

Abstract
Ceramides are known to be a key component of the stratum corneum, the outermost protective layer of the skin that controls barrier function. In this work, molecular dynamics simulations are used to examine the behavior of ceramide bilayers, focusing on non-hydroxy sphingosine (NS) and non-hydroxy phytosphingosine (NP) ceramides. Here, we propose a modified version of the CHARMM force field for ceramide simulation, which is directly compared to the more commonly used GROMOS-based force field of Berger (Biophys. J. 1997, 72); while both force fields are shown to closely match experiment from a structural standpoint at the physiological temperature of skin, the modified CHARMM force field is better able to capture the thermotropic phase transitions observed in experiment. The role of ceramide chemistry and its impact on structural ordering is examined by comparing ceramide NS to NP, using the validated CHARMM-based force field. These simulations demonstrate that changing from ceramide NS to NP results in changes to the orientation of the OH groups in the lipid headgroups. The arrangement of OH groups perpendicular to the bilayer normal for ceramide NP, verse parallel for NS, results in the formation of a distinct hydrogen bonding network, that is ultimately responsible for shifting the gel-to-liquid phase transition to higher temperature, in direct agreement with experiment.
Keywords: ceramides, force field comparison, phase transition behavior, head group chemistry
I. INTRODUCTION
The ability of the skin to regulate bodily functions and act as an effective barrier to chemical penetrants is controlled by the thin, outermost layer, known as the stratum corneum. The stratum corneum consists of dead, impenetrable skin cells (corneocytes) that are surrounded by a rich lipid matrix arranged in a brick-and-mortar like configuration.1 While the lipids of the stratum corneum are known to be predominantly composed of ceramides (CER), cholesterol, and free fatty acids, the molecular level organization of the skin lipids and their role in maintaining the barrier function of the skin is not well understood.2 It is, however, known that CERs play a crucial role in maintaining the physiological function of the stratum corneum3 as a number of skin diseases1, 4 are known to be related to reduced CER levels in this layer.
Experimentally, twelve distinct CERs have been observed in the stratum corneum of human skin.1, 2, 5–7 CERs are a class of double-tailed lipids consisting of hydrophobic acyl and sphingosine tails joined together by a hydrophilic head group. Based on the functional group attached to the sphingosine tail, CERs can be classified into sphingosines (S), phytosphingosines (P) and hydroxysphingosines (H). Sphingosines have an alkene bond in the sphinogsine tail; phytosphingosines have the alkene group replaced by a hydroxyl group; and in hydroxysphingosines a α-hydroxyl group is connected to the alkene bond in the corresponding tail. The structure of the sphingosine and phytosphingosine CERs studied in this work are shown in figure 1a and b, respectively. CERs can be further classified as either non-hydroxy (N) or α-hydroxy (A) CERs based on whether a hydroxyl group is attached to the carbon beside the carbonyl bond of the fatty acid tail. The subtle differences in molecular structure have been shown to influence phase transition temperatures and other characteristic properties of CER based bilayers.7–13 Furthermore, while the length of the sphingoid tail is fairly constant at 16 carbons, the fatty acid tail exhibits considerable polydispersity in length; in human stratum corneum the fatty acid tails range from 24 to 34 carbons2, 3, 5 resulting is considerable asymmetry between the lengths of the two tails.
Figure 1.
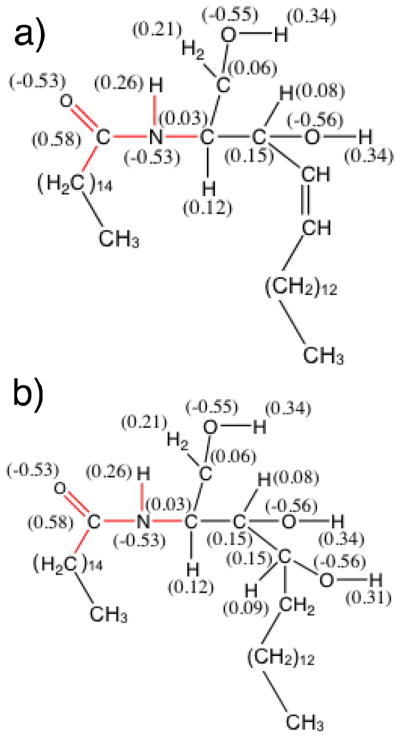
The molecular structure of (a) CER NS C16, which belongs to the non-hydroxy sphingosine class of ceramides, and (b) CER NP C16, a non-hydroxy phytosphingosine. The notation C16 is used to indicate the length of the CER acyl chain. The red in the figure indicates the bonded interactions of the CER molecule for which CHARMM parameters are proposed in this work. Numerical label in parenthesis represents partial charges determined from ab initio calculations.
The diversity of CERs that are found in the stratum corneum makes the investigation of their structure and behavior at the molecular scale challenging. As a result, many experiments have focused on establishing the core behavior of CER-based systems by studying simplified “model” systems, e.g., bilayers composed of a single CER component or simple, well-defined mixtures. 14–19
While molecular simulation has been used extensively to study phospholipid bilayers and has provided a wealth of structural information,20–22 in comparison far less is known about the atomic level structure and interactions of bilayers involving CERs. This is, in part, related to the fact that neither GROMOS nor CHARMM – two of the most commonly used force fields for lipid simulations – have been optimized specifically for CERs. Recent efforts have been made to adapt these force fields for CERs23–25 by combining them with other force field parameter sets to handle missing interactions; however, these adapted parameters have not been extensively validated against experiment or compared. Of the limited simulation studies on CER bilayers reported in the literature, only CER NS has been considered.24–31 In the first such study, Pandit and Scott25 performed 20 ns simulations of a preassembled bilayer of CER NS C16 (where the subscript indicates the number of carbons in the CER acyl chain) at ~368K using an adapted version of a force field developed by Chiu et al.32 for sphingomyelin (SM), which has a large polar phosphorylcholine head group in place of the central hydroxyl group. From these relatively short simulations, it was found that intermolecular hydrogen bonding in CER NS was significantly altered as compared to that seen in a C18 SM bilayer; in CER NS lipids, the absence of a large polar head group significantly reduced the ordering of the water molecules at the lipid/water interface. Subsequently, Imai et al.24 studied the residence time of water on the surface of preassembled CER NS bilayers using a fully atomistic model based on the CHARMM force field, finding that SM bilayers were capable of retaining water molecules longer than CER bilayers; although a series of CER NS bilayers were studied with acyl chain lengths ranging from C12 – C20, the simulations were limited in timescale, capturing only 20 ns, which may not be sufficient for a preassembled bilayer to fully relax itself from its initial state. Additionally, the authors did not provide the parameters used for the functional groups in the CER lipids that are absent from the CHARMM lipid topology. In more recent work, Noro and coworkers,26, 27, 33 using the united atom model adapted from the GROMOS force field of Berger,23 studied the interaction of dimethylsulfoxide (DMSO) with gel-phase bilayers of CER NS C24 and calculated the free energy of pore formation in both CER NS and CER NS/DMSO systems. Again, however, we note that the timescale considered was rather limited, with run times of only 20 ns after initial relaxation. Subsequently, the same authors studied a preassembled bilayer composed of CER NS, cholesterol and a saturated free fatty acid 24 carbons in length, finding that cholesterol caused bilayer compression and interdigitation of the CER tails in the two opposing bilayer leaflets.28–30
From the extensive simulation studies of phospholipid bilayers, it has been well established that the GROMOS34, 35 and CHARMM36, 37 force fields are robust, and thus a certain level of transferability to CERs is expected; however, small, but non-negligible, differences between the two force fields and with experiment are found for phospholipid systems with regards to properties such as the electron density profile and APL34, 35, 38, and considerable efforts have been made to further optimize these parameters to provide better agreement with experiment. For example, Chandrasekhar et al.34 tested the GROMOS96 aliphatic alkane parameter set 45A3 with simulations of a dipalmitoyl phosphatidylcholine (DPPC) bilayer at 323 K. When comparing the average APL with experimental values, they found that GROMOS96 predicted an APL below the lowest reported experimental value, however a later version of the GROMOS force field (G53A6 parameter set)35 was found to greatly improve the fluidity of the bilayers, reducing this deviation. Similarly, Sonne et al.37 modified the original CHARMM27 topology (named CHARMM27r) by proposing new partial charges for the atoms in the DPPC head group. Using the modified force field, a DPPC bilayer was studied and an APL of ~60 Å2 (compared to 48 Å2 with CHARMM27) was obtained, which is ~4–5 Å2 smaller than the experimental value.39 Motivated by the fact that CHARMM27r systematically yielded values of the APL that were smaller than experimental estimates and predicted the formation of gel-like bilayers well above the experimentally determined gel transition temperature, Klauda, et al.36 proposed a new lipid parameter set (which was subsequently included in the most recent update to the CHARMM force field, CHARMM36). Select torsional, Lennard-Jones and partial charge parameters were modified by using, as reference, both quantum mechanical and experimental data; the changes were validated through the study of six phospholipids, including DPPC. The modifications were shown to provide improvements to properties such as the APL at zero tension, structure factor, NMR order parameters, and dipole electrostatic potential, as compared to the CHARMM27r parameter set. To evaluate the accuracy of the available simulation models, Piggot, et al.38 simulated DPPC and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers using five freely available force fields that have been commonly used to model lipid bilayers, including force fields based upon GROMOS23 and CHARMM3636. This work demonstrated that some bilayer properties have pronounced force field dependence, while others are less sensitive. For example, the volume and APL, as well as bilayer height were found to be accurately reproduced in comparison with experimental data by most of the force fields studied, while lipid diffusion showed striking differences depending on the force field used.
