

Abstract
Membrane fusion is a key step in the life cycle of all envelope viruses, but this process is energetically unfavorable; the transmembrane fusion subunit (TM) of the virion-attached glycoprotein actively catalyzes the membrane merger process. Retroviral glycoproteins are the prototypical system to study pH-independent viral entry. In this study, we determined crystal structures of extramembrane regions of the TMs from Mason-Pfizer monkey virus (MPMV) and xenotropic murine leukemia virus-related virus (XMRV) at 1.7-Å and 2.2-Å resolution, respectively. The structures are comprised of a trimer of hairpins that is characteristic of class I viral fusion proteins and now completes a structural library of retroviral fusion proteins. Our results allowed us to identify a series of intra- and interchain electrostatic interactions in the heptad repeat and chain reversal regions. Mutagenesis reveals that charge-neutralizing salt bridge mutations significantly destabilize the postfusion six-helix bundle and abrogate retroviral infection, demonstrating that electrostatic stapling of the fusion subunit is essential for viral entry. Our data indicate that salt bridges are a major stabilizing force on the MPMV and XMRV retroviral TMs and likely provide the key energetics for viral and host membrane fusion.
INTRODUCTION
The fusion of the viral lipid bilayer with the host membrane is indispensable for the obligate intracellular life cycle of all enveloped viruses. Viral-host cell fusion is typically catalyzed by one or more virion-associated surface envelope glycoproteins, which are structurally organized into three classes: classes I, II, and III (reviewed in references 1 and 2). Regardless of the class, the glycoprotein is displayed on the surface of the virus in a metastable, prefusion conformation, and depending on the virus, the glycoprotein is activated for fusion through one of three principal mechanisms: (i) receptor binding in a pH-independent fashion, (ii) low pH, or (iii) a combination of both receptor binding and low pH (reviewed in references 2 and 3).
Retroviruses are a distinct family of enveloped RNA viruses that are classified into seven genera: alpha-, beta-, delta-, epsilon-, and gammaretroviruses; lentivirus; and spumavirus. The retroviral envelope glycoproteins (termed Env) belong to the class I viral fusion protein family. Env is synthesized as a single-chain polypeptide precursor and posttranslationally cleaved to yield a receptor-binding surface (SU) domain and a fusion-active transmembrane (TM) domain. Mature Env is incorporated as a metastable trimer of SU-TM heterodimers on the surface of the virus. Initial attachment to host cells is mediated through the interaction of the SU domain with a host cell receptor. In many retroviruses, this is thought to trigger a conformational change that allows the TM subunit to form an extended prehairpin conformation that facilitates the insertion of its hydrophobic fusion peptide into the host plasma membrane. Subsequently, two α-helical heptad repeat regions (HR1 and HR2) in the TM fold back together in a structural rearrangement that brings the two bilayers together. The mixing of the outer leaflets of the viral and host membranes forms a hemifusion stalk. The hemifused bilayers further open into a fusion pore as the TM stabilizes into its final postfusion six-helix bundle conformation. Although membrane fusion is a thermodynamically feasible process, it has a large activation barrier. The formation of the highly stable six-helix bundle is thought to provide the free energy required for membrane fusion.
The beta-, delta-, and gammaretroviruses are the prototypical system for understanding pH-independent entry. Crystal structures of two deltaretrovirus fusion subunits, human T-lymphotropic virus type 1 (HTLV-1) (4) and bovine leukemia virus (BLV) (5), were determined previously, and both TM subunits were shown to adopt a similar trimeric coiled-coil structure in the postfusion conformation. Surprisingly, there are not any complete fusion subunit structures for beta- and gammaretroviruses, as the previously reported Moloney murine leukemia virus (MoMLV) TM structure displays only HR1 and chain reversal regions of the fusion core; its entire C-terminal HR2 region is missing (6). Additional structural information from beta- and gammaretrovirus fusion glycoproteins will complete a representative structural library for the retrovirus family and allow for a detailed cross-comparison with other viral fusion proteins to identify common features important for pH-independent entry.
Here, we determined the structure of the complete extramembrane region of the fusion protein from both beta- and gammaretroviral genera. Moreover, comparison of the Mason-Pfizer monkey virus (MPMV) and xenotropic murine leukemia virus-like virus (XMRV) TM structures unveiled the presence of key electrostatic interactions, clustered at two unique regions of the postfusion six-helix bundle structure. Site-directed mutagenesis of these electrostatic staples abrogated viral entry and reduced the overall stability of the fusion proteins. Our findings propose a common biochemical strategy utilized by pH-independent retroviruses to stabilize their postfusion glycoprotein conformation to overcome the energy barrier for fusion.
MATERIALS AND METHODS
Expression and protein purification.
DNAs corresponding to MPMV TM (residues 412 to 513) (GenBank accession number NP_056894.1) and XMRV TM (residues 472 to 568) (GenBank accession number ACY30460.1) were codon optimized and synthesized with a 6-histidine tag followed by a tobacco etch virus (TEV) protease cleavage site into a pJexpress414 vector (DNA2.0 Inc.). Cysteine-to-serine mutations (MPMV, C483S; XMRV, C538S) were engineered to abrogate nonspecific intermolecular disulfide bond-mediated aggregation. All wild-type (WT) or mutant MPMV and XMRV fusion glycoproteins were expressed in Escherichia coli BL21(DE3) cells. Cell cultures were grown to an optical density at 600 nm (OD600) of 0.6 at 37°C and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 18 h at 25°C. Cells were lysed by using a hydraulic cell disruption system (Constant Systems). Protein purification was performed stepwise by using Ni-nitrilotriacetic acid (NTA) metal affinity followed by size-exclusion chromatography on a prep-grade Superdex-75 10/300 column equilibrated in 1× Tris-buffered saline (TBS) (10 mM Tris-HCl [pH 7.5] and 150 mM NaCl). Protein concentration was quantified by determining the A280.
Crystallization and structure determination.
