

Abstract
Helicobacter pylori colonizes the human stomach and confers an increased risk for the development of peptic ulceration, noncardia gastric adenocarcinoma, and gastric lymphoma. A secreted H. pylori toxin, VacA, can cause multiple alterations in gastric epithelial cells, including cell death. In this study, we sought to identify host cell factors that are required for VacA-induced cell death. To do this, we analyzed gene trap and short hairpin RNA (shRNA) libraries in AZ-521 human gastric epithelial cells and selected for VacA-resistant clones. Among the VacA-resistant clones, we identified multiple gene trap library clones and an shRNA library clone with disrupted expression of connexin 43 (Cx43) (also known as gap junction protein alpha 1 [GJA1]). Further experiments with Cx43-specific shRNAs confirmed that a reduction in Cx43 expression results in resistance to VacA-induced cell death. Immunofluorescence microscopy experiments indicated that VacA did not colocalize with Cx43. We detected production of the Cx43 protein in AZ-521 cells but not in AGS, HeLa, or RK-13 cells, and correspondingly, AZ-521 cells were the most susceptible to VacA-induced cell death. When Cx43 was expressed in HeLa cells, the cells became more susceptible to VacA. These results indicate that Cx43 is a host cell constituent that contributes to VacA-induced cell death and that variation among cell types in susceptibility to VacA-induced cell death is attributable at least in part to cell type-specific differences in Cx43 production.
INTRODUCTION
Helicobacter pylori is a Gram-negative bacterium that persistently colonizes about 50% of the world's population (1, 2). H. pylori colonization causes gastric inflammation in all infected individuals and is a risk factor for the development of peptic ulcer disease, gastric adenocarcinoma, and gastric lymphoma (3, 4). Gastric cancer is one of the most common infection-related cancers and is the second leading cause of cancer-related death worldwide (5, 6). One of the important virulence factors produced by H. pylori is a secreted pore-forming toxin known as VacA (7–14). VacA is produced as a 140-kDa protoxin, which undergoes proteolytic processing to yield a signal peptide, passenger domain, and β-barrel domain. The 88-kDa toxin is secreted through a type V, or autotransporter, pathway (15–19).
Multiple types of cells are susceptible to VacA, including gastric epithelial cells and cells of the immune system (1, 2, 7–14, 20). As a first step in VacA intoxication, the toxin binds to host cell receptors (7, 9). Multiple potential receptors have been identified, including sphingomyelin (21, 22), receptor protein-tyrosine phosphatases (RPTP-α and RPTP-β) (23, 24), and low-density lipoprotein receptor-related protein-1 (LRP1) (25) in gastric epithelial cells and integrin-β2 receptor (CD18) in T cells (26). After binding to cells, VacA can be internalized into cells through a pinocytotic process (27). Internalized VacA first accumulates in early endosomes and then traffics to late endosomes (27–29) and mitochondria (30, 31). There are many possible consequences of VacA interactions with epithelial cells, including cell vacuolation, disruption of endosomal and lysosomal function, depolarization of the plasma membrane potential, permeabilization of epithelial monolayers, detachment of epithelial cells from the basement membrane, autophagy, and cell death (7–14, 20, 32–34). VacA can cause death of gastric epithelial cells through both apoptosis and programmed cell necrosis (14, 20, 35–37).
The mechanisms by which VacA causes cell death are not yet completely understood but are thought to be dependent on localization of VacA to mitochondria (30, 38–40). Effects of VacA on mitochondria include reduction in mitochondrial transmembrane potential, cytochrome c release, and mitochondrial network fragmentation (30, 38–40, 41–43), which can lead to poly(ADP-ribose) polymerase (PARP) cleavage, reduction of cellular ATP content, and impaired cell cycle progression (9, 35, 41–43). The proapoptotic factors BAX and BAK, as well as dynamin-related protein 1 (Drp1), have roles in VacA-mediated cell death (31, 42, 44). VacA can cause cell death in several cell lines, including HeLa (30, 38, 39, 45), AGS (20, 36, 37, 41, 46), and AZ-521 cells (25, 35, 42, 44, 47), but among these cell types, AZ-521 cells are the most susceptible to VacA-mediated killing (35). The molecular mechanisms underlying this enhanced susceptibility of AZ-521 cells are not understood.
