

Abstract
The adenovirus immediate early gene E1A initiates the program of viral gene transcription and reprograms multiple aspects of cell function and behavior. For adenoviral (Ad) vector-mediated gene transfer and therapy approaches, where replication-defective (RD) gene transfer is required, E1A has thus been the primary target for deletions. For oncolytic gene therapy for cancer, where replication-competent (RC) Ad viral gene expression is needed, E1A has been either mutated or placed under tumor-specific transcriptional control. A novel Ad vector that initially infected target tumor cells in an RD manner for transgene expression but that could be “switched” into an RC, oncolytic state when needed might represent an advance in vector technology. Here, we report that we designed such an Ad vector (proAdΔ24.GFP), where initial Ad replication is silenced by a green fluorescent protein (GFP) transgene that blocks cytomegalovirus (CMV)-mediated transcription of E1A. This vector functions as a bona fide E1A-deleted RD vector in infected tumor cells. However, because the silencing GFP transgene is flanked by FLP recombination target (FRT) sites, we show that it can be efficiently excised by Flp recombinase site-specific recombination, either when Flp is expressed constitutively in cells or when it is provided in trans by coinfection with a second RD herpes simplex virus (HSV) amplicon vector. This switches the RD Ad, proAdΔ24.GFP, into a fully RC, oncolytic Ad (rAdΔ24) that lyses tumor cells in culture and generates oncolytic progeny virions. In vivo, coinfection of established flank tumors with the RD proAdΔ24.GFP and the RD Flp-bearing HSV1 amplicon leads to generation of RC, oncolytic rAdΔ24. In an orthotopic human glioma xenograft tumor model, coinjection of the RD proAdΔ24.GFP and the RD Flp-bearing HSV1 amplicon also led to a significant increase in animal survival, compared to controls. Therefore, Flp-FRT site-specific recombination can be applied to switch RD Ad into fully oncolytic RC Ad for tumor therapy and is potentially applicable to a variety of gene therapy approaches.
INTRODUCTION
Adenoviruses (Ads) are nonenveloped double-stranded DNA viruses containing a linear genome of about 36 kb. The process of viral DNA replication is initiated by expression of the viral immediate early gene E1A, which triggers the transcription of other viral genes to produce progeny virions (1). E1A is critical not only for activation of early viral gene transcription but also for forcing the host quiescent cell to enter the cell cycle for efficient viral DNA synthesis, because it possesses a moiety that binds pRB (2, 3). In fact, mutation of the pRB-binding region of E1A (these mutant viruses have been designated AdΔ24 and dl922-947 virus in the literature) restricts Ad replication to Rb pathway-disrupted cells (such as cancer cells), since these mutants cannot force quiescent cells to enter the cell cycle (4–6).
This critical function of E1A has been exploited to engineer replication-defective (RD) Ad vectors in which this gene is deleted. E1A-deleted, RD Ads can express heterologous transgenes under the control of constitutive promoters in target cells and are severely impaired in expressing other endogenous Ad genes (7). The critical function of E1A has also been exploited to engineer replication-competent (RC), oncolytic Ad through deletion of the pRB-binding moiety, thereby restricting replication to pRB-deficient cells (i.e., cancer cells) (5, 6, 8–11). Another oncolytic strategy has been to regulate E1A transcription using tumor-specific promoters (12) or to combine this with other viral gene modifications (such as E1B and receptors) (13–16). Both RD and RC strategies have been extensively utilized for biological mechanistic studies, for preclinical studies in animal models of disease, and finally in human clinical trials for a variety of illnesses, including cancer (17, 18).
In the context of Ad use in cancer therapy, we wondered if a “dual-purpose” Ad could be engineered to be initially RD when it infects cancer cells but to be switched into an RC state upon provision of a defined signal. Such a dual-purpose Ad could provide anticancer gene expression in its RD state (for instance, by expression of immunostimulatory molecules) but also direct cytotoxicity when conversion into an RC Ad was desired. It potentially could also be “safer” in case the RD Ad infected normal cells, since the switch to RC would not be provided. In this report, we provide proof-of-principle experiments that an RD Ad can be engineered to infect glioma cells in vitro and in vivo and express a transgene such as green fluorescent protein (GFP). Upon provision of a switch, the RD converts to RC and produces RC viral progeny in vitro and in vivo. This results into a statistically significant increase in survivorship of animals bearing intracranial glioma xenografts. This technology thus could be potentially applicable to a variety of experimental therapy strategies for cancer.
MATERIALS AND METHODS
DNA construction. (i) Adenoviral vectors.
Ad5 E1 region (NCBI AC_000008, nucleotides [nt] 541 to 3530) was amplified from the Ad5 genomic DNA (VR-5; ATCC, Manassas, VA) by PCR and cloned in pCR4blunt-TOPO (Invitrogen), and then CR2 domain-deleted E1A (E1AΔ24, nt 923 to 946) was constructed as described elsewhere (5), and finally pcDNA-E1Rd24 was made by inserting the blunt-ended EcoRI (EcoRIb) fragment of the E1 region into pcDNA3.1 (Invitrogen, Grand Island, NY). pFRT-CMV was made by inserting cytomegalovirus (CMV) promoter/enhancer (P/E) from pcDNA3.1 into pBS2FRT, which has an FLP recombination target (FRT) site at the HindIII site of pBluescript II SK− (Stratagene, La Jolla, CA), and then the SalIb/SacIIb fragment (CMV P/E and FRT region) was inserted into BamHIb/EcoRV sites of pENTR2B (Invitrogen). pIShuttle and pRIShuttle vector were based on pShuttle vector (Stratagene) (15) but were constructed by adding synthesized I-SceI recognition sequences besides both PacI sites and modified by replacing the plasmid replication origin from ColE1 to R6Kγ origin derived from pSM2 (Open Biosystems, Lafayette, CO) (for pRIShuttle). The EcoRIb/PvuII fragment (CMV P/E, FRT site) from pENTR-FRTCMV was inserted into a KpnIb site of pRIShutte vector, and then an E1 region (SalIb/XhoI) of pcDNA-E1Rd24 was inserted in the BglIIb/XhoI sites of the former construct to make pR6I-FCMV-E1Rd24pA. After adding the FRT site upstream of the enhanced GFP (eGFP) gene in pBS2FRT as pFRT-EGFR, the shuttle vector pR6I-CMVF2EGFP-E1d24pA was made by inserting the ClaIb and SpeIb fragment of pFRT-EGFR into a NotIb site of pR6I-FCMV-E1Rd24pA. pIShuttle-CMV-EGFP was constructed by ligation between EcoRV of pIShuttle and the AflIIIb site of pEGFP-C1 (Clontech, Mountain View, CA). For retrofitting to create proAdΔ24.GFP and E1-deleted AdΔE1.GFP vectors, pAdEasy-1 DNA (Stratagene) and the respective PmeI-digested pR6I-CMVF2EGFP-E1d24pA and pIShuttle-CMV-EGFP vectors were cotransformed in Escherichia coli strain DH5α/pir, which maintained Red recombinase plasmid pKD46 (Gene Bridges, Heidelberg, Germany) derivative vector on kanamycin-containing LB agar.
