

ABSTRACT
Hepatitis C virus (HCV) is a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma in humans. The life cycle of HCV is closely associated with the metabolism of lipids, especially very-low-density lipoprotein (VLDL) in hepatocytes. Hepatocyte nuclear factor 4α (HNF4α), the most abundant transcription factor in the liver, regulates the VLDL secretory pathway. However, the effects of HNF4α on the HCV life cycle are unclear. In this study, we investigated the regulatory effects of HNF4α on HCV assembly and secretion. HCV in HNF4α-deficient hepatocytes showed reduced assembly and secretion but unchanged entry and RNA replication. Bezafibrate, a chemical inhibitor of HNF4α, suppressed HCV assembly and secretion. HNF4α downregulation resulted in rearrangement of cytosolic lipid droplets (LDs), as evidenced by the aggregation of large LDs and distorted cytosolic distribution. Phospholipase A2 GXIIB (PLA2GXIIB), an HNF4α-regulated factor involved in VLDL secretion, was found to be crucial in HCV secretion. PLA2GXIIB expression was upregulated in hepatocytes harboring HCV subgenomic replicons or in HCV-infected hepatocytes. This upregulation was transcriptionally controlled in an HNF4α-dependent manner after HCV infection. Furthermore, PLA2GXIIB combined with microsomal triglyceride transfer protein was found to be responsible for the regulation of HNF4α-induced HCV infectivity. These results suggest that HNF4α and its downstream PLA2GXIIB are important factors affecting the late stage of the HCV life cycle and may serve as potential drug targets for the treatment of HCV infection.
IMPORTANCE
The assembly and secretion of hepatitis C virus (HCV) are closely correlated with the very-low-density lipoprotein (VLDL) secretory pathway. However, the molecular mechanism by which HCV cooperates with VLDL to facilitate assembly and release is unclear. In this study, we report the function of hepatocyte nuclear factor 4α (HNF4α), the most abundant liver-enriched transcription factor, in HCV assembly and release. HNF4α is crucial in VLDL-mediated lipid transport. We found that HNF4α abundance was increased upon HCV infection and that HNF4α downregulation impaired the assembly and secretion of HCV. We also determined that PLA2GXIIB, a direct target factor of HNF4α involved in VLDL secretion, was upregulated upon HCV infection. These results suggest that PLA2GXIIB is required in the late stage of the HCV life cycle and that the VLDL secretory pathway is important for HCV assembly and secretion. This study provides insights into the mechanism by which HCV cooperates with HNF4α to be assembled into and released together with VLDL particles.
INTRODUCTION
Hepatitis C virus (HCV) is a major pathogen of liver diseases such as hepatic steatosis, cirrhosis, and hepatocellular carcinoma (1). The current standard treatment for such diseases involves alpha interferon plus ribavirin. However, this treatment is limited by its low efficiency in some HCV genotypes, severe side effects, and high cost (2). The direct antiviral agents recently available have shown excellent efficacy against a broad range of HCV genotypes (3). However, these compounds target viral proteins and induce the emergence of drug-resistant viruses. Thus, novel treatment regimens for HCV infection are needed.
The host lipid metabolism is known to regulate the life cycle of HCV. HCV particles circulating in the blood exist as HCV lipoviroparticles that are associated with very-low-density lipoproteins (VLDLs)/low-density lipoproteins (LDLs) (4). Cellular factors, including LDL receptor (LDLR), scavenger receptor class B type I (SR-BI), and Niemann-Pick C1-like 1 (NPC1L1), are required for both HCV entry and lipid uptake (5). Chemical compounds that block these factors show potent anti-HCV activities (6–8). HCV RNA replication occurs in specialized cellular membranes and shares cellular factors, such as microRNA miR-122, with lipid metabolism (9, 10). HCV assembly involves nucleocapsid formation in an endoplasmic reticulum (ER)-related compartment in close vicinity to cytosolic lipid droplets (LDs) (11). HCV maturation requires the addition of lipid and/or apolipoproteins, possibly in a post-ER compartment (12, 13). Finally, similar to VLDL particles, HCV particles are secreted out of cells through exocytosis. The VLDL secretory pathway is hijacked to facilitate HCV assembly and secretion (14, 15). Cellular factors, such as microsomal triglyceride transfer protein (MTP) and apolipoprotein E (ApoE), are required by both HCV and VLDL (12, 16).
Hepatocyte nuclear factor 4α (HNF4α) is a hepatocyte-enriched transcription factor belonging to the nuclear receptor superfamily. HNF4α has important functions in the regulation of lipid metabolism, especially lipid transport. Mice with conditional knockout of the liver HNF4α gene (HNF4αLiv KO mice) show disrupted lipid metabolism and transport because of the deficiency in VLDL assembly and secretion (17). Many HNF4α-regulated factors, including NPC1L1 (18), miR-122 (19), MTP (20), and ApoE (21), modulate the life cycle of HCV. In addition, HNF4α expression and activity are upregulated in hepatocytes that harbor genotype 1b subgenomic replicon (22). These findings suggest that HNF4α is involved in the HCV life cycle. However, direct evidence to prove these results remains elusive.
The expression of phospholipase A2 GXIIB (PLA2GXIIB), an atypical member of the phospholipase A2 family, is regulated by HNF4α. We have previously established a PLA2GXIIB-null mouse model that developed severe hepatic steatosis and hypolipidemia, indicating that PLA2GXIIB is involved in VLDL secretion (23). Other studies have also reported that PLA2GXIIB expression is remarkably elevated in HCV-related liver tumor tissues (24). However, whether or not PLA2GXIIB is involved in the life cycle and the mechanism of HCV remains unknown.
This study is the first to investigate the regulatory effects of HNF4α and PLA2GXIIB on the HCV life cycle. Our results demonstrated that HNF4α downregulation by small interfering RNA (siRNA) or bezafibrate reduced HCV assembly and secretion. PLA2GXIIB was upregulated by HCV infection; PLA2GXIIB knockdown impaired HCV secretion. These observations provide direct evidence that HNF4α and its downstream PLA2GXIIB are required in the assembly and secretion of HCV.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
Huh7 and derivative Huh7.5.1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; HyClone, Beijing, China) and 1% penicillin-streptomycin. Huh7-sgHCV1b (genotype 1b [GT1b]) and Huh7.5.1-sgJFH (GT2a) cells harboring genotype 1b and 2a subgenomic (sg) replicons were maintained in the aforementioned medium supplemented with 400 μg/ml G418 and 2 μg/ml puromycin, respectively. Huh7.5.1 cured cells were obtained by treating Huh7.5.1-sgJFH cells with 270 ng/ml of pegylated alpha interferon 2a (Pegasys) for 2 weeks. The cured cells were confirmed by quantifying intracellular HCV RNA copies via quantitative reverse transcription-PCR (qRT-PCR) and by detecting NS3 protein via Western blotting. Huh7.5.1 cells infected with HCV-Jc1EGFP (where EGFP is enhanced green fluorescent protein) at a multiplicity of infection (MOI) of 0.02 were maintained for more than 2 weeks and used as persistently infected cells in assembly and secretion experiments.
The plasmid pFL-J6/JFH/Jc1 used to generate infectious HCV-Jc1 was provided by Apath, LLC. HCV-Jc1 virus was generated by transfecting Huh7.5.1 cells with 10 μg of Jc1 RNA by electroporation. The viral supernatants were collected at 8 days to 10 days posttransfection. The EGFP gene was inserted into the C terminus of NS5A-encoding sequence within the HCV-Jc1 genome to generate an HCV reporter virus termed HCV-Jc1EGFP. Huh7.5.1 cells were transfected with HCV-Jc1EGFP RNA, and the viral supernatants were serially passaged to obtain a cell culture-adapted virus with robust propagation efficiency. HCV-Jc1EGFP virus stocks were produced by infecting Huh7.5.1 cells with the adapted HCV-Jc1EGFP at an MOI of 0.02. At 8 days postinfection, supernatants were collected for the following experiments. An adapted HCV-JFH1 virus (designated HCV-mutJFH1) from ChinaWave Co., Ltd., grows more rapidly in a cell culture system and exhibits higher specific infectivity than HCV-Jc1.
Plasmids that encode human HNF4α (NCBI accession number NP_000448), MTP (tagged with Flag; NM_000253), ApoE (tagged with HA; NP_000032), and PLA2GXIIB (tagged with V5; NP_115951) proteins were generated by inserting genes into pcDNA3.1(+), pcDNA3.1(+), lentiviral vector pRlenti, and pcDNA4-V5-His C, respectively. siRNA-resistant HNF4α- and PLA2GXIIB-encoding plasmids were cloned into pcDNA3.1(+) and pRlenti, respectively.