In the current study, we analyzed gene trap and shRNA libraries in AZ-521 cells, selected for VacA-resistant clones, and thereby sought to identify host cell factors that are required for VacA-induced death of these cells. We report here that connexin 43 (Cx43) is a host cell factor that contributes to VacA-induced cell death in AZ-521 cells. Connexins are components of gap junctions, which form intercellular channels between adjacent cells. These channels provide a route for diffusion of low-molecular-weight molecules from cell to cell and play an important role in cell-cell communication (48). Therefore, connexins regulate many physiological processes. Cx43 is the most common connexin isoform and is expressed by many different cell types, including gastric and intestinal epithelial cells (49–51), ventricular myocytes, astrocytes, muscle cells, endothelial cells (52–54), and several types of immune cells (including T and B cells, neutrophils, mast cells, monocytes, and macrophages) (55–60). We report that Cx43 is produced by AZ-521 cells but not by several other cell types that are commonly used for studies of VacA and provide evidence that variation among cell types in susceptibility to VacA-induced cell death is attributable at least in part to cell type-specific differences in Cx43 protein production.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori wild-type (WT) strain 60190 (ATCC 49503) and an isogenic mutant expressing VacA-G14A, a mutant toxin that is defective in membrane channel formation and does not cause cell death (35, 61), were grown on Trypticase soy agar plates containing 5% sheep blood at 37°C in ambient air containing 5% CO2. H. pylori liquid cultures were grown in Brucella broth supplemented with 5% fetal bovine serum (FBS) (Atlanta Biologicals) or 0.5% activated charcoal.
H. pylori broth culture supernatants and purification of VacA.
VacA was purified from culture supernatant of H. pylori strain 60190 as described previously (62), and purified VacA was acid activated before adding it to cultured cells (62). For experiments using H. pylori broth culture supernatant (derived from cultures in Brucella broth containing FBS), supernatants were concentrated 30-fold by ultrafiltration with a 30-kDa-cutoff membrane. The relative concentrations of VacA in broth culture supernatants from WT and mutant H. pylori strains were determined by Western blotting using rabbit anti-VacA antiserum (33), and the concentrations of VacA in individual preparations were then normalized. A relative VacA concentration of 1 corresponds to a 1:1,600 dilution of the concentrated WT H. pylori broth culture supernatant. The culture supernatants containing normalized concentrations of VacA were diluted in cell culture medium, and serial dilutions were added to cultured cells.
Cell culture.
AZ-521 human gastric adenocarcinoma cells (derived from human gastric cancer tissue; Culture Collection of Health Science Resource Bank, Japan Health Science Foundation), HeLa cells (derived from a human cervical adenocarcinoma; ATCC CCL-2), and RK13 rabbit kidney cells (derived from normal rabbit kidney; ATCC CCL-37) were grown in minimal essential medium supplemented with 10% FBS (Atlanta Biologicals) and 1 mM nonessential amino acids, and AGS human gastric epithelial cells (derived from a human gastric adenocarcinoma; ATCC CRL1739) were grown in RPMI medium containing 10% FBS, 2 mM l-glutamine, and 10 mM HEPES buffer.
Generation of a gene trap library.
To construct a gene trap library, the U3neoSV1 retrovirus vector (63) was obtained from Zirus, Inc. (Buford, GA), and AZ-521 cells were infected for 1 h with U3neoSV1 at a multiplicity of infection (MOI) of 0.1 in the presence of 4 μg/ml of Polybrene (Sigma), a cationic polymer used to increase the infection efficiency. The medium was subsequently changed, and the cells were grown overnight. One day later, cells were exposed to the antibiotic G418 (0.375 mg/ml) (Research Products International Corp.) to select for cells containing the U3neo cassette.
Generation of an shRNA library.
To generate an shRNA library in AZ-521 cells, a pooled Decode RNAi-GIPZ annotated gene screening library (positive selection kit; Open Biosystems) packaged in a lentivirus vector was added to AZ-521 cells at an MOI of 0.3. Cells were incubated for 3 days and then exposed to puromycin (5 μg/ml) to select for cells containing the shRNA.
Analysis of gene trap library and identification of targeted genes.
Gene trap library cells were incubated 2 times with 10 μg/ml of purified acid-activated VacA (twice the concentration required to kill AZ-521 cells [35]) in the presence of 10 mM ammonium chloride for 24 h each, and surviving cells were picked as single-cell clones. Surviving cells were subsequently analyzed in a secondary screen to test whether they exhibited resistance to VacA-containing H. pylori broth culture supernatant in comparison to parental control cells. Clones that exhibited significantly increased survival compared to parental control cells were then analyzed further.