(ii) HSV-1 amplicon vectors.
For the herpes simplex virus 1 (HSV-1) amplicon system, pHnR was constructed from the ligation of NotI and NruI sites of pHG (gift from Y. Saeki) and pEHHnR (H. Nakashima, unpublished data). Flp-transducing HnR-CF vector was made by inserting PvuII and BamHIb fragment of pCAGGS-FLPe (Gene Bridges) into NotIb of pHnR.
Cell lines.
Vero 2-2 and G16-9 cells were used for HSV-1 amplicon packaging and for calculating the transducing unit (TU), respectively. 293A (Invitrogen) and human glioma cell lines U87MG, U87ΔEGFR, U251, U373, LN229, and Gli36 and its derivatives were maintained in Dulbecco's modified eagle medium (DMEM) supplemented with 2% or 10% fetal bovine serum (FBS), 10 mM HEPES, and penicillin-streptomycin (Invitrogen). The Gli36Flp cell line was established by stable transfection of pCAGGS-FLPe DNA into Gli36 cells and selection of the puromycin-resistant clones.
Adenoviral packaging.
Adenovirus vector DNA was cotransfected with pCBASce (a gift from M. Jasin, Cornell University, Ithaca, NY) (19) in 293A cells on 6-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfected cells were overlaid with 0.9% agarose containing growth medium. Ten to 14 days postinfection, GFP-positive plaques were isolated and viruses were expanded in 293A cells. The titers were determined by the standard 50% tissue culture infectious dose (TCID50) method.
Packaging of HSV-1 amplicon.
The amplicon vector packaging into HSV-1 virions was described previously (20). Briefly, Vero 2-2 cells were seeded on 100-mm dishes the day before transfection. fHSVΔpacΔ27 0+ and pEBHICP27 DNA were cotransfected with the amplicon vector using Lipofectamine reagent (Invitrogen). Three days later, cells and media were harvested, and viruses were recovered in 450 mM NaCl–Hanks' balanced salt solution (HBSS) from cells. HSV-1 amplicon viruses were concentrated by ultracentrifugation at 75,000 × g for 3 h and stored in HBSS at −80°C until use. TU was calculated by counting red fluorescent protein (RFP)-positive cell numbers, using G16-9 cells.
Infection assay using Gli36 cells.
Prior to Ad infection, 2 × 105 Gli36 cells were exposed to the HSV amplicon for 4 h, followed by a washout with glycine-saline buffer, pH 3, and phosphate-buffered saline (PBS), before reculturing in fresh medium. The following day, Ad viruses were used to infect the same cells for 1 h, followed by a washout with glycine-saline buffer and PBS. For Ad titration, cells were harvested and lysed by sonication and then titrated on 293A cells.
Quantification of Ad DNA and RNA.
Cells were lysed in 0.1 M Tris-Cl, pH 8.0, and 0.5% Na-deoxycholate, and then cellular DNA and unpackaged viral DNA were digested with DNase I (Roche, Indianapolis, IN). The packaging adenoviral DNA was recovered in Tris-EDTA (TE) buffer following protease K treatment and ethanol precipitation. Total mRNA was isolated from cells or tissues using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Copy numbers of genes were determined using a ABI real-time PCR detection system (Invitrogen). The plasmid pProAdΔ24.GFP DNA was used as the template in the copy number standard curve.
Immunoblotting.
Briefly, cells were lysed by incubation in SDS sample buffer directly. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes, and then membranes were blotted with anti-E1A (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), antifiber (1:500; Thermo, Rockford, IL), anti-GFP (1:2,000; MBL, Woburn, MA), or antitubulin (1:3,000; Sigma, St. Louis, MO). Blots were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse IgG secondary antibodies (1:25,000; GE Health Care, Pittsburgh, PA). Detection of proteins was performed by incubating blots with ECL Plus reagent and subsequently exposing them to film.
In vivo animal experiments.
All animal studies were performed in accordance with guidelines issued by both The Ohio State University Institutional and the Harvard Medical School Animal Care and Use Committees, utilizing approved animal protocols. U87ΔEGFR xenograft tumors were established in the flanks of athymic mice to an average size of 100 to 250 mm3. For direct viral injection, proAdΔ24.GFP (107 PFU) and HnR-CF amplicon (106 TU) were prepared in 10 μl HBSS. For some experiments, Gli36 cells were infected with proAdΔ24.GFP the day before injection into animals. The following day, these cells were infected by HSV-1 amplicon for 4 h and then trypsinized and resuspended in HBSS. The cells were injected intratumorally with 1 × 106 cells (10-μl total injection volume). For survival experiments, Gli36Flp or U87 xenograft tumors were established into the right hemisphere of athymic mice brains for 7 or 9 days, prior to intratumoral injection of of proAdΔ24.GFP at 5 × 108 or 2.5 × 108 PFU, respectively. In addition, HnR-CR or HnR amplicon vectors at a dose of 1× 106 TU were coinjected with proAdΔ24.GFP virus for the experiments with U87 glioma-bearing mice. Kaplan-Meier survival analyses were conducted using JMP10 (SAS Institute Inc.) software. For titration, Ad particles were recovered by semigradient CsCl purification following 0.5% Na-deoxycholate and DNase I treatment of dispersed tumor cells. For immunohistochemistry analysis, after intracardiac perfusion with 4% paraformaldehyde in PBS, tumors were harvested and frozen in O.C.T. compound (Sakura Finetek, Torrance, CA). Twenty-micrometer-thick sections were treated with 1.5% H2O2 in methanol and immunostained using the following primary antibodies: anti-E1A and anti-GFP. Sections were then incubated with HRP-labeled IgGs (GE Health Care, Pittsburgh, PA) followed by detection with Pierce DAB substrates (Thermo, Rockford, IL) and counterstained with hematoxylin. For real-time reverse transcription (RT)-PCR analysis, tumors were harvested and dipped into RNAlater RNA Stabilization reagent (Qiagen, Valencia, CA) overnight before homogenization with pestles, followed by resuspension with syringes and needle in lysis buffer, following the manufacturer's kit instructions.