Antibodies and reagents.
Western blot analysis, immunofluorescence assay, and flow cytometry (FCM) were performed using primary antibodies against the following proteins: HCV core (C7-50; Abcam), HCV NS3 (H23; Abcam), HNF4α (3113S [Cell Signaling] and Sc-8987X [Santa Cruz]), CD81 (MAB4615; R&D Systems), ApoE (10817-RP02; Sino Biological), and PLA2GXIIB (SAB1103079 [Sigma] and ab85069 [Abcam]). The monoclonal antibody against albumin was provided by Miguel A. Esteban. Monoclonal antibodies against β-tubulin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Flag, V5, and hemagglutinin (HA) tags were used according to the manufacturer's instructions. siRNA specifically targeting HNF4α (25), ApoE (16), CD81 (26), HCV (27), and phosphatidylinositol 4-kinase III alpha (PI4KIIIα) (28) were previously reported. The target sequence of the PLA2GXIIB siRNA is AUA UCG CUG UGA UGC AAA A. Primers used for qRT-PCR are available upon request.
Bezafibrate (B7273; Sigma) was reconstituted in dimethyl sulfoxide (DMSO). Cytotoxicity was examined using alamarBlue reagent (AbD Serotec, Oxford, United Kingdom) in accordance with the manufacturer's instructions.
HCV life cycle experiments.
The effect of HNF4α or PLA2GXIIB on HCV entry was determined using an siRNA-mediated knockdown assay according to previously described methods (29). Briefly, Huh7.5.1 cells were plated into a six-well plate and then transfected with HNF4α or PLA2GXIIB siRNA. HCV-Jc1 was inoculated at an MOI of 0.05 at 48 h posttransfection. Subsequently, the cells were collected to quantify intracellular HCV RNA copy numbers at 24 h postinfection. Nonspecific siRNA and CD81 siRNA were used as negative and positive controls, respectively.
GT2a- or GT1b-harboring cells were transfected with specific siRNAs to investigate the effect of HNF4α or PLA2GXIIB on HCV RNA replication. HCV RNA copy numbers and NS3 expression were quantified at the time points postinfection indicated on Fig. 2D to G. HCV- and PI4KIIIα-specific siRNAs were used as positive controls.
FIG 2.

HNF4α is not required for HCV entry or RNA replication. (A) Huh7.5.1 cells were transfected with HNF4α siRNA (siHNF4α), CD81 siRNA (siCD81), or a scrambled siRNA (siCtrl), followed by inoculation of HCV-Jc1 (MOI of 0.05) at 48 h posttransfection. HNF4α expression at 48 h posttransfection was detected via Western blot analysis. (B) Cellular membrane CD81 expression was measured by flow cytometry in Huh7.5.1 cells at 48 h posttransfection, and the mean fluorescence intensity of CD81 was calculated. (C) The infected Huh7.5.1 cells were collected at 24 h postinfection, and relative quantification of the change in intracellular HCV RNA copy numbers was calculated after normalization by 18S RNA. (D) Huh7.5.1-sgJFH cells were transfected with siRNA-targeted HNF4α, phosphatidylinositol 4-kinase III alpha (PI4KIIIα), or HCV. Cell lysates were harvested at the indicated time points posttransfection. Relative quantification of the change in HCV RNA copies was calculated by normalization with 18S RNA. The data are presented as means ± SEM (n = 3; *, P < 0.05;**, P < 0.01; ***, P < 0.001). (E) Huh7.5.1-sgJFH cell lysates were harvested at the indicated time points posttransfection to detect HNF4α and NS3 protein expression via Western blot analysis. The ratios for the relative band intensities of each target protein normalized with GAPDH were calculated. (F) Huh7-sgHCV1b cells were transfected with siHNF4α and collected to quantify HCV RNA copy numbers. siPI4KIIIα and siHCV were used as positive controls. Relative quantification of the change in HCV RNA copy numbers was calculated at the indicated time points posttransfection after normalization with 18S RNA. The results are shown as means ± SEM (n = 3; **, P < 0.01; ***, P < 0.001). (G) Detection of HNF4α and NS3 in Huh7-sgHCV1b cells caused by siHNF4α, siPI4KIIIα, and siHCV transfection via Western blot analysis. The ratios for relative band intensities normalized by GAPDH were calculated.
Huh7.5.1 cells or persistently infected Huh7.5.1 cells were used for HCV assembly and secretion assays. Huh7.5.1 cells were plated on six-well plates at 3 × 105 cells/well. After 24 h, the cells were transfected with specific siRNAs at a final concentration of 100 nM or treated with different doses of bezafibrate. At 3 h posttransfection, the transfected Huh7.5.1 cells were infected with HCV-Jc1 or HCV-Jc1EGFP at an MOI of 0.02. Viral supernatants and cell pellets were collected at 72 h postinfection for virus titration, Western blotting, and qRT-PCR. Huh7.5.1 cells that were persistently infected with HCV-Jc1EGFP were alternatively used for HCV assembly and secretion assays. The cells were transfected with specific siRNAs or treated with 300 μg/ml of bezafibrate, followed by the quantification of HCV infectivity in the supernatants or cell lysates at 72 h posttreatment.
Virus titration assay.
HCV titers in the cell lysates or viral supernatants were quantified as previously described (29). Two assays were used for virus titration. For the titration of supernatant HCV-Jc1 titers, viral supernatants were clarified and inoculated on Huh7.5.1 cells by endpoint dilution. At 72 h postinfection, Huh7.5.1 cells were fixed with 4% paraformaldehyde and then immunostained for HCV core antigen. Cell lysates with DMEM containing 10% FBS were subjected to four cycles of freezing and thawing to determine intracellular HCV-Jc1 infectivity. Cell debris was removed by low-speed centrifugation. The supernatants were collected to determine infectivity as described above. An FCM assay was also used for HCV-Jc1EGFP titration. Approximately 50 μl to 200 μl of clarified supernatant or cell lysate was inoculated on Huh7.5.1 cells. The infected cells were collected for FCM at 48 h postinfection. The relative infectivity was then normalized and calculated as a percentage.
Promoter reporter analysis.
The PLA2GXIIB promoter reporter plasmid was previously reported (23). Huh7.5.1 cells were plated on 96-well plates with 2 × 104 cells/well and then transfected with the PLA2GXIIB promoter reporter plasmid and an HNF4α-encoding plasmid to investigate the regulatory effect of HNF4α on the PLA2GXIIB promoter reporter. A pSV40-Rluc plasmid that encodes Renilla luciferase (Rluc) was cotransfected to monitor transfection efficiency. An HNF4α-binding-deficient promoter reporter plasmid was used as a negative control. At 24 h posttransfection, the cells were lysed with Dual-Glo luciferase assay reagent (E2920; Promega) and firefly and Renilla luciferase activities were measured according to the manufacturer's instructions. Huh7.5.1 cells were transfected with the PLA2GXIIB promoter reporter and pSV40-Rluc plasmid with or without siRNA targeting HNF4α (siHNF4α) to evaluate the regulatory effect of PLA2GXIIB promoter reporter on HCV infection. At 3 h posttransfection, the cells were infected with HCV-Jc1EGFP at an MOI of 0.2. The promoter activities were analyzed at the time points postinfection indicated in the legends to Fig. 6G and H and on Fig. 6F.
FIG 6.

HNF4α is required for the HCV-induced upregulation of PLA2GXIIB expression. (A) Relative quantification of the change in PLA2GXIIB transcript normalized to GAPDH in Huh7.5.1-sgJFH cells compared with Huh7.5.1 cells. The data are presented as means ± SEM (n = 3; ***, P < 0.001). (B) Relative quantification of the change in PLA2GXIIB transcript after normalization to GAPDH in Huh7-sgHCV1b cells compared with Huh7 cells. The data are presented as means ± SEM (n = 3; **, P < 0.01). (C) PLA2GXIIB and HCV NS3 proteins were detected in Huh7.5.1-sgJFH or Huh7-sgHCV1b cells via Western blot analysis. (D) Huh7-sgHCV1b cells were transfected with siPI4KIIIα or siHCV, with siHNF4α as a positive control. The abundance of PLA2GXIIB protein was measured by antibodies against NS3, HNF4α, and PLA2GXIIB at 72 h posttransfection. (E) PLA2GXIIB protein was measured in HCV-Jc1EGFP-infected Huh7.5.1 cells (MOI of 0.02) compared with mock-infected cells in a time course analysis. (F) Activities of the PLA2GXIIB promoter reporter in Huh7.5.1 cells infected with HCV-Jc1EGFP (MOI of 0.2) compared with those of mock-infected cells were quantified in a time course analysis. (G) Huh7.5.1 cells were transfected with the wild-type and mutant PLA2GXIIB promoter reporter plasmids and then inoculated with HCV-Jc1EGFP (MOI of 0.2) at 3 h posttransfection. Activities of the promoter reporter were quantified at 48 h post-HCV infection. (H) Huh7.5.1 cells were cotransfected with PLA2GXIIB promoter reporter plasmids and siHNF4α, and activities of the promoter reporter were analyzed in Huh7.5.1 cells either noninfected or infected with HCV-Jc1EGFP (MOI of 0.2) at 48 h postinfection. psi, postinfection.