To identify the genes that were disrupted in VacA-resistant gene trap library clones that passed the secondary screen, genomic DNA was isolated from the cells using a QIAamp DNA Blood Maxi kit (Qiagen). Shuttle vectors and genomic DNA flanking the integration site were then recovered by digesting the genomic DNA with EcoRI or BamHI, self-ligating, transforming into Escherichia coli, and selecting on LB agar containing 100 μg/ml of carbenicillin (Sigma). Individual colonies were amplified, and recovered genomic DNAs were sequenced using primers that anneal to the gene trap shuttle vector. Sequences were analyzed by the RepeatMasker web server followed by nucleotide-nucleotide BLAST searches against the NCBI databases.
Analysis of shRNA library and identification of targeted genes.
shRNA library cells were incubated 2 times with 10 μg/ml of purified acid-activated VacA in the presence of 10 mM ammonium chloride for 24 h each, and surviving cells were picked as single-cell clones. Surviving cells were subsequently analyzed in a secondary screen to test whether they exhibited resistance to VacA-containing H. pylori broth culture supernatant in comparison to parental control cells. Clones that exhibited a significantly increased survival compared to that of parental control cells were subsequently analyzed to identify the targeted genes. To identify the shRNA that was expressed in VacA-resistant shRNA library clones that passed the secondary screen, genomic DNA was isolated from the cells using the Wizard genomic DNA purification kit (Promega), and the shRNA was PCR amplified and sequenced.
Cell viability assays.
For cell viability assays, cells (AZ-521, HeLa, RK13, and AGS) were seeded at 4 × 104 cells/well into 96-well plates and incubated overnight. Cells were then incubated with serial dilutions of H. pylori broth culture supernatants containing WT VacA or VacA-G14A in the presence of 10 mM ammonium chloride. Cell viability was assessed using the CellTiterAQueousOne Solution cell proliferation assay (Promega) or the ATPlite 1-step assay (PerkinElmer) according to the manufacturer's instructions.
Western blot analysis of Cx43.
Cells (AZ-521, HeLa, RK13, and AGS) were seeded at 7 × 105 cells/well into 6-well plates and incubated overnight. Cells were lysed in CelLyticM cell lysis reagent (Sigma). Expression of Cx43 was assessed by Western blotting using a rabbit anti-Cx43 antibody (1:1,000; abcam) followed by a horseradish peroxidase-conjugated secondary antibody (1:10,000; Promega). Expression of β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was monitored using anti-β-actin and anti-GAPDH antibodies (1:1,000; abcam) to ensure equal loading. Proteins were visualized by incubation with a chemiluminescent substrate solution (Pierce) and exposure to film.
Inhibition of Cx43 expression.
AZ-521 cells were transduced with lentivirus particles expressing either nontargeting (NT) shRNA or two different Cx43-specific shRNAs (Cx43 shRNA 83010 and Cx43 shRNA 83012) (Open Biosystems) at an MOI of 12. Cells were incubated for 3 days and then exposed to puromycin (5 μg/ml) to select for cells containing the shRNA. Cx43 protein expression was analyzed by Western blotting.
Analysis of VacA localization and Cx43 localization.
To analyze the interactions of VacA with control AZ-521 cells compared to results with cells that expressed Cx43-specific shRNA, purified VacA was labeled with Alexa 488 (Molecular Probes) according to the manufacturer's instructions. AZ-521 cells expressing NT shRNA, Cx43 shRNA 83010, or Cx43 shRNA 83012 were seeded at 4 × 104 cells/well into 8-well culture slides (BD Biosciences) and incubated overnight. Cells were then incubated with 5 μg/ml of acid-treated Alexa 488-labeled VacA in the presence of 10 mM ammonium chloride at 37°C for 1 h. Cells were washed, fixed in 4% formaldehyde, and permeabilized in phosphate-buffered saline (PBS) containing 0.25% Triton X-100. 7-Amino-actinomycin D (7-AAD) (Pharmingen) was used for nuclear staining, and cells were viewed with an LSM 510 confocal microscope (Carl Zeiss).