RESULTS
Restoring E1A and Ad gene expression in an RD Ad.
The strategy that we pursued is illustrated in Fig. 1a. An RD Ad was engineered (proAdΔ24.GFP), in which mutant E1A (E1AΔ24) transcription (5) from the CMV promoter was silenced by insertion of an eGFP gene flanked by FRT sites. We postulated that in the presence of the site-specific recombinase Flp, excisional recombination of eGFP would happen between the flanking FRT sites (21). This would relieve the “silencing” of transcription of E1AΔ24, thus starting the cascade of adenoviral gene expression and replication and generating progeny oncolytic rAdΔ24 in Flp-expressing cells.
FIG 1.

Switching to a replication-competent (RC) Ad from a replication-defective (RD) proAdΔ24.GFP via FRT recombination. (a) Schematic representation of the designed adenoviral vectors: proAdΔ24.GFP and AdΔE1.GFP are based on pAdEasy-1, which lacks the E1 and E3 regions (ΔE1 and ΔE3, respectively) and exposes two inverted terminal repeats (ITRs) after linearization by I-SceI enzyme. After Flp-mediated excision of the enhanced green fluorescence protein (eGFP) transgene based on the two flanking FRT sites in proAdΔ24.GFP (upper), the CMV promoter relocates upstream of the E1 region, which contains a CR2 domain-deleted E1AΔ24 gene, and native E1B promoter and gene, to transform into RC rAdΔ24 (middle). The RD AdΔE1.GFP vector lacks the E1 region and expresses the GFP gene downstream of the CMV promoter. (b) Parental Gli36 or constitutively Flp-expressing Gli36Flp cells were infected with proAdΔ24.GFP (MOI of 10), and GFP expression was analyzed 24 h postinfection (hpi). Scale bar, 200 μm. (c) Immunoblot analyses of Ad E1A, fiber, and GFP in Gli36 (Flp-negative) and Gli36Flp (Flp-positive) cells 24 hpi with wild-type Ad (Ad5wt), proAdΔ24.GFP, or AdΔE1.GFP (MOI = 10). (d) PCR detection of the FRT sites flanking the GFP gene locus using genomic DNA isolated from Gli36 or Gli36Flp cell lysate after proAdΔ24.GFP infection at an MOI of 10. Pre- and post-FLP-mediated recombination PCR products are indicated by sizes of 1.6 kb and 0.45 kb, respectively, and correspond to the sequences designated CGE and CE in panel a. proAdΔ24.GFP plasmid DNA was employed as the control.
In order to test the feasibility of this approach, a cell line that stably expressed Flp recombinase was generated in human Gli36 glioma cells (Gli36Flp; Fig. 1b). We proceeded to verify if infection of Gli36Flp cells would lead to adenoviral gene expression and replication. As expected, infection of Gli36 cells with proAdΔ24.GFP led to widespread expression of GFP. In contrast, infection of Gli36Flp cells with proAdΔ24.GFP led to a visible decrease in the number and amount of GFP-expressing cells (Fig. 1b). Western blot analysis confirmed that expression of E1A did not occur in Gli36 cells infected with proAdΔ24.GFP, similar to the lack of expression of E1A upon RD AdΔE1.GFP infection (Fig. 1c). However, there was robust expression of GFP. Conversely, E1A and fiber expression was restored in Gli36Flp cells infected with proAdΔ24.GFP, similar to the level of expression of E1A and fiber that occurred upon infection with wild-type Ad. Genomic PCR was also utilized to confirm the excision of the FRT-flanked GFP locus in proAdΔ24.GFP-infected Gli36Flp cells (Fig. 1d). In Gli36 cells infected with proAdΔ24.GFP, the GFP-silencing cassette locus was present (this PCR product in Fig. 1a is designated CGE), but it was absent in Gli36Flp cells infected with proAdΔ24.GFP. As expected, in Gli36 cells infected with proAdΔ24.GFP, the single FRT postrecombination locus (this PCR product in Fig. 1a is designated CE) was absent, but it was present in Gli36Flp cells infected with proAdΔ24.GFP (Fig. 1d). These results thus showed that expression of E1A and Ad genes, which were absent in RD proAdΔ24.GFP, could be restored by Flp-FRT mediated, site-specific recombinatorial excision of the GFP-silencing cassette.
In vitro cytotoxicity and viral yields of RD Ad as a function of Flp.
We next sought to determine if proAdΔ24.GFP was cytotoxic to Gli36 cells. Figure 2a shows that there was no significant effect on Gli36 proliferative ability upon infection with RD proAdΔ24.GFP at multiplicities of infection (MOI) of up to 30. At higher MOIs, there was an effect on proliferation (at MOI of 100, P = 0.025; at MOI of 300, P = 0.0001) for the first few days, but then proliferation resumed at the same rate as that of cells infected with the lower MOI. A decrease in proliferative ability was evident at an MOI of 1,000 (3 days postinfection [dpi], P = 0.00066), but this type of cytotoxicity is well known to occur at such high MOIs with RD Ad (19, 22). Ad DNA copy numbers in Gli36 cells at MOIs of 10 and 30 showed a decrease in Gli36 cell proliferation (Fig. 2b). These results thus indicated that in the absence of Flp, proAdΔ24.GFP resulted in a nonproductive infection of Gli36 cells without a reduction of cell proliferation and viability, except at very high MOIs. In contrast, Gli36Flp cells were sensitive to proAdΔ24.GFP infection at MOIs as low as 3 (Fig. 2c and d). Titers of progeny Ad in proAdΔ24.GFP-infected Gli36Flp cells were 91.9-fold (± standard error of the mean [SEM] of 30.4) more than those in proAdΔ24.GFP-infected Gli36 cells, while there was no difference in recovered Ad after infection of Gli36 versus GliFlp cells with an RD Ad that lacked the E1 gene (AdΔE1.GFP), as expected (Fig. 2e). We also tested whether Flp expression somehow could complement E1 function. Figure 2f shows that this was not the case, since there was no increase in GFP DNA copy number in proAdΔ24.GFP-infected cells as a function of Flp. Conversely and expectedly, there was a significant increase in E1A copy number in proAdΔ24.GFP-infected Gli36Flp cells, as a function of Flp. These results thus confirmed that while the RD proAdΔ24.GFP vector was not cytotoxic to cells and did not result in progeny Ad production, it became cytotoxic/oncolytic and led to progeny Ad production in the presence of Flp. Therefore, Flp was able to efficiently convert the RD proAdΔ24.GFP into an RC rAdΔ24.