Cytosolic LDs and supernatant ApoE quantification.
Huh7.5.1 cells were transfected with HNF4α or PLA2GXIIB siRNA or treated with bezafibrate and then inoculated with HCV-mutJFH1 for 72 h. These cells were then trypsinized and collected for Nile Red (lot 22190; AAT Bioquest, Inc., CA) staining and FCM. To measure the rearrangement of LDs, HCV-mutJFH1-infected cells were fixed for LD staining with Nile Red. HCV core protein was monitored by specific antibody and examined with a Zeiss LSM 710 NLO confocal microscope. Supernatants were collected for virus titration. Twenty-five microliters of clarified supernatant was resolved by 10% SDS-PAGE, and the levels of secreted ApoE and albumin were quantified through Western blotting.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM) and analyzed by t test or two-way analysis of variance (ANOVA).
RESULTS
HNF4α regulates HCV assembly and secretion. (i) HNF4α is increased by HCV replication.
HNF4α is a key mediator of cellular lipid metabolism, particularly VLDL assembly and secretion. HNF4α expression and activity were increased in HCV subgenomic replicon-replicating Huh.8 cells (genotype 1b) (22). To validate this observation, transcripts of HNF4α and its downstream factors MTP, PLA2GXIIB, ApoB, and ApoE were quantified in Huh7.5.1-sgJFH cells (Huh7.5.1 cells with a replicating genotype 2a subgenomic replicon) and Huh7.5.1 cured cells (produced by treating Huh7.5.1-sgJFH cells with Pegasys for 2 weeks to eliminate the replicon). As shown in Fig. 1A, the transcripts of HNF4α, PLA2GXIIB, MTP, and ApoE increased in Huh7.5.1-sgJFH cells compared with those in Huh7.5.1 cells. When the replicon was eliminated, transcripts of these factors returned to levels comparable to those of Huh7.5.1 cells (Fig. 1A). A time course analysis was performed to monitor the expression of HNF4α protein in HCV infection. Huh7.5.1 cells were infected with HCV-Jc1EGFP at an MOI of 0.02. The expression of HNF4α protein was detected at the time points postinfection indicated on Fig. 1B. Compared with the noninfected cells, the HCV-infected Huh7.5.1 cells showed increased HNF4α protein expression (Fig. 1B, compare lanes 2, 4, and 6 with lanes 1, 3, and 5, respectively). When treated with Pegasys, the HCV-Jc1EGFP-infected Huh7.5.1 cells showed reduced HNF4α abundance (Fig. 1C, compare lanes 2 and 3). These results suggest that the upregulated HNF4α expression is dependent on HCV replication.
FIG 1.
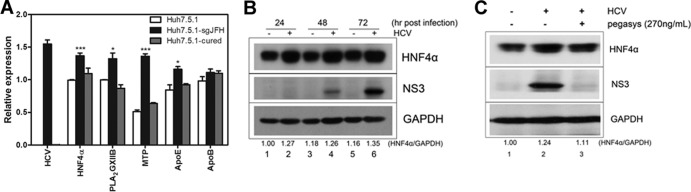
HNF4α expression is induced by HCV infection. (A) Transcripts of HNF4α and its downstream factors were quantified in Huh7.5.1-sgJFH cells (Huh7.5.1 cells with replicating genotype 2a subgenomic replicon) and Huh7.5.1 cured cells after normalization by GAPDH. Huh7.5.1 cured cells were obtained by treating Huh7.5.1-sgJFH cells with pegylated interferon α-2a (Pegasys) for 2 weeks. The data are presented as means ± SEM (n = 3; *, P < 0.05; ***, P < 0.001). (B) HNF4α and HCV NS3 proteins were detected in HCV-Jc1EGFP-infected Huh7.5.1 cells (MOI of 0.02) using specific antibodies and compared with mock-infected cells in a time course analysis. The ratio for the relative band intensity of HNF4α normalized with GAPDH was calculated. (C) Huh7.5.1 cells infected with HCV-Jc1EGFP (MOI of 0.02) were treated with or without Pegasys at 270 ng/ml and then cultured for 72 h. Abundances of HNF4α and NS3 proteins were detected by Western blotting using specific antibodies.
These observations suggest that HNF4α is crucial in HCV life cycle. To test this hypothesis, we evaluated the effect of HNF4α on different stages of HCV life cycle.
(ii) HNF4α is not required for HCV entry or RNA replication.
To determine whether HNF4α is required for HCV entry, Huh7.5.1 cells were transfected with HNF4α-specific siRNA (siHNF4α) and then incubated for 48 h for efficient knockdown. These cells were then infected with HCV-Jc1, followed by the quantification of intracellular HCV RNA copy numbers at 24 h postinfection. siHNF4α efficiently decreased HNF4α protein expression (Fig. 2A, compare lanes 1 and 3) with no considerable effect on cell viability (data not shown). As a positive control, a CD81-specific siRNA (siCD81) reduced CD81 expression by ∼55% and ∼30% at the transcript (data not shown) and protein (Fig. 2B) levels, respectively. No significant decrease in HCV entry efficacy was observed upon HNF4α silencing, whereas siCD81 suppressed HCV entry efficacy by ∼70% (Fig. 2C). Evaluation of the effects of HNF4α overexpression on HCV entry revealed no significant changes compared with pcDNA-transfected cells (data not shown). CD81, claudin-1, SR-BI, occludin, LDLR, and NPC1L1 are cellular factors required for HCV entry. Transcripts of these factors were indistinguishable when HNF4α was knocked down or overexpressed (data not shown). Collectively, these results indicate that HNF4α is not required for HCV entry.
To determine whether HNF4α is required for HCV RNA replication, a time course analysis was performed on Huh7.5.1-sgJFH cells. After the cells were transfected with siHNF4α, HCV RNA copy numbers and NS3 protein expression were quantified at the time points posttransfection indicated on Fig. 2D to G. HCV- and PI4KIIIα-specific siRNAs were used as positive controls (27, 28). HNF4α expression was efficiently reduced (Fig. 2E). However, no significant decrease in HCV RNA copy numbers (Fig. 2D) or NS3 expression (Fig. 2E) was observed. In contrast, siPI4KIIIα and siHCV significantly reduced HCV RNA copy numbers and NS3 protein expression. Similar results were observed in Huh7-sgHCV1b cells (Fig. 2F and G). HNF4α overexpression in Huh7.5.1-sgJFH or Huh7-sgHCV1b cells did not affect RNA replication (data not shown). Collectively, these results indicate that HNF4α is not required for HCV RNA replication.
(iii) HNF4α is required for HCV assembly and secretion.
To investigate whether HNF4α is involved in the late stage of the HCV life cycle, Huh7.5.1 cells were transfected with siHNF4α (Fig. 3A and B show the effect of knockdown) and then infected with HCV-Jc1. ApoE reportedly affects the assembly and secretion of HCV (29). Thus, ApoE siRNA (siApoE) was used as a positive transfection control. The infected cells and culture supernatants were collected at 72 h postinfection. Since HNF4α was not required for HCV entry or RNA replication, the reduced intracellular HCV RNA copy numbers in the multicycle infection system (Fig. 3C) argued the effect of HNF4α on the late stage of the HCV life cycle.
FIG 3.