Despite the improvements in the GROMOS and CHARMM force fields for phospholipid bilayer simulations, it is unclear which force field is most appropriate for CERs, as neither has been specifically optimized for these systems and a direct side-by-side comparison of the force fields, both with each other and with experiments, is not yet available. Furthermore, while the literature does contain results of CER simulations using a CHARMM-based force field, the full parameter set used was not defined, making it impossible to use and evaluate this force field. In an effort to better understand the structural role of CERs in lipid bilayers and to understand the role of head group chemistry, we propose additional force field parameters for CHARMM to enable the simulation of CER NS and NP bilayers and compare the simulation results to those obtained from simulations with the Berger23 modified version of the GROMOS force field. CER NS was chosen for this study because it is the most abundant ceramide species in human stratum corneum, has been previously examined in simulation, and has also been studied extensively in experiment40–42 providing information regarding structural ordering and phase transitions. CER NP, the phytosphingosine counterpart of CER NS, is studied to assess, in a broad sense, the role of head group chemistry on bilayer properties. The remainder of the paper is organized as follows. In Section II, the force fields, simulation methodology, and analysis methods are presented, including a detailed description of the head group partial charges and missing parameters for CHARMM-based simulation of CERs. In Section III, results for bilayers composed of CER NS are reported, comparing the structural behavior and phase transitions predicted by both the CHARMM- and GROMOS-based force fields with experiment. Additionally, section III reports the properties of the bilayers formed by CER NP and compared to CER NS and experiment, with an emphasis on changes to hydrogen bonding resulting from head group structure. In section IV we provide concluding remarks.
II. SIMULATION METHOD
2.1 Force fields
Here, models based on the all-atom CHARMM force field36, 37, 43–45 and the united-atom GROMOS force field23, 34, 35 have been applied to simulate the CER lipid bilayers. Specifically, two systems have been studied: CER NS C16 and CER NP C16, where both the fatty acid tail and the sphingosine/phytosphingosine tails are 16 carbons in length (note that the sphingosine/phytosphingosine tail is technically composed of 18 carbons, but the first 2 carbons are considered to be part of the head group region). Since both CHARMM and GROMOS are incomplete, in that they do not contain all the parameters required to study CERs, for clarity we first provide details of the extended force fields used in this work.
All-atom CHARMM-based force field
Schematics of the all-atom CER NS are shown in Figures 1a and 2a, and CER NP in Figures 1b and 2c. The parameters for the lipid tails and OH groups are taken from the CHARMM36 topology files36. Although the CHARMM force field is extensive, it has not been parameterized specifically for lipids containing an amide group. In order to determine the missing parameters, ab initio calculations were performed to obtain the force constants and equilibrium distances/angles of atomistic CER NS and NP molecules. The electron structure was also obtained in order to determine the atomic partial charges. A detailed description of the procedure employed is included in the supplemental information. The partial charges of the head groups are summarized graphically in Figure 1a and 1b, for CER NS and NP, respectively. The missing dihedral parameters and van der Waals parameters for the CER amide group were taken from the peptide bond parameters of protein force field44; bonded parameters were validated against the ab initio calculations, the details of which are also provided in the supplemental information. The TIP3P46 water model was used. Throughout the rest of this paper, we will refer to this extended version of the force field simply as CHARMM-CER.
Figure 2.
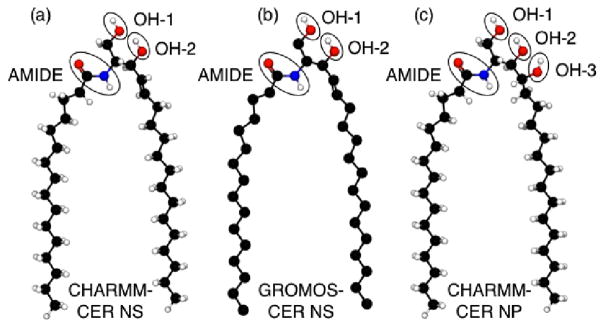
Schematic of the (a) CHARMM-CER NS model; (b) GROMOS-CER NS model and (c) CHARMM-CER NP model. We note that even though GROMOS is a united-atom force field, the GROMOS-CER model includes explicit hydrogens on the amide, OH-1 and OH-2 groups. Note gray atoms represent carbon atoms in a double bonded configuration.
United-atom GROMOS-based force field
Like CHARMM, GROMOS parameters have not been derived purposely for ceramide head group atoms. Specifically, there are no parameters explicitly stated for the amide group shown in Figure 2b. The force field used in this work comes from Berger23 who adapted the united atom GROMOS force field for the study of CERs; we briefly summarize this forcefield below. Note, in the GROMOS force field, the aliphatic tails are treated as united atoms (i.e., no explicit hydrogens) while the hydrogens bonded to oxygens and nitrogen in the head group are accounted for explicitly. Bonded and non-bonded interaction parameters used for atoms in the amide group were taken from the peptide nitrogen in the GROMOS87 parameter set and the hydrocarbon tails employ a Ryckaert-Bellemans potential,47 which was used to better reproduce the experimental APL in DPPC bilayer simulations.23 The partial charges for the headgroup atoms were taken from the side chain of serine, as in Mombelli et al.,48 while the united atom carbons in the tail are charge-neutral. The atoms were broken into small, neutral charge groups to prevent discontinuities in the potential at the cutoff. The charge groups were taken to be as small as possible, e.g., the nitrogen and hydrogen in the amide group make up a charge group and each united atom carbon in the tail represents a single charge group. The simple point charge (SPC) water model49 was used. For simplicity, henceforth this extended version of the GROMOS force field will be referred to as GROMOS-CER.