Prior to crystallization, the polyhistidine tag was removed by using proteases. For MPMV TM, TEV protease (1:100 protease-to-protein molar ratio), purified as previously described (7), was used to cleave the protein at 22°C for 24 h. For XMRV TM, TEV protease did not efficiently cleave the tag; thus, a chymotrypsin digest (1:3,000 protease-to-protein molar ratio) was performed at 22°C for 1.5 h. Cleavage reactions were stopped with a final concentration of 1 mM phenylmethylsulfonyl fluoride (PMSF), and the mixtures were subsequently purified on a Superdex-75 10/300 column equilibrated in 1× TBS. MPMV and XMRV TMs were concentrated to 15 mg/ml and crystallized by hanging-drop vapor diffusion. Crystals for MPMV TM were obtained in 18% (wt/vol) polyethylene glycol 3400 (PEG 3400) and 0.2 M sodium thiocyanate, while plate-like crystals were grown for XMRV TM in a solution containing 23% (wt/vol) PEG 3350, 0.1 M Bis-Tris (pH 5.5), 0.2 M NaCl, and 4% (vol/vol) 2,2,2-trifluoroethanol. Both MPMV and XMRV TM crystals were soaked in a cryoprotectant containing the mother liquor components plus 25% (vol/vol) glycerol, prior to being flash cooled in liquid nitrogen. Data for XMRV and MPMV TMs were collected on a Rigaku FR-E Superbright X-ray generator and a Saturn A200 HD charge-coupled-device (CCD) detector. All data were reduced and scaled by using d*TREK (8). The MPMV and XMRV TM structures were determined by molecular replacement using the program Phaser (9) and human syncytin-2 (PDB accession number 1Y4M) (10) and Moloney murine leukemia virus TM (PDB accession number 1MOF) (6) as the initial search models, respectively. Iterative rounds of model rebuilding were performed by using the program Coot (11), followed by simulated annealing torsion angle refinement using the program PHENIX.refine (12). Data collection and refinement statistics are presented in Table 1.
TABLE 1.
Data collection and refinement statistics
| Parameterc | Value |
|
|---|---|---|
| MPMV TM | XMRV TM | |
| Data collection statistics | ||
| Space group | P3 | P21212 |
| Unit cell dimensions (Å) | ||
| a = | 44.0 | 100.0 |
| b = | 44.0 | 148.4 |
| c = | 81.7 | 58.1 |
| α = β = | 90° | 90° |
| γ = | 120° | 90° |
| Wavelength (Å) | 1.54 | 1.54 |
| Resolution range (Å) | 34.5–1.7 | 47.6–2.2 |
| Total no. of reflections | 101,292 | 295,524 |
| No. of unique reflections (no. of reflections in outer-resolution shell) | 18,731 (1,694) | 44,752 (4,394) |
| Completeness (%) (completeness in outer-resolution shell) | 96.2 (85.4) | 99.9 (99.5) |
| Rmerge (%)a (Rmerge in outer-resolution shell) | 4.6 (26.5) | 8.8 (50.3) |
| Multiplicity (multiplicity in outer-resolution shell) | 5.4 (4.1) | 6.6 (5.7) |
| I/σ(I) [I/σ(I) in outer-resolution shell] | 19.3 (4.4) | 9.0 (2.2) |
| Refinement statistics | ||
| No. of molecules in asymmetric unit | 2 | 9 |
| No. of residues | 182 | 757 |
| No. of waters | 197 | 301 |
| No. of chloride ions | 3 | 3 |
| Rwork/Rfree (%)b | 15.4/19.3 | 21.4/26.6 |
| Avg B factor (Å2) | ||
| Overall | 17.1 | 31.4 |
| Protein | 15.8 | 31.3 |
| Solvent | 26.4 | 32.9 |
| RMSD bonds (Å) | 0.010 | 0.013 |
| RMSD angles (°) | 1.1 | 1.3 |
| Ramachandran plot statistics (%) | ||
| Most favored region | 100.0 | 99.7 |
| Additional allowed region | 0.0 | 0.3 |
| Disallowed region | 0.0 | 0.0 |
Rmerge = ΣΣj|Ij − 〈I〉|/Σ|〈I〉|, where Ij and 〈I〉 represent the diffraction intensity values of the individual measurements and the corresponding mean values, respectively. The summation is over all unique measurements. Values given in parentheses refer to reflections in the outer-resolution shell: 1.76 to 1.70 Å for MPMV and 2.28 to 2.20 Å for XMRV.
, where Fobs and Fcalc are the observed and calculated structure factors, respectively. For Rfree, the sum is extended over a subset of reflections (5%) excluded from all stages of refinement.
RMSD, root mean square deviation.
Circular dichroism spectroscopy and thermal melts.
Circular dichroism (CD) spectral scans and thermal melts of fusion proteins (concentrations ranging from 0.2 to 2 mg/ml) were acquired on a Jasco J-810 spectropolarimeter using 1-mm quartz cuvettes (Helma). CD wavelength scans were collected at between 190 and 250 nm and averaged over five accumulations. Thermal denaturation assays were carried out at a single wavelength (222 nm) by increasing the temperature from 20°C to 99°C in 2°C/min increments. The heating of all retroviral fusion proteins to 99°C in a buffer containing 10 mM potassium phosphate (pH 7.5) and 150 mM NaCl was insufficient to denature the predominantly α-helical fusion proteins. A total of 1 M guanidine-HCl was added to all samples in order to fully denature MPMV TM and XMRV TM within a temperature range of 20°C to 99°C. The data were baseline corrected and plotted as a function of folded and unfolded states versus temperature. Melting temperature (Tm) values were calculated from thermal melt curves fitted by using a biphasic sigmoidal nonlinear regression algorithm in GraphPad.
Viral entry assays. (i) Cells and expression constructs.
All cell culture media, additives, and fetal bovine serum (FBS) were purchased from Life Technologies/Gibco. Human embryonic kidney HEK 293T cells (ATCC CRL-11268) were cultured in 1× Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (vol/vol) heat-inactivated FBS plus 1× antibiotic-antimycotic and propagated at 37°C in a 5% CO2 incubator. A single-cycle murine leukemia virus (MLV) pseudovirus was generated by using a multiplasmid expression system consisting of a shuttle vector for MLV, a MLV gag-pol-carrying plasmid, and a plasmid encoding the appropriate XMRV or MPMV envelope glycoprotein (13, 14). The MLV shuttle vectors are self-inactivating, packageable plasmids containing an enhanced green fluorescent protein (eGFP) reporter gene and a spleen focus-forming virus 5′ long terminal repeat (LTR) promoter. The MLV gag-pol plasmid contains the full gene (14, 15). The XMRV env gene was synthesized and cloned into pDISPLAY (Invitrogen). The expression plasmids for MPMV Env (pTMO) (16) and the MLV backbone were kindly provided by Eric Hunter (Emory University) and Marc-André Langlois (University of Ottawa). XMRV and MPMV Envs were fused with a hemagglutinin A (HA) epitope tag at their N and C termini, respectively, for detection. Viral glycoprotein mutants were generated by using the QuikChange II site-directed mutagenesis protocol.
(ii) Pseudovirus production.