FIG 2.

(a) Gli36 cells were cultured for 5 days, following proAdΔ24.GFP infection at MOIs that ranged between 10 and 1,000. Viable cells were counted by trypan blue exclusion assay. (b) Real-time PCR analysis of proAdΔ24.GFP DNA was performed, using genomic DNA isolated from cultured Gli36 cells, at indicated times after infection with proAdΔ24.GFP at an MOI of 10 or 30. (c) Gli36 or Gli36Flp cells, grown on 6-well plates to a density of 5 × 104, were infected with RD AdΔE1.GFP or proAdΔ24.GFP at MOIs of 0.1 to 30. Crystal violet staining was carried out 7 days later. (d) Cell viability of Gli36 and Gli36Flp cells, 3 dpi with proAdΔ24.GFP (MOI, 10). Viable cells were counted using the trypan blue exclusion assay. (e) AdΔE1 or proAdΔ24.GFP viral titers were assayed 2 dpi of Gli36 and Gli36Flp cells (MOI, 10). Data are presented as means ± SEM, based on independently performed triplicate samples. (f) Real-time quantitative PCR analysis of E1A and GFP gene copy numbers from viral DNA recovered from proAdΔ24.GFP-infected Gli36 (Flp−) or Gli36Flp (Flp+) cell lysates treated with DNase I, followed by proteinase K. Data are presented as means ± standard deviations (SD) of triplicate samples. The experiment is representative of multiply performed experiments.
Oncolysis of glioma cells from Flp-treated RD Ad.
We next sought to determine if adenoviral progeny generated from RD proAdΔ24.GFP-infected Gli36Flp cells functioned as effective oncolytic agents. Supernatants from infected Gli36 or control, noninfected Gli36Flp cells were harvested and employed to infect target human glioma cell lines U87ΔEGFR and U251 (Fig. 3a). There was a significant increase of viral DNA in the U87ΔEGFR and U251 cells infected with supernatants from proAdΔ24.GFP-infected Gli36Flp cells compared to supernatants from noninfected Gli36Flp cells, indicative of ongoing oncolytic adenovirus replication in the former cells. Adenoviral gene expression analyses showed a significant time-dependent increase in E1A, hexon, and penton gene expression in glioma cells infected with supernatants from RD proAdΔ24.GFP-infected Gli36Flp cells, while the expression of these adenoviral gene products was negligible in glioma cells infected with supernatants from proAdΔ24.GFP-infected Gli36 cells (Fig. 3b). Finally, there was a significant prolongation in the survivorship of mice with established intracranial human Gli36Flp gliomas injected with the RD proAdΔ24.GFP vector (Fig. 3c). These results thus further confirmed that expression of Flp recombinase in Gli36Flp cells led to the generation of effective oncolytic RC adenoviruses from the RD viral genome that can lead to significant biologic effects in vivo.
FIG 3.

Sonicated Gli36 or Gli36Flp cell lysates, 2 dpi with proAdΔ24.GFP, were incubated on U87ΔEGFR or U251 cells for 1 or 3 days and then analyzed using viral genomic DNA from Ad particles released from cells (a) or mRNA present from U87ΔEGFR or U251 cell lysates (b). Data are presented as the relative fold increases measured against viral DNA or mRNA isolated from Gli36 lysate. Data are presented as means ± SD of triplicate samples. P values are based on Student's t test. (c) Kaplan-Meier survival curves for athymic nude mice with intracranial Gli36Flp tumors treated 7 days after implantation. Gli36Flp cells (2 × 105) were inoculated (2 mm lateral and 0.5 mm anterior from bregma and 2.5-mm depth) into the brain on day zero. The RD proAdΔ24.GFP was intratumorally injected using the same stereotactic coordinates at day 7 (n = 6). Control animals were injected with vehicle HBSS (n = 4).
Flp can be delivered by a second vector system to switch RD Ad to RC Ad.
Next, we attempted to determine if Flp could be delivered exogenously by a second vector system in order to reactivate oncolytic virus (OV) from the RD proAdΔ24.GFP-infected Gli36 cells (Fig. 4a). We thus engineered an Flp recombinase gene into a replication-defective herpes simplex virus 1 (HSV-1) vector (HSV-1 amplicon) (20) under transcriptional control of a constitutive CAG promoter, thus generating HnR-CF (Fig. 4b). Gli36 cells were infected either with the parental amplicon, HnR, or with HnR-CF, followed by proAdΔ24.GFP infection. Because HnR and HnR-CF both express nuclear RFP and proAdΔ24.GFP expresses cytosolic GFP, single or double infections can be monitored. In addition, double infection with HnR-CF and proAdΔ24.GFP would be expected to reduce GFP expression, since it would be excised by Flp, leading to RC Ad generation. In fact, Fig. 4c shows that in the control, HnR- and proAdΔ24.GFP-coinfected Gli36 cells, there were several cells expressing cytoplasmic GFP only (single proAdΔ24.GFP infection) or nuclear RFP only (HnR infection only) and some expressing colocalized cytosolic GFP/nuclear RFP and/or an overall cellular yellow fluorescence (doubly infected with HnR and proAdΔ24.GFP). In contrast, in HnR-CF, proAdΔ24.GFP-infected Gli36 cells, there was a visible reduction in green and yellow cells, with mostly red cells remaining on the dish. This would be in agreement with loss of GFP fluorescence in doubly infected (HnR-CF- and proAdΔ24.GFP-infected) cells, due to the expected Flp-mediated excision of the GFP-silencing transgene cassette from proAdΔ24.GFP. Indeed, E1A and fiber expression were observed only in HnR-CF- and proAdΔ24.GFP-coinfected cells (Fig. 4d). Adenoviral replication was further confirmed by showing a significant increase in Ad DNA copy number in HnR-CF- and proAdΔ24.GFP-coinfected cells compared to cells infected with HnR and proAdΔ24.GFP or with proAdΔ24.GFP alone (Fig. 4e). Finally, we determined if oncolytic rAdΔ24 was produced by proAdΔ24.GFP-loaded Gli36 cells infected by HnR or HnR-CF. Lysates from these cells were added to target U87ΔEGFR, U373, LN229, or U251 glioma cells. Figure 4f shows evidence of visible cytotoxicity in these target glioma cells after addition of lysate from Gli36 cells infected with HnR-CF + proAdΔ24.GFP, with cytotoxicity increasing as function of the percentage of added cell lysate. In contrast, there was no obvious target cell cytotoxicity after addition of lysate from the HnR + proAdΔ24.GFP-infected Gli36 cells. Taken together, these data thus showed that an HSV-1 amplicon could be employed to deliver the Flp function into cells and that the combination of this amplicon with the RD Ad resulted in productive adenoviral replication, generating oncolytic rAdΔ24 virus.