2.2 Simulation details
CHARMM-CER
Atomistic models of hydrated CER bilayers were simulated using the LAMMPS50, 51 simulation engine. For all CHARMM-CER systems, 100 molecules were placed in each leaflet of a preassmbled bilayer and then solvated with 10,000 water molecules. In order to relax each system, the temperature was increased from 0 K to 305 K within the NVE ensemble using velocity rescaling; the timestep was gradually increased to 1.0 fs in order to avoid energetic problems due to atomistic overlap. Simulations were run for 30,000 steps at which point the simulations were switched to the NPT anisotropic ensemble (Nose-Hoover style non-Hamiltonian equations of motion from Shinoda, et al.52) at a pressure of 1 atm, where pressure control is applied independently in each direction. The long-range electrostatic interactions were calculated through a particle-particle particle-mesh (PPPM) solver with a cutoff of 12 Å.53 A timestep of 1.0 fs was used during the NPT stage, where all simulations were run for at least 100 ns to ensure sufficient sampling within the steady state regime; specifically, CER NS was run for 107 ns and CER NP for 105 ns. Data for the APL, bilayer height, and system energy were collected during the last 30 ns to ensure a steady state had been reached.
To examine the thermotropic behavior (i.e., determine phase transitions as a function of temperature), an equilibrated bilayer structure at 305 K (run previously for >100 ns) was heated at a rate of 5 K/ns until bilayer rupture using the NPT ensemble with the same parameters described above. Cooling curves were started from a temperature just below the gel-to-liquid transition and cooled at a rate of 5 K/ns. As discussed in detail below, good agreement is found in the measured properties between the simulations run for >100 ns at a constant temperature of 305 K and the systems cooled to 305 K from just below the gel-to-liquid transition.
GROMOS-CER
The GROMOS-CER model was simulated using the GROMACS simulation engine.54 The initial structure was constructed in the same manner as the CHARMM-CER bilayer, again using 100 lipids/leaflet. A steepest descent energy minimization run was performed to relax the initial configuration, followed by a 100 ps NVT equilibration at 305 K. The systems were then simulated for 100 ns in the NPT ensemble at 305 K and 1 atm using a Berendsen thermostat with anisotropic pressure coupling. A 2 fs timestep was used. All interactions were cutoff at 12 Å and all bonds were constrained using the SHAKE algorithm. The simulation procedure employed replicates the reported procedures used in prior united atom CER studies by Notman et al.27 Again, data was collected during the final 30 ns of simulation to ensure a steady state had been reached. Similar to simulations with the CHARMM-CER force field, thermotropic behavior was examined by heating an equilibrated bilayer structure at a rate of 5 K/ns from 305 K until bilayer rupture in the NPT ensemble. Cooling simulations were started from a temperature just below the gel-to-liquid transition and cooled at a rate of 5 K/ns, again good agreement is found between systems that were run at a constant temperature of 305 K for 100 ns and those that were cooled from higher temperature.
It is important to note that both the SPC and TIP3P water models employed here have previously been shown to agree well with experiment with regards to density and heat of vaporization, even at the boiling point and our simulations show no evidence of anomalous behavior associated with the water models at elevated temperatures55.
2.3 Analysis
To provide a robust understanding of the lipid bilayers, several different metrics were used to quantify the structural ordering, determine phase transitions, and compare directly with experiment. The APL, a commonly reported quantity describing the compactness of the bilayer, was calculated by dividing the interfacial area of the bilayer (i.e., the xy plane) by the number of lipids in each leaflet. The bilayer thickness, also commonly reported in experimental and simulation studies, was calculated as the difference in z-values (bilayer normal direction) where the water density drops from its bulk value to 1/e of its bulk value.27 The nematic order parameter, S2,56 of the tails was calculated to quantify the global orientational order of the tails, allowing a clear determination of gel vs. fluid bilayers. A value of S2 approaching unity indicates that the tails are in an ordered crystalline nematic structure suggesting a gel-like bilayer and, in this case, a value of S2 < 0.8 (determined via visual inspection for the systems at hand) tends to indicate a more liquid-like ordering of the tails suggesting a fluid-like bilayer. The 2-d global hexagonal order parameter57, 58 (i.e., global in that it quantifies long range hexagonal order) was also calculated to quantify the in-plane ordering of the tails. The hexagonal order parameter lies in the interval [0,1], where the upper bound corresponds to perfect hexagonal arrangement, and the magnitude of the value relates proportionally to the quality of the ordering. The tilt angle of the lipid tails relative to the bilayer normal was also quantified, where a tilt angle of 0° indicates that the tails are parallel to the bilayer normal. Additionally, the heat capacity, Cp, was calculated from the simulations performed as a function of temperature by taking the derivative of enthalpy with respect to temperature. A detailed description of the calculation of each of these metrics is provided in the supplementary information.
III. RESULTS AND DISCUSSION
Comparison of CHARMM-CER and GROMOS-CER force fields for C16 CER NS at 305K
As a first comparison of the CHARMM-CER and GROMOS-CER force fields, the bilayer structural properties are examined at a constant temperature of 305K, as this is the physiologically relevant temperature of skin. Simulation snapshots of CER NS after 100 ns of simulation time are plotted for the CHARMM-CER force field and the GROMOS-CER force field in Figure 3. Visually, similar results are observed, where both force fields demonstrate stable, well ordered bilayer structures, with clear orientational ordering along the molecular axis of the tails and hexagonal ordering between neighboring tails in the planar direction.
Figure 3.


Snapshots of the observed phases for CER NS along the order-disorder transition. The horizontal dotted line demarkates gel phase bilayers (above) from liquid-like bilayer phases (below). Note water is removed for clarity and hydrogens removed from tail groups. In all cases the figure at left is the side view of the bilayer and the figure at right is oriented to highlight the in-plane ordering of the tails segments
To provide a quantitative comparison of the behavior, the APL, bilayer thickness and chain tilt angle relative to the bilayer normal are calculated and summarized in Table 1. CHARMM-CER results in bilayers with a larger APL, smaller thickness and a larger tilt angle than the GROMOS-CER force field. The difference in the bilayer thickness predicted from each force field can be attributed to the difference in the tilt angles, as a system with a larger tilt angle and identical tail length will demonstrate a smaller bilayer thickness.
Table 1.
APL, bilayer height and tilt angle of CER bilayers from the constant temperature simulations.
| APL (Å2) | Bilayer thickness (Å) | Tilt angle (°) | |
|---|---|---|---|
| CHARMM-CER | 42.4±0.2 | 42.5 ± 0.5 | 24.3 ± 0.9 |
| GROMOS-CER | 39.8 ± 0.2 | 43.7 ± 0.4 | 17 ± 1 |
Based on X-ray diffraction experiments of pure, hydrated CER NS, Shah et al.40 report a lamellar structure with a bilayer repeat distance of 46.9 Å at 299K. In this regard, it would appear that both force fields underestimate the bilayer height slightly, however it is important to note that the bilayer thickness can be calculated in multiple ways (see supplemental information), and thus the small differences seen here may instead be related to the methodological choice rather than a deficiency in the models. Using surface pressure-potential-molecular area isotherms, Scheffer et al.59 calculated the APL of a CER NS monolayer to be 40 Å2, Brockman et al.60 calculated 37.8 Å2, and Löfgren and Pascher13 calculated 42 Å2, which are all consistent with the values obtained from the two force fields. We also note that in the simulations performed by Metcalf and Pandit31 at 323K, an APL of ~44 Å2 for pure CER and a tilt angle ~26 degrees for a mixture of 90% CER NS and 10% SM were reported, which are consistent with our results from the CHARMM-CER force field.
