

Abstract
Echovirus 7 enters polarized Caco-2 intestinal epithelial cells by a clathrin-mediated endocytic process and then moves through the endosomal system before releasing its genome into the cytoplasm. We examined the possible role in virus entry of core components of the autophagy machinery. We found that depletion of Beclin-1, Atg12, Atg14, Atg16, or LC3 with specific small interfering RNAs inhibited echovirus 7 infection upstream of uncoating but had little or no effect on virus attachment to the cell surface. These data indicate that multiple autophagy-related proteins are important for one or more events that occur after the virus has bound its receptor on the cell surface but before RNA is released from the virus capsid. Although we have not determined the mechanism by which each protein contributes to virus entry, we found that stable depletion of Atg16L1 interfered with virus internalization from the cell surface rather than with intracellular trafficking. Autophagy gene products may thus participate in the endocytic process that moves virus into polarized Caco-2 cells.
INTRODUCTION
Picornaviruses are nonenveloped viruses with a positive-sense single-stranded RNA genome. A number of picornaviruses—polioviruses, coxsackieviruses, echoviruses, other enteroviruses, and rhinoviruses—are human pathogens (1, 2). Echoviruses, including echovirus 7 (EV7), are a common cause of febrile illnesses and aseptic meningitis in the United States, particularly during the summer and fall (3).
Because many human picornaviruses are believed to invade their hosts by crossing the intestinal epithelium, we have been interested in the interaction between these viruses and intestinal epithelial cells. We recently observed that EV7 enters polarized Caco-2 epithelial cells by clathrin-mediated endocytosis and moves through early endosomes and then to late endosomes before RNA is released from the capsid into the cytoplasm (a process referred to as “uncoating”) (4); we also found that virus entry and uncoating require the function of Rab7 (4), a GTPase critical for the maturation of late endosomes and for endosome-lysosome fusion (5). However, unlike other viruses that move to late endosomes and/or require Rab7 during the entry process (6–10), EV7 does not depend on endosomal acidification for its entry (4). We therefore considered the possibility that Rab7 might play a role in entry other than virus delivery to acidic endosomes.
Because Rab7 has been reported to play a role in autophagy (11, 12), a cellular process in which cytoplasmic contents are delivered to lysosomes for degradation (reviewed in references 13 and 14), we examined whether host factors important for autophagy are required for EV7 entry and infection. We present evidence that autophagy-related gene products are important for EV7 entry into polarized Caco-2 cells. Surprisingly, at least one of these proteins, Atg16L1, is important for virus internalization from the cell surface rather than for intracellular trafficking.
MATERIALS AND METHODS
Cells and viruses.
Caco-2 cells (ATCC HTB-37) were cultured in minimal essential medium (MEM) containing 20% fetal bovine serum, sodium pyruvate, nonessential amino acids, and penicillin-streptomycin. EV7, coxsackievirus B3 (CVB3)-RD, and vesicular stomatitis virus (VSV), preparation of neutral red-loaded virus, and virus labeling with Alexa Fluor 594 (AF-594) have been described (4).
Antibodies, chemicals, and small interfering RNAs (siRNAs).
Rabbit polyclonal antibody specific for Beclin-1 was purchased from Abgent (Am1818a). Rabbit polyclonal anti-UVRAG (U7508) antibody was from Sigma. Rabbit polyclonal antibodies specific for LC3 (PM036), Atg16L1 (PM040), and Atg14 (PD026) were from MBL International. Rabbit polyclonal anti-Atg12 antibody was purchased from Cell Signaling Technology (catalog no. 2010). The rabbit anti-LC3 antibody used for immunoblotting was provided by Ravi Amaravadi (University of Pennsylvania). Mouse anti-FLAG antibody was from Agilent (catalog no. 200472). 3-Methyladenine (3-MA) was from Sigma (M9281), as were pepstatin A (P-4245) and E64d (E-8640).
Control siRNA (15) was from Ambion, and siRNAs pools targeting LC3 and Atg12 (16) were from Dharmacon. Atg16L1 (17), UVRAG (18), and Atg14 siRNAs 1 (18) were synthesized using previously reported sequences. Beclin-1 siRNAs 1 and 2 were synthesized using sequences published in references 18 and 19, respectively. Transfection of Caco-2 cells with siRNAs (20 nM concentration) was performed as described previously (4).
Virus infection assay.
Caco-2 cells transfected with siRNAs or treated with 3-MA were plated in collagen-coated eight-well chamber slides (BD Biosciences) at 5 × 104 cells per well and used for infection after 2 days, when morphological polarization had been achieved. To quantify viral infection, viruses (2 PFU/cell) in binding buffer (MEM containing 20 mM HEPES) were incubated with polarized monolayers for 1 h at 4°C. Unbound virus was then washed away, complete medium was added, and cells were incubated for 6 h at 37°C. Infected cells were fixed with a 3:1 mixture of ice-cold methanol-acetone for 2 min. Cells were stained with anti-VP1 antibody (Ncl-Entero; Novocastra) and anti-mouse secondary antibody conjugated to fluorescein isothiocyanate (FITC). Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). Images were captured with a fluorescence microscope (Olympus BX51) using a ×20 objective lens. Three to four fields (700 to 1,000 cells) were captured for each well. Infected cells and total DAPI-stained cells were counted using ImageJ software (http://rsbweb.nuh.gov/ij/).
Neutral red infectious center (NRIC) assays.
Monolayers of siRNA-transfected cells, or monolayers exposed to 3-MA for 1 h, were exposed to neutral-red-loaded virus for 1 h at 4°C in the dark; buffer was removed, infection was initiated with prewarmed medium, and monolayers were incubated in the dark at 37°C to permit entry to proceed. At 90 min, monolayers were exposed to white light for 10 min, with duplicate control monolayers kept in the dark as unilluminated controls. Cells were detached with trypsin-EDTA and replated onto fresh monolayers in the absence of siRNAs or inhibitors. An agarose overlay was added after 3 h, and plaques were counted after 2 days at 37°C.