HNF4α is required for HCV assembly and secretion. Huh7.5.1 cells were transfected with siHNF4α and then inoculated with HCV-Jc1 at an MOI of 0.02 at 3 h posttransfection. The viral supernatants and cells were collected at 72 h postinfection. (A) Detection of HNF4α caused by siHNF4α transfection via Western blot analysis. siCtrl and ApoE siRNA (siApoE) were used as negative and positive controls, respectively. (B) Relative change in transcript of HNF4α and ApoE upon HNF4α or ApoE silencing in Huh7.5.1 cells at 72 h post-HCV infection. (C) Intracellular HCV RNA copy numbers were quantified after normalization by 18S RNA, induced by siHNF4α or siApoE. (D) Clarified viral supernatants were titrated by endpoint dilution on Huh7.5.1 cells, and the infectious HCV titers were calculated as the number of focus-forming units (FFU)/ml. (E) Detection of supernatant HCV RNA copies (calculated as genome equivalents [GE]/ml) via qRT-PCR. (F) Specific infectivity of HCV particles after each siRNA treatment was calculated using the extracellular HCV titer divided by the extracellular viral RNA copy number (FFU/GE). (G) Infectious particles in the cell lysates were released from cells by four cycles of freezing and thawing, and the supernatants were clarified for titration (calculated as FFU). (H) The ratios of the infectivity in the supernatants to that in the cell lysates upon siHNF4α or siApoE treatment were calculated. (I) Huh7.5.1 cells were cotransfected with siHNF4α and siRNA-resistant HNF4α DNA, and HCV-Jc1EGFP was inoculated at an MOI of 0.02 at 3 h posttransfection. The cell lysates were collected at 72 h postinfection to detect HNF4α expression. (J) Viral supernatants upon HNF4α reconstitution in HNF4α-silenced cells were collected to quantify HCV infectivity using flow cytometry. Triple independent experiments were performed, and the results are presented as means ± SEM (n = 3; *, P < 0.05;**, P < 0.01;***, P < 0.001). Intra, intracellular; extra, extracellular.
Intracellular and supernatant viruses were titrated to determine the effect of siHNF4α on the late stage of the HCV life cycle. The infectious titers in the viral supernatant from the HNF4α-silenced cells were reduced by approximately 70% (Fig. 3D). The HCV RNA copy numbers in the supernatant were slightly reduced because of HNF4α silencing (Fig. 3E). Thus, the specific infectivity (extracellular HCV titer divided by extracellular viral RNA copy number) was significantly reduced by siHNF4α (Fig. 3F). The infectious titers in the cell lysates were significantly reduced upon siHNF4α transfection (Fig. 3G). The reduced infectivity in the supernatants and cell lysates indicated that the assembly efficiency of HCV was impaired in the HNF4α-silenced cells. Moreover, the ratio of the titers in the supernatant to those in the cell lysate was significantly reduced upon siHNF4α transfection (Fig. 3H), suggesting that the secretion of virion particles was also inhibited upon HNF4α silencing. These results indicate that HNF4α affects both HCV assembly and secretion, similar to the effect of ApoE. Huh7.5.1 cells were cotransfected with siRNA-resistant HNF4α-encoding plasmids and siHNF4α to rule out any off-target effect. HCV-Jc1EGFP was infected at 3 h posttransfection. Viral supernatants at 72 h postinfection were harvested for FCM assay. HNF4α protein expression was rescued upon pHNF4α-resistant transfection (Fig. 3I, compare lanes 3 and 4). Predictably, the reduced HCV infectivity caused by HNF4α silencing was recovered upon siHNF4α-resistant plasmid transfection (Fig. 3J). Collectively, these results show that HNF4α is required for HCV assembly and secretion.
(iv) HNF4α antagonist bezafibrate suppresses HCV assembly and secretion.
Bezafibrate is an antagonist of HNF4α (30). Huh7.5.1 cells were infected with HCV-Jc1EGFP in the presence of bezafibrate to confirm the effect of HNF4α on the HCV life cycle. At 72 h postinfection, HNF4α expression was downregulated by bezafibrate in a dose-dependent manner (Fig. 4A, compare lane 1 with lanes 2 to 4) with no apparent cellular cytotoxicity (data not shown). MTP transcript was also downregulated accordingly (data not shown), indicating that the transcriptional activity of HNF4α was reduced upon bezafibrate treatment.
FIG 4.

HNF4α antagonist bezafibrate suppresses HCV assembly and secretion. Huh7.5.1 cells were infected with HCV-Jc1EGFP at an MOI of 0.02 in the presence of different concentrations of bezafibrate (0 μg/ml to 300 μg/ml). DMSO was used as a vehicle, and the final concentration in the medium was 2‰. (A) At 72 h postinfection, the cell lysates were collected to detect HNF4α, HCV NS3, and core proteins via Western blotting. (B) Intracellular HCV RNA copies were quantified after normalization by 18S RNA at 72 h postinfection upon different concentrations of bezafibrate treatment. (C) Viral supernatant infectivity (calculated as FFU/ml) upon bezafibrate treatment was determined on Huh7.5.1 cells by endpoint dilution. (D) Measurement of supernatant HCV RNA copies (calculated as GE/ml) upon bezafibrate treatment. (E) Calculation of specific infectivity (described as FFU/GE) upon bezafibrate treatment. (F) Quantification of HCV titers in the cell lysates (calculated as FFU) in Huh7.5.1 cells upon bezafibrate treatment. (G) The ratios of the supernatant infectivity to that in the cell lysates treated with different concentrations of bezafibrate were calculated. (H) Huh7.5.1 cells persistently infected with HCV-Jc1EGFP were inoculated with DMSO or 300 μg/ml bezafibrate. The relative HCV infectivity (percentage of the value of the DMSO-treated condition) in the supernatants and the cell lysates in persistently infected Huh7.5.1 cells at 72 h postinoculation was determined using flow cytometry. The results are presented as means ± SEM (n = 3; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
To measure the efficiency of HCV RNA replication in bezafibrate-treated cells, intracellular HCV RNA copies were quantified. A dose-dependent decrease was observed (Fig. 4B). Bezafibrate did not affect HCV entry or RNA replication (data not shown). Thus, the reduction in intracellular HCV RNA copy numbers indicated that HCV assembly and secretion were inhibited.
To evaluate the effect of bezafibrate on HCV assembly and secretion, infectious titers in the supernatant and cell lysate were quantified. As shown in Fig. 4C, bezafibrate suppressed the infectivity of the secreted HCV, with a maximal inhibition level of approximately 90%. HCV RNA copies in the supernatant (Fig. 4D) and specific infectivity (Fig. 4E) were both reduced upon bezafibrate treatment. Intracellular virus titers were quantified. Bezafibrate at a concentration of 300 μg/ml showed a maximal inhibition level of approximately 75% (Fig. 4F). These results indicated that the assembly efficiency of HCV was impaired upon bezafibrate treatment. The ratio of the supernatant infectivity to that in the cell lysate significantly decreased with increasing bezafibrate concentration (Fig. 4G). High concentrations of bezafibrate inhibited HCV assembly and secretion, whereas low concentrations (e.g., 100 μg/ml) of bezafibrate mainly impaired HCV assembly. Cells persistently infected with HCV were alternatively used for HCV assembly and secretion experiments. Huh7.5.1 cells persistently infected with HCV-Jc1EGFP were treated with 300 μg/ml bezafibrate. Viral supernatants and cell lysates were collected at 72 h posttreatment. As shown in Fig. 4H, intracellular and supernatant infectious titers were reduced upon bezafibrate treatment by approximately 30% and 50%, respectively. Collectively, these results reveal that bezafibrate impairs HCV assembly and secretion.
PLA2GXIIB, a downstream factor regulated by HNF4α, is required for HCV secretion. (i) HNF4α regulates PLA2GXIIB expression.
Given that HCV is assembled into and released together with VLDLs, we hypothesize that certain HNF4α-regulated VLDL biogenesis-related factors are responsible for the HNF4α-induced regulation of HCV production. We previously reported that PLA2GXIIB functions in VLDL biogenesis/secretion and that HNF4α regulates PLA2GXIIB expression in HepG2 and HeLa cells (23). Thus, PLA2GXIIB might function in the HCV life cycle. To test this possibility, we first evaluated whether HNF4α regulates PLA2GXIIB expression in Huh7.5.1 cells. A PLA2GXIIB promoter reporter plasmid (pGL3-PLA2GXIIBprom) was cotransfected into Huh7.5.1 cells with an HNF4α-encoding plasmid. HNF4α induced the PLA2GXIIB promoter activities by more than 3-fold compared with the pGL3-basic reporter (Fig. 5A). A mutant PLA2GXIIB promoter reporter plasmid (pGL3-PLA2GXIIBprom mutB) with a mutated HNF4α-responsive binding site was used as a negative control, which was not activated by HNF4α (Fig. 5A).
FIG 5.

HNF4α regulates PLA2GXIIB expression in Huh7.5.1 cells. (A) Huh7.5.1 cells were transfected with three plasmids including the wild-type or mutant PLA2GXIIB promoter reporter plasmid, an HNF4α-encoding plasmid, and a plasmid encoding Renilla luciferase for transfection efficiency measurement. At 48 h posttransfection, reporter activities were analyzed by a Dual-Glo luciferase kit. The wild-type and the mutant construct of the PLA2GXIIB promoter reporter lacking the HNF4α-responsive binding site are described in the schematic. (B) The relative change in PLA2GXIIB transcript in Huh7.5.1 cells transfected with an HNF4α-encoding plasmid at 72 h posttransfection was determined compared with the value in mock-transfected cells. The results are presented as means ± SEM (n = 3; *, P < 0.05). (C) The relative change in HNF4α and PLA2GXIIB transcripts in HNF4α-silenced Huh7.5.1 cells at 72 h posttransfection was quantified. The results are presented as means ± SEM (n = 3; *, P < 0.05). (D) Huh7.5.1 cells were cotransfected with siHNF4α and siRNA-resistant HNF4α-encoding plasmids. The cell lysates at 72 h posttransfection were collected to detect HNF4α and PLA2GXIIB protein expression via Western blot analysis.