Phase transitions for CER NS C16
As further comparison/validation of the two force fields, temperature scans are employed to examine the phase transitions as they compare to experiment.8, 40–42, 61, 62 Additionally, it is important to note that when modeling the stratum corneum, mixtures of different CERs, cholesterol and free fatty acids with a range of tail lengths are present, which is likely to result in some level of spatial hetereogeneity, where, for example, gel and fluid-like tail segments exist simultaneously in different regions of the bilayer.7, 16, 19, 63, 64 To be confident a given force field will provide dependable results for mixtures, it is important to investigate the thermotropic behavior, as temperature scans will enable sampling of different structural conformations of the tails. For both force fields, equilibrated bilayer structures were heated from 305 K until bilayer rupture; similarly, systems were cooled from below the main phase transition to 305 K.
Figure 3 presents simulation snapshots of CER NS bilayers as a function of temperature for heating runs conducted with both force fields. Visually, a clear transition from an ordered gel bilayer to a fluid-like bilayer occurs at ~390K and ~430K for simulations performed with the CHARMM-CER and GROMOS-CER force fields, respectively. This transition from gel- to liquid-like behavior is consistent with the trends reported in the prior simulation studies of Notman et al.,27 where an ordered hexagonal arrangement of the lipid tails was observed at lower temperature state points of 283 K and 323 K and liquid-like ordering at a higher temperature of 363 K, although we note that this prior work considered CER NS with a longer fatty acid tail (24 carbons) than studied here.
To provide a clearer picture of the behavior, the nematic order parameter, hexagonal order parameter, APL, tilt angle, bilayer height, and the heat capacity, Cp, are calculated and plotted in Figure 4 as a function of temperature. Focusing first on the Cp, two distinct peaks are found at 358K and 383K in the heating curve for CHARMM-CER system, and a peak is seen at ~348K in the cooling curve. Evidence of only a single transition (i.e., a single peak in Cp) at 428 K is found for the GROMOS-CER force field. These peaks appear to correlate well with changes in the various structural metrics, discussed below, and the estimated transitions are highlighted as vertical lines in figure 4. Additionally, we note that at 305K, both heating and cooling data closely agree, providing additional validation of the robustness of the simulations presented prior (i.e., the initial structure did not strongly influence bilayer properties for the 100 ns runs).
Figure 4.

Structural and thermodynamic properties for CER NS bilayer along the order-disorder transition. (a) S2 order parameter. (b) Hexagonal order parameter. (c) APL. (d) Tilt angle. (e) Bilayer height. (f) Heat capacity. The left column presents results from the CHARMM-CER force field and right column presents results from the GROMOS-CER force field. The solid lines represent the estimated gel-to-liquid phase transition temperature and dotted lines highlight the secondary peak in the heat capacity. The red lines correspond to the heating curve and the blue to the cooling curve.
For the CHARMM-CER force field, both the S2 and hexagonal order parameters for the lipid tails show a clear drop at temperatures higher than ~380K. This corresponds to the loss of orientational and in-plane ordering of the tails and the formation of a bilayer with a fluid-like tail structure, consistent with visual inspection (see figure 3); this transition is closely coupled to the large peak in the heat capacity that occurs above ~380K. Similar behavior is seen in the GROMOS-CER simulations, however, at an elevated temperature of ~420K.
The tilt angle for the CHARMM-CER system appears to demonstrate a hysteresis bounded by the two smaller peaks in heat capacity (348 K and 358 K, shown as dashed lines). Typically, a hysteresis of ±15 K is reported when passing through the transitions of measurable quantities in simulation,65 consistent with these results; other bilayer systems, such as phospholipids and bolaamphiphiles, have also been reported to have significant temperature hysteresis when comparing the heating and cooling scan.66–68 As discussed in the literature,66–68 the main reason for the hysteresis is that the reformation of the more ordered structure is kinetically limited. Performing the same examination of the GROMOS-CER simulations (figure 4, right column), only a single transition appears evident at ~420K when considering the order parameters, heat capacity, and visual inspection. Only the tilt angle appears to show any temperature dependence prior to the gel-to-fluid bilayer transition, although at temperatures where both experiment and CHARMM-CER do not predict gel-like bilayers.
Comparing with experiment, the CHARMM-CER results appear to agree quite well with the calorimetric work of Chen et al.,61 who performed Fourier transform infrared (FTIR) spectroscopy and differential scanning calorimetry (DSC) studies of CER NS. The main order-disorder transition temperature was reported to be 366.1 ±1 K, in closer agreement with CHARMM-CER (~380 K) than GROMOS-CER (~420 K). We note that it is expected that the simulated heating scans will predict slightly elevated transition temperatures (e.g., by as much as 15 K compared to cooling65), especially given the rather “fast” heating rates used in simulations compared to experimental studies; taking this into account brings the CHARMM-CER force field in very close agreement with experiment. CHARMM-CER also provides good agreement with the work of Shah and Atienza,42 who reported two transitions within the ordered regime at ~350 K and ~355 K for experiments of non-hydroxy fatty acid CERs; these transitions are in close agreement with the peaks observed at ~348 and ~358 K in Cp and the hysteresis in tilt angle in the CHARMM-CER simulations. Similarly, by examining the methylene stretching and scissoring modes, Chen et al.61 observed a solid-solid phase transition at similar temperatures (~340 – 345 K) resulting from chain packing alterations. However, in their work, the nature of the transition was associated with a change from orthorhombic (higher temperature) to hexagonal (lower temperature) subcells; in our simulations, no evidence of a hexagonal to orthorhombic transition is observed, although, we note that CHARMM-CER does predict alterations to the tail tilt-angle.
While there is consensus in the literature that the CER lipid tails are well ordered, the packing pattern is not completely agreed upon.8, 41, 61, 62, 64 For example, at physiologically relevant temperatures, there is evidence of hexagonal41 and orthorhombic packing62, as well as mixtures thereof69. While these results seem to disagree, it is important to consider that the orthorhombic phase is hexagonal in nature (i.e., hexagonal, but “compressed” in one direction) and for CER NS, it is suggested that the characteristic length scales in the orthorhombic phase differ by only ~10%7. As such, the differences between these phases may ultimately be very sensitive to slight differences between experimental procedures and/or simulation models/procedures.
Properties of CER NP C16
As CHARMM-CER appears to more accurately model the behavior of CER NS, it was chosen for examination of the behavior of CER NP. To first provide a validation of CHARMM-CER for CER NP, a temperature scan was performed to determine the order-disorder transition for comparison with experiment. Similar to the CER NS temperature scan, the thermotropic behavior was investigated by heating from an equilibrated bilayer structure at 305K (run for 105 ns), until bilayer rupture. The nematic order parameter, hexagonal order parameter, APL, tilt angle, bilayer height, and the heat capacity, Cp, are calculated and plotted in Figure 5. From these measurements, it appears that CER NP transitions from a gel to liquid bilayer state is at ~390–395K, which is roughly 10–15 K higher than CER NS. This shift in the transition temperature is consistent with experiment; specifically, Garidel et al.9 reported a main solid-liquid phase transition at 383 K and Rerek, et al.8 reported chain melting at 388K; recall, the main transition temperature of CER NS was reported to be 366 K.61
Figure 5.

Structural and thermodynamic properties for CER NP bilayer with CHARMM-CER force field along order-disorder transition.
