at 37°C with the VLP-immunized sera at different dilutions as indicated. After three washes, the treated cells were grown at 37°C for 72 h and then observed for CPE or subjected to an MTT assay for cell viability evaluation as described in Materials and Methods. (A) Representative images of cells treated with the indicated antisera at a 1:8 dilution. (B) Viability of the antisera treated cells measured by the MTT assay. The data are means ± the SEM of the OD490 readings of four replicate wells. The dash line indicates the cutoff, which is 50% of the OD490 reading of the uninfected cells.
DISCUSSION
The primary objective of the present study was to develop a broadly protective vaccine against EV71 infections. Previously, two conserved linear neutralizing epitopes, SP55 and SP70, were identified (10). Here, we tested whether incorporating these two epitopes into heterologous VLPs can achieve an enhanced protective immunity. We demonstrated that the fusions of HBc with SP55 or SP70 could self-assemble into VLPs with the epitope displayed on the surface (Fig. 2). Immunization of mice with three doses of 10 μg of HBcSP70 VLPs (containing ∼1 μg of SP70) in the presence of alum adjuvant induced neutralizing antibodies at titers of 64 to 128 (Fig. 3). Previously, Foo et al. showed that 50 μg of synthetic SP70 peptide conjugated to a carrier protein, diphtheria toxoid (Dkp), when codelivered with the stronger (yet unapproved for human use) Freund adjuvant, elicited only low titers (16 to 32) of neutralizing antibodies (10). Together, these results suggest that peptide epitopes on the VLPs are more immunogenic than their soluble form.
Recently, several research groups have produced EV71 VLPs in insect cells by coexpressing P1 and 3CD proteins (18, 27). Each EV71-VLP contains 60 copies each of the VP0, VP1, and VP3 capsid subunit proteins (28). Compared to the HBcSP55 and HBcSP70 VLPs, EV71 VLPs appeared to elicit higher titers of neutralizing antibodies (18, 27). For example, Ku et al. reported that a geometric mean neutralizing titer of 9,190 was induced in mouse sera after three immunizations with EV71 VLP (equivalent to 5 μg of VP0) plus alum adjuvant (18). This is as expected because, presumably, an EV71 VLP possesses all of the identified and unidentified linear neutralizing epitopes (10, 29, 30) and unidentified conformational neutralizing epitopes that are present on an authentic EV71 virion. In contrast, in the present study, each HBcSP55 or HBcSP70 fusion contains only one EV71 epitope. It is possible to create fusion proteins containing tandem-linked multiple copies of the same epitope or different epitopes of EV71, so that the total copy number of EV71 neutralizing epitopes displayed on chimeric HBc VLPs can increase significantly, which may lead to greatly enhanced immunogenicity and the ability to induce EV71-neutralizing antibodies. This strategy has proven successful for an HBc-M2e-based influenza vaccine (31, 32). Compared to the insect cell-derived EV71 VLPs, chimeric VLPs expressed in E. coli clearly have an advantage in the speed of vaccine production. It is likely that chimeric HBc VLPs presenting a new epitope can be designed and produced in large quantity within a 2-week time frame. This advantage is therefore particularly important in a rapid response to outbreaks caused by EV71 mutants which escape from current EV71 inactivated whole virus and VLP candidate vaccines.
EV71 can be classified into 11 subtypes. It has been shown that antibodies to SP55 or SP70 were able to cross-neutralize subtypes B2, B4, B5, C2, and C4 (10). In our study, we found that anti-SP70 neutralized both A and C4 subtypes and that anti-SP55 neutralized only C4 but not A subtype (Table 1). Sequence comparison analysis shows that SP70 (YPTFGEHKQEKDLEY) is completely identical among all subtypes, whereas SP55 of the C4 subtype (PDSRESLAWQTATNP), which was used for immunization in this study, shares only 86.7% homology with that (PDSRDSLAWPTATNP) of the EV71/BrCr strain, the sole member of the A subtype. This result suggests that the two amino acid mutations are responsible for EV71/BrCr escape from neutralization with anti-SP55. Collectively, our data indicate that SP70 is a better epitope than SP55 in terms of inducing broadly neutralizing antibodies.
The in vivo protective efficacy of the chimeric VLP vaccines were assessed in two passive immunization/virus challenge models. Passive transfer of anti-HBcSP70 sera protected all mice from death, while that of anti-HBcSP55 only provide partial protection (1 of 12 mice died) (Fig. 5). Similar results were also obtained from another challenge experiment in which neutralizing antibodies were transferred from dams to newborn pups (Fig. 5). Therefore, we conclude that SP70 is superior to SP55 in inducing protective immunity.