LC3 lipidation.
Caco-2 cells (3 × 105 cells per well) were plated on collagen-coated six-well plates and incubated 2 days at 37°C. Cell monolayers were incubated with EV7 (300 PFU/cell, purified by sucrose gradient centrifugation) in binding buffer for 1 h at 4°C. For 0 min, cells were harvested and remaining cells were changed to complete medium and incubated for the indicated times at 37°C. In some experiments, cells were pretreated with pepstatin A (10 μg/ml) and E64d (2.5 μg/ml) or to 10 mM 3-MA beginning 1 h before cells were exposed to virus. Cells were lysed as described previously (20), and the protein concentration of each sample was measured with a BCA protein assay kit (Thermo Scientific). Equal amounts of protein were electrophoresed in 15% sodium dodecyl sulfate (SDS)-polyacrylamide gels (Bio-Rad) and transferred to polyvinylidene difluoride membranes. Membranes were probed with anti-LC3 antibody and developed with horseradish peroxidase-conjugated secondary antibody and chemiluminescence reagents (West Dura; Thermo Scientific). Images were captured, and band intensities were measured using a Gel Logic 4000 PRO imaging system and software (Carestream Health). Ratios of LC3-II to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) intensity were normalized to that observed in the no-virus sample at 0 min, which was set as 1.
EV7 colocalization with LC3, EEA1, and Rab5.
Caco-2 cells were incubated with red fluorescence-labeled EV7 (AF594-EV7) for 1 h at 4°C at 300 PFU/cell. After a washing step with phosphate-buffered saline (PBS), complete medium was added, and the cells were incubated at 37°C to allow entry to proceed. At intervals, the cells were fixed with 4% paraformaldehyde, permeabilized with 100 μg of digitonin/ml for 15 min at room temperature, stained with anti-LC3 or anti-EEA1 antibodies, followed by fluorophore-conjugated secondary antibodies, and examined in a fluorescence microscope. In some experiments, cells were transfected with a plasmid encoding Rab5-GFP (provided by George Bloom, University of Virginia) 2 days before infection. Colocalization of red and green signal was determined for the fields shown in Fig. 5, using the Colocalization Test plugin in the WCIF ImageJ bundle to calculate the Pearson correlation coefficient.
FIG 5.
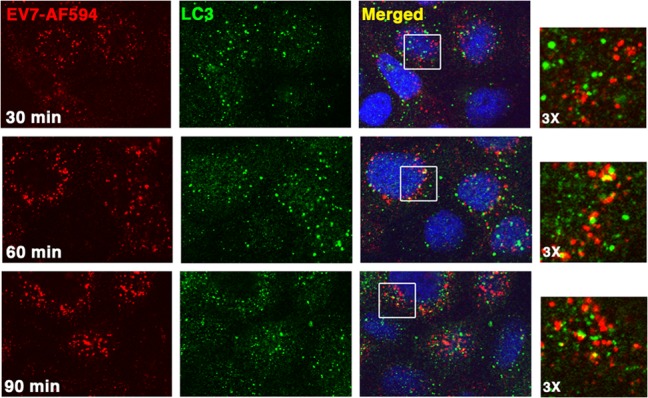
EV7 shows little colocalization with LC3 during entry. Fluorescence-labeled EV7 (EV7-AF594, red) was permitted to bind Caco-2 monolayers in the cold; then, unbound virus was removed, and monolayers were incubated at 37°C to allow entry to occur. At intervals, monolayers were fixed and stained with anti-LC3 antibody (green). Higher-magnification images show only limited colocalization of EV7 and LC3 in perinuclear vesicles at 60 and 90 min.
Virus binding assay.
EV7 was radiolabeled with [35S]methionine-cysteine and purified on sucrose gradients as described previously (21). Caco-2 cells transfected with siRNAs, transduced with lentivirus vectors, or pretreated with 10 mM 3-MA for 3 h at 37°C were plated in collagen-coated 24-well plates (1.5 × 105 cells per well). Confluent monolayers were exposed to 35S-labeled virus (30,000 cpm per well) for 1 h at room temperature and then washed three times to remove unbound virus; the cell-bound radioactivity was measured in a scintillation counter (Perkin-Elmer).
Stable knockdown of Atg16L1 in Caco-2 cells using lentiviral shRNA, and characterization of Atg16L1-depleted cells.
The siRNA sequence targeting Atg16L1 (17) was inserted into the lentiviral shRNA expression plasmid pSIH1-H1-puro (System Bioscience, Mountain View, CA) (provided by Shengbing Huang, Mayo Clinic), and virus was produced as described previously (22) by using a self-inactivating HIV-based vector plasmid pCMV-Delta.8.9.1 and pVSV-G encoding the VSV G protein (provided by Miroslaw Kozlowski and Marcus Davey, Children's Hospital of Philadelphia). For the transduction of Caco-2 cells, supernatant containing virus was added to cells in the presence of with 8 μg of Polybrene (Sigma)/ml. Puromycin (12.5 μg/ml; Sigma) was added at 24 h after transduction, and puromycin-resistant colonies were isolated and tested for target gene knockdown.
Expression of DAF on Atg16L1-depleted cells and control cells (transduced with a lentivirus vector with a nontargeting shRNA insert) was measured by flow cytometry with monoclonal antibody IA10. To determine whether there was a postentry block to virus replication in knockdown cells, cell monolayers were transfected with EV7 RNA, transcribed in vitro with T7 RNA polymerase from full-length EV7 cDNA cloned into the pSport vector, essentially as described for the generation of CVB3 RNA (23). Monolayers in 12-well plates were exposed to 2 μg of viral RNA in Lipofectamine 2000 for 4 h at 37°C, and then the medium was replaced, and infection was permitted to proceed for 6 more hours. Monolayers were frozen and thawed, and lysate titers were determined by plaque assay on HeLa cells.