Comparisons of the structural differences between head groups: CER NS versus NP
Table 2 plots various structural properties of CER NS and NP at 305K. We find that CER NP predicts a less tilted, but thicker bilayer than CER NS, although both phases have similar APLs and values of the order parameters. These differences in tilt angle are surprising, given that CER NP and NS differ only in the replacement of an alkene functional group by a third hydroxy group (see Figures 1 and 2). Although fewer experimental measurements of the structural properties exist for CER NP compared to CER NS, surface pressure-potential-molecular area isotherms of CER NP monolayers conducted by Löfgren and Pascher13 calculated the APL to be 48 Å2 at zero surface tension, suggesting CHARMM-CER underpredicts this spacing (note, in these experiments the APL drops rapidly to 42 Å2 when the applied surface tension is increased to 30 dynes/cm). However, given that the APL for both simulated systems is roughly the same but CER NP has a larger bilayer thickness, this indicates that CER NP tail segments have a larger volume per lipid (i.e., larger accessible space per tail) than CER NS, consistent with the conclusion from IR spectroscopy studies that phytosphingosines pack less tightly than sphingosines.8
Table 2.
APL, bilayer thickness, tilt angle, nematic order parameter, and hexagonal order parameter of CER NS and NP bilayers from simulations run at 305K.
| APL (Å2) | Bilayer thickness (Å) | Tilt angle (°) | S2 | Hex OP | |
|---|---|---|---|---|---|
| CER NS C16 | 42.4±0.2 | 42.5 ± 0.5 | 24.3 ± 0.9 | 0.980 | 0.68 ± 0.02 |
| CER NP C16 | 42.1 ± 0.2 | 44.1 ± 0.5 | 20.5 ± 0.8 | 0.980 | 0.68 ± 0.02 |
To further investigate the differences between CER NS and NP, the hydrogen bonds formed by the AMIDE and OH groups between lipids (see labels given in Figure 2), as well as between the lipids and water, have been determined and are reported in Table 3. As might be anticipated, due to the additional OH group, CER NP is found to have more hydrogen bonds than CER NS. This is consistent with the work of Rerek et al.,8 who found that phytosphingosines have more significant hydrogen bonding than their sphingosine counterparts. In the 200-lipid bilayer system studied here, CER NP has, in total, 15% more hydrogen bonds than the CER NS bilayer of the same size and under the same thermodynamic conditions. With respect to the total number of hydrogen bonds formed by OH groups, there are 25% more hydrogen bonds in the CER NP bilayer than in the CER NS bilayer. This indicates that the additional OH group in CER NP plays a significant role in the formation of hydrogen bonds with surrounding hydrogen donors/acceptors, whereas the alkene group in CER NS is more stable in a hydrophobic environment.
Table 3.
Number of hydrogen bonds among the functional groups in the head region and water for CER NS and NP bilayers from simulation run at 305 K, as calculated from VMD70 using a cutoff distance of 3 Å and an angle criteria of 60°.
| Group | CER NS C16 | CER NP C16 | ||
|---|---|---|---|---|
|
| ||||
| Lipid-lipid | Lipid-water | Lipid-lipid | Lipid-water | |
| AMIDE | 14 ± 2.4 | 103 ± 9.1 | 15 ± 3.3 | 87 ± 7.1 |
| OH | 89 ± 7.2 | 422 ± 22.7 | 244 ± 11.7 | 361 ± 18.6 |
| Total | 103 ± 9.6 | 525 ± 31.8 | 259 ± 15.0 | 448 ± 25.7 |
Notably, the distribution between lipid-lipid hydrogen bonds and lipid-water hydrogen bonds presents opposite trends for the two CERs studied. In CER NS, the most hydrogen bonds are formed with water (80%), with only 20% of the hydrogen bonds formed between lipids; while in CER NS this ratio becomes 40% versus 60%. The lipid-lipid hydrogen bonding snapshots for CER NS and NP are shown in Figure 6, from which we can see that the CER NP head groups form a tight and cohesive hydrogen bonding network, while CER NS shows more sparse and discontinuous intermolecular hydrogen bonds.
Figure 6.

Lipid-lipid hydrogen bonding formed by AMIDE and OH groups. (a) CER NS leaflet; (b) CER NP leaflet. Blue - nitrogen; red – oxygen; white – hydrogen.
Experimentally, Garidel et al.63 studied the microstructures of CER NS and CER NP via FTIR spectra and found that CER NS is characterized by weaker hydrogen bonding interactions between the head groups than CER NP, which is consistent with our observations. When comparing CERs of the same chain length, or chain length distribution, experimentally one typically observes that the gel-liquid phase transition is higher for phytosphingosines than sphingosines.9, 41, 61, 62, 71 Garidel et al.63 hypothesized that this may result from the stronger interactions between the polar head groups. The simulations performed in this study support this hypothesis, where CER NP (phytosphingosine) is able to form a stronger hydrogen bond network compared to CER NS (sphingosine) and exhibits a slightly augmented order-disorder transition temperature.
Another notable point from Table 3 is that the simulations demonstrate that the AMIDE group in CER NS forms more hydrogen bonds than in CER NP, with ~20% more AMIDE groups participating in the formation of the hydrogen bonding network. This may be due to the strong head group hydrogen bonding network that forms among the three OH groups in CER NP, making it less advantageous for the AMIDE to participate in the formation of hydrogen bonds.
In Figure 7, the distributions of the angle between the hydroxyl oxygen and connecting carbon bonds (C-O) and the bilayer normal in both the CER NS and CER NP systems are plotted for the OH-1 and OH-2 groups (see labels given in Figure 2). From figure 7a, it is observed that the C-O angle for CER NS is evenly distributed at 25°, 90° and 155°, suggesting two thirds of the C-O bonds with OH-1 align perpendicularly while only one third are in the horizontal direction. In comparison, the curves for CER NP are significantly more peaked with a higher intensity value at ~90°. Thus, in CER NP systems, the C-O bond with OH-1 tends to be more horizontal relative to the bilayer normal. The C-O bond with the OH-2 group in both systems shows similar trends: C-O in CER NS distributes evenly at 60° and 120°, while for CER NP a single peak is seen at 95°. These results suggest that in CER NP, the C-O bonds adjacent to the OH groups adopt a more horizontal orientation, resulting in a flatter head group region at the lipid-water interface. This change in position appears to be the cause for the augmented hydrogen bonding network seen for CER NP.
Figure 7.

Distribution of angle between hydroxyl oxygen and connecting carbon relative to bilayer normal for CER NS and CER NP head groups. (a) C-O bond with OH-1; (b) C-O bond with OH-2. Red lines represent CER NS and blue lines represent CER NP at T = 305 K
The radial distribution function (RDF) has also been calculated to further compare the structural characteristics and head group arrangement in CER NS and NP bilayers. The RDFs of the nitrogen in the AMIDE group and oxygen in the OH-1 and OH-2 group for CER NS and NP are shown in figure 8.
Figure 8.

Head group RDFs for CER NS C16 and CER NP C16. (a) Nitrogen-nitrogen RDF. (b) Oxygen-oxygen RDF for OH-1. (c) Oxygen-oxygen RDF for OH-2. Red lines represent CER NS and blue lines represent CER NP.