A total of 1.5 × 106 HEK 293T cells were seeded into each 10-cm cell culture dish (Corning) and incubated overnight at 37°C with 5% CO2. At 40% confluence, growth medium was replaced with serum-free 1× DMEM. Expression vectors (2.4 μg packaging, 1.8 μg gag-pol, and 1.8 μg env plasmids) were mixed with Genejuice transfection agent (Millipore) at a 3:1 ratio of transfection agent to plasmid DNA. The transfection mixture was added dropwise onto HEK 293T cells and incubated for 6 h. Subsequently, cells were washed with 1× phosphate-buffered saline (PBS) and overlaid with 10 ml of 1× DMEM with 10% (vol/vol) FBS. Supernatants containing infectious viral particles were collected at 48 h posttransfection, filtered through 0.45-μm-pore-size polyvinylidene difluoride (PVDF) syringe filters, and concentrated 20-fold by using Amicon Ultra-15 centrifugal concentrators (100-kDa-molecular-mass cutoff; Millipore).
(iii) Virus titration.
An MLV core antigen enzyme-linked immunosorbent assay (ELISA) kit (Cell Biolabs) was used, according to the manufacturer's protocols, to determine the number of viral particles in the supernatant. Mutant pseudotyped viruses were measured by ELISA for viral particle amounts only and normalized to WT levels for infectivity assays. In addition, for WT Env pseudotyped viruses, biological titration assays were performed to measure the number of transducing viral particles. Briefly, 3 × 105 HEK 293T cells were seeded per well into a 6-well plate 24 h prior to infection. The concentrated virus-containing supernatants were serially diluted 1-, 10−1-, 10−2-, 10−3-, and 10−4-fold. Fifty microliters of XMRV or 100 μl of MPMV pseudovirus-containing samples was mixed with 500 μl of 1× DMEM, added to HEK 293T cells, and incubated for 90 min. Cells were then washed with 1× PBS and supplemented with and additional 2 ml of 1× DMEM with 10% (vol/vol) FBS. The percentage of eGFP-positive cells was analyzed by flow cytometry (FACSCalibur; BD Biosciences) at 24 h postinfection. The titer (in transducing units/ml) was calculated as [(% eGFP-positive cells/100) × number of cells transduced]/volume of virus (17).
(iv) Detection of GP expression on producer cells.
The expression levels of WT and mutant viral glycoproteins on HEK 293T producer cells were analyzed by immunoblotting. Here, Env-transfected cell monolayers were washed with PBS, and cells were lysed with NP-40 lysis buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, and 0.5% [vol/vol] NP-40) supplemented with 2× protease inhibitor cocktail (Roche). Cell lysates were normalized with Bradford assays and analyzed by anti-HA immunoblotting.
(v) Detection of Env incorporation into pseudotyped viruses.
The incorporation of WT or mutant viral Env into pseudotyped viruses was analyzed by immunoprecipitation (IP) and immunoblots. The amount of pseudotyped virus particles in the supernatant was estimated by an MLV p30 ELISA as described above. Equivalent amounts of WT and mutant pseudotyped virus particles were mixed with anti-HA antibody and protein A-agarose beads at 4°C for 2 h. Subsequently, the beads were washed five times with 1 ml TNEN buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% [vol/vol] NP-40) and subjected to immunoblot analysis, as described above.
(vi) Viral infectivity assay.
HEK 293T cells (3.0 × 105) were seeded into six-well plates and grown overnight at 37°C in 5% CO2. These target cells were overlaid with 0.5 ml of pseudovirus-containing DMEM (normalized titers, as described above) and incubated for 90 min. Subsequently, cells were washed with 1× PBS and overlaid with 2 ml of 1× DMEM with 10% (vol/vol) FBS. Samples were analyzed at 24 h postinfection by flow cytometry (FACSCalibur; BD Biosciences). Flow cytometry data were processed by using the FlowJo software package. Expression of eGFP was used to determine the efficiency of viral fusion. Experimental results are shown as the number of cells infected by virions containing the mutant viral glycoprotein divided by the number of cells infected by virions containing the WT glycoprotein, multiplied by 100 to obtain percentages. Each experiment was done in triplicate and repeated twice.
Accession numbers.
Atomic coordinates and structure factors for MPMV and XMRV TMs have been deposited in the Protein Data Bank (PDB) under accession numbers 4JF3 and 4JGS, respectively. All plasmids, along with sequences, have been deposited in the Addgene database under accession numbers 49087 (MPMV) (http://www.addgene.org/49087) and 49088 (XMRV) (http://www.addgene.org/49088).
RESULTS AND DISCUSSION
Structure determination.
We determined the crystal structures of the fusion core regions of the transmembrane proteins of Mason-Pfizer monkey virus (MPMV), a betaretrovirus, and xenotropic murine leukemia virus-like virus (XMRV), a gammaretrovirus. The MPMV and XMRV fusion cores lack the hydrophobic N-terminal fusion peptide and the membrane-associated transmembrane anchor (Fig. 1a). In addition, to prevent nonspecific disulfide bond formation, the third cysteine in the CX6CC motif was mutated to serine (MPMV, C483S; XMRV, C538S). This third cysteine is thought to mediate a disulfide linkage between the SU and TM subunits, similar to Ebola virus glycoprotein (GP) (18). Both MPMV and XMRV TMs purify as a clear trimer by size-exclusion chromatography. The resulting MPMV and XMRV TM structures (Fig. 1b) were refined against data at 1.7-Å and 2.2-Å resolution, respectively (Table 1). Electron density maps revealed clear density for residues 421 to 512 and 476 to 563 in MPMV and XMRV TMs, respectively (Fig. 1c). Weak electron density was observed for the first 16 residues in XMRV TM. MPMV has a predicted N-linked glycosylation site at Asn487 in its fusion subunit. However, our crystallized MPMV TM was expressed in bacteria and therefore lacks N-linked glycosylation. This is not expected to affect the overall structure of MPMV TM given that Asn487 resides in the tether loop region rather than the more structured HR1 or HR2 region. The MPMV TM described here is the first fusion protein structure from the betaretrovirus family, and the XMRV TM structure now provides a complete picture of fusion cores from gammaretroviruses, as the previously determined MoMLV TM structure (6) was missing the entire C-terminal region of the molecule.
FIG 1.