To determine whether expression of shRNA-resistant Atg16L1 restored susceptibility of knockdown cells to infection of shRNA, cells were transfected with a plasmid expression vector encoding FLAG-tagged murine Atg16L1 (Addgene 24302) and selected in neomycin. Pooled neomycin-resistant cells were exposed to EV7. Cells expressing murine Atg16L1 were identified by FLAG staining, and infected cells were identified by staining for VP1 at 6 h; the percentage of cells infected was determined for cells that did and did not express murine Atg16L1.
EV7 entry assay in Caco-2 cells expressing Atg16L1 shRNA.
Caco-2 monolayers were incubated with red fluorescence-labeled EV7-AF594 for 1 h at 4°C at 300 PFU/cell. After a PBS wash, complete medium was added, and the cells were placed at 37°C to allow virus entry. At 0, 60, or 90 min, cells were fixed with 4% paraformaldehyde (PFA) for 12 min, and excess PFA was removed by using PBS containing 50 mM NH4Cl. Surface virus was stained with anti-VP1 antibody (Vector Laboratories, Inc., catalog no. VP-E603) in PBS containing 3% bovine serum albumin (Sigma), followed by FITC-conjugated goat anti-mouse secondary antibody in PBS containing 10% normal goat serum. Images of five to eight fields per time point were captured in a fluorescence microscope using a ×63 objective (∼50 cells). Fluorescence intensity in the red channel (Ftotal) and the green channel (Fsurface) was measured using ImageJ software. The intensity in the green channel was adjusted for all time points by a factor sufficient to render the red and green intensities equal at 0 min, when all virus was localized to the cell surface. The extent of internalization was calculated as (Ftotal − Fsurface)/Ftotal.
Analysis of experimental variation.
In all figures, error bars indicate the means and standard deviations of three or more independent samples. Single asterisks indicate P values of <0.05, as determined by using the Student t test; double asterisks indicate P values of <0.01.
RESULTS
EV7 infection and entry depend on PI3-kinase, Beclin-1, and Atg14.
Autophagy is a process in which cytoplasmic contents are delivered to lysosomes for degradation. It begins with formation of a membranous precursor, the isolation membrane or phagophore, which elongates and fuses, capturing cytoplasm within a double-membrane vesicle referred to as an autophagosome. Ultimately, fusion of the autophagosome with lysosomes (or earlier components of the endosomal pathway) delivers autophagosome contents to lysosomes for degradation and recycling. Because Rab7—which we recently found to be important for EV7 entry (4)—is required for autophagosome fusion with lysosomes and endosomes (11, 12), we considered the possibility that the autophagy and EV7 entry pathways might intersect. In a recent report, adenovirus 2 was observed in vesicles with both early endosome markers and autophagosome markers, and it was suggested that early endosomes carrying this virus may fuse with autophagosomes during the entry process (24).
Formation of the autophagosome is a complex process involving multiple cellular proteins, many of which were first identified as the products of autophagy-related genes (Atg) in yeast, and most of which function in multiprotein complexes (13, 14). Formation of the isolation membrane is initiated by a complex of the class III phosphatidylinositol 3-kinase (PI3-kinase) Vps34 with Beclin-1 (also known as Atg6) and Atg14. Formation of the isolation membrane also depends on a complex of Atg12, Atg5, and Atg16, which is further required for elongation and closure of the membrane, and which regulates the conjugation of LC3 (also known as Atg8) to phosphatidyl-ethanolamine, permitting LC3 to associate with the membrane of the maturing autophagosome. Autophagosomal membranes have been proposed to arise from a variety of sources, including the endoplasmic reticulum, the Golgi apparatus, the mitochondria, and the plasma membrane (25). To test for a possible role for autophagy in EV7 infection, we treated cells with 3-methyladenine (3-MA), an inhibitor of Vps34 and other PI3-kinases commonly used to suppress autophagy (26), exposed them to EV7 or CVB3 (2 PFU per cell), and then determined the numbers of infected cells at 6 h by staining for the viral capsid protein VP1; under these conditions, input virus is not detected (not shown), and VP1 staining is a marker of viral protein synthesis. 3-MA had a marked inhibitory effect on EV7 infection but had no effect on infection by CVB3-RD (Fig. 1A and B).
FIG 1.

PI3-kinase, Beclin-1, and Atg14 are important for EV7 entry and infection. (A) 3-MA inhibits infection by EV7 but not by CVB3. Caco-2 cells in polarized monolayers were exposed to 3-MA or to control medium and exposed to EV7 or CVB3-RD as described in Materials and Methods. Infected cells were identified by staining with anti-VP1 antibody, and nuclei were identified by staining with DAPI. (B) For each virus, results are shown as the percentages of cells infected, normalized to the result with control siRNA, ± the standard deviations (SD) for triplicate samples. (C) 3-MA inhibits EV7 entry in the neutral red infectious center (NRIC) assay. Monolayers pretreated with 3-MA or control medium were exposed to neutral red-labeled EV7 and CVB3-RD, and neutral red infectious center assays were performed at 90 min as described in Materials and Methods. The results are normalized to those obtained with unilluminated controls exposed to control siRNA. (D) 3-MA does not inhibit CVB3-RD entry in the NRIC assay. (E) siRNAs targeting Beclin-1 inhibit EV7 infection but not CVB3 infection; the inset shows an immunoblot to measure Beclin-1 depletion. (F) Quantitation of results shown in panel E. (G) Beclin-1 siRNA inhibits EV7 entry in the NRIC assay. To understand why an siRNA may inhibit infection in the routine assay (as in panels D and F) but still may inhibit only in illuminated cells in the NRIC assay (as in panels E and G), it is important to remember that these assays are configured quite differently. In the infection assay, cells are stained at 6 h to detect VP1; if an siRNA inhibits replication only partially during the 6-h period, it may still prevent the synthesis of sufficient VP1 for the cell to be scored positive. However, in the NRIC assay, cells are infected and then resuspended and replated on a fresh monolayer for 2 days. If an siRNA permits even a single infectious virion to escape the cell, that virion may well have time, over the 2-day course of the assay, to infect the monolayer, replicate, and form a plaque. However, if the siRNA has kept the virus from uncoating at 90 min, the light flash effectively kills the virus, and there is no escape. In this and all subsequent figures, single asterisks indicate P values of <0.05, and double asterisks indicate P values of <0.01.