Huh7.5.1 cells were transfected with the HNF4α-encoding plasmid to directly evaluate the regulatory effect of HNF4α on PLA2GXIIB. The PLA2GXIIB transcript was quantified at 48 h posttransfection. Ectopic HNF4α expression significantly increased PLA2GXIIB transcript (Fig. 5B). PLA2GXIIB protein abundance was also increased (Fig. 5D, compare lanes 1 and 2). The effect of downregulated HNF4α expression on PLA2GXIIB expression in Huh7.5.1 cells was also evaluated. HNF4α silencing downregulated PLA2GXIIB transcript (Fig. 5C) and protein expression (Fig. 5D, compare lanes 1 and 3). The downregulated PLA2GXIIB expression was recovered when HNF4α expression was rescued upon siHNF4α-resistant plasmid transfection (Fig. 5D, compare lanes 3 and 4). These results indicate that PLA2GXIIB expression is regulated by HNF4α in Huh7.5.1 cells.
(ii) PLA2GXIIB expression is upregulated by HCV infection.
A previous study reported that PLA2GXIIB is upregulated in HCV-related liver tumor tissues (24). Here, three experiments were performed to determine whether or not PLA2GXIIB expression is induced by HCV infection. First, PLA2GXIIB transcript was increased in Huh7.5.1-sgJFH and Huh7-sgHCV1b cells compared with those in Huh7.5.1 and Huh7 cells, respectively (Fig. 6A and B). PLA2GXIIB protein expression was also upregulated in Huh7.5.1-sgJFH and Huh7-sgHCV1b cells (Fig. 6C, compare lanes 1 and 2 and lanes 3 and 4). Huh7-sgHCV1b cells were transfected with siPI4KIIIα or siHCV to confirm the regulatory effect of HCV RNA replication on PLA2GXIIB expression. siHNF4α was used as a positive control. At 72 h posttransfection, PLA2GXIIB expression was reduced upon siPI4KIIIα or siHCV silencing compared with the level with a scrambled control siRNA (siCtrl), concomitant with decreased NS3 protein expression (Fig. 6D). This result suggests that PLA2GXIIB is specifically upregulated in replicon cells. Second, Huh7.5.1 cells were infected with HCV-Jc1EGFP at an MOI of 0.02. PLA2GXIIB expression was quantified in a time course analysis. PLA2GXIIB protein expression compared with levels in mock-infected cells was increased concomitant with HCV RNA replication (Fig. 6E). Third, the PLA2GXIIB promoter was activated upon HCV infection (Fig. 6F). These results demonstrate that PLA2GXIIB expression is upregulated by HCV infection.
(iii) HNF4α is required for HCV-induced upregulation of PLA2GXIIB.
To investigate whether HNF4α is required for HCV-induced upregulation of PLA2GXIIB, Huh7.5.1 cells were transfected with the wild-type or mutant construct of a PLA2GXIIB promoter reporter and then infected with HCV-Jc1EGFP. As shown in Fig. 6G, HCV infection effectively activated the wild-type PLA2GXIIB promoter reporter but failed to activate the mutant reporter. This result confirmed the importance of HNF4α in the activation of the PLA2GXIIB promoter. Cotransfection of siHNF4α with the PLA2GXIIB promoter reporter reduced PLA2GXIIB promoter activity by 2.6-fold (Fig. 6H). A comparable reduction was also observed in the cells without HCV infection (Fig. 6H). Collectively, these results indicate that HNF4α is required for HCV-induced upregulation of PLA2GXIIB expression.
(iv) PLA2GXIIB is required for HCV secretion.
We investigated the susceptibility of HCV entry or HCV RNA replication to PLA2GXIIB silencing to determine whether or not PLA2GXIIB is required in the HCV life cycle. No inhibition of HCV entry or RNA replication was observed upon PLA2GXIIB knockdown (data not shown). Thus, we speculated that PLA2GXIIB possibly functions in the late stage of the HCV life cycle. Huh7.5.1 cells were transfected with siPLA2GXIIB and then infected with HCV-Jc1EGFP to investigate the effect of PLA2GXIIB on HCV assembly and secretion. siPLA2GXIIB efficiently reduced PLA2GXIIB expression (Fig. 7A) with no measurable effect on cell viability (data not shown). Infectious HCV titers in the supernatant were reduced by approximately 50% upon siPLA2GXIIB transfection (Fig. 7B), whereas the released HCV RNA copy numbers remained unchanged (Fig. 7C). Thus, the specific infectivity (Fig. 7D) was reduced upon PLA2GXIIB silencing. When the HCV titers in the cell lysates were quantified, a 2-fold accumulation was observed (Fig. 7E). HCV RNA replication remained unchanged, regardless of PLA2GXIIB downregulation (Fig. 7F). These results indicated that PLA2GXIIB interfered with HCV secretion but not assembly. A rescue experiment was performed to confirm this conclusion. Huh7.5.1 cells were cotransfected with siPLA2GXIIB and siRNA-resistant PLA2GXIIB-encoding plasmids (Fig. 7G shows the knockdown and rescue effects). HCV-Jc1EGFP was then inoculated at 3 h posttransfection. The viral supernatant infectivity was determined by FCM. The reduced HCV infectivity by siPLA2GXIIB was completely recovered by the reconstitution of PLA2GXIIB expression (Fig. 7H), underscoring the effect of PLA2GXIIB on infectious HCV production. Collectively, these findings show that PLA2GXIIB is required for HCV secretion.
FIG 7.

PLA2GXIIB is required for HCV secretion. (A) Huh7.5.1 cells were transfected with siPLA2GXIIB and then infected with HCV-Jc1EGFP at an MOI of 0.02 at 3 h posttransfection. The cell lysates were collected to detect PLA2GXIIB and HCV NS3 proteins using Western blot analysis at 72 h post-HCV infection. (B) Infectious HCV titers in the supernatants (FFU/ml) induced by siPLA2GXIIB were quantified compared with values of the siCtrl-treated condition at 72 h postinfection. (C) Quantification of HCV RNA copies (GE/ml) in the supernatants induced by siPLA2GXIIB at 72 h postinfection. (D) Specific infectivity (FFU/GE) of the virus in the supernatants upon siPLA2GXIIB treatment was calculated at 72 h postinfection. (E) The intracellular HCV titers induced by siPLA2GXIIB compared with those induced by siCtrl were quantified at 72 h postinfection. (F) Relative quantification of the change in intracellular HCV RNA copy numbers in siPLA2GXIIB-transfected Huh7.5.1 cells. (G) Huh7.5.1 cells were transfected with siPLA2GXIIB and siRNA-resistant PLA2GXIIB-encoding plasmids and then inoculated with HCV-Jc1EGFP at 3 h posttransfection. PLA2GXIIB protein abundance in PLA2GXIIB-silenced Huh7.5.1 cells upon rescue was detected at 72 h postinfection. (H) The relative infectious HCV titers in the supernatants were quantified by flow cytometry in PLA2GXIIB-rescued Huh7.5.1 cells compared with the PLA2GXIIB-silenced condition. Experiments were performed at least three times, and the results are shown as means ± SEM (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
PLA2GXIIB and MTP reconstitutions recover the reduced effect of HNF4α silencing on HCV infectivity.
To address which factors downstream of HNF4α are attributed to the HNF4α-induced regulation of HCV infectivity, the effects of PLA2GXIIB overexpression were first investigated. Huh7.5.1 cells were transfected with plasmids encoding PLA2GXIIB and then were infected with HCV-Jc1 at an MOI of 0.01 at 24 h posttransfection. As shown in Fig. 8A, PLA2GXIIB protein overexpression was readily detected at all time points postinfection. The infectious titers in the supernatants increased by more than 2-fold compared with the mock-transfected condition at 72 h postinfection (Fig. 8B), coinciding with the result shown in Fig. 7H. However, MTP (Fig. 8C) or ApoE (Fig. 8E) overexpression did not increase HCV infectivity in the supernatants compared with the mock-transfected condition (Fig. 8D and F). These results show that MTP and ApoE are sufficient in Huh7.5.1 cells for HCV production and that ectopic PLA2GXIIB expression enhances HCV infectivity, consistent with the results under the condition of HNF4α overexpression (Fig. 3J).