Structures of retroviral fusion glycoproteins. (a) General diagram of a CX6CC-containing retroviral envelope glycoprotein. MPMV and XMRV Envs are synthesized as a single polypeptide precursor that is posttranslationally cleaved to form the SU and TM subunits. The SU subunit of MPMV and XMRV typically contains 6 and 10 N-linked glycans (red Y symbols), respectively, and is the subunit responsible for attachment to the host receptor. The MPMV Env N-linked glycosylation pattern is shown on the schematic. The TM subunit contains a single N-linked glycan, a hydrophobic fusion peptide (FP), and two α-helical heptad repeat regions (HR1 and HR2). The two heptad repeat regions are separated by a chain-reversing CX6CC motif and a short tether region. The first and second cysteines in the CX6CC motif form an intrasubunit disulfide bond, and the third cysteine forms an intersubunit disulfide linkage with a cysteine in the SU subunit. The carboxy terminus contains a transmembrane (TM) anchor and a cytoplasmic tail (CT) that interacts with the retroviral Gag protein. Fusion subunit regions included in the construct for structural studies are shown in color. (b) Ribbon representation of the beta- and gammaretrovirus fusion cores. The intrasubunit disulfide bonds are indicated as sticks. (c) Representative σA-weighted 2Fo − Fc electron density map contoured at 1σ, superimposed with the final MPMV TM and XMRV TM refined model (the N-terminal HR1 portion is shown). Overall, the electron density is well defined throughout the HR1, HR2, and chain reversal regions. (d) Structural similarity of retroviral fusion subunits. Shown is a superimposition of the fusion subunits from HTLV-1 (PDB accession number 1MG1), MPMV (PDB accession number 4JF3), and XMRV (PDB accession number 4JGS).
Structure of retroviral fusion glycoproteins.
All retroviral fusion glycoprotein subunits that have been biochemically characterized adopt a trimeric structure. Each monomer contains one long central α-helical region and a second shorter α-helix at its C terminus (Fig. 1b). These two helices are termed HR1 and HR2, respectively, and each helix consists of a repeating pattern of 7 amino acids (a, b, c, d, e, f, and g) with hydrophobic residues at the first and fourth positions (underlined). Hydrophobic leucine and isoleucine residues on the HR1 α-helical region align in regular knobs-into-hole packing to form a long, central trimeric core. In all of the retroviral TMs, a single asparagine layer in HR1 disrupts the leucine/isoleucine knob-in-hole packing (19) to make contact with a putative chloride ion that may act as a switch to allow conformational changes necessary for fusion with the host cell membrane. In the deltaretroviral TMs, a second asparagine layer coordinates an ordered water molecule (4). At the base of the HR1 core, a short 310 helix and a disulfide bond-stabilized, single-turn α-helix form the chain reversal region, where the polypeptide chain completes a 180° turn (Fig. 1b). The polypeptide chain then extends into a random coil that tethers the HR2 helix. The helical content of HR2 varies from six turns in MPMV TM to five turns in XMRV TM. The HR2 helix packs into the grooves formed between two HR1 helices to form the six-helix bundle that is widely considered to be the postfusion conformation. The packing of HR2 into HR1 brings the amino and carboxy termini together, positioning the virus and the host membrane-interacting segments of the full-length viral glycoprotein into close proximity. The MPMV and XMRV TMs have a high degree of overall structural similarity to the HTLV-1 and BLV fusion subunits (root mean squared deviation = ∼1.6 Å) (4, 5) (Fig. 1d).
Identification of intra- and intersubunit salt bridges.
Structural comparison of the MPMV and XMRV TMs revealed an intricate network of intra- and interchain electrostatic interactions (Fig. 2a). These interactions were not previously identified, as the HR2 regions in previously crystallized retroviral TMs were missing. Retrovirus electrostatic interactions cluster into two distinct regions on the TM fusion core: (i) the heptad repeat and (ii) chain reversal regions.
FIG 2.

Intra- and complex intermolecular retroviral salt bridges. (a) Ribbon diagram of MPMV and XMRV TMs. Electrostatic interactions are shown in the insets. Residues labeled with asterisks are contributed by a neighboring subunit. (b) Relative infectivities of wild-type and salt bridge mutant pseudotyped retroviruses. Data represent the averages of data from at least six independent experiments. (c) Thermal denaturation of the indicated wild-type and mutant retroviral fusion subunits. Data points were fitted to a nonlinear biphasic sigmoidal curve in GraphPad.
(i) Heptad repeat region.
The binding of HR2 to HR1 is stabilized through multiple single and complex-type salt bridges (Fig. 2a). In MPMV TM, an intramolecular electrostatic interaction (D435-R506) is formed at the top of the fusion subunit, and the central HR region is anchored by a series of complex, interchain electrostatic interactions. In the central HR1, three sets of complex salt bridges are formed (K495-D445-K488, D445-K488-D448, and K488-D448-R493). In XMRV TM, three separate pairs of salt bridges span the heptad repeat region (D490-R557, E495-R555, and E502-R548); it lacks the complex salt bridge in the middle of the HR1 core observed for the MPMV TM. In all of the retroviral fusion proteins, the HR1 trimer is rich in negatively charged aspartate or glutamate residues, and these residues cluster to form layers of anionic charge (Fig. 3). On the counterpart HR2 helical surface in retroviral TMs, a cationic surface complementary to the negative charge is found. These positively and negatively charged residues are highly conserved within their own retroviral family.
FIG 3.
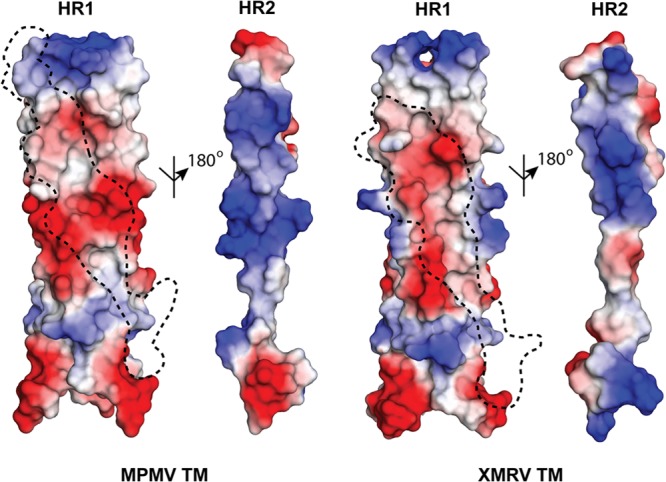
Surface electrostatic potential of the HR1-HR2 interface. A negatively charged surface is displayed on the MPMV and XMRV HR1 trimers and is complementary to a positively charged surface on HR2. The footprint of the HR2 helix on the HR1 trimeric core is shown by a dashed line. Red and blue regions denote negative and positive charges, respectively.
(ii) Chain reversal region.
Both MPMV and XMRV fusion proteins contain a single, structurally conserved, intersubunit arginine-glutamate electrostatic pair (MPMV TM, R462-E480; XMRV TM, R517-E535) in the chain reversal region (Fig. 2a). The arginine residue is present at the base of the HR1 region, whereas a glutamate resides on a loop after the single-turn α-helix in the CX6CC motif.
Electrostatic stapling of HR1-HR2 is necessary for retroviral entry.