We then tested whether 3-MA inhibited virus entry, using the previously described neutral red infectious center (NRIC) assay to measure virus uncoating (4, 27). When virus is grown in the presence of neutral red (NR), dye is concentrated inside the virion, in close proximity to the RNA. When exposed to light, NR damages the RNA and inactivates the virus; however, once RNA has been released from the capsid, exposure to light has no effect. We previously found that plaque formation by neutral-red-loaded EV7 (NR-EV7) is inhibited when Caco-2 monolayers are exposed to light at 30 and 60 min postinfection, whereas the majority of NR-EV7 becomes insensitive to light by 90 min (4), indicating that EV7 uncoating largely occurs by 90 min. We pretreated Caco-2 cells with 3-MA, exposed them to NR-EV7, and allowed infection to proceed in the dark for 90 min. Cells were then exposed to light, harvested, replated on cell monolayers, overlaid with agar, and incubated to permit the appearance of infectious centers. To control for nonspecific effects of the inhibitor, parallel assays were performed in which the illumination step was omitted. If a chemical inhibitor or an siRNA prevents or significantly delays cellular events upstream of RNA release, we expect it to reduce the formation of infectious centers in illuminated—but not in unilluminated—cells. 3-MA inhibited infectious center formation in illuminated samples, with little inhibitory effect in unilluminated samples (Fig. 1C) and with no effect on infectious center formation by CVB3-RD (Fig. 1D). These results indicate that 3-MA inhibits EV7 infection upstream of uncoating and suggest that PI3-kinase activity is needed for EV7 entry.
This suggested a possible role for the Vps34–Beclin-1–Atg14 complex in EV7 infection. Depletion of Beclin-1 with specific siRNAs inhibited infection by EV7 but not by CVB3 (Fig. 1E and F). Further, depletion of Beclin-1 inhibited EV7 upstream of uncoating, as determined by the NRIC assay (Fig. 1G), indicating a role for Beclin-1 in some aspect of virus entry. Similarly, depletion of Atg14 specifically inhibited infection by EV7, but not CVB3 (Fig. 2A), and inhibited EV7 entry, as determined by the NRIC assay (Fig. 2B). In contrast, depletion of UVRAG (UV radiation resistance-associated gene protein), which is involved in a separate Vps34–Beclin-1 complex that does not include Atg14 (18) had no effect on infection by EV7 (Fig. 2C). These results suggest that the Vps34–Beclin-1–Atg14 complex, which functions in initiation of autophagosome formation, is specifically required for EV7 entry and infection.
FIG 2.

Atg14, but not UVRAG, is involved in EV7 infection and entry. (A) Atg14 siRNA inhibits EV7 infection. (B) Atg14 siRNA inhibits EV7 entry in the NRIC assay. (C) UVRAG siRNA does not inhibit EV7 infection.
EV7 infection and entry depend on Atg12, Atg16L1, and LC3.
Elongation and closure of the isolation membrane depends on the complex involving Atg5-Atg12-Atg16L1, as well as on LC3. Depletion of Atg12, Atg16L1, or LC3 with siRNAs inhibited EV7 infection (Fig. 3A, C, and E) and inhibited upstream of uncoating, as measured by the NRIC assay at 90 min (Fig. 3B, D, and F). These results provided additional evidence that EV7 entry depends specifically on the core autophagy machinery.
FIG 3.

Atg12, Atg16L1, and LC3 are important for EV7 infection and entry. (A) Atg12 siRNA inhibits EV7 infection. (B) Atg12 siRNA inhibits EV7 entry in the NRIC assay. (C) AtgL1 siRNA inhibits infection by EV7 but not by CVB3-RD or VSV. (D) Atg16L1 siRNA inhibits EV7 entry in the NRIC assay. (E) LC3 siRNA inhibits EV7 infection. (F) LC3 siRNA inhibits EV7 entry in the NRIC assay.
Autophagic signaling is constitutively active in Caco-2 cells and shows little increase during EV7 entry.
During the induction of autophagy, LC3 protein is conjugated to phosphatidylethanolamine, which permits its association with the developing autophagosome (28, 29), and mature autophagosomes can be identified by immunofluorescence staining for LC3 (30). In uninfected Caco-2 cells, staining for LC3 revealed multiple punctate structures, which were not seen when LC3 was depleted with siRNA (Fig. 4A); similar numbers of punctate structures were seen after starvation or exposure to rapamycin (data not shown), two treatments commonly used to induce autophagy (30). These results were consistent with a high constitutive level of autophagy in Caco-2 cells, as has been previously reported for some Caco-2 cell clones (31).
FIG 4.