Figure 8a shows that CER NP has a higher intensity of the first peak in the nitrogen-nitrogen RDF than CER NS, while the peak position for the two lipids is very similar. This suggests that for CER NP, the nitrogen atoms have, on average, more nearest neighbors than the nitrogen atoms in the CER NS system. Since the APL for both CER NP and CER NS bilayers is ~42 Å2, we can infer that the CER NP AMIDE groups tend to assemble together and form a more open polar interface than the CER NS system. This can also be seen by examining the position of the secondary peaks; CER NS has a minor peak at 6.25 Å while CER NP peaks around 7.4 Å. This measurement is consistent with the observation by Rerek et al.,8 who proposed that the more open polar interface allows phytosphingosine CERs to form a stronger hydrogen bond network compared to sphingosine CERs.
While the RDF measurements in figure 8b for OH-1 are similar for both systems, the measurements for OH-2 shown in figure 8c differ significantly for NS and NP. Although the spacing of the first two peaks is similar, the magnitude of the first peak in CER NS is approximately double that of CER NP. This suggests that without the influence of the OH-3 group in the phytosphigosine tail, the OH-2 group in CER NS possesses more order than its counterpart in CER NP; the latter tends to form a loosely arranged structure. This is supported by the distribution of C-O angles reported in figure 7, where OH-2 in CER NP adopts a more parallel arrangement, influencing this packing.
IV. CONCLUSIONS
The structure and thermotropic properties of CER NS bilayers have been compared using force fields based upon the CHARMM force field (CHARMM-CER), developed as part of this work, and the GROMOS force field (GROMOS-CER) modified by Berger. We find that at physiological conditions, both force fields demonstrate similar behavior for CER NS, consistent with the available experimental measurements; however, subtle differences between the force fields are seen with respect to bilayer height and tilt angle, where GROMOS-CER tends to predict more compact, less tilted structures than CHARMM-CER. Examination of thermotropic properties and comparison with experimental observations shows more significant differences between the two force fields. GROMOS-CER predicts a main gel-liquid phase transition at ~420 K whereas CHARMM-CER predicts a transition at ~380 K, more consistent with experiment. CHARMM-CER also resolves a subtle order-order phase transition at ~350 K, consistent with experimental measurements, while this transition is absent in the GROMOS-CER simulations. Given that the stratum corneum is composed of more than a dozen different lipids, it is important that a force field that is capable of resolving the subtle structural differences and phase transitions in order to accurately model this layer. However, it is important to note that the increased accuracy of the proposed CHARMM-based force field comes with additional computational cost, which may place practical limits on the system size and timescale that can be efficiently examined. As such, the GROMOS-based force field will no doubt still play a vital role regarding the investigation of CERs, particularly given that it could be optimized to better capture thermotropic behavior.
When examining the role of head group chemistry by comparing CER NS to NP, our results find that the substitution of OH-3 in place of the alkene bond changes not only the neighboring OH-2 from a perpendicular to a horizontal orientation, but also induces the formation of a more significant hydrogen bonding network, resulting in much stronger intermolecular head group interactions compared with that seen in CER NS. These structural differences lead to a higher gel-liquid phase transition temperature for the phytosphingosines (CER NP) compared to sphingosines (CER NS), consistent with experiment. Slight changes in the CER head group chemistry appear to have a dramatic influence on the morphological features and thermodynamic properties of the lipid bilayers, underscoring the importance of performing computational investigations of the molecular-level behavior of the different classes of CERs found in the stratum corneum.
Supplementary Material
Acknowledgments
This work was supported by grant number R01 AR057886-01 from the National Institute of Arthritis and Muscoskeletal and Skin Diseases. Computational resources were provided by the National Energy Research Scientific Computing Center, supported by the Office of Science of the Department of Energy under Contract No. DE-AC02-05CH11231. We also acknowledge Annette Bunge and Joke Bouwstra for useful discussions.
Footnotes
A detailed description of the procedure employed to develop the CHARMM-CER force field with ab initio calculations is included in the supplemental information. The parameters used for the amide group in CERs, including the force constants and equilibrium distances/angles as well as the atomic partial charges are provided. A detailed description of the calculation of each of the bilayer metrics is also provided. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schaefer H, Redelmeier TE. Skin Barrier: Principles of Percutaneous Absorption; Karger; New York: 1996. pp. 31–49. [Google Scholar]
- 2.Downing DT, Stewart ME, Wertz PW, Colton SW, VI, Strauss JS. Skin Lipids. Comp Biochem Physiol, Part B: Biochem Mol Biol. 1983;76:673–678. doi: 10.1016/0305-0491(83)90376-0. [DOI] [PubMed] [Google Scholar]
- 3.Robson KJ, Stewart ME, Michelsen S, Lazo ND, Downing DT. 6-Hydroxy-4-Sphingenine in Human Epidermal Ceramides. J Lipid Res. 1994;35:2060–2068. [PubMed] [Google Scholar]
- 4.Bleck O, Abeck D, Ring J, Hoppe U, Vietzke JP, Wolber R, Brandt O, Schreiner V. Two Ceramide Subfractions Detectable in Cer(AS) Position by HPTLC in Skin Surface Lipids of Non-Lesional Skin of Atopic Eczema. J Invest Dermatol. 1999;113:894–900. doi: 10.1046/j.1523-1747.1999.00809.x. [DOI] [PubMed] [Google Scholar]
- 5.Stewart ME, Downing DT. A New 6-Hydroxy-4-Sphingenine-Containing Ceramide in Human Skin. J Lipid Res. 1999;40:1434–1439. [PubMed] [Google Scholar]
- 6.Wertz PW. Lipids and Barrier Function of the Skin. Acta Derm-Venereol. 2000:7–11. doi: 10.1080/000155500750042790. [DOI] [PubMed] [Google Scholar]
- 7.Pilgram GSK, Vissers DCJ, van der Meulen H, Pavel S, Lavrijsen SPM, Bouwstra JA, Koerten HK. Aberrant Lipid Organization in Stratum Corneum of Patients with Atopic Dermatitis and Lamellar Ichthyosis. J Invest Dermatol. 2001;117:710–717. doi: 10.1046/j.0022-202x.2001.01455.x. [DOI] [PubMed] [Google Scholar]
- 8.Rerek ME, Chen, Markovic B, Van Wyck D, Garidel P, Mendelsohn R, Moore DJ. Phytosphingosine and Sphingosine Ceramide Headgroup Hydrogen Bonding: Structural Insights through Thermotropic Hydrogen/Deuterium Exchange. J Phys Chem B. 2001;105:9355–9362. [Google Scholar]
- 9.Garidel P. Calorimetric and Spectroscopic Investigations of Phytosphingosine Ceramide Membrane Organisation. Phys Chem Chem Phys. 2002;4:1934–1942. [Google Scholar]
- 10.Raudenkolb S, Hubner W, Rettig W, Wartewig S, Neubert RHH. Polymorphism of Ceramide 3. Part 1: An Investigation Focused on the Head Group of N-Octadecanoylphytosphingosine. Chem Phys Lipids. 2003;123:9–17. doi: 10.1016/s0009-3084(02)00127-5. [DOI] [PubMed] [Google Scholar]
- 11.Raudenkolb S, Wartewig S, Neubert RHH. Polymorphism of Ceramide 3. Part 2: A Vibrational Spectroscopic and X-Ray Powder Diffraction Investigation of N-Octadecanoyl Phytosphingosine and the Analogous Specifically Deuterated D(35) Derivative. Chem Phys Lipids. 2003;124:89–101. doi: 10.1016/s0009-3084(03)00045-8. [DOI] [PubMed] [Google Scholar]