The role of each retroviral salt bridge was quantified in a cell-based viral entry assay. Single-cycle infectious viral particles containing the MPMV or XMRV full-length wild-type or mutant envelope glycoprotein were produced. Wild-type MPMV and XMRV Env pseudovirions were tested against human embryonic kidney HEK 293T cells. HEK 293T cells exhibited high infectivity levels, consistent with previous studies (20, 21), and were used for all viral infectivity studies. Immunoprecipitation and immunoblot analyses of the producer cells and pseudotyped viruses revealed that mutations of the full-length retroviral glycoproteins did not affect processing or incorporation into virions (Fig. 4). Electron microscopy of the pseudovirions revealed a typical retrovirus particle structure approximately 100 nm in diameter and proper formation of the retroviral capsid and envelope (data not shown). Infectivity levels measured by flow cytometry demonstrated that alanine point mutations in any of the salt bridges in the central parts of the HR or chain reversal regions completely abolished the entry of retroviral particles into host cells or reduced it to very low levels (Fig. 2b and Table 2). Mutations of salt bridges located at the membrane-proximal ends of HR1 and HR2 (MPMV TM, D435-R506) inhibited but did not completely block entry. Inter- and intrasubunit salt bridges that anchor the membrane-distal and central portions of the heptad repeats and those within the chain reversal region were highly sensitive to mutagenic disruption, and thus, these electrostatic interactions are critical to the viral entry mechanism. The striking all-or-none effect suggests that the loss of a single electrostatic interaction, which typically contributes 3 to 5 kcal/mol of energy for binding (22, 23), may be enough to prevent crossing of the activation barrier threshold for the formation of the postfusion conformation.
FIG 4.

Characterization of MPMV- and XMRV-pseudotyped virions. WT and mutant MPMV or XMRV pseudovirions were normalized by an MLV p30 ELISA and separated by SDS-PAGE. Expression of WT or mutant Env was detected in pseudovirions and producer cells by immunoblot analysis using an anti-HA primary antibody. All pseudovirion samples were nonreduced, and the mature covalently attached MPMV and XMRV Envs (MPMV, gp86; XMRV, gp85) were detected. In the producer cell lysate, the MPMV and XMRV Envs do not dissociate into the SU and TM subunits under reducing conditions, suggesting the presence of the uncleaved precursor glycoprotein (MPMV, pr86; XMRV, pr85). Mock is a separate experiment consisting of cotransfection of the MLV backbone and gag-pol vectors; no envelope glycoprotein vector was transfected. β-Actin was used as a loading control.
TABLE 2.
Summary of retroviral fusion protein mutations
| Virus and mutation | Protein expression/viral incorporationb | Relative infectivity (%) | Tm1 (°C) | Tm2 (°C) | Location |
|---|---|---|---|---|---|
| MPMV (betaretrovirus) | |||||
| Wild type | +++++/+++++ | 100.0 | n/ac | 78 | |
| D435A | +++++/+++++ | 12.7 | 55a | 78 | HR1 top |
| D445A | +++++/+++++ | 0.0 | 54a | 74 | HR1 central |
| D448A | +++++/+++++ | 0.0 | 53a | 73 | HR1 bottom |
| R462A | +++++/+++++ | 0.0 | n/a | 71 | Chain reversal region |
| E480A | ++++/++++ | 0.0 | 51a | 70 | Chain reversal region |
| K488A | +++++/+++++ | 0.0 | 51a | 70 | HR2 bottom |
| R493A | +++++/+++++ | 0.0 | n/a | 69 | HR2 bottom |
| K495A | +++++/+++++ | 0.0 | n/a | 78 | HR2 central |
| R506A | +++++/+++++ | 10.0 | n/a | 73 | HR2 top |
| XMRV (gammaretrovirus) | |||||
| Wild type | +++++/+++++ | 100.0 | n/a | 93 | |
| D490A | +++++/+++++ | 0.0 | 62 | 89 | HR1 top |
| E495A | +++++/+++++ | 0.0 | 66 | 89 | HR1 central |
| E502A | +++++/+++++ | 30.5 | 64 | 89 | HR1 bottom |
| R517A | +++++/+++++ | 0.0 | n/a | 83 | Chain reversal region |
| E535A | +++++/+++++ | 5.4 | n/a | 83 | Chain reversal region |
| R548A | +++++/+++++ | 0.0 | 67 | 85 | HR2 bottom |
| R555A | +++++/+++++ | 0.0 | 67 | 86 | HR2 central |
| R557A | +++++/+++++ | 0.0 | n/a | 86 | HR2 top |
The Tm value is an estimate.
+ denotes the level of expression.
n/a, not applicable.
Electrostatic staples stabilize the structure of the heptad repeat regions.
Circular dichroism (CD) spectroscopy is an excellent tool to investigate protein secondary structure and the folding and unfolding of macromolecules as a function of temperature. α-Helices display negative CD spectral peaks at 208 nm and 222 nm, whereas the β-sheet or random-coil conformation is detected as single minimum at 215 nm or below 210 nm, respectively. CD spectra of the wild-type and mutant MPMV and XMRV fusion proteins exhibited the typical double minima at 208 nm and 222 nm, characteristic of their predominantly α-helical secondary structures. The CD signatures for all retroviral fusion mutants were similar (data not shown), suggesting that the relative α-helical content did not change as a result of the targeted mutations.
The stabilities of retroviral fusion glycoproteins and mutants were determined by monitoring the decrease in helical content at 222 nm as a function of temperature over the range from 20°C to 99°C (Table 2). The thermal unfolding of all retroviral fusion proteins was irreversible. Although thermodynamic properties cannot be obtained from an irreversible thermal melt, the estimation of an apparent melting temperature (Tm) serves as a simple measure of protein stability. All wild-type retroviral fusion subunits unfolded cooperatively and were highly stable, with apparent Tm values above 75°C. The high melting temperatures are consistent with the established stabilities of other viral fusion proteins (24–26).
Mutations of salt bridges in the heptad repeat regions in XMRV TM resulted in pronounced biphasic transitions in the thermal denaturation curves, whereas wild-type proteins and the chain reversal region salt bridge mutations displayed only single transitions. We hypothesize that in the first transition, Tm1, the HR1 trimeric core remained intact, and the loss of CD signal resulted from unfolding of the HR2 helix (Fig. 2c). The second, higher-temperature (>80°C) transition (Tm2) is likely due to unfolding of the HR1 helix core. In XMRV TM, salt bridge mutations in the HR region resulted in 27°C to 31°C decreases in Tm1 compared to the wild type. Specifically, mutations of residues on the cation pocket (R548, R555, and R557 in XMRV TM) revealed a decrease of ∼30% in the CD signal, which corresponded well with the total α-helical content in the HR2 region. In addition, this was consistent with the structural models, as these residues were predicted only to destabilize HR2 binding to HR1. It is interesting to note that mutations in the HR1 anion pocket (XMRV TM D490A, E495A, and E502A) resulted in a larger loss in the CD signal (50 to 60%). This suggests that mutations of the negatively charged aspartate or glutamate residues in the HR1 helix destabilize the binding of both HR2 and also, likely, the HR1 trimeric core. In MPMV TM, the two-phase transitions of HR mutants were not as pronounced as those seen in XMRV TM mutants (Fig. 2c); however, there was a decrease in the CD signal resulting in a downward shift of the apparent Tm. Given the poorly defined melting transition, we were not able to accurately determine the apparent Tm1 and Tm2 values for MPMV TM. The lack of a clear biphasic transition in MPMV TM was likely due to the higher number of interchain complex salt bridges in the HR region than in XMRV TM. Overall, our results suggest that extensive ion pair networks in the HR region provide a general strategy to influence the thermal stability of retroviral fusion subunits.