FIG 8.

PLA2GXIIB and MTP reconstitutions recover the reduced HCV infectivity by siHNF4α. Huh7.5.1 cells were transfected with different doses of plasmids encoding PLA2GXIIB, MTP, or ApoE and then inoculated with HCV-Jc1 (MOI of 0.01) at 24 h posttransfection. Cells and supernatants were collected at the indicated time points postinfection. (A) Overexpressed PLA2GXIIB and HCV core proteins were detected with antibodies against PLA2GXIIB and core protein at the indicated time points postinfection. (B) HCV infectivity in the supernatants upon PLA2GXIIB overexpression were titrated and calculated as FFU/ml. (C) Exogenous MTP-Flag fusion proteins were detected at the indicated time points post-HCV infection with flow cytometry by using intracellular staining with an anti-Flag antibody. (D) The supernatant infectious titers were quantified under the condition of MTP overexpression compared with mock-transfected cells. (E) HCV core and HA-tagged ApoE proteins were detected via Western blot analysis with specific antibodies against core protein and HA tag, respectively. (F) Infectious HCV titers in the supernatants under the condition of ApoE overexpression were quantified. (G) Plasmids encoding PLA2GXIIB, MTP, and ApoE alone or in combination were cotransfected with siHNF4α into Huh7.5.1 cells and then inoculated with HCV-Jc1EGFP at an MOI of 0.02 at 3 h posttransfection. The cell lysates were collected to quantify NS5A-GFP frequency (infection efficiency) at 72 h postinfection. (H) The supernatants were collected and inoculated on Huh7.5.1 cells to quantify HCV titers. The relative HCV infectivity in the supernatants after normalization to the siCtrl-transfected condition was calculated as a percentage. The data are presented as means ± SEM (n = 3; *, P < 0.05). “ph” indicates a plasmid that encodes a human protein.
Reconstitution experiments were performed in HNF4α-silenced Huh7.5.1 cells to identify the factors responsible for the HNF4α-regulated HCV infectivity. Huh7.5.1 cells were cotransfected with siHNF4α and plasmids encoding PLA2GXIIB, MTP, or ApoE alone or in combination. HCV-Jc1EGFP was inoculated at 3 h posttransfection at an MOI of 0.02. The infection efficiency was determined by monitoring NS5A-GFP expression (Fig. 8G) in transfected cells at 72 h postinfection. The supernatant infectivity at 72 h postinfection was quantified by FCM assay. In HNF4α-silenced Huh7.5.1 cells, HCV infectivity in the supernatants was reduced by ∼50% compared with the level in mock-silenced cells (Fig. 8H). PLA2GXIIB, MTP, or ApoE overexpression alone did not recover the reduced HCV infectivity upon siHNF4α transfection (Fig. 8H). The impaired HCV infectivity was completely rescued when PLA2GXIIB was reconstituted in combination with MTP (Fig. 8H). Reconstituted PLA2GXIIB and ApoE expression, as well as reconstituted MTP and ApoE, partially but not significantly recovered the reduced HCV infectivity by siHNF4α transfection (Fig. 8H). The reconstituted combination of PLA2GXIIB, MTP, and ApoE did not completely rescue the impaired HCV infectivity (Fig. 8H), indicating the presence of a complicated interaction among these factors. Identical results were also obtained on persistently infected Huh7.5.1 cells (data not shown). These results indicate that PLA2GXIIB and MTP are sufficient to rescue the reduced HCV infectivity in HNF4α-deficient cells.
HNF4α downregulation induces rearrangement of cytosolic LDs.
ApoE is the most abundant apolipoprotein on HCV particles; it is crucial for HCV assembly and infectivity (29, 31). To determine whether HNF4α regulates HCV assembly and secretion in an ApoE-dependent manner, Huh7.5.1 cells were transfected with siHNF4α or treated with bezafibrate and then infected with HCV-mutJFH1 infection. ApoE abundance and HCV titers in the viral supernatants were quantified at 72 h postinfection. Consistent with earlier results, HNF4α downregulation remarkably suppressed the infectious HCV titers in the supernatants (Fig. 9A). However, siHNF4α or bezafibrate treatment did not remarkably decrease ApoE secretion (normalized to albumin) (Fig. 9B and C). The abundance of ApoE in the cell lysates upon bezafibrate treatment was unchanged compared with DMSO treatment (data not shown). These results indicate that HNF4α affects HCV assembly and secretion in an ApoE-independent manner.
FIG 9.

HNF4α downregulation induces rearrangement of cytosolic LDs. (A) Huh7.5.1 cells were transfected with specific siRNAs and then infected with HCV-mutJFH1 at 3 h posttransfection in the absence or presence of 300 μg/ml bezafibrate. The viral supernatant infectivity was quantified at 72 h postinfection. The data are shown as means ± SEM (n = 3; **, P < 0.01). (B) The viral supernatants at 72 h post-HCV infection were subjected to low-speed centrifugation; 25 μl of clarified supernatants was resolved by 10% SDS-PAGE, and the levels of secreted ApoE and albumin were quantified through Western blotting. (C) The ratios for the relative band intensity of ApoE after normalization by albumin were calculated (n = 3). (D) Huh7.5.1 cells were plated on chambered coverglass and then infected the following day with HCV-mutJFH1 (MOI of 2) in the presence of bezafibrate (300 μg/ml). Cells were fixed at 72 h postinfection to stain lipid droplets (red) with Nile Red (0.1 μg/ml) and the nucleus (blue) with 4′,6′-diamidino-2-phenylindole (1 μg/ml). A change in LD number (E) and LD size (F) per cell in bezafibrate or DMSO-treated HCV-mutJFH1-infected Huh7.5.1 cells was quantified. Quantification was performed in approximately 20 individual cell images from bezafibrate- or DMSO-treated conditions using ImageJ software. (G) Huh7.5.1 cells were transfected with siHNF4α or siPLA2GXIIB, infected with HCV-mutJFH1 (MOI of 2), and then cultured for an additional 72 h. Cells were fixed and stained for lipid droplets. (H) Huh7.5.1 cells at 72 h post-HCV infection in the presence of bezafibrate (300 μg/ml) were costained with lipid droplets (red) with Nile Red and HCV core protein (blue) with specific antibody. Colocalization of LDs and HCV core protein in HCV-infected Huh7.5.1 cells upon bezafibrate treatment was detected with confocal analysis. Scale bar, 10 μm.
Cytosolic LDs are also crucial for HCV assembly and infectious virus production. The rearrangement of LDs reportedly impairs HCV infectivity (32–34). Huh7.5.1 cells were infected with HCV-mutJFH1 in the presence of bezafibrate and then observed under a confocal microscope at 72 h postinfection to investigate whether or not HNF4α regulates the rearrangement of LDs. Bezafibrate decreased the number but increased the size of LDs in the HCV-infected cells (Fig. 9D to F). However, the total fluorescence intensity of LDs did not significantly increase (data not shown). Similar to bezafibrate, siHNF4α transfection yielded large aggregates of LDs and distorted cytosolic distributions in the HCV-infected Huh7.5.1 cells (Fig. 9G). This result coincides with the observations in specific HNF4α-null Drosophila or mice (17, 35). We then analyzed the location of the HCV core protein on LDs upon bezafibrate treatment. As shown in Fig. 9H, bezafibrate reduced core protein expression and perturbed the typical perinuclear distribution pattern observed in the HCV-infected cells treated with DMSO. However, the HCV core protein remained localized to the periphery of LDs. Identical results were observed in HNF4α silencing (data not shown). This result suggests that HNF4α downregulation affects core protein abundance and cytosolic distribution but not the location of the core protein on LDs. Therefore, the rearrangement of LDs can be attributed to the effect of HNF4α on infectious HCV production.
DISCUSSION
Biophysical and ultrastructural studies indicate that the incorporation of HCV particles with VLDLs determines HCV infectivity, aids in HCV entry and release from cells, and helps the virus escape from host immune surveillance. HCV hijacks the VLDL secretory pathway for its own assembly and secretion (12, 15). However, the molecular mechanism remains elusive. In this study, the importance of HNF4α, a central nuclear receptor controlling cellular lipid transport, in HCV assembly and secretion was illustrated. We showed that HCV replication induced HNF4α-mediated upregulation of genes involved in the VLDL pathway. We also demonstrated that HNF4α downregulation impaired HCV assembly and secretion, whereas HNF4α overexpression promoted infectious HCV production.