To compare the localization of VacA and Cx43, AZ-521 cells were incubated with 5 μg/ml of purified acid-activated VacA in the presence of 10 mM ammonium chloride at 37°C for 1 h. Cells were then washed, fixed in 4% formaldehyde, and permeabilized in PBS containing 0.25% Triton X-100. Cells were incubated with anti-VacA serum (1:1,000) and Cx43 antibody (1:500; abcam), followed by Alexa 488- and Alexa 555-conjugated anti-rabbit and anti-mouse secondary antibodies (each at 1:500; from Molecular Probes). 4′,6-Diamidino-2-phenylindole, dihydrochloride (DAPI) (BD Pharmingen), was used for nuclear staining. Cells were viewed with a 710 confocal laser scanning microscope (Carl Zeiss). Confocal laser scanning micrographs were collected at 0.66-μm intervals in the Z plane, and the image stacks were displayed using the ortho slice function of the Zen 2010 software program. The Z-stack images were also used to digitally render three-dimensional (3D) and 4D images using the Zen 2010 software package. The localization of Cx43 in HeLa cells was analyzed using the same approach.
Expression of Cx43 in HeLa cells.
HeLa cells were transduced with lentivirus particles encoding human Cx43 or with negative-control Lentifect lentivirus particles (GeneCopoeia) at an MOI of 5. Cells were incubated for 3 days and then exposed to 5 μg/ml puromycin to select for transduced cells.
RESULTS
Analysis of gene trap and shRNA libraries.
In this study, we sought to identify cellular factors that are required for VacA-induced death of gastric epithelial cells. To accomplish this goal, we generated a gene trap library in AZ-521 cells by infecting cells with a replication-deficient retrovirus vector containing a promoterless neomycin resistance gene. Insertion of the gene trap vector into an actively transcribed region of the genome results in a neomycin-resistant phenotype. Selection of neomycin-resistant cells results in a library of cells containing random insertions of the gene trap vector in the host genome and disrupted expression of the genes into which the vector has inserted. We incubated the AZ-521 gene trap library with purified VacA, as described in Materials and Methods. Surviving cells were picked as single-cell clones, expanded, and tested in secondary screens to confirm that they were resistant to VacA. We then analyzed the sites of gene trap vector insertion in 65 VacA-resistant clones to determine which genes had been disrupted. There was a high level of redundancy among the VacA-resistant clones that were identified (i.e., many of the same clones were detected multiple times), which provided evidence that a reasonably complete set of the VacA-resistant clones in this library had been identified. In total, we identified 17 distinct VacA-resistant clones (Table 1). In three cases, the same gene was disrupted by gene trap vector insertion into multiple distinct sites; thus, 14 different genes were disrupted in the VacA-resistant cells (Table 1).
TABLE 1.
Genes identified in gene trap library screena
| Gene symbolb | Gene or product name | Alternate name(s) |
|---|---|---|
| DLGAP1 | Discs, large (Drosophila) homolog-associated protein 1 | DAP-1, GKAP, SAPAP1 |
| EFHA2 | EF-hand domain family, member A2 | MICU3 |
| EIF1 | Eukaryotic translation initiation factor 1 | A121, EIF-1, EIFIA, ISO1, SUI1 |
| FMN1 | Formin 1 | DKFZP686C2281, MGC125288, MGC125289 |
| GAS5 | Growth arrest-specific 5 | NCRNA00030, SNHG2 |
| GJA1 | Gap junction protein alpha 1, 43 kDa | Cx43, ODD, ODOD, SDTY3 |
| KITL | Kit ligand | FPH2, KL-1, SCF, SF |
| KLHL14 | Kelch-like 14 | KIAA1384 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 | NCRNA00047, NEAT2, PRO1073 |
| PARM1 | Prostate androgen-regulated transcript 1 | CIPAR1, DKFZP64O0823, WSC4 |
| RCC1 | Regulator of chromosome condensation 1 | None |
| SNORD58B | Small nucleolar RNA, C/D box 58B | U58B |
| SNORD76 | Small nucleolar RNA, C/D box 76 | U76 |
| TGFβ2 | Transforming growth factor β2 | None |
Multiple independent clones, each with a different site of gene trap vector insertion into the same gene, were identified in three cases. There were 4 insertions into RCC1, 2 insertions into GAS5, and 3 insertions into Cx43/GJA1.