In the chain reversal region, all salt bridge mutations had a single transition thermal denaturation profile (Fig. 2c). A single transition was expected, as mutations of the chain reversal region salt bridges should not significantly affect the folding or assembly of HR2 onto the HR1 core. In general, mutations of chain reversal salt bridges had more modest effects on the melting temperature (ΔTm of ∼10°C), with the exception of the MPMV TM E480A mutation, than mutations in the HR regions (ΔTm of >25°C). Our results suggest that the chain reversal ion pair plays a role in stabilizing the fusion subunit but not as large an extent as the electrostatics found in the HR1 or HR2 region.
Implications of electrostatic interactions for retroviral fusion.
Retroviral envelope glycoproteins may be divided into two types: ones that contain a disulfide bond between the SU and TM subunits and ones that are noncovalently associated (27). Covalent-type retroviral Env contains a conserved immunosuppressive domain (ISD) and a CX6CC motif in the chain reversal region of its fusion subunit. CX6CC-containing glycoproteins are commonly observed within the alpha-, delta-, and gammaretroviral genera but not in lentiviruses or spumaviruses (Fig. 5). The betaretrovirus genus is unique from the others, as examples of both covalent-type and non-covalent-type viral glycoproteins are observed (27). For instance, MPMV Env, which contains a CX6CC motif, is thought to be derived from a gammaretrovirus through a recombination event. On the other hand, mouse mammary tumor virus (MMTV) and Jaagsiekte sheep retrovirus (JSRV) belong to the betaretrovirus genus, but the SU and TM subunits are noncovalently associated (27), similar to the lentiviral envelope glycoproteins (HIV-1 gp160).
FIG 5.

Sequence alignment of retroviral fusion subunits. Sequence alignments are separated into covalent-type (a) and non-covalent-type (b) retroviral envelope glycoproteins. Secondary structural definitions and residue numbering shown above the alignments are based on HTLV-1 gp21 and HIV-1 gp41 for the covalently and noncovalently attached Envs, respectively. Residues involved in electrostatic interactions are labeled with an asterisk, and those that are conserved across and within each beta-, delta- and gammaretroviral family are highlighted in green and yellow, respectively. Abbreviations are as follows: HTLV-1/2, human T-lymphotropic virus type 1/2; STLV-1/2/3, simian T-cell leukemia virus type 1/2/3; BLV, bovine leukemia virus; MPMV, Mason-Pfizer monkey virus; SMRV, squirrel monkey retrovirus; SRV-1/2, simian retrovirus; RD114, feline endogenous retrovirus; REV, reticuloendotheliosis virus; SNV, spleen necrosis virus; GALV, gibbon ape leukemia virus; KORV, koala retrovirus; PERV, porcine endogenous retrovirus; XMRV, xenotropic murine leukemia virus-like virus; MoMLV, Moloney murine leukemia virus; KiMSV, Kirsten murine sarcoma virus; FeLV, feline leukemia virus; Ga-FeSV, Gardner-Arnstein feline sarcoma virus; ASLV, avian sarcoma leukosis virus; RSV, Rous sarcoma virus; HIV-1, human immunodeficiency virus type 1; SIV, simian immunodeficiency virus; MMTV, mouse mammary tumor virus; JSRV, Jaagsiekte sheep retrovirus.
Electrostatic interactions are of fundamental importance for protein folding and function. The MPMV and XMRV retroviral fusion subunits are rich with salt bridges that form “electrostatic staples.” Each retroviral fusion subunit contains at least four sets of salt bridges, and each electrostatic interaction plays an important role in the formation of the postfusion state. Our sequence alignment of retroviral TMs demonstrated that all covalent-type fusion subunits contain a high degree of sequence similarity, in particular residues at the C-terminal end of the HR1 helix and those within the chain reversal region (Fig. 5a). Sequence alignments with the non-covalent-type Env, such as HIV-1, JSRV, and MMTV, reveal poor overall sequence conservation (Fig. 5b). For instance, MMTV TM has 30% sequence identity with JSRV TM but only ∼6% sequence identity with the covalent-type MPMV TM. Moreover, the non-covalent-type fusion subunits are different in size and physiochemical properties from the CX6CC-containing fusion subunits. Our crystal structure of MPMV TM is a representative model for other covalent-type betaretrovirus TM proteins, but it is difficult to predict the position or roles of salt bridges from non-covalent-type betaretroviral TMs without a crystal structure.
The intricate network of intra- and intersubunit electrostatic interactions found at the HR1/HR2 and chain reversal regions of MPMV and XMRV TMs are strictly conserved within their respective retroviral families. However, an electrostatic salt bridge formed at the membrane-proximal end of the HR1/HR2 core (HTLV-1 TM, D353-R416; MPMV TM, D435-R506; XMRV TM, D490-R557) and an intersubunit electrostatic pair at the chain reveal region (HTLV-1 TM, R380-E398; MPMV TM, R462-E480; XMRV TM, R517-E535) are structurally conserved in all covalent-type beta-, delta-, and gammaretrovirus fusion subunits (Fig. 5b). Given the structural conservation, this suggests that electrostatic interactions may be required at both ends of the fusion protein for entry.