As a key transcription factor for VLDL-associated genes, HNF4α affected HCV assembly and secretion by regulating downstream genes, including MTP and PLA2GXIIB. In normal Huh7.5.1 cells, the overexpression of PLA2GXIIB and not MTP increased HCV infectivity. In HNF4α-deficient cells, ectopic PLA2GXIIB or MTP alone did not rescue the reduced HCV infectivity. However, the reduced HCV infectivity was completely recovered by reconstituted PLA2GXIIB in combination with MTP. The synergistic effect can be well understood because these two factors function in different stages of the HCV life cycle. Previous studies showed that inhibition of MTP activity reduces HCV assembly (12). The current study showed that PLA2GXIIB knockdown reduced HCV infectivity in the supernatant and accumulated intracellular HCV infectivity. These results suggest that PLA2GXIIB is required for HCV secretion. Indeed, VLDL secretion in PLA2GXIIB-null mice is also reduced (23). The unchanged expression levels of MTP and ApoB suggest that VLDL assembly cannot be distinguished in PLA2GXIIB-null mice. These results emphasize further the tight interaction between VLDL and HCV assembly and secretion. Although ApoE transcript was upregulated by ectopic HNF4α expression, the abundance of ApoE in the cell lysates and supernatants was not affected by HNF4α downregulation. In normal Huh7.5.1 cells, ApoE silencing reduced both HCV assembly and secretion, supporting previous reports (29). Meanwhile, ectopic ApoE expression did not increase HCV infectivity in the supernatants. This result suggests that ApoE expression is sufficient for HCV life cycle in normal cells. ApoE overexpression alone or in combination with PLA2GXIIB and MTP did not rescue the reduced HCV infectivity in HNF4α-deficient cells. These results suggest that ApoE may not be a critical downstream factor affecting the regulatory activity of HNF4α on HCV infectivity. ApoE possibly functioned differently from HNF4α.
Therefore, we propose a model to illustrate the interaction between HNF4α and the HCV life cycle (Fig. 10). HCV infection upregulates HNF4α expression, which in turn transactivates PLA2GXIIB and MTP. MTP is required in HCV assembly, whereas PLA2GXIIB facilitates the secretion of HCV lipoviroparticles. Reduced HNF4α expression downregulates MTP and PLA2GXIIB, accompanied with cytosolic LD rearrangement, which in turn impairs HCV assembly and secretion. This model emphasizes the importance of the VLDL secretory pathway for HCV assembly and secretion and suggests a possible mechanism by which HCV cooperates with HNF4α to be assembled into and released together with VLDL particles.
FIG 10.

Proposed model of HNF4α regulation in the HCV life cycle. HCV infection upregulates HNF4α expression, which in turn transactivates numerous genes (PLA2GXIIB and MTP) involved in VLDL biogenesis. MTP is required in HCV assembly, whereas PLA2GXIIB facilitates secretion of HCV lipoviroparticles. Reduced HNF4α expression downregulates MTP and PLA2GXIIB expression levels, concomitant with impaired HCV assembly and secretion. luLD, luminal lipid droplets.
Lipoprotein components, such as ApoE, triglyceride, and cholesterol, associate with HCV particles and therefore determine HCV infectivity (36–38). Among these components, ApoE is reportedly trans-activated by HNF4α and is an abundant component of HCV particles. However, several lines of evidence suggest that ApoE is not sufficient to endow HCV particles with infectivity. Shimizu et al. reported that the HCV RNA associated with ApoE can be detected in eluted fractions of gel filtration chromatography irrespective of infectivity (39). In addition, ApoE can be detected preferentially in fractions with higher density but not in fractions containing the majority of the total HCV infectivity (40). A recent study has revealed that the abundance of the incorporated ApoE was indistinguishable in HCV particles with different sizes although more than 40% of the total infectivity is contained in a narrow fraction with a density of ∼1.13 g/ml (41). The levels of secreted triglyceride and cholesterol are reduced in HNF4αLiv KO mice (17). However, whether these two lipids of lipoproteins are responsible for the HNF4α-induced regulation of HCV specific infectivity must be explored.
Cytosolic LDs are dynamic organelles for cellular lipid storage and transport. In the present study, HNF4α or PLA2GXIIB downregulation induced abnormal aggregation of large LDs and reduced the number of cytosolic LDs. Such LD rearrangement was accompanied by the reduced expression and distorted cytosolic distribution of HCV proteins, similar to that of nordihydroguaiaretic acid and cyclophilin inhibitors (33, 34). These results echo the notion that cytosolic LDs are important organelles for HCV assembly and secretion (11). As another line of supporting evidence, the HNF4α-specific knockout mice accumulated aggregated cytosolic LDs in the hepatocytes (17).
The development of direct-acting antiviral agents that target HCV viral proteins has updated the optimal treatment regimens of chronic HCV infection. However, a high risk of drug resistance has become a major concern. A chemical agent targeting a cellular factor possibly has the potential to overcome the resistance as well as provide broad genotype coverage. In this study, we evaluated the effect of HNF4α antagonist on HCV treatment. Bezafibrate, an antagonist of HNF4α, is widely used for treating dyslipidemia. Bezafibrate is also an agonist of peroxisome proliferator-activated receptor α (PPARα). However, we found that parallel with HNF4α downregulation, bezafibrate efficiently inhibited HCV assembly and secretion. Such activity was not parallel with the increased PPARα activity (data not shown). These observations demonstrate that the anti-HCV activity of bezafibrate is due to the inhibited HNF4α expression and not the increased PPARα activity. These findings provide a mechanism by which bezafibrate enhances a sustained virological response in patients with single or combination treatment with alpha interferon plus ribavirin (42, 43). In addition, other clinically approved hypolipidemia drugs, such as ezetimibe and lovastatin, also display excellent antiviral effects against HCV (6, 44). Thus, whether or not hypolipidemia drugs are viable options for HCV treatment warrants further investigation.
PLA2GXIIB is a secreted phospholipase A2 lacking enzymatic activity, and a high level of PLA2GXIIB expression can be found in the liver (45). Supporting previous clinical reports, the current study indicated that HCV infection or viral RNA replication can induce PLA2GXIIB expression. Whether or not PLA2GXIIB can serve as a novel serum biomarker of HCV infection deserves further study.
In the present study, PLA2GXIIB was transcriptionally upregulated by HCV in an HNF4α-dependent manner. Although the underlying molecular mechanism is not clear, HCV possibly promotes HNF4α activity in an indirect manner. Oxidative stress induced by HCV replication is reportedly responsible for the increased HNF4α activity in Huh.8 cells (22). In addition, HCV infection enhances the activities of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) (46) and CREB-binding protein (47), which work as coactivators of HNF4α to promote HNF4α activity.
The results of this study provided new insights into the mechanisms underlying the pathogenesis of HCV infection. Cellular gluconeogenesis genes regulated by HNF4α, including phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, are upregulated in HCV replicating cells (48). This upregulation contributes to HCV-related insulin resistance and type 2 diabetes mellitus. In addition, HNF4α is associated with oxidative stress and liver inflammation, two elements closely related to chronic hepatitis (22, 49). Numerous novel and direct targets regulated by HNF4α have been recently identified through protein-binding microarrays (50). Given the importance of HNF4α in the immune response, apoptosis, cell cycle control, and carcinogenesis, further mechanistic studies should focus on the function of HNF4α in HCV infection and subsequent metabolic disorders and hepatocellular carcinoma.
ACKNOWLEDGMENTS
We thank all members in the laboratory of viral immunology for helpful suggestions, Li Zhaotao, Pang Hongwen, and Zheng Yi for technical assistance, and Xing Yifei for proofreading the manuscript.
We declare that we have no competing interests.
This work was supported by the National Basic Research Program (973) (grant numbers 2009CB522300 and 2010CB530100).