The secondary aim of the present study was to explore the mechanism of neutralization by antibodies against SP55 or SP70. We showed that anti-SP70 was able to inhibit infection at both pre- and postattachment steps, whereas anti-SP55 could not block virus attachment but could neutralize infection after virus binding to cell surface. This may partially explain why anti-SP55 was less protective in vivo. To understand why anti-SP55 could not block virus attachment, we performed binding ELISA with different EV71 capture antigens. We found that anti-SP55 could recognize E. coli-expressed VP1 protein but could not bind to E-particles, F-particles or recombinant VLPs (Fig. 7). In contrast, anti-SP70 reacted with all of the capture antigens (Fig. 7). Therefore, the ability of antibodies to bind authentic EV71 particles dictates their attachment-blocking effect.
It is intriguing that SP55-specific antibodies could indeed inhibit infection at a postattachment step. It was previously suggested that the SP55 was a linear neutralizing epitope independent of conformation (10). Epitope mapping using overlapping peptides indicates that 13 amino acid residues (PDSRESLAWQTAT) of SP55 are required for recognition by anti-SP55, whereas a shorter sequence (FGEHKQEKDL) is sufficient for anti-SP70 binding (Fig. 8). Given the fact that SP55-specific antibodies can bind synthetic peptides and the VP1 protein, we speculate that the essential residues of the SP55 epitope are not sufficiently exposed on empty EV71 particles (E-particles) or native mature virions (F-particles) for antibody recognition but are more exposed and accessible probably due to the conformational change triggered by receptor binding after virus attachment onto target cells. In support of this speculation, a recent cryoEM study demonstrated that EV71 “A-particle,” an intermediate particle form in the uncoating process, had an expanded, altered structure (33). It is therefore possible that anti-SP55 may bind to altered EV71 intermediate particles (such as the “A-particle”) and, by doing so, arrest further conformation change required for genome release. However, whether this is true remains to be determined.
In conclusion, we demonstrate here that chimeric VLPs displaying SP55 and SP70 epitopes are promising vaccine candidates against a broad spectrum of EV71 subtypes and strains. We also demonstrated distinct mechanisms of neutralization by the SP55- and SP70-targeted antibodies. Our results should have important implications for the development of a safe and effective universal EV71 vaccine.
ACKNOWLEDGMENTS
We thank Bing Sun and Qibin Leng for providing EV71 and CA16 strains. We also thank Bo Wang for the technical support. We are grateful to Xia Jin and Paul Zhou for scientific discussions and Andy Tsun for excellent editorial contributions.
This study was supported by grants from the National Natural Science Foundation of China (grant 31370930), the Science and Technology Commission of Shanghai Municipality (grant 13431900601), and the Chinese Academy of Sciences “100 Talents” program. Y.C. and L.K. were supported by the 973 program of the Ministry of Science and Technology of China (2013CB910401) and by the National Natural Science Foundation of China (31270771 and 31222016).
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev. 26:91–107. 10.1111/j.1574-6976.2002.tb00601.x [DOI] [PubMed] [Google Scholar]
- 2.Wong SS, Yip CC, Lau SK, Yuen KY. 2010. Human enterovirus 71 and hand, foot and mouth disease. Epidemiol. Infect. 138:1071–1089. 10.1017/S0950268809991555 [DOI] [PubMed] [Google Scholar]
- 3.Lee TC, Guo HR, Su HJ, Yang YC, Chang HL, Chen KT. 2009. Diseases caused by enterovirus 71 infection. Pediatr. Infect. Dis. J. 28:904–910. 10.1097/IFN.0b013e3181a41d63 [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Qian Y, Wang S, Serrano JM, Li W, Huang Z, Lu S. 2010. EV71: an emerging infectious disease vaccine target in the Far East? Vaccine 28:3516–3521. 10.1016/j.vaccine.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 5.Lee MS, Chang LY. 2010. Development of enterovirus 71 vaccines. Expert Rev. Vaccines 9:149–156. 10.1586/erv.09.152 [DOI] [PubMed] [Google Scholar]