- 12.Raudenkolb S, Wartewig S, Neubert RHH. Polymorphism of Ceramide 6: A Vibrational Spectroscopic and X-Ray Powder Diffraction Investigation of the Diastereomers of N-(Alpha-Hydroxyoctadecanoyl)-Phytosphingosine. Chem Phys Lipids. 2005;133:89–102. doi: 10.1016/j.chemphyslip.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Löfgren H, Pascher I. Molecular Arrangements of Sphingolipids. The Monolayer Behaviour of Ceramides. Chem Phys Lipids. 1977;20:273–284. doi: 10.1016/0009-3084(77)90068-8. [DOI] [PubMed] [Google Scholar]
- 14.Bouwstra JA, Gooris GS. The Lipid Organization in Human Stratum Corneum and Model Systems. Open Derm J. 2010;4:10–13. [Google Scholar]
- 15.Bouwstra JA, Gooris GS, Dubbelaar FER, Ponec M. Phase Behavior of Stratum Corneum Lipid Mixtures Based on Human Ceramides: The Role of Natural and Synthetic Ceramide 1. J Invest Dermatol. 2002;118:606–617. doi: 10.1046/j.1523-1747.2002.01706.x. [DOI] [PubMed] [Google Scholar]
- 16.Groen D, Gooris GS, Bouwstra JA. Model Membranes Prepared with Ceramide EOS, Cholesterol and Free Fatty Acids Form a Unique Lamellar Phase. Langmuir. 2010;26:4168–4175. doi: 10.1021/la9047038. [DOI] [PubMed] [Google Scholar]
- 17.Engelbrecht T, Hauss T, Suss K, Vogel A, Roark M, Feller SE, Neubert RHH, Dobner B. Characterisation of a New Ceramide EOS Species: Synthesis and Investigation of the Thermotropic Phase Behaviour and Influence on the Bilayer Architecture of Stratum Corneum Lipid Model Membranes. Soft Matter. 2011;7:8998–9011. [Google Scholar]
- 18.Engelbrecht TN, Schroeter A, Hauss T, Deme B, Scheidt HA, Huster D, Neubert RH. The Impact of Ceramides NP and AP on the Nanostructure of Stratum Corneum Lipid Bilayer. Part I: Neutron Diffraction and 2H NMR Studies on Multilamellar Models Based on Ceramides with Symmetric Alkyl Chain Length Distribution. Soft Matter. 2012;8:6599–6607. [Google Scholar]
- 19.Mao G, VanWyck D, Xiao X, Mack Correa MC, Gunn E, Flach CR, Mendelsohn R, Walters RM. Oleic Acid Disorders Stratum Corneum Lipids in Langmuir Monolayers. Langmuir. 2013;29:4857–4865. doi: 10.1021/la4002384. [DOI] [PubMed] [Google Scholar]
- 20.Lyubartsev AP, Rabinovich AL. Recent Development in Computer Simulations of Lipid Bilayers. Soft Matter. 2011;7:25–39. [Google Scholar]
- 21.Alwarawrah M, Dai J, Huang J. Modification of Lipid Bilayer Structure by Diacylglycerol: A Comparative Study of Diacylglycerol and Cholesterol. J Chem Theory Comput. 2012;8:749–758. doi: 10.1021/ct200790q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava A, Voth GA. Hybrid Approach for Highly Coarse-Grained Lipid Bilayer Models. J Chem Theory Comput. 2013;9:750–765. doi: 10.1021/ct300751h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger O, Edholm O, Jahnig F. Molecular Dynamics Simulations of a Fluid Bilayer of Dipalmitoylphosphatidylcholine at Full Hydration, Constant Pressure, and Constant Temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai Y, Liu XL, Yamagishi J, Mori K, Neya S, Hoshino T. Computational Analysis of Water Residence on Ceramide and Sphingomyelin Bilayer Membranes. J Mol Graphics Modell. 2010;29:461–469. doi: 10.1016/j.jmgm.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Pandit SA, Scott HL. Molecular-Dynamics Simulation of a Ceramide Bilayer. J Chem Phys. 2006;124:7. doi: 10.1063/1.2140689. [DOI] [PubMed] [Google Scholar]
- 26.Notman R, Anwar J, Briels WJ, Noro MG, den Otter WK. Simulations of Skin Barrier Function: Free Energies of Hydrophobic and Hydrophilic Transmembrane Pores in Ceramide Bilayers. Biophys J. 2008;95:4763–4771. doi: 10.1529/biophysj.108.138545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Notman R, den Otter WK, Noro MG, Briels WJ, Anwar J. The Permeability Enhancing Mechanism of DMSO in Ceramide Bilayers Simulated by Molecular Dynamics. Biophys J. 2007;93:2056–2068. doi: 10.1529/biophysj.107.104703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das C, Noro MG, Olmsted PD. Simulation Studies of Stratum Corneum Lipid Mixtures. Biophys J. 2009;97:1941–1951. doi: 10.1016/j.bpj.2009.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das C, Olmsted PD, Noro MG. Water Permeation through Stratum Corneum Lipid Bilayers from Atomistic Simulations. Soft Matter. 2009;5:4549–4555. [Google Scholar]
- 30.Hoopes MI, Noro MG, Longo ML, Faller R. Bilayer Structure and Lipid Dynamics in a Model Stratum Corneum with Oleic Acid. J Phys Chem B. 2011;115:3164–3171. doi: 10.1021/jp109563s. [DOI] [PubMed] [Google Scholar]
- 31.Metcalf R, Pandit SA. Mixing Properties of Sphingomyelin Ceramide Bilayers: A Simulation Study. J Phys Chem B. 2012;116:4500–4509. doi: 10.1021/jp212325e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiu SW, Vasudevan S, Jakobsson E, Mashl RJ, Scott HL. Structure of Sphingomyelin Bilayers: A Simulation Study. Biophys J. 2003;85:3624–3635. doi: 10.1016/S0006-3495(03)74780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.den Otter WK, Notman R, Anwar J, Noro MG, Briels WJ. Modulating the Skin Barrier Function by DMSO: Molecular Dynamics Simulations of Hydrophilic and Hydrophobic Transmembrane Pores. Chem Phys Lipids. 2008;154(Supplement):S2–S3. [Google Scholar]
- 34.Chandrasekhar I, Kastenholz M, Lins RD, Oostenbrink C, Schuler LD, Tieleman DP, van Gunsteren WF. A Consistent Potential Energy Parameter Set for Lipids: Dipalmitoylphosphatidylcholine as a Benchmark of the Gromos96 45a3 Force Field. Eur Biophys J. 2003;32:67–77. doi: 10.1007/s00249-002-0269-4. [DOI] [PubMed] [Google Scholar]
- 35.Poger D, Van Gunsteren WF, Mark AE. A New Force Field for Simulating Phosphatidylcholine Bilayers. J Comput Chem. 2010;31:1117–1125. doi: 10.1002/jcc.21396. [DOI] [PubMed] [Google Scholar]
- 36.Klauda JB, Venable RM, Freites JA, O‚ÄôConnor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sonne J, Jensen M, Hansen FY, Hemmingsen L, Peters GH. Reparameterization of All-Atom Dipalmitoylphosphatidylcholine Lipid Parameters Enables Simulation of Fluid Bilayers at Zero Tension. Biophys J. 2007;92:4157–4167. doi: 10.1529/biophysj.106.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piggot TJ, Pineiro A, Khalid S. Molecular Dynamics Simulations of Phosphatidylcholine Membranes: A Comparative Force Field Study. J Chem Theory Comput. 2012;8:4593–4609. doi: 10.1021/ct3003157. [DOI] [PubMed] [Google Scholar]
- 39.Nagle JF, Tristram-Nagle S. Structure of Lipid Bilayers. Biochim Biophys Acta, Rev Biomembr. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah J, Atienza J, Dong ZX, Shipley GG. X-Ray-Diffraction and Calorimetric Studies of Non-Hydroxy Fatty-Acid (16-0) Ceramide. Biophys J. 1994;66:A288–A288. [Google Scholar]
- 41.Shah J, Atienza JM, Duclos RI, Rawlings AV, Dong ZX, Shipley GG. Structural and Thermotropic Properties of Synthetic C16-0 (Palmitoyl) Ceramide - Effect of Hydration. J Lipid Res. 1995;36:1936–1944. [PubMed] [Google Scholar]
- 42.Shah J, Atienza JM, Rawlings AV, Shipley GG. Physical Properties of Ceramides Effect of Fatty Acid Hydroxylation. J Lipid Res. 1995;36:1945–1955. [PubMed] [Google Scholar]
- 43.Feller SE, MacKerell AD. An Improved Empirical Potential Energy Function for Molecular Simulations of Phospholipids. J Phys Chem B. 2000;104:7510–7515. [Google Scholar]
- 44.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 45.Pastor RW, MacKerell AD. Development of the CHARMM Force Field for Lipids. J Phys Chem Lett. 2011;2:1526–1532. doi: 10.1021/jz200167q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 47.Ryckaert JP, Bellemans A. Molecular Dynamics of Liquid Alkanes. Farady Discussion. Chem Soc. 1978;66:95–106. [Google Scholar]
- 48.Mombelli E, Morris R, Taylor W, Fraternali F. Hydrogen-Bonding Propensities of Sphingomyelin in Solution and in a Bilayer Assembly: A Molecular Dynamics Study. Biophys J. 2003;84:1507–1517. doi: 10.1016/S0006-3495(03)74963-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans WF. Intermolecular Forces. D. Reidel Publishing; Dordrecht, The Netherlands: 1981. Interaction Models for Water in Relation to Protein Hydration; pp. 331–342. [Google Scholar]
- 50. [accessed March 08, 2012];Lammps Molecular Dynamics Simulator. Http://Lammps.Sandia.Gov.