Autophagy is constitutively active in Caco-2 cells. (A) Caco-2 monolayers were treated with siRNA targeting LC3, or with control siRNA, and then stained to detect endogenous LC3. (B) Caco-2 monolayers were treated with siRNA targeting Atg16L1, and cell lysates were immunoblotted to detect the unlipidated (LC3-I) and lipidated (LC3-II) forms of LC3. (C) Caco-2 monolayers were exposed to EV7 or to medium alone. At the indicated times, monolayers were lysed and immunoblots were performed to detect the LC3-I and LC3-II. Similar results were obtained in three separate experiments. (D) Quantification of the LC3-II/GAPDH ratios (determined as in Materials and Methods), normalized to the 0-min no-virus control, ± the SD for three independent experiments.
The lipidated form of LC3 (LC3-II) migrates more rapidly than unmodified LC3 protein (LC3-I) in SDS-PAGE, and the conversion of LC3-I to LC3-II is commonly used as a marker for the induction of autophagy (30). Strong LC3-II bands and weak LC3-I bands were seen on immunoblots of lysates of uninfected monolayers (Fig. 3E and 4B, control siRNA), again consistent with a high baseline level of autophagic signaling in the absence of specific stimuli. In 293T cells LC3-I and LC3-II bands were of equal intensity (data not shown), demonstrating that the antibody was capable of recognizing LC3-I, and both bands were reduced in intensity in Caco-2 cells treated with LC3 siRNA, confirming their identity (see Fig. 3E, inset). Further, the intensity of the LC3-II band was reduced, and the intensity of the LC3-I band slightly increased, in cells treated with siRNA targeting Atg16L1 (Fig. 4B), which is required for LC3 lipidation (32).
As we had observed a relationship between autophagy proteins and EV7 entry, we measured whether autophagic signaling increased early in infection. Caco-2 monolayers were exposed to EV7 (or mock infected) at 4°C and then shifted to 37°C to allow infection to proceed; at intervals, lysates were prepared, and LC3 was detected by immunoblotting (Fig. 4C). The amount of LC3-II, measured relative to GAPDH, showed little, if any increase (Fig. 4E). Taken together, these results indicate that virus infection does not further increase the already high baseline level of autophagic signaling in these cells.
EV7 shows little association with autophagosomes during entry.
To determine whether EV7 associates with autophagosomes during its entry process, cells were exposed to EV7 conjugated with red fluorescent dye (EV7-AF594) at 4°C and then incubated at 37°C for 30, 60, or 90 min. After fixation and permeabilization, cells were stained with anti-LC3 antibody and fluorescein isothiocyanate (FITC)-labeled secondary antibody (green) (Fig. 5). Although some virus-containing vesicles were associated with vesicles expressing LC3 (Fig. 5, 3-fold-enlarged images), very little colocalization was observed (Pearson coefficient 0.15 at 30 min, 0.16 at 60 min, and 0.15 at 90 min). Although we cannot exclude the possibility that virus- and LC3-associated vesicles associate at times we did not observe, these results provide no evidence that virus moves through LC3-expressing vesicles during entry.
EV7 binding to cells is not significantly affected by 3-MA or by siRNAs targeting autophagy proteins.
EV7 binds to a receptor, DAF, on the apical surface of Caco-2 cells, and we considered the possibility that depletion of autophagy proteins might affect virus entry by altering receptor recycling and virus binding. To test this, we measured the attachment of 35S-labeled EV7 to Caco-2 cells treated with 3-MA or specific siRNAs (Fig. 6). 3-MA, or siRNAs targeting Beclin-1, LC3, or Atg16L1, had no effect on EV7 attachment, and siRNAs targeting Atg12 and Atg14 had small effects that were not statistically significant. In contrast, depletion of DAF with siRNA markedly reduced virus attachment, as expected. These results indicate that autophagy proteins are important for events that occur after virus has bound to DAF on the cell surface. To identify the specific entry events in which autophagy gene products are involved, we attempted to examined entry of fluorescent virions into Caco-2 cells treated with 3-MA or siRNAs.
FIG 6.
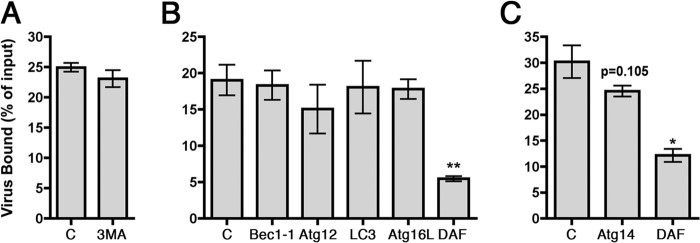
Autophagy gene products are not required for virus attachment to Caco-2 cells. Monolayers pretreated with 3-MA (A) or with siRNAs targeting Beclin-1, Atg12, LC3, and Atg16L1 (B) or Atg14 (C), or a positive-control siRNA targeting DAF (B and C) were incubated with radiolabeled EV7, and virus binding was measured as described in Materials and Methods.
3-MA interferes with endosome maturation.
In control Caco-2 cells, EV7 accumulated at 60 min in perinuclear vesicles marked by the early endosome antigen EEA1, a Rab5 effector molecule important for endosome fusion events (Fig. 7). In contrast, in cells treated with 3-MA, virus concentrated at 60 min in perinuclear vesicles that were abnormally large and devoid of the early endosomal protein EEA1, a Rab5 effector important for endosome fusion events (33); these vesicles expressed Rab5, as indicated by association with green fluorescent protein (GFP)-tagged Rab5 (Fig. 7, lower panels). PI3-kinase activity is known to be important for recruitment of EEA1 to Rab5-positive early endosomes, and our observations were consistent with a previous report that wortmannin, another inhibitor of PI3-kinase, causes rapid dissociation of EEA1 from endosomal membranes (34). These results indicate that 3-MA inhibits normal endosome maturation in these cells, independently of any effects on autophagosome formation.