- 6.Foo DG, Alonso S, Chow VT, Poh CL. 2007. Passive protection against lethal enterovirus 71 infection in newborn mice by neutralizing antibodies elicited by a synthetic peptide. Microbes Infect. 9:1299–1306. 10.1016/j.micinf.2007.06.002 [DOI] [PubMed] [Google Scholar]
- 7.Wu CN, Lin YC, Fann C, Liao NS, Shih SR, Ho MS. 2002. Protection against lethal enterovirus 71 infection in newborn mice by passive immunization with subunit VP1 vaccines and inactivated virus. Vaccine 20:895–904. 10.1016/S0264-410X(01)00385-1 [DOI] [PubMed] [Google Scholar]
- 8.Chen HF, Chang MH, Chiang BL, Jeng ST. 2006. Oral immunization of mice using transgenic tomato fruit expressing VP1 protein from enterovirus 71. Vaccine 24:2944–2951. 10.1016/j.vaccine.2005.12.047 [DOI] [PubMed] [Google Scholar]
- 9.Chen HL, Huang JY, Chu TW, Tsai TC, Hung CM, Lin CC, Liu FC, Wang LC, Chen YJ, Lin MF, Chen CM. 2008. Expression of VP1 protein in the milk of transgenic mice: a potential oral vaccine protects against enterovirus 71 infection. Vaccine 26:2882–2889. 10.1016/j.vaccine.2008.03.041 [DOI] [PubMed] [Google Scholar]
- 10.Foo DG, Alonso S, Phoon MC, Ramachandran NP, Chow VT, Poh CL. 2007. Identification of neutralizing linear epitopes from the VP1 capsid protein of enterovirus 71 using synthetic peptides. Virus Res. 125:61–68. 10.1016/j.virusres.2006.12.005 [DOI] [PubMed] [Google Scholar]
- 11.Sette A, Fikes J. 2003. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr. Opin. Immunol. 15:461–470. 10.1016/S0952-7915(03)00083-9 [DOI] [PubMed] [Google Scholar]
- 12.Dudek NL, Perlmutter P, Aguilar MI, Croft NP, Purcell AW. 2010. Epitope discovery and their use in peptide based vaccines. Curr. Pharm. Des. 16:3149–3157. 10.2174/138161210793292447 [DOI] [PubMed] [Google Scholar]
- 13.Pumpens P, Grens E. 1999. Hepatitis B core particles as a universal display model: a structure-function basis for development. FEBS Lett. 442:1–6. 10.1016/S0014-5793(98)01599-3 [DOI] [PubMed] [Google Scholar]
- 14.Schodel F, Peterson D, Hughes J, Wirtz R, Milich D. 1996. Hybrid hepatitis B virus core antigen as a vaccine carrier moiety: I. presentation of foreign epitopes. J. Biotechnol. 44:91–96. 10.1016/0168-1656(95)00118-2 [DOI] [PubMed] [Google Scholar]
- 15.Pumpens P, Borisova GP, Crowther RA, Grens E. 1995. Hepatitis B virus core particles as epitope carriers. Intervirology 38:63–74 [DOI] [PubMed] [Google Scholar]
- 16.Oliveira GA, Wetzel K, Calvo-Calle JM, Nussenzweig R, Schmidt A, Birkett A, Dubovsky F, Tierney E, Gleiter CH, Boehmer G, Luty AJ, Ramharter M, Thornton GB, Kremsner PG, Nardin EH. 2005. Safety and enhanced immunogenicity of a hepatitis B core particle Plasmodium falciparum malaria vaccine formulated in adjuvant Montanide ISA 720 in a phase I trial. Infect. Immun. 73:3587–3597. 10.1128/IAI.73.6.3587-3597.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nardin EH, Oliveira GA, Calvo-Calle JM, Wetzel K, Maier C, Birkett AJ, Sarpotdar P, Corado ML, Thornton GB, Schmidt A. 2004. Phase I testing of a malaria vaccine composed of hepatitis B virus core particles expressing Plasmodium falciparum circumsporozoite epitopes. Infect. Immun. 72:6519–6527. 10.1128/IAI.72.11.6519-6527.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ku Z, Ye X, Huang X, Cai Y, Liu Q, Li Y, Su Z, Huang Z. 2013. Neutralizing antibodies induced by recombinant virus-like particles of enterovirus 71 genotype C4 inhibit infection at pre- and post-attachment steps. PLoS One 8:e57601. 10.1371/journal.pone.0057601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Yan K, Feng Y, Huang X, Ku Z, Cai Y, Liu F, Shi J, Huang Z. 2012. A virus-like particle vaccine for coxsackievirus A16 potently elicits neutralizing antibodies that protect mice against lethal challenge. Vaccine 30:6642–6648. 10.1016/j.vaccine.2012.08.071 [DOI] [PubMed] [Google Scholar]