Gene nomenclature is based on recommendations of the HUGO Gene Nomenclature Committee at the European Bioinformatics Institute (http://www.genename.org).
As a complementary approach, we introduced an shRNA library into AZ-521 cells. To generate the shRNA library, pools of shRNAs targeting human genes were introduced into the cells by lentivirus transduction. The shRNAs integrate randomly into the genome, and small interfering RNAs (siRNAs) targeting human genes are continually produced, resulting in inhibited expression of specific genes. We incubated a portion of the library with purified VacA, as described in Materials and Methods. Surviving cells were picked as single-cell clones, expanded, tested in secondary screens to confirm that they were resistant to VacA, and then analyzed to identify the shRNAs present in the resistant clones. A total of 25 VacA-resistant shRNA library clones were identified, and there was no redundancy in the genes that were targeted by the shRNAs in these clones.
We identified only one gene, the gap junction protein alpha 1 gene (GJA1 or Cx43; here designated Cx43), that was targeted in both the VacA-resistant gene trap clones and the VacA-resistant shRNA clones. Secondary screens confirmed that these clones originating with the gene trap library or the shRNA library exhibited increased resistance to VacA in comparison to results for control parental cells (Fig. 1B). Among the VacA-resistant gene trap clones, there were three independent sites of gene trap vector insertion into the Cx43 gene (Fig. 1A), and all three insertions occurred within the Cx43 exon. Therefore, we conducted further experiments to investigate a potential role of Cx43 in VacA-induced cell death.
FIG 1.
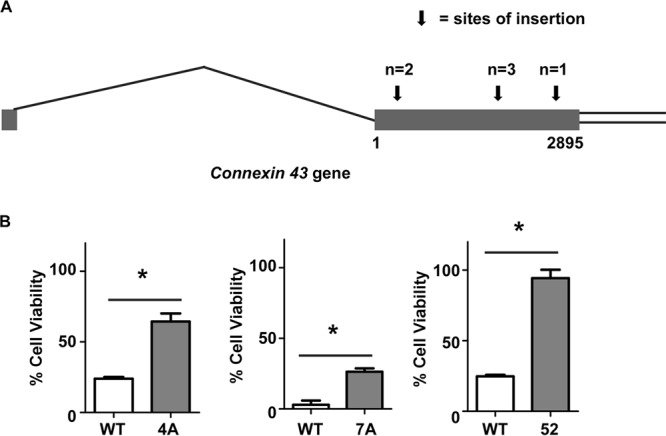
Analysis of VacA-resistant shRNA library clones and gene trap library clones in which Cx43 was targeted. An AZ-521 gene trap library and an AZ-521 shRNA library were each incubated with purified VacA. (A) Six VacA-resistant clones isolated from the gene trap library contained disruptions in Cx43, and these arose from three independent insertions of the gene trap vector into the gene encoding Cx43. The diagram shows the Cx43 gene, which contains only one exon (denoted by the gray rectangle) (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=mouse&c=Gene&l=Gja1), along with the sites of gene trap vector insertion, marked by arrows. All insertions occurred within the Cx43 exon. (B) Two of the gene trap library clones (4A and 7A, each with a vector insertion in a different site within Cx43) and the shRNA library clone containing Cx43-specific shRNA (clone 52, which expressed shRNA 5′-GGTGCATGTTGGTATTTAA) were tested in a secondary screen (CellTiterAQueousOne Solution cell proliferation assay) that compared the susceptibilities of these library clones and WT cells to H. pylori broth culture supernatants containing VacA. Asterisks indicate P < 0.05 (Student's t test). The results represent means and standard errors based on triplicate determinations.
Role of Cx43 in VacA-induced cell death.
To further validate a role of Cx43 in VacA-induced cell death, we transduced parental AZ-521 cells with lentivirus particles expressing either nontargeting (NT) shRNA or two different Cx43-specific shRNAs (Cx43 shRNA 83010 and Cx43 shRNA 83012), and cells stably expressing the shRNAs were selected, as described in Materials and Methods. The sequence of Cx43 shRNA 83012 (5′-GGTGCATGTTGGTATTTAA) was identical to that which was present in the VacA-resistant clone selected from the shRNA library, but the sequence of shRNA 83010 (5′-CCAATATGGTGTTTACATT) was distinct. Western blot analysis showed reduced production of Cx43 in cells expressing Cx43 shRNA compared to that in cells expressing NT shRNA (Fig. 2A). We then incubated the AZ-521 cells expressing either Cx43-specific shRNA (83010 and 83012) or NT shRNA with culture supernatants containing wild-type (WT) VacA or VacA-G14A, a mutant toxin that is defective in membrane channel formation (61). Cells expressing either Cx43 shRNA 83010 or Cx43 shRNA 83012 exhibited resistance to WT VacA compared to cells expressing NT shRNA (Fig. 2B to E). These experiments confirmed that a reduction in Cx43 protein expression confers resistance to VacA-induced cell killing.