FIG 7.
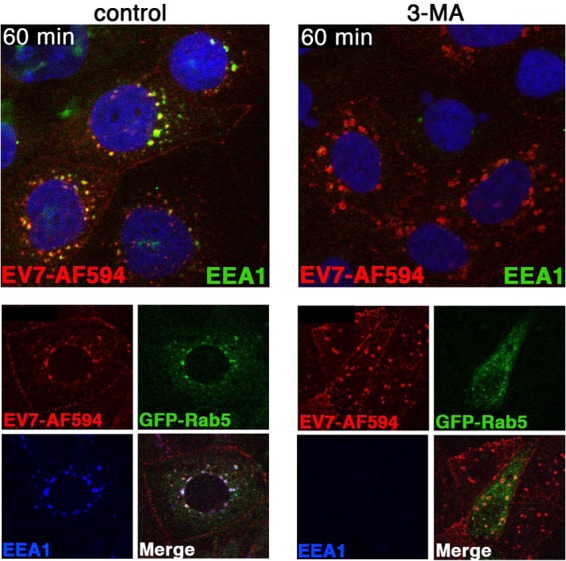
3-MA interferes with endosome maturation. In the top panels, cells were treated with 3-MA, then exposed to EV7-AF549 (red), incubated at 37°C for 60 min, and stained to detect EEA1 (green). In the lower panels, cells transfected with Rab5-GFP (green) were infected with EV7-AF549 and stained to detect EEA1 (blue).
Stable depletion of Atg16L1 impairs virus internalization from the cell surface.
In cells treated with siRNAs targeting specific autophagy gene products we found considerable cell-to-cell variation, making it difficult to determine how virus entry and trafficking were perturbed (data not shown). This variation most likely reflected heterogeneous transfection and knockdown efficiency, which was evident when we stained monolayers for one of the targeted proteins, Atg16L1 (not shown).
Therefore, to examine the mechanism by which an autophagy protein might contribute to virus entry, we generated a Caco-2 cell line stably expressing an shRNA targeting Atg16L1. As expected, stable depletion of Atg16L1 inhibited infection by EV7 (Fig. 8A); the fact that inhibition was only partial suggests that, although Atg16L1 is important for EV7 infection, it may not be indispensable. Notably, depletion of Atg16 did not prevent infection by VSV (Fig. 8A), another virus that depends on traffic through the endosomal system, indicating that these cells had no gross defect in endosomal maturation or traffic.
FIG 8.

EV7 internalization from the cell surface is inhibited in cells stably depleted of Atg16L1. A stable Atg16L1-depleted Caco-2 cell line was obtained by transduction with a lentivirus encoding Atg16L1 shRNA; the control cell line was transduced with a lentivirus encoding a control shRNA. (A) Infection is inhibited in the Atg16L1-depleted cell line, but not in a cell line transduced with a control shRNA. (B) Virus binding to Atg16-depleted and control cells. (C) Atg16L1-depleted and control monolayers were transfected with EV7 RNA and virus production at 10 h was determined by plaque assay. (D) Susceptibility of Atg16L1-depleted cells to EV7 infection is restored by transfection with mouse Atg16L1. Atg16L1-depleted cells were transfected with a plasmid encoding mAtg16L1, and then exposed to EV7; after 6 h, virus-infected cells and mAtg16-expressing cells were as described in Materials and Methods, and the percentage of cells infected was determined in both the mAtg16+ and mAtg16− cell populations. (E) Atg16-depleted and control monolayers were exposed to EV7-AF594 (red) for 1 h in the cold; then, unbound virus was removed, and monolayers were incubated at 37°C to permit entry to proceed. At 0, 60, and 90 min, monolayers were fixed without permeabilization and then stained with anti-VP1 and FITC (green)-labeled secondary antibody to detect virus still on the cell surface. In control cells, red intracellular virus is evident at 60 and 90 min, whereas in Atg16-depleted cells virus is retained on the cell surface, as evident from colocalized red and green signal. (F) Quantification of virus internalization, calculated from three separate experiments, performed as described in Materials and Methods.
Atg16-depleted cells had no defect in expression of surface DAF (compared to control cells by flow cytometry with anti-DAF antibody, not shown) and showed no defect in the binding of 35S-labeled EV7 (Fig. 8B). Further, the Atg16L1-depleted cells showed no defect in virus production when transfected with EV7 RNA, confirming that infection was blocked upstream of uncoating. To eliminate the possibility of off-target effects of the Atg16 shRNA, we restored Atg16L1 by transfection with murine Atg16L1, which is not susceptible to knockdown by the shRNA targeting human Atg16L1. A significantly higher level of infection was noted in cells that expressed murine Atg16L1 than in those that did not (Fig. 8C), indicating that the shRNA effect depended specifically on Atg16L1 knockdown.
To examine virus entry in the Atg16-depleted cells, we permitted EV7-AF594 (red) to bind to cells in the cold, and then incubated the cells at 37°C to permit entry to proceed. At intervals, cells were fixed without permeabilization and stained with anti-VP1 antibody and FITC (green)-tagged secondary antibody (Fig. 8D). Under these conditions, virus that has entered the cell is inaccessible to antibody and appears red, whereas virus on the cell surface shows both red and green fluorescence and appears yellow in merged images. In control cells, the majority of virus appeared to have entered the cells by 60 min, whereas in Atg16L1-depleted cells most virus appeared to be retained on the cell surface. To quantify these results, we collected images of multiple microscopic fields, measured the total red and green fluorescence intensity and calculated an internalization index (as described in Materials and Methods). The results (Fig. 8E) confirmed that virus entered control cells by 60 min, whereas internalization was significantly reduced in Caco-2 cells stably depleted of Atg16L1.