- 20.Liu CC, Guo MS, Lin FH, Hsiao KN, Chang KH, Chou AH, Wang YC, Chen YC, Yang CS, Chong PC. 2011. Purification and characterization of enterovirus 71 viral particles produced from Vero cells grown in a serum-free microcarrier bioreactor system. PLoS One 6:e20005. 10.1371/journal.pone.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Huang X, Ku Z, Wang T, Liu F, Cai Y, Li D, Leng Q, Huang Z. 2013. Characterization of enterovirus 71 capsids using subunit protein-specific polyclonal antibodies. J. Virol. Methods 187:127–131. 10.1016/j.jviromet.2012.09.024 [DOI] [PubMed] [Google Scholar]
- 22.Huang Z, Santi L, Lepore K, Kilbourne J, Arntzen CJ, Mason HS. 2006. Rapid, high-level production of hepatitis B core antigen in plant leaf and its immunogenicity in mice. Vaccine 24:2506–2513. 10.1016/j.vaccine.2005.12.024 [DOI] [PubMed] [Google Scholar]
- 23.Haq TA, Mason HS, Clements JD, Arntzen CJ. 1995. Oral immunization with a recombinant bacterial antigen produced in transgenic plants. Science 268:714–716. 10.1126/science.7732379 [DOI] [PubMed] [Google Scholar]
- 24.Ku Z, Shi J, Liu Q, Huang Z. 2012. Development of murine monoclonal antibodies with potent neutralization effects on enterovirus 71. J. Virol. Methods 186:193–197. 10.1016/j.jviromet.2012.06.025 [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Huang X, Liu Q, Huang Z. 2013. Identification of conserved neutralizing linear epitopes within the VP1 protein of coxsackievirus A16. Vaccine 31:2130–2136. 10.1016/j.vaccine.2013.02.051 [DOI] [PubMed] [Google Scholar]
- 26.Vanlandschoot P, Cao T, Leroux-Roels G. 2003. The nucleocapsid of the hepatitis B virus: a remarkable immunogenic structure. Antivir. Res. 60:67–74. 10.1016/j.antiviral.2003.08.011 [DOI] [PubMed] [Google Scholar]
- 27.Chung YC, Ho MS, Wu JC, Chen WJ, Huang JH, Chou ST, Hu YC. 2008. Immunization with virus-like particles of enterovirus 71 elicits potent immune responses and protects mice against lethal challenge. Vaccine 26:1855–1862. 10.1016/j.vaccine.2008.01.058 [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Peng W, Ren J, Hu Z, Xu J, Lou Z, Li X, Yin W, Shen X, Porta C, Walter TS, Evans G, Axford D, Owen R, Rowlands DJ, Wang J, Stuart DI, Fry EE, Rao Z. 2012. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 19:424–429. 10.1038/nsmb.2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirk K, Poh CL, Fecondo J, Pourianfar H, Shaw J, Grollo L. 2012. Cross-reactive neutralizing antibody epitopes against enterovirus 71 identified by an in silico approach. Vaccine 30:7105–7110. 10.1016/j.vaccine.2012.09.030 [DOI] [PubMed] [Google Scholar]
- 30.Liu CC, Chou AH, Lien SP, Lin HY, Liu SJ, Chang JY, Guo MS, Chow YH, Yang WS, Chang KH, Sia C, Chong P. 2011. Identification and characterization of a cross-neutralization epitope of enterovirus 71. Vaccine 29:4362–4372. 10.1016/j.vaccine.2011.04.010 [DOI] [PubMed] [Google Scholar]
- 31.De Filette M, Fiers W, Martens W, Birkett A, Ramne A, Lowenadler B, Lycke N, Jou WM, Saelens X. 2006. Improved design and intranasal delivery of an M2e-based human influenza A vaccine. Vaccine 24:6597–6601. 10.1016/j.vaccine.2006.05.082 [DOI] [PubMed] [Google Scholar]
- 32.De Filette M, Min Jou W, Birkett A, Lyons K, Schultz B, Tonkyro A, Resch S, Fiers W. 2005. Universal influenza A vaccine: optimization of M2-based constructs. Virology 337:149–161. 10.1016/j.virol.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 33.Shingler KL, Yoder JL, Carnegie MS, Ashley RE, Makhov AM, Conway JF, Hafenstein S. 2013. The enterovirus 71 A-particle forms a gateway to allow genome release: a cryoEM study of picornavirus uncoating. PLoS Pathog. 9:e1003240. 10.1371/journal.ppat.1003240 [DOI] [PMC free article] [PubMed] [Google Scholar]