- 51.Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J Comput Phys. 1995;117:1–19. [Google Scholar]
- 52.Shinoda S, Mikami Lammps Npt Barostat. Phys Rev B. 2004;69:134103. [Google Scholar]
- 53.Hockney, Eastwood . Computer Simulation Using Particles. Adam Hilger; New York: 1989. pp. 121–142. [Google Scholar]
- 54.Kutzner B, van der Spoel D, Lindahl E. Gromacs 4: Algorithms for Highly Efficient, Load–Balanced, and Scalable Molecular Simulation Hess. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen WL, Jenson C. Temperature Dependence of TIP3P, SPC, and TIP4P Water from NPT Monte Carlo Simulations: Seeking Temperatures of Maximum Density. J Comput Chem. 1998;19:1179–1186. [Google Scholar]
- 56.Wilson MR. Determination of Order Parameters in Realistic Atom-Based Models of Liquid Crystal Systems. J Mol Liq. 1996;68:23–31. [Google Scholar]
- 57.Keys AS, Iacovella CR, Glotzer SC. Characterizing Structure through Shape Matching and Applications to Self-Assembly. In: Langer JS, editor. Annual Review of Condensed Matter Physics. Vol. 2. Vol. 2. 2011. pp. 263–285. [Google Scholar]
- 58.Keys AS, Iacovella CR, Glotzer SC. Characterizing Complex Particle Morphologies through Shape Matching: Descriptors, Applications, and Algorithms. J Comput Phys. 2011;230:6438–6463. [Google Scholar]
- 59.Scheffer L, Solomonov I, Jan Weygand M, Kjaer K, Leiserowitz L, Addadi L. Structure of Cholesterol/Ceramide Monolayer Mixtures: Implications to the Molecular Organization of Lipid Rafts. Biophys J. 2005;88:3381–3391. doi: 10.1529/biophysj.104.051870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brockman HL, Momsen MM, Brown RE, He LL, Chun J, Byun HS, Bittman R. The 4,5-Double Bond of Ceramide Regulates Its Dipole Potential, Elastic Properties, and Packing Behavior. Biophys J. 2004;87:1722–1731. doi: 10.1529/biophysj.104.044529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen HC, Mendelsohn R, Rerek ME, Moore DJ. Fourier Transform Infrared Spectroscopy and Differential Scanning Calorimetry Studies of Fatty Acid Homogeneous Ceramide 2. Biochim Biophys Acta: Biomembr. 2000;1468:293–303. doi: 10.1016/s0005-2736(00)00271-6. [DOI] [PubMed] [Google Scholar]
- 62.Moore DJ, Rerek ME, Mendelsohn R. FTIR Spectroscopy Studies of the Conformational Order and Phase Behavior of Ceramides. J Phys Chem B. 1997;101:8933–8940. [Google Scholar]
- 63.Garidel P, Foelting B, Schaller I, Kerth A. The Microstructure of the Stratum Corneum Lipid Barrier: Mid-Infrared Spectroscopic Studies of Hydrated Ceramide:Palmitic Acid:Cholesterol Model Systems. Biophys Chem. 2010;150:144–156. doi: 10.1016/j.bpc.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 64.Moore DJ, Rerek ME, Mendelsohn R. Lipid Domains and Orthorhombic Phases in Model Stratum Corneum: Evidence from Fourier Transform Infrared Spectroscopy Studies. Biochem Biophys Res Commun. 1997;231:797–801. doi: 10.1006/bbrc.1997.6189. [DOI] [PubMed] [Google Scholar]
- 65.Waheed Q, Tjornhammar R, Edholm O. Phase Transitions in Coarse-Grained Lipid Bilayers Containing Cholesterol by Molecular Dynamics Simulations. Biophys J. 2012;103:2125–2133. doi: 10.1016/j.bpj.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holopainen JM, Lemmich J, Richter F, Mouritsen OG, Rapp G, Kinnunen PKJ. Dimyristoylphosphatidylcholine/C16:0-Ceramide Binary Liposomes Studied by Differential Scanning Calorimetry and Wide- and Small-Angle X-Ray Scattering. Biophys J. 2000;78:2459–2469. doi: 10.1016/S0006-3495(00)76790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohler K, Forster G, Hauser A, Dobner B, Heiser UF, Ziethe F, Richter W, Steiniger F, Drechsler M, Stettin H, Blume A. Temperature-Dependent Behavior of a Symmetric Long-Chain Bolaamphiphile with Phosphocholine Headgroups in Water: From Hydrogel to Nanoparticles. J Am Chem Soc. 2004;126:16804–16813. doi: 10.1021/ja046537k. [DOI] [PubMed] [Google Scholar]
- 68.Garidel P, Blume A. Interaction of Alkaline Earth Cations with the Negatively Charged Phospholipid 1,2-Dimyristoyl-Sn-Glycero-3-Phosphoglycerol: A Differential Scanning and Isothermal Titration Calorimetric Study. Langmuir. 1999;15:5526–5534. [Google Scholar]
- 69.Ongpipattanakul B, Francoeur ML, Potts RO. Polymorphism in Stratum Corneum Lipids. Biochim Biophys Acta: Biomembr. 1994;1190:115–122. doi: 10.1016/0005-2736(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 70.Humphrey W, Dalke A, Schulten K. Vmd: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 71.Garidel P. Structural Organisation and Phase Behaviour of a Stratum Corneum Lipid Analogue: Ceramide 3A. Phys Chem Chem Phys. 2006;8:2265–2275. doi: 10.1039/b517540b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.