DISCUSSION
The results we report here indicate that several core components of the autophagy machinery—Beclin-1, Atg12, Atg14, Atg16L1, and LC3—are important for EV7 entry into polarized Caco-2 cells. Depletion of Beclin-1, Atg12, Atg14, Atg16, or LC3 with specific siRNAs inhibited EV7 infection upstream of uncoating but had little or no effect on virus attachment to the cell surface. These data indicate that multiple autophagy-related proteins are important for one or more events that occur after virus has bound DAF on the cell surface and before RNA is released from the virus capsid.
We undertook these studies because we suspected that autophagy gene products might be related to the traffic of virus to the intracellular site where uncoating occurs. Instead, we found that depletion of Atg16L1 interfered with internalization of virus particles from the cell surface, suggesting a role for Atg16L1 in the endocytic process that brings virus into the cell. Our previous work indicates that EV7 entry depends on factors associated with clathrin-mediated endocytosis (CME), including clathrin heavy chain, dynamin II, and the clathrin-adaptor protein eps15 (4). Although autophagy gene products are not generally considered to function in CME, Atg16L1 has been found to interact directly with clathrin heavy chain, and clathrin- and dynamin-dependent uptake of plasma membrane has been found to contribute to the formation of preautophagosomal vesicles marked by Atg16L1, Atg5, and Atg12 (35). These vesicles, often located at the cell periphery, subsequently recruit LC3 and evolve into mature autophagosomes (35). It has been suggested that the plasma membrane, by serving as a large reservoir for rapid synthesis of precursor vesicles, may be particularly important when autophagy is highly active (35). We found that autophagy is constitutively active in Caco-2 cells (and in two other polarized intestinal cell lines [not shown]), as indicated by multiple LC3-positive vesicles at baseline and high levels of LC3-II. In these cells, the rapid uptake of plasma membrane to form autophagosome precursors may facilitate internalization of membrane-associated virus, providing a possible explanation for why EV7 internalization involves Atg16L1 in addition to clathrin and dynamin. We do not yet know whether autophagy gene products are involved in EV7 entry in other cell types.
In addition, we do not yet know whether other autophagy gene products function during virus internalization from the cell surface, as is the case for Atg16L1. We have not determined the precise functions of Beclin-1, Atg12, Atg14, or LC3, and our conclusion that they are involved in the entry process is based on the NRIC assay, in which specific siRNAs prevented formation of infectious centers only in samples exposed to light before uncoating had occurred. In our previous experience, reversible inhibitors of clathrin-mediated endocytosis—which block virus internalization from the cell surface—and siRNAs targeting the same endocytic pathway, both had specific effects in the NRIC assay (4); similarly, siRNAs that trap CVB3 on the cell surface inhibit infectious center formation only in illuminated samples (Kim and Bergelson, unpublished). We are therefore confident that the NRIC data indicate a role for multiple autophagy proteins in EV7 entry—either for internalization or for other events in the entry process, such as intracellular trafficking or the destabilization of the virus capsid. We can speculate that, because the PI3-kinase-Beclin-Atg14 complex, as well as Atg5, Atg12, and Atg16, are—like Atg16—essential for initiation of phagophore formation, these factors may be required for recruitment of plasma membrane during autophagosome generation and thus may also be involved in virus internalization. However, we are unaware of any evidence linking LC3 to the internalization of ligands from the cell surface. Further, the observation that 3-MA did not prevent virus from accumulating in perinuclear vesicles suggests that the PI3-kinase activity of Vps34 is not essential for internalization from the cell surface and that the Vps34–Beclin-1–Atg14 complex may be involved in some other entry event.
Autophagy has been implicated in several steps in picornavirus life cycles (36), but our results provide the first direct evidence that autophagy-related proteins promote entry by a picornavirus. Infection by poliovirus stimulates LC3 activation and autophagosome formation by a mechanism that involves the direct effects of viral nonstructural proteins (16, 37); inhibition of autophagy interferes with poliovirus replication and replication of other enteroviruses in vitro (16, 38–40), and autophagy is involved in the pathogenesis of coxsackievirus-induced pancreatitis in vivo (41). Another picornavirus, foot-and-mouth disease virus (FMDV), stimulates autophagosome formation early in infection, before viral nonstructural proteins are synthesized, and FMDV colocalizes with LC3, presumably in autophagosomes, within 2 h of infection (42). These results suggest a role for autophagy during FMDV entry, although this has not yet been demonstrated directly.
Viruses have evolved to make use of normal host functions, and studies of virus entry have been instrumental in our understanding of normal endocytic processes. The unexpected observation that Atg16L1 and other autophagy gene products are involved in EV7 entry may lead to new insights about the cellular factors that regulate endocytosis in polarized cells.
ACKNOWLEDGMENTS
We are grateful to Kunal Patel for the suggestion that we examine the role of autophagy in EV7 entry, to Sara Cherry for comments on the work, and to Carolyn Coyne for helpful criticism of the manuscript. We thank Miroslaw Kozlowski, Marcus Davey, George Bloom, and Shengbing Huang for plasmids, Ravi Amaravadi for LC3 antibody, and Andrea Stout and Jasmine Zhao of the University of Pennsylvania Cell and Developmental Biology Microscopy Core for assistance with confocal imaging.
This study was supported by National Institutes of Health grant R01AI072490 and by the Plotkin Endowed Chair at the Children's Hospital of Philadelphia.