FIG 2.

Inhibition of Cx43 expression increases cellular resistance to VacA toxicity. (A) AZ-521 cells were transduced with lentivirus particles encoding nontargeting (NT) shRNA or two different Cx43-specific shRNAs (83010 and 83012), and cells stably expressing the shRNAs were selected. Expression of the Cx43 protein was analyzed by Western blotting using an anti-Cx43 antibody. Expression of β-actin was monitored to ensure equal loading. The effect of shRNA treatment on Cx43 production was evaluated by densitometry. Cx43 expression was decreased in Cx43 shRNA-expressing cells compared to that in cells expressing NT shRNA. (B to E) Control AZ-521 cells (NT) and cells expressing Cx43-specific shRNA (83010 or 83012) were incubated with serial dilutions of H. pylori broth culture supernatants containing WT VacA (B and D) or the inactive mutant protein VacA-G14A (C and E). After 24 h, cell viability was determined by the ATPlite assay. The Cx43-specific shRNA conferred a protective effect in the experiments shown in panels B and D but not in the experiments shown in panels C and E. A low level of cell death in response to high concentrations of supernatants containing VacA-G14A is attributed to nonspecific effects. Error bars represent means plus standard errors from combined results of two independent experiments, each performed in triplicate. Asterisks indicate P < 0.05, Student's t test.
To investigate whether there were detectable differences in the interactions of VacA with cells expressing NT shRNA, Cx43 shRNA 83010, or Cx43 shRNA 83012, we analyzed VacA localization by immunofluorescence microscopy. VacA bound to cells expressing Cx43-specific shRNA, similar to the binding of VacA to cells expressing NT shRNA (Fig. 3A to C). To determine whether VacA colocalized with Cx43, we analyzed the localization of both proteins using confocal laser scanning microscopy. As expected, Cx43 was detected mainly at the junctions between cells, and this result was confirmed by optical sectioning and analysis of image stacks using the ortho slice function, as well as by analysis of 3D images (data not shown). VacA was also detected on the surfaces of cells but was not detected primarily at cell junctions. Moreover, we did not detect colocalization of VacA with Cx43 (Fig. 3D and E). Collectively, the results shown in Fig. 3 provide evidence that Cx43 is not the primary receptor for VacA in these cells.
FIG 3.

Analysis of VacA and Cx43 localization. (A to C) AZ-521 cells expressing nontargeting shRNA (NT), Cx43-specific shRNA 83010, or Cx43 shRNA 83012 were incubated with 5 μg/ml of acid-activated Alexa 488-labeled VacA in the presence of 10 mM ammonium chloride at 37°C for 1 h. Cells were imaged as described in Materials and Methods. The nucleus is shown in red, and VacA localization is shown in green. (D and E) AZ-521 cells were incubated with 5 μg/ml of acid-activated VacA in the presence of 10 mM ammonium chloride at 37°C for 1 h, and control cells were not treated with VacA. Cells were fixed, permeabilized, and stained to detect VacA and Cx43, as described in Materials and Methods. Panel D shows AZ-521 control cells that were not treated with VacA, and panel E shows AZ-521 cells treated with VacA for 1 h. The nucleus is shown in blue, Cx43 is shown in red, and VacA is shown in green. Bars, 20 μm.
Analysis of Cx43 protein expression in multiple gastric cell lines.