Footnotes
Published ahead of print 30 October 2013
REFERENCES
- 1.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463. 10.1038/nrmicro1645 [DOI] [PubMed] [Google Scholar]
- 2.Feld JJ, Hoofnagle JH. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972. 10.1038/nature04082 [DOI] [PubMed] [Google Scholar]
- 3.Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444. 10.1002/hep.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 80:2418–2428. 10.1128/JVI.80.5.2418-2428.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ploss A, Evans MJ. 2012. Hepatitis C virus host cell entry. Curr. Opin. Virol. 2:14–19. 10.1016/j.coviro.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1–like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285. 10.1038/nm.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F. 2011. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 54:48–55. 10.1016/j.jhep.2010.06.024 [DOI] [PubMed] [Google Scholar]
- 8.Wong-Staal F, Syder AJ, McKelvy JF. 2010. Targeting HCV entry for development of therapeutics. Viruses 2:1718–1733. 10.3390/v2081718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. 2006. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 3:87–98. 10.1016/j.cmet.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 10.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. 10.1126/science.1113329 [DOI] [PubMed] [Google Scholar]
- 11.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097. 10.1038/ncb1631 [DOI] [PubMed] [Google Scholar]
- 12.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129. 10.1128/JVI.02053-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki T. 2012. Morphogenesis of infectious hepatitis C virus particles. Front. Microbiol. 3:38. 10.3389/fmicb.2012.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye J. 2012. Hepatitis C virus: a new class of virus associated with particles derived from very low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 32:1099–1103. 10.1161/ATVBAHA.111.241448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853. 10.1073/pnas.0700760104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793. 10.1128/JVI.01091-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. 2001. Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 21:1393–1403. 10.1128/MCB.21.4.1393-1403.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwayanagi Y, Takada T, Suzuki H. 2008. HNF4α is a crucial modulator of the cholesterol-dependent regulation of NPC1L1. Pharm. Res. 25:1134–1141. 10.1007/s11095-007-9496-9 [DOI] [PubMed] [Google Scholar]
- 19.Li ZY, Xi Y, Zhu WN, Zeng C, Zhang ZQ, Guo ZC, Hao DL, Liu G, Feng L, Chen HZ, Chen F, Lv X, Liu DP, Liang CC. 2011. Positive regulation of hepatic miR-122 expression by HNF4α. J. Hepatol. 55:602–611. 10.1016/j.jhep.2010.12.023 [DOI] [PubMed] [Google Scholar]
- 20.Sheena V, Hertz R, Nousbeck J, Berman I, Magenheim J, Bar-Tana J. 2005. Transcriptional regulation of human microsomal triglyceride transfer protein by hepatocyte nuclear factor-4α. J. Lipid Res. 46:328–341. 10.1194/jlr.M400371-JLR200 [DOI] [PubMed] [Google Scholar]
- 21.Dang Q, Walker D, Taylor S, Allan C, Chin P, Fan J, Taylor J. 1995. Structure of the hepatic control region of the human apolipoprotein E/C-I gene locus. J. Biol. Chem. 270:22577–22585. 10.1074/jbc.270.38.22577 [DOI] [PubMed] [Google Scholar]
- 22.Qadri I, Iwahashi M, Kullak-Ublick GA, Simon FR. 2006. Hepatocyte nuclear factor (HNF) 1 and HNF4 mediate hepatic multidrug resistance protein 2 up-regulation during hepatitis C virus gene expression. Mol. Pharmacol. 70:627–636. 10.1124/mol.106.023499 [DOI] [PubMed] [Google Scholar]
- 23.Guan M, Qu L, Tan W, Chen L, Wong C-W. 2011. Hepatocyte nuclear factor-4 alpha regulates liver triglyceride metabolism in part through secreted phospholipase A2 GXIIB. Hepatology 53:458–466. 10.1002/hep.24066 [DOI] [PubMed] [Google Scholar]
- 24.Smith MW, Yue ZN, Geiss GK, Sadovnikova NY, Carter VS, Boix L, Lazaro CA, Rosenberg GB, Bumgarner RE, Fausto N, Bruix J, Katze MG. 2003. Identification of novel tumor markers in hepatitis C virus-associated hepatocellular carcinoma. Cancer Res. 63:859–864. [PubMed] [Google Scholar]
- 25.Misawa K, Horiba T, Arimura N, Hirano Y, Inoue J, Emoto N, Shimano H, Shimizu M, Sato R. 2003. Sterol regulatory element-binding protein-2 interacts with hepatocyte nuclear factor-4 to enhance sterol isomerase gene expression in hepatocytes. J. Biol. Chem. 278:36176–36182. 10.1074/jbc.M302387200 [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Randall G, Higginbottom A, Monk P, Rice CM, McKeating JA. 2004. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 78:1448–1455. 10.1128/JVI.78.3.1448-1455.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu S, Pei R, Guo M, Han Q, Lai J, Wang Y, Wu C, Zhou Y, Lu M, Chen X. 2012. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J. Virol. 86:13025–13037. 10.1128/JVI.01785-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:7577–7582. 10.1073/pnas.0902693106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53. 10.1002/hep.23278 [DOI] [PubMed] [Google Scholar]
- 30.Hertz R, Bishara-Shieban J, Bar-Tana J. 1995. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J. Biol. Chem. 270:13470–13475 [DOI] [PubMed] [Google Scholar]
- 31.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 84:12048–12057. 10.1128/JVI.01063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olmstead AD, Knecht W, Lazarov I, Dixit SB, Jean F. 2012. Human subtilase SKI-1/S1P is a master regulator of the HCV Lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 8:e1002468. 10.1371/journal.ppat.1002468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Syed GH, Siddiqui A. 2011. Effects of hypolipidemic agent nordihydroguaiaretic acid on lipid droplets and hepatitis C virus. Hepatology 54:1936–1946. 10.1002/hep.24619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson LJ, Lin K, Compton T, Wiedmann B. 2011. Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 8:329. 10.1186/1743-422X-8-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palanker L, Tennessen JM, Lam G, Thummel CS. 2009. Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 9:228–239. 10.1016/j.cmet.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MM, Miyamura T, Wakita T, Suzuki T. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 82:5715–5724. 10.1128/JVI.02530-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu Y, Hishiki T, Sugiyama K, Ogawa K, Funami K, Kato A, Ohsaki Y, Fujimoto T, Takaku H, Shimotohno K. 2010. Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology 407:152–159. 10.1016/j.virol.2010.08.011 [DOI] [PubMed] [Google Scholar]
- 38.Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. 2010. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 84:12048–12057. 10.1128/JVI.01063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu Y, Hishiki T, Ujino S, Sugiyama K, Funami K, Shimotohno K. 2011. Lipoprotein component associated with hepatitis C virus is essential for virus infectivity. Curr. Opin. Virol. 1:19–26. 10.1016/j.coviro.2011.05.017 [DOI] [PubMed] [Google Scholar]
- 40.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286:3018–3032. 10.1074/jbc.M110.175018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. U. S. A. 110:9505–9510. 10.1073/pnas.1307527110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujita N, Kaito M, Kai M, Sugimoto R, Tanaka H, Horiike S, Konishi M, Iwasa M, Watanabe S, Adachi Y. 2006. Effects of bezafibrate in patients with chronic hepatitis C virus infection: combination with interferon and ribavirin. J. Viral Hepat. 13:441–448. 10.1111/j.1365-2893.2005.00718.x [DOI] [PubMed] [Google Scholar]
- 43.Fujita N, Kaito M, Tanaka H, Horiike S, Adachi Y. 2004. Reduction of serum HCV RNA titer by bezafibrate therapy in patients with chronic hepatitis C. Am. J. Gastroenterol. 99:2280. 10.1111/j.1572-0241.2004.40695_3.x [DOI] [PubMed] [Google Scholar]
- 44.Ye J, Wang C, Sumpter R, Jr, Brown MS, Goldstein JL, Gale M., Jr 2003. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. U. S. A. 100:15865–15870. 10.1073/pnas.2237238100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouault M, Bollinger JG, Lazdunski M, Gelb MH, Lambeau G. 2003. Novel mammalian group XII secreted phospholipase A2 lacking enzymatic activity. Biochemistry 42:11494–11503. 10.1021/bi0349930 [DOI] [PubMed] [Google Scholar]
- 46.Shlomai A, Rechtman MM, Burdelova EO, Zilberberg A, Hoffman S, Solar I, Fishman S, Halpern Z, Sklan EH. 2012. The metabolic regulator PGC-1α links hepatitis C virus infection to hepatic insulin resistance. J. Hepatol. 57:867–873. 10.1016/j.jhep.2012.06.021 [DOI] [PubMed] [Google Scholar]
- 47.Gomez-Gonzalo M, Benedicto I, Carretero M, Lara-Pezzi E, Maldonado-Rodriguez A, Moreno-Otero R, Lai MM, Lopez-Cabrera M. 2004. Hepatitis C virus core protein regulates p300/CBP co-activation function. Possible role in the regulation of NF-AT1 transcriptional activity. Virology 328:120–130. 10.1016/j.virol.2004.06.044 [DOI] [PubMed] [Google Scholar]
- 48.Deng L, Shoji I, Ogawa W, Kaneda S, Soga T, Jiang DP, Ide YH, Hotta H. 2011. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 85:8556–8568. 10.1128/JVI.00146-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikolaidou-Neokosmidou V, Zannis VI, Kardassis D. 2006. Inhibition of hepatocyte nuclear factor 4 transcriptional activity by the nuclear factor κB pathway. Biochem. J. 398:439–450. 10.1042/BJ20060169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, Jiang T, Sladek FM. 2010. Integrated approach for the identification of human hepatocyte nuclear factor 4α target genes using protein binding microarrays. Hepatology 51:642–653. 10.1002/hep.23357 [DOI] [PMC free article] [PubMed] [Google Scholar]