Due to the long-range attractive effects of electrostatic interactions, we suggest that the retroviral salt bridges help locate, position, and stabilize the interactions of HR1 and HR2, acting in a manner analogous to a zipper. Cryo-electron microscopy (EM) reconstructions of the prefusion Moloney murine leukemia virus Env revealed a tripod-like structure that implies the HR2 regions of the fusion subunit are splayed out from the central glycoprotein core (28). In order to form the postfusion six-helix bundle, the HR2 region must undergo significant conformational changes to allow its packing into the HR1 trimeric core. The chain reversal region is the point at which the fusion subunit polypeptide chain reverses direction. Thus, after triggering of the retroviral glycoprotein, it is logical that the CR region is important in initiating the conformational changes to form the postfusion state. We hypothesize that the CR region salt bridge (MPMV, R462-E480; XMRV, R517-E535) directs the initial alignment and anchors the turn in order to facilitate HR2 to pack into the prehairpin HR1 intermediate. In addition, the CR region salt bridge is intersubunit in nature; thus, the formation of one salt bridge will have the potential to confer a concerted conformational change to the other subunits. The role of the retroviral CR region salt bridge is analogous to the role of a slider in a zipper in fastening the base and aligning the teeth. This interpretation accounts for the comparably low infectivity levels of both the mutated CR and HR1/HR2 core pseudotyped virions despite significant differences in thermal stability. Thus, stabilization of the chain reversal region during the fusion event appears to be just as important to infectivity as stabilization of the final postfusion conformation. Previous work on HTLV-1 gp21 also suggested that the R380-E398 chain reversal salt bridge may stabilize the disulfide-bonded chain reversal region (29). The electrostatic interactions located between the HR1 and HR2 helices are equivalent to the teeth of the zipper to fasten and stabilize the postfusion six-helix bundle once HR2 packs into the HR1 trimeric core.
The extensive use of electrostatic interactions to stabilize the postfusion structure is consistent with the biology of retroviruses. For retroviruses entering the plasma membrane at neutral pH, electrostatic interactions would have a large effect, as acidic and basic residues would be fully ionized to stabilize the six-helix bundle. Notably, a conserved salt bridge (Lys574-Asp632) between the N- and C-terminal heptad repeat regions of HIV-1 gp41 was shown to be critical for stability and viral entry (30, 31). Moreover, synthetic peptides and small-molecule inhibitors targeting the gp41 salt bridge demonstrated potent antiviral activity against HIV-1-mediated membrane fusion (32, 33). Many members of the paramyxovirus family, such as respiratory syncytial virus and parainfluenza virus, are also thought to undergo a pH-independent mechanism of entry (34, 35). The postfusion structures of paramyxovirus family F glycoproteins also reveal the presence of electrostatic interactions in their heptad repeat regions (36, 37), supporting a trend that electrostatic interactions may be an important feature in fusion subunits that function in a pH-independent manner.
In contrast, the role of electrostatic interactions in class I fusion proteins from viruses that enter through an endosomal pathway is not clearly defined. In influenza A virus (IAV) and lymphocytic choriomeningitis virus (LCMV), a number of salt bridges are observed in the fusion protein subunit (38, 39). It is likely that these salt bridges play a role in viral entry, as mutation of the LCMV-equivalent salt bridge in another arenavirus (Junin virus) resulted in reduced viral infectivity (40). No thermal denaturation studies on these electrostatic interactions are available, and thus, its role in stabilizing the postfusion six-helix bundle is unclear. However, recent work on avian sarcoma leukosis virus (ASLV) TM may provide insights into the role of fusion protein salt bridges from viruses that are low-pH dependent (41). ASLV is an alpharetrovirus that undergoes a two-step entry mechanism with receptor binding occurring at the plasma membrane followed by trafficking and fusion at the endosome (42). ALSV TM is lined with at least three pairs of salt bridges; however, alanine-scanning mutations failed to significantly reduce postfusion TM stability. Instead, hydrophobic residues were identified as the key to stabilizing the ASLV TM six-helix bundle. The use of nonionizable residues to stabilize the ASLV postfusion glycoprotein makes biochemical sense, as hydrophobic interactions will be unaffected by pH changes when the virus migrates to the endosome, whereas electrostatic interactions will have reduced effects at acidic pH. It is tempting to speculate that electrostatic interactions in fusion subunits from pH-dependent viruses are not involved in stabilizing the postfusion state; the precise role of these interactions await further studies.
Conclusions.
In this study, we determined the structures of the beta- and gammaretrovirus fusion subunits, thus completing a structural library of pH-independent retrovirus fusion proteins. Structural analysis of the fusion proteins reveals an abundance of electrostatic interactions in the heptad repeat and chain reversal regions. The conserved nature of the ion pairs suggests that electrostatic stapling is critical to stabilize the postfusion six-helix bundle. The elimination of the MPMV and XMRV TM salt bridges destabilized the fusion core and fully blocked entry in a cell-based assay. Disruption of these electrostatic staples presumably imbalance the energetics required to overcome the large activation barrier to fuse the viral and host lipid bilayers. Our results describe a key feature of the class I retroviral fusion machinery and improves our overall understanding of mechanisms of viral entry.
ACKNOWLEDGMENTS
This work was supported by a Canadian Institutes of Health Research (CIHR) operating grant (MOP-115066), a Canada Research Chair in Structural Virology, and a CIHR New Investigator award (MSH-113554) to J.E.L. Moreover, support for stipends were provided by University of Toronto graduate fellowships to H.A. and J.D.C. and an Ontario graduate scholarship to J.D.C.
We thank the Molecular Structure and Function program at the Hospital for Sick Children Research Institute (Toronto, ON, Canada) for access to a circular dichroism spectrometer; Aiping Dong, Cheryl Arrowsmith, and Aled Edwards for access to the Structural Genomics Consortium X-ray diffraction facility; and Farshad Azimi for technical discussions and critical reading of the manuscript. In addition, we are indebted for the generosity of Eric Hunter (Emory University) for the MPMV Env expression construct and Marc-André Langlois (University of Ottawa) for the MLV backbone.
We declare that we have no competing financial interests.