FIG 4.

An HSV-1 amplicon expressing Flp leads to oncolytic Ad production after proAdΔ24.GFP infection of Flp-negative cells. (a) Schematic of the strategy of oncolytic activation through Flp-mediated excision of the GFP gene from the RD proAdΔ24.GFP, leading to its switch to an RC Ad in infected cells. Flp is delivered and expressed from an HSV-1 amplicon vector. (b) HSV-1 amplicon vectors are based on an HnR vector, in which an RFP gene fused to histone H2B (to localize it to nucleus) is transcriptionally regulated from an HSV-1 IE4/5 promoter. The HSV-1 origin of DNA replication (oriS) and HSV-1 DNA cleavage/packaging signal (pac) are also shown. The HnR-CF vector includes a CAG promoter-driven Flp gene (FLPe) and internal ribosome entry site (IRES)-mediated puromycin-resistant gene (puroR) expression cassette in HnR. (c) The HSV-1 amplicon HnR or HnR-CF (MOI, 3) was used to infect Gli36 cells, along with proAdΔ24.GFP (MOI, 10). Nuclear RFP (red) and cytosolic GFP (green) fluorescence indicates infection and transgene expression mediated by HSV-1 amplicon and proAdΔ24.GFP, respectively. Scale bar, 100 μm. (d) Immunoblot analyses of viral proteins using anti-E1A, fiber, GFP, and antitubulin (αtub) antibodies. (e) Real-time quantitative PCR analyses of purified Ad DNA using primer sets to detect the E1A locus. Data are presented as means ± SD of triplicate samples. One of two independent experiments is shown. P values are based on Student's t test. (f) Sonicated Gli36 cell lysates and media after HnR or HnR-CF coinfection with proAdΔ24.GFP was added to the indicated glioma cell lines cultured on 96-well plates at different ratios (0 to 50%). Pictures of plates were obtained after crystal violet staining.
In vivo evidence of generation of RC from RD Ad after Flp delivery.
Finally, we determined if the RD Ad would generate an RC oncolytic rAdΔ24 virus in vivo, after Flp delivery. First, RD proAdΔ24.GFP was injected alone or coinjected with HnR-CF directly into a subcutaneous (s.c.) tumor composed of U87ΔEGFR glioma cells. Ad yields were then assayed from these tumors, 6 days after injection (Fig. 5a). There was a significant increase in Ad titers in tumors coinjected with HnR-CF and proAdΔ24.GFP compared to those injected with proAdΔ24.GFP alone, suggestive of viral replication enhanced by the Flp function from the HnR-CF amplicon in s.c. tumor cells. E1A mRNA levels in tumors injected with Gli36 cells infected with HnR-CF + proAdΔ24.GFP approached levels that were similar to E1A mRNA levels after infection of Gli36Flp with proAdΔ24.GFP (Fig. 5b). Immunohistochemical analysis also revealed that E1A expression was visualized in Gli36 tumors injected with both HnR-CF and proAdΔ24.GFP but not in control-infected tumors (Fig. 5c). As expected, GFP immunohistochemistry was present in tumors injected with HnR-CF + proAdΔ24.GFP and control HnR + proAdΔ24.GFP or proAdΔ24.GFP alone. These results thus suggested that in vivo production of RC Ad occurred from tumor cells infected with HnR-CF + proAdΔ24.GFP.
FIG 5.

(a) ProAdΔ24.GFP (107 PFU) (proAdΔ24) with or without 106 TU of HnR-CF (106 TU) was intratumorally injected into subcutaneous (s.c.) U87ΔEGFR tumors established in the flanks of athymic mice. Ad particles were purified from wholly harvested U87ΔEGFR tumors by cesium chloride gradient centrifugation 6 days postinjection, and titers were measured as TCID50 using 293 cells. (b and c) Gli36 or Gli36Flpcells (106) were infected with proAdΔ24.GFP (MOI, 30) and with or without HnR or HnR-CF (MOI, 3) and then intratumorally injected into s.c. U87ΔEGFR tumors. Real time-qPCR analyses of E1A expression in subcutaneous tumors, 7 days postinjection of the proAdΔ24.GFP-infected Gli36Flp or Gli36 cells with or without HnR-CF (b). Data are presented as means ± SD of triplicate samples. P values were calculated with Student's t test. (c) Tissues were fixed at the 5-day time point and analyzed by immunohistochemistry using E1A and GFP antibodies. Bar, 200 μm.
Codelivery of RD Ad and amplicon expressing Flp into established human glioma xenografts leads to long-term survival of mice.
On the basis of these results, we sought to evaluate the virotherapeutic effects of proAdΔ24.GFP combined with Flp delivery by HnR-CF in a human glioma xenograft model in athymic mice brains. Figure 6 shows that the coadministering of HnR-CF with proAdΔ24.GFP significantly prolonged the survivorship of human U87 glioma-bearing mice compared to control administrations (vehicle alone, or HnR + proAdΔ24.GFP). Together with previous results, these data indicate that the RD proAdΔ24.GFP does not lead to an anticancer effect in vivo unless Flp is provided exogenously, thus leading to generation of RC, oncolytic rAdΔ24 viruses that are cytotoxic to tumors. This results in the observed prolongation of animal survivorship.