Footnotes
Published ahead of print 23 October 2013
REFERENCES
- 1.Racaniello VR. 2007. Picornaviridae: the viruses and their replication, p 795–834 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 1 Lippincott/Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Pallansch M, Roos R. 2007. Enteroviruses: poliovirus, coxsackieviruses, echoviruses, and newer enteroviruses, p 839–894 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 1 Lippincott/Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3.Khetsuriani N, Lamonte-Fowlkes A, Oberste S, Pallansch MA. 2006. Enterovirus surveillance–United States, 1970-2005. MMWR Surveill. Summ. 55:(SS-8)1–20 http://www.cdc.gov/mmwr/PDF/ss/ss5508.pdf [PubMed] [Google Scholar]
- 4.Kim C, Bergelson JM. 2012. Echovirus 7 entry into polarized intestinal epithelial cells requires clathrin and Rab7. mBio 3:e00304–11. 10.1128/mBio.00304-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Somsel Rodman J, Wandinger-Ness A. 2000. Rab GTPases coordinate endocytosis. J. Cell Sci. 113:183–192 http://jcs.biologists.org/content/113/2/183.long [DOI] [PubMed] [Google Scholar]
- 6.Prchla E, Kuechler E, Blaas D, Fuchs R. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 68:3713–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieczkarski SB, Whittaker GR. 2003. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343. 10.1034/j.1600-0854.2003.00090.x [DOI] [PubMed] [Google Scholar]
- 8.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 4:e1000244. 10.1371/journal.ppat.1000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harbison CE, Lyi SM, Weichert WS, Parrish CR. 2009. Early steps in cell infection by parvoviruses: host-specific differences in cell receptor binding but similar endosomal trafficking. J. Virol. 83:10504–10514. 10.1128/JVI.00295-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lozach P-Y, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, Helenius A. 2010. Entry of bunyaviruses into mammalian cells. Cell Host Microbe 7:488–499. 10.1016/j.chom.2010.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. 2004. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117:4837–4848. 10.1242/jcs.01370 [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez MG, Munafo DB, Beron W, Colombo MI. 2004. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117:2687–2697. 10.1242/jcs.01114 [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Klionsky DJ. 2009. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335:1–32. 10.1007/978-3-642-00302-8_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coyne CB, Voelker T, Pichla SL, Bergelson JM. 2004. The coxsackievirus and adenovirus receptor interacts with the multi-PDZ domain protein-1 (MUPP-1) within the tight junction. J. Biol. Chem. 279:48079–48084. 10.1074/jbc.M409061200 [DOI] [PubMed] [Google Scholar]
- 16.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. 2010. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16:90–97. 10.1038/nm.2069 [DOI] [PubMed] [Google Scholar]
- 18.Itakura E, Kishi C, Inoue K, Mizushima N. 2008. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19:5360–5372. 10.1091/mbc.E08-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. 2009. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11:468–476. 10.1038/ncb1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. 10.1016/j.cell.2005.10.035 [DOI] [PubMed] [Google Scholar]
- 21.Bergelson JM, St John N, Kawaguchi S, Chan M, Stubdal H, Modlin J, Finberg RW. 1993. Infection by echoviruses 1 and 8 depends on the a2 subunit of human VLA-2. J. Virol. 67:6847–6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davey MG, Zoltick PW, Todorow CA, Limberis MP, Ruchelli ED, Hedrick HL, Flake AW. 2012. Jaagsiekte sheep retrovirus pseudotyped lentiviral vector-mediated gene transfer to fetal ovine lung. Gene Ther. 19:201–209. 10.1038/gt.2011.83 [DOI] [PubMed] [Google Scholar]
- 23.Pan J, Narayanan B, Shah S, Yoder JD, Cifuente JO, Hafenstein S, Bergelson JM. 2011. Single amino acid changes in the virus capsid permit coxsackievirus B3 to bind decay-accelerating factor. J. Virol. 85:7436–7443. 10.1128/JVI.00503-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng X, Carlin CR. 2013. Host cell autophagy modulates early stages of adenovirus infections in airway epithelial cells. J. Virol. 87:2307–2319. 10.1128/JVI.02014-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mari M, Tooze SA, Reggiori F. 2011. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep. 3:25. 10.3410/B3-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seglen PO, Gordon PB. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 79:1889–1892. 10.1073/pnas.79.6.1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol. 5:e183. 10.1371/journal.pbio.0050183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. 2004. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279:36268–36276. 10.1074/jbc.M401461200 [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakiyama T, Musch MW, Ropeleski MJ, Tsubouchi H, Chang EB. 2009. Glutamine increases autophagy under Basal and stressed conditions in intestinal epithelial cells. Gastroenterology 136:924–932. 10.1053/j.gastro.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. 2008. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 19:2092–2100. 10.1091/mbc.E07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature 397:621–625. 10.1038/17618 [DOI] [PubMed] [Google Scholar]
- 34.Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394:494–498. 10.1038/28879 [DOI] [PubMed] [Google Scholar]
- 35.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. 2010. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 12:747–757. 10.1038/ncb2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein KA, Jackson WT. 2011. Picornavirus subversion of the autophagy pathway. Viruses 3:1549–1561. 10.3390/v3091549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor MP, Kirkegaard K. 2007. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 81:12543–12553. 10.1128/JVI.00755-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143–9153. 10.1128/JVI.00641-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon SY, Ha YE, Choi JE, Ahn J, Lee H, Kweon HS, Lee JY, Kim DH. 2008. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J. Virol. 82:11976–11978. 10.1128/JVI.01028-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS. 2009. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 81:1241–1252. 10.1002/jmv.21502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alirezaei M, Flynn CT, Wood MR, Whitton JL. 2012. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe 11:298–305. 10.1016/j.chom.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berryman S, Brooks E, Burman A, Hawes P, Roberts R, Netherton C, Monaghan P, Whelband M, Cottam E, Elazar Z, Jackson T, Wileman T. 2012. Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J. Virol. 86:12940–12953. 10.1128/JVI.00846-12 [DOI] [PMC free article] [PubMed] [Google Scholar]