Footnotes
Published ahead of print 16 October 2013
REFERENCES
- 1.Plemper RK. 2011. Cell entry of enveloped viruses. Curr. Opin. Virol. 1:92–100. 10.1016/j.coviro.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219. 10.1080/10409230802058320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. 10.1038/nsmb.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobe B, Center RJ, Kemp BE, Poumbourios P. 1999. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc. Natl. Acad. Sci. U. S. A. 96:4319–4324. 10.1073/pnas.96.8.4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamb D, Schuttelkopf AW, van Aalten DM, Brighty DW. 2011. Charge-surrounded pockets and electrostatic interactions with small ions modulate the activity of retroviral fusion proteins. PLoS Pathog. 7:e1001268. 10.1371/journal.ppat.1001268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fass D, Harrison SC, Kim PS. 1996. Retrovirus envelope domain at 1.7 angstrom resolution. Nat. Struct. Biol. 3:465–469. 10.1038/nsb0596-465 [DOI] [PubMed] [Google Scholar]
- 7.Tropea JE, Cherry S, Waugh DS. 2009. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol. Biol. 498:297–307. 10.1007/978-1-59745-196-3_19 [DOI] [PubMed] [Google Scholar]
- 8.Pflugrath JW. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55:1718–1725 [DOI] [PubMed] [Google Scholar]
- 9.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renard M, Varela PF, Letzelter C, Duquerroy S, Rey FA, Heidmann T. 2005. Crystal structure of a pivotal domain of human syncytin-2, a 40 million years old endogenous retrovirus fusogenic envelope gene captured by primates. J. Mol. Biol. 352:1029–1034. 10.1016/j.jmb.2005.07.058 [DOI] [PubMed] [Google Scholar]
- 11.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 12.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58:1948–1954. 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 13.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. 10.1016/S0092-8674(03)00423-9 [DOI] [PubMed] [Google Scholar]
- 14.Langlois MA, Beale RC, Conticello SG, Neuberger MS. 2005. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33:1913–1923. 10.1093/nar/gki343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 13:2009–2013. 10.1016/j.cub.2003.10.034 [DOI] [PubMed] [Google Scholar]
- 16.Brody BA, Kimball MG, Hunter E. 1994. Mutations within the transmembrane glycoprotein of Mason-Pfizer monkey virus: loss of SU-TM association and effects on infectivity. Virology 202:673–683. 10.1006/viro.1994.1389 [DOI] [PubMed] [Google Scholar]
- 17.Barde I, Salmon P, Trono D. 2010. Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. Chapter 4:Unit 4.21. 10.1002/0471142301.ns0421s53 [DOI] [PubMed] [Google Scholar]
- 18.Lee JE, Fusco MH, Hessell AJ, Oswald WB, Burton DR, Saphire EO. 2008. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454:177–182. 10.1038/nature07082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walshaw J, Woolfson DN. 2003. Extended knobs-into-holes packing in classical and complex coiled-coil assemblies. J. Struct. Biol. 144:349–361. 10.1016/j.jsb.2003.10.014 [DOI] [PubMed] [Google Scholar]
- 20.Stieler K, Schulz C, Lavanya M, Aepfelbacher M, Stocking C, Fischer N. 2010. Host range and cellular tropism of the human exogenous gammaretrovirus XMRV. Virology 399:23–30. 10.1016/j.virol.2009.12.028 [DOI] [PubMed] [Google Scholar]
- 21.Diehl WE, Stansell E, Kaiser SM, Emerman M, Hunter E. 2008. Identification of postentry restrictions to Mason-Pfizer monkey virus infection in New World monkey cells. J. Virol. 82:11140–11151. 10.1128/JVI.00269-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson DE, Becktel WJ, Dahlquist FW. 1990. pH-induced denaturation of proteins: a single salt bridge contributes 3-5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 29:2403–2408. 10.1021/bi00461a025 [DOI] [PubMed] [Google Scholar]
- 23.Fersht AR. 1972. Conformational equilibria in -and -chymotrypsin. The energetics and importance of the salt bridge. J. Mol. Biol. 64:497–509 [DOI] [PubMed] [Google Scholar]
- 24.Harrison JS, Higgins CD, Chandran K, Lai JR. 2011. Designed protein mimics of the Ebola virus glycoprotein GP2 alpha-helical bundle: stability and pH effects. Protein Sci. 20:1587–1596. 10.1002/pro.688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison JS, Koellhoffer JF, Chandran K, Lai JR. 2012. Marburg virus glycoprotein GP2: pH-dependent stability of the ectodomain alpha-helical bundle. Biochemistry 51:2515–2525. 10.1021/bi3000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weissenhorn W, Wharton SA, Calder LJ, Earl PL, Moss B, Aliprandis E, Skehel JJ, Wiley DC. 1996. The ectodomain of HIV-1 env subunit gp41 forms a soluble, alpha-helical, rod-like oligomer in the absence of gp120 and the N-terminal fusion peptide. EMBO J. 15:1507–1514 [PMC free article] [PubMed] [Google Scholar]
- 27.Henzy JE, Coffin JM. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J. Virol. 87:1937–1946. 10.1128/JVI.01442-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forster F, Medalia O, Zauberman N, Baumeister W, Fass D. 2005. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc. Natl. Acad. Sci. U. S. A. 102:4729–4734. 10.1073/pnas.0409178102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maerz AL, Center RJ, Kemp BE, Kobe B, Poumbourios P. 2000. Functional implications of the human T-lymphotropic virus type 1 transmembrane glycoprotein helical hairpin structure. J. Virol. 74:6614–6621. 10.1128/JVI.74.14.6614-6621.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Y, Liu S, Jing W, Lu H, Cai D, Chin DJ, Debnath AK, Kirchhoff F, Jiang S. 2007. Conserved residue Lys574 in the cavity of HIV-1 Gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J. Biol. Chem. 282:25631–25639. 10.1074/jbc.M703781200 [DOI] [PubMed] [Google Scholar]
- 31.He Y, Liu S, Li J, Lu H, Qi Z, Liu Z, Debnath AK, Jiang S. 2008. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J. Virol. 82:11129–11139. 10.1128/JVI.01060-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. U. S. A. 95:15613–15617. 10.1073/pnas.95.26.15613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. 1999. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–115. 10.1016/S0092-8674(00)80066-5 [DOI] [PubMed] [Google Scholar]
- 34.Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 35.Srinivasakumar N, Ogra PL, Flanagan TD. 1991. Characteristics of fusion of respiratory syncytial virus with HEp-2 cells as measured by R18 fluorescence dequenching assay. J. Virol. 65:4063–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 85:7788–7796. 10.1128/JVI.00555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293. 10.1073/pnas.0503989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Igonet S, Vaney MC, Vonhrein C, Bricogne G, Stura EA, Hengartner H, Eschli B, Rey FA. 2011. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc. Natl. Acad. Sci. U. S. A. 108:19967–19972. 10.1073/pnas.1108910108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rachakonda PS, Veit M, Korte T, Ludwig K, Bottcher C, Huang Q, Schmidt MF, Herrmann A. 2007. The relevance of salt bridges for the stability of the influenza virus hemagglutinin. FASEB J. 21:995–1002. 10.1096/fj.06-7052hyp [DOI] [PubMed] [Google Scholar]
- 40.York J, Agnihothram SS, Romanowski V, Nunberg JH. 2005. Genetic analysis of heptad-repeat regions in the G2 fusion subunit of the Junin arenavirus envelope glycoprotein. Virology 343:267–274. 10.1016/j.virol.2005.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aydin H, Smrke BM, Lee JE. 13 September 2013. Structural characterization of a fusion glycoprotein from a retrovirus that undergoes a hybrid 2-step entry mechanism. FASEB J. 10.1096/fj.13-2323-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mothes W, Boerger AL, Narayan S, Cunningham JM, Young JA. 2000. Retroviral entry mediated by receptor priming and low pH triggering of an envelope glycoprotein. Cell 103:679–689. 10.1016/S0092-8674(00)00170-7 [DOI] [PubMed] [Google Scholar]