FIG 6.
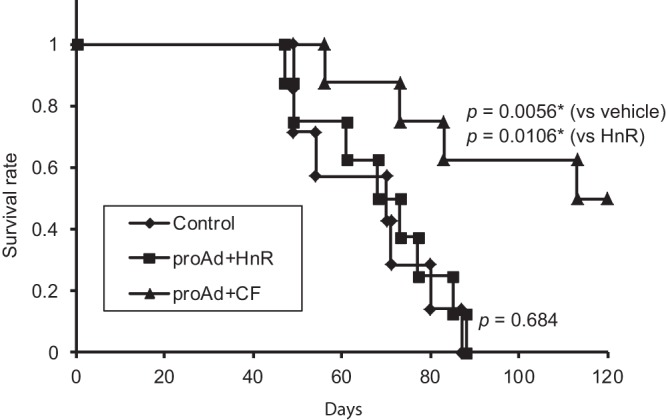
Kaplan-Meier survival curves for athymic nude mice treated when intracranial U87 tumors were 9 days old. Human U87 cells (2 × 105) were inoculated (2 mm lateral and 0.5 mm anterior from bregma and 2.5 mm deep) into the brain of mice on day zero. The RD proAdΔ24.GFP along with either HnR (n = 8) or Flp-bearing HnR-CF (n = 8) was intratumorally injected using the same stereotactic coordinates 9 days later. Control mice (n = 7) were injected with vehicle HBSS.
DISCUSSION
In this report, we show for the first time that an RD Ad can be “switched” into an RC, oncolytic Ad in vitro and in vivo. In particular, the technology that we present consists of utilizing FRT site-specific recombination to excise a silencing GFP transgene that prevents E1A gene expression in a RD adenoviral vector. Initially, we showed that this was effective when the RD proAdΔ24.GFP was used to infect cells that stably expressed Flp. Then, we were able to show that this also functioned as expected when a second replication-defective vector was utilized to express Flp in trans and coinfected/coinjected with RD proAdΔ24.GFP vector in vitro and in vivo. In fact, Fig. 5a shows that direct injection of the RD adenoviral vector together with the Flp-expressing amplicon vector increased intratumoral RC Ad titers by one-half of a logarithmic unit. Figure 6 shows that this led to a significantly increased survivorship in mice with orthotopic human gliomas. Therefore, direct coadministration of an RD vector together with an Flp-bearing vector is a feasible approach against tumors. Thus, these experiments provide proof of principle to show that RD adenoviral vectors can be converted into effective RC oncolytic Ad.
The reported strategy could be useful in multiple settings. For instance, an RD vector could be utilized first to deliver an immunostimulatory gene (in lieu of the GFP transgene in proAdΔ24.GFP) in a tumor, and this immunostimulation could then be amplified by delivery of the Flp vector to generate virus-mediated oncolysis. As an example of this approach, we recently published a clinical trial in which human subjects afflicted by malignant glioma were treated with peritumoral injections of an RD adenoviral vector expressing the HSV-tk (thymidine kinase) transgene (23). The transgene product provides chemosensitivity to the prodrug valacyclovir but also is highly immunogenic, leading to an immune response against HSV tk-expressing cancer cells (22). Therefore, one could envision a strategy whereby initial HSV-tk gene delivery mediated by the RD adenoviral vector could be utilized to start an immune response against the tumor, followed at a later stage by injection of the Flp vector to switch to the RC adenovirus, which could further biodistribute in tumor and amplify oncolysis but also increase immunostimulation. The delivery of the Flp vector could also be delayed until tumors recur and be used as a salvage therapy to convert the tumor RD vector into an RC vector. In this strategy, an RD integrating retroviral vector could be injected into tumors. Tumors would undergo standard-of-care treatment (radiation and/or chemotherapy). However, at a later stage, when the tumor recurs, the presence of the RD vector in the tumor genome could be exploited by injection of the Flp-expressing vector, leading to conversion of the tumor-killing RC retrovirus. While the findings reported in this paper are the first proof of RD-to-RC switching, the above strategies will require additional experimentation to determine their feasibility and to assess whether our reported system is useful in these aforementioned contexts. However, we can preliminarily report that the reported strategy is particularly useful in one application (H. Nakashima and E. A. Chiocca, unpublished data): carrier-cell-mediated delivery has been practiced as a means to shield oncolytic viruses from deleterious host factors, such as complement. One limitation is that carrier cells themselves can be lysed by the OV before they distribute to tumor. Data that we are preparing for a follow-up manuscript show that the RD-to-RC switch can be employed to deliver silent, RD vectors within carrier cells to tumors with minimal lysis or cytotoxicity to the carrier cells. Once in tumor, the RD-to-RC switch can then be triggered to obtain the biologic, anticancer effect of the RC. We believe that this will provide proof of the utility of the reported application. We do not claim that the efficacy of our approach is superior to that of direct injection of an oncolytic virus but rather that our approach will be useful in different contexts of anticancer therapy, as discussed above.
The vectors employed in this study consisted of an RD Ad and an Flp-expressing HSV amplicon vector. However, other vectors should be just as suitable. We also attempted to engineer an RD Ad that also expressed the Flp gene in order to provide all needed functions for RD-to-RC transformation in a single construct. However, difficulties in restricting Flp expression to when it was desired as well as in undesirable recombination events led us to separate the Flp function into a second vector. It is possible that a more desirable single-vector construct could be engineered not only by trying to transcriptionally restrict Flp expression but also by adding appropriate microRNA target sequences to the 3′-untranslated region (UTR) of Flp (24–26). For instance, the CAG promoter in HnR-CF could be replaced by other promoters that are regulated by the microenvironment and/or pharmacologic agents for temporal regulation of Flp gene transcription (27–29). Flp mRNA and protein could be regulated by appropriate microRNAs and/or ligands (fused with the estrogen receptor domain) (30, 31). This would allow even more highly specific expression of Flp to cancer cells. In addition, to avoid recombination events in a single-vector construct, additional experimentation with appropriate flanking sequences may be needed.