We previously reported that AZ-521 cells are more susceptible to VacA-induced cell death than are AGS cells (35). Cx43 expression is often lost in tumor cells during tumor development (64–67), and therefore, we hypothesized that the differential sensitivity of AZ-521 and AGS cells might correlate with differences in Cx43 expression. To investigate the expression of Cx43 in cell lines that are commonly used for studies of VacA and determine whether levels of Cx43 protein expression correlated with increased VacA susceptibility, we analyzed Cx43 protein expression in several epithelial cell lines of diverse tissue origin by Western blotting. Specifically, we tested AZ-521 cells (derived from human gastric cancer tissue), AGS cells (derived from a human gastric adenocarcinoma), HeLa cells (derived from a human cervical adenocarcinoma), and RK13 cells (derived from normal rabbit kidney). We detected Cx43 production in AZ-521 cells but did not detect Cx43 production in AGS, HeLa, or RK13 cells (Fig. 4A). Correspondingly, among these cell types, AZ-521 cells were the most susceptible to VacA-induced killing (Fig. 4B to E).
FIG 4.
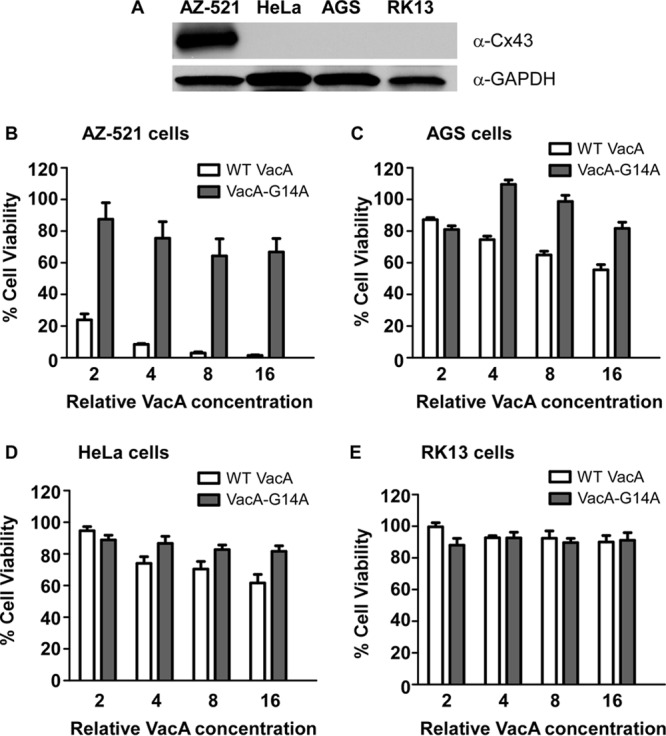
AZ-521 cells express Cx43 and are more susceptible to VacA than are cell lines that do not express Cx43. (A) Cx43 expression in AZ-521, HeLa, AGS, and RK13 cells was evaluated by Western blotting using an anti-Cx43 antibody. Production of GAPDH was monitored to ensure equal loading. (B to E) AZ-521, HeLa, AGS, and RK13 cells were incubated with serial dilutions of H. pylori broth culture supernatants containing WT VacA or VacA-G14A (an inactive mutant protein) for 24 h, and cell viability was determined by the ATPlite assay. AZ-521 cells were the most susceptible to VacA-induced killing. Error bars represent means plus standard errors from combined results of three independent experiments, each performed in triplicate.
We hypothesized that cells not expressing Cx43 might become more susceptible to VacA-induced cell death if they produced Cx43. To test this hypothesis, we transduced HeLa cells with lentivirus particles containing Cx43 or a control lentivirus, and we selected for transduced cells, as described in Materials and Methods. Western blot analysis showed that Cx43 was produced by HeLa-Cx43 cells but not HeLa-control cells (Fig. 5A). The level of Cx43 protein produced by HeLa-Cx43 cells was substantially lower (about 6-fold less based on densitometry) than that produced by AZ-521 cells. Despite the relatively low level of Cx43 production in the HeLa-Cx43 cells, these cells exhibited an increased susceptibility to VacA-induced cell death compared to that of HeLa-control cells (Fig. 5B and C). We also examined the localization of Cx43 in HeLa-Cx43 cells compared to that in HeLa-control cells and AZ-521 cells by confocal microscopy. In agreement with the Western blot results, confocal microscopy showed that Cx43 was produced by AZ-521 and HeLa-Cx43 but not HeLa-control cells, and the level of Cx43 protein production was lower in HeLa-Cx43 cells than in AZ-521 cells (Fig. 6A to C). The localization of Cx43 was similar in both cells types. Collectively, these results demonstrate that the susceptibility of cells to VacA-induced cell death correlates with the levels of Cx43 protein production.
FIG 5.