The significance of the reported strategy may also relate to safety. Although most clinical trials employing RC viruses have not shown evidence of undesirable toxic side effects from virus replication, the possibility still exists. In this context, the ability to initially administer/use an RD vector that would be much less toxic and provide it with replication competence only when or where needed by provision of Flp may provide an avenue for making cancer gene and viral therapy safer. In addition, we employed the FRT site-specific recombination approach, but others (e.g., Cre-loxP) might also be workable (18). In fact, Ng et al. showed that these two approaches were directly comparable and equivalent as a means to generate “gutless” adenoviral vectors in 293 cells coinfected with helper virus and the adenoviral vector (32–34). The engineering of gutless adenoviral vectors utilizes an RC helper virus and RD vector to package and generate an RD “gutless” vector. The site-specific recombination has also been used to eliminate helper vector contamination for packaging of helper-dependent (HD) gutless adenoviruses that are devoid of all viral coding sequences (34, 35). Immune and inflammatory responses to HD Ads are greatly reduced, allowing for enhanced transgene expression in target tissues (36, 37). In comparison, here, we employ a similar engineering strategy to achieve the opposite result: we use an RD vector and RD helper to generate an RC virus utilizing the Flp-mediated recombinatorial approach (21). The site-specific recombinase Flp from Saccharomyces cerevisiae binds the two halves of target FRT sites to excite FRT-flanked sequences in a manner similar to that of the Cre-loxP system (38). FLP has been widely used for genomic manipulations in mammalian cells and animal models (39), and this report provides another example of the power of this technology.
In conclusion, the reported advance provides a framework for switching RD vectors into their oncolytic counterparts for tumor therapy. Further experimentation will provide knowledge related to its utility in a variety of applications.
ACKNOWLEDGMENTS
This project was supported by National Institutes of Health grants R21NS0632901, 1P01CA163205, and P01CA069246.
We acknowledge Yoshinaga Saeki for HSV-1 materials and advice, Kazue Kasai and Tran Nguyen for lab assistance, and Maria Jasin for the plasmid DNA.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Flint J, Shenk T. 1989. Adenovirus E1A protein paradigm viral transactivator. Annu. Rev. Genet. 23:141–161. 10.1146/annurev.ge.23.120189.001041 [DOI] [PubMed] [Google Scholar]
- 2.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124–129. 10.1038/334124a0 [DOI] [PubMed] [Google Scholar]
- 3.Howe JA, Mymryk JS, Egan C, Branton PE, Bayley ST. 1990. Retinoblastoma growth suppressor and a 300-kDa protein appear to regulate cellular DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 87:5883–5887. 10.1073/pnas.87.15.5883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frisch SM, Mymryk JS. 2002. Adenovirus-5 E1A: paradox and paradigm. Nat. Rev. Mol. Cell Biol. 3:441–452. 10.1038/nrm827 [DOI] [PubMed] [Google Scholar]
- 5.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. 2000. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19:2–12. 10.1038/sj.onc.1203251 [DOI] [PubMed] [Google Scholar]
- 6.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, Hawkins L, Kirn D. 2000. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 6:1134–1139. 10.1038/80474 [DOI] [PubMed] [Google Scholar]
- 7.Danthinne X, Imperiale MJ. 2000. Production of first generation adenovirus vectors: a review. Gene Ther. 7:1707–1714. 10.1038/sj.gt.3301301 [DOI] [PubMed] [Google Scholar]
- 8.Bauerschmitz GJ, Kanerva A, Wang M, Herrmann I, Shaw DR, Strong TV, Desmond R, Rein DT, Dall P, Curiel DT, Hemminki A. 2004. Evaluation of a selectively oncolytic adenovirus for local and systemic treatment of cervical cancer. Int. J. Cancer 111:303–309. 10.1002/ijc.20217 [DOI] [PubMed] [Google Scholar]
- 9.Delgado-Enciso I, Cervantes-Garcia D, Martinez-Davila IA, Ortiz-Lopez R, Alemany-Bonastre R, Silva-Platas CI, Lugo-Trampe A, Barrera-Saldana HA, Galvan-Salazar HR, Coronel-Tene CG, Sanchez-Santillan CF, Rojas-Martinez A. 2007. A potent replicative delta-24 adenoviral vector driven by the promoter of human papillomavirus 16 that is highly selective for associated neoplasms. J. Gene Med. 9:852–861. 10.1002/jgm.1071 [DOI] [PubMed] [Google Scholar]
- 10.Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, Moeniralm S, Saeki Y, Carette JE, Weissleder R, Vandertop WP, van Beusechem VW, Dirven CM, Chiocca EA. 2006. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol. Ther. 14:779–788. 10.1016/j.ymthe.2006.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamfers ML, Idema S, Bosscher L, Heukelom S, Moeniralm S, van der Meulen-Muileman IH, Overmeer RM, van der Valk P, van Beusechem VW, Gerritsen WR, Vandertop WP, Dirven CM. 2007. Differential effects of combined Ad5-delta 24RGD and radiation therapy in in vitro versus in vivo models of malignant glioma. Clin. Cancer Res. 13:7451–7458. 10.1158/1078-0432.CCR-07-1265 [DOI] [PubMed] [Google Scholar]
- 12.Curiel DT, Siegal GP, Rosenthal EL, Rivera AA, Ji S, Wang M, Lu B, Makhija SK, Mathis JM, Zhu ZB. 2007. Targeting of a conditionally replicative adenovirus agent to human squamous cell carcinomas of the head and neck. Int. J. Oncol. 31:1213–1222 http://www.spandidos-publications.com/ijo/31/5/1213. [PubMed] [Google Scholar]
- 13.Reid T, Galanis E, Abbruzzese J, Sze D, Wein LM, Andrews J, Randlev B, Heise C, Uprichard M, Hatfield M, Rome L, Rubin J, Kirn D. 2002. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): phase II viral, immunologic, and clinical endpoints. Cancer Res. 62:6070–6079 http://cancerres.aacrjournals.org/content/62/21/6070.full.pdf [PubMed] [Google Scholar]
- 14.Wang Y, Hallden G, Hill R, Anand A, Liu TC, Francis J, Brooks G, Lemoine N, Kirn D. 2003. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 21:1328–1335. 10.1038/nbt887 [DOI] [PubMed] [Google Scholar]
- 15.Fukuda K, Abei M, Ugai H, Seo E, Wakayama M, Murata T, Todoroki T, Tanaka N, Hamada H, Yokoyama KK. 2003. E1A, E1B double-restricted adenovirus for oncolytic gene therapy of gallbladder cancer. Cancer Res. 63:4434–4440 http://cancerres.aacrjournals.org/content/63/15/4434.long [PubMed] [Google Scholar]
- 16.Nakashima H, Kaur B, Chiocca EA. 2010. Directing systemic oncolytic viral delivery to tumors via carrier cells. Cytokine Growth Factor Rev. 21:119–126. 10.1016/j.cytogfr.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathis JM, Stoff-Khalili MA, Curiel DT. 2005. Oncolytic adenoviruses-selective retargeting to tumor cells. Oncogene 24:7775–7791. 10.1038/sj.onc.1209044 [DOI] [PubMed] [Google Scholar]
- 18.Toth K, Dhar D, Wold WS. 2010. Oncolytic (replication-competent) adenoviruses as anticancer agents. Expert Opin. Biol. Ther. 10:353–368. 10.1517/14712590903559822 [DOI] [PubMed] [Google Scholar]
- 19.Richardson C, Moynahan ME, Jasin M. 1998. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 12:3831–3842. 10.1101/gad.12.24.3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saeki Y, Fraefel C, Ichikawa T, Breakefield XO, Chiocca EA. 2001. Improved helper virus-free packaging system for HSV amplicon vectors using an ICP27-deleted, oversized HSV-1 DNA in a bacterial artificial chromosome. Mol. Ther. 3:591–601. 10.1006/mthe.2001.0294 [DOI] [PubMed] [Google Scholar]
- 21.O'Gorman S, Fox DT, Wahl GM. 1991. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251:1351–1355. 10.1126/science.1900642 [DOI] [PubMed] [Google Scholar]
- 22.Aguilar LK, Guzik BW, Aguilar-Cordova E. 2011. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J. Cell. Biochem. 112:1969–1977. 10.1002/jcb.23126 [DOI] [PubMed] [Google Scholar]
- 23.Chiocca EA, Aguilar LK, Bell SD, Kaur B, Hardcastle J, Cavaliere R, McGregor J, Lo S, Ray-Chaudhuri A, Chakravarti A, Grecula J, Newton H, Harris KS, Grossman RG, Trask TW, Baskin DS, Monterroso C, Manzanera AG, Aguilar-Cordova E, New PZ. 2011. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 29:3611–3619. 10.1200/JCO.2011.35.5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly EJ, Hadac EM, Greiner S, Russell SJ. 2008. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 14:1278–1283. 10.1038/nm.1776 [DOI] [PubMed] [Google Scholar]
- 25.Ylosmaki E, Hakkarainen T, Hemminki A, Visakorpi T, Andino R, Saksela K. 2008. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific microRNA. J. Virol. 82:11009–11015. 10.1128/JVI.01608-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugio K, Sakurai F, Katayama K, Tashiro K, Matsui H, Kawabata K, Kawase A, Iwaki M, Hayakawa T, Fujiwara T, Mizuguchi H. 2011. Enhanced safety profiles of the telomerase-specific replication-competent adenovirus by incorporation of normal cell-specific microRNA-targeted sequences. Clin. Cancer Res. 17:2807–2818. 10.1158/1078-0432.CCR-10-2008 [DOI] [PubMed] [Google Scholar]
- 27.Hernandez-Alcoceba R, Pihalja M, Nunez G, Clarke MF. 2001. Evaluation of a new dual-specificity promoter for selective induction of apoptosis in breast cancer cells. Cancer Gene Ther. 8:298–307. 10.1038/sj.cgt.7700304 [DOI] [PubMed] [Google Scholar]
- 28.Sektas M, Szybalski W. 1998. Tightly controlled two-stage expression vectors employing the Flp/FRT-mediated inversion of cloned genes. Mol. Biotechnol. 9:17–24. 10.1007/BF02752694 [DOI] [PubMed] [Google Scholar]
- 29.Fechner H, Wang X, Pico AH, Wildner J, Suckau L, Pinkert S, Sipo I, Weger S, Poller W. 2007. A bidirectional Tet-dependent promotor construct regulating the expression of E1A for tight control of oncolytic adenovirus replication. J. Biotechnol. 127:560–574. 10.1016/j.jbiotec.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 30.Nichols M, Rientjes JM, Logie C, Stewart AF. 1997. FLP recombinase/estrogen receptor fusion proteins require the receptor D domain for responsiveness to antagonists, but not agonists. Mol. Endocrinol. 11:950–961. 10.1210/me.11.7.950 [DOI] [PubMed] [Google Scholar]
- 31.Matsuda T, Cepko CL. 2007. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. U. S. A. 104:1027–1032. 10.1073/pnas.0610155104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitani K, Graham FL, Caskey CT, Kochanek S. 1995. Rescue, propagation, and partial purification of a helper virus-dependent adenovirus vector. Proc. Natl. Acad. Sci. U. S. A. 92:3854–3858. 10.1073/pnas.92.9.3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kochanek S, Clemens PR, Mitani K, Chen HH, Chan S, Caskey CT. 1996. A new adenoviral vector: replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proc. Natl. Acad. Sci. U. S. A. 93:5731–5736. 10.1073/pnas.93.12.5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng P, Cummings DT, Evelegh CM, Graham FL. 2000. Yeast recombinase FLP functions effectively in human cells for construction of adenovirus vectors. Biotechniques 29:524–526, 528 http://www.biotechniques.com/multimedia/archive/00011/00293st04_11792a.pdf [DOI] [PubMed] [Google Scholar]
- 35.Umana P, Gerdes CA, Stone D, Davis JR, Ward D, Castro MG, Lowenstein PR. 2001. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 19:582–585. 10.1038/89349 [DOI] [PubMed] [Google Scholar]
- 36.Vetrini F, Ng P. 2010. Gene therapy with helper-dependent adenoviral vectors: current advances and future perspectives. Viruses 2:1886–1917. 10.3390/v2091886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dindot S, Piccolo P, Grove N, Palmer D, Brunetti-Pierri N. 2011. Intrathecal injection of helper-dependent adenoviral vectors results in long-term transgene expression in neuroependymal cells and neurons. Hum. Gene Ther. 22:745–751. 10.1089/hum.2010.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauer B, Henderson N. 1988. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U. S. A. 85:5166–5170. 10.1073/pnas.85.14.5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buchholz F, Angrand PO, Stewart AF. 1998. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat. Biotechnol. 16:657–662. 10.1038/nbt0798-657 [DOI] [PubMed] [Google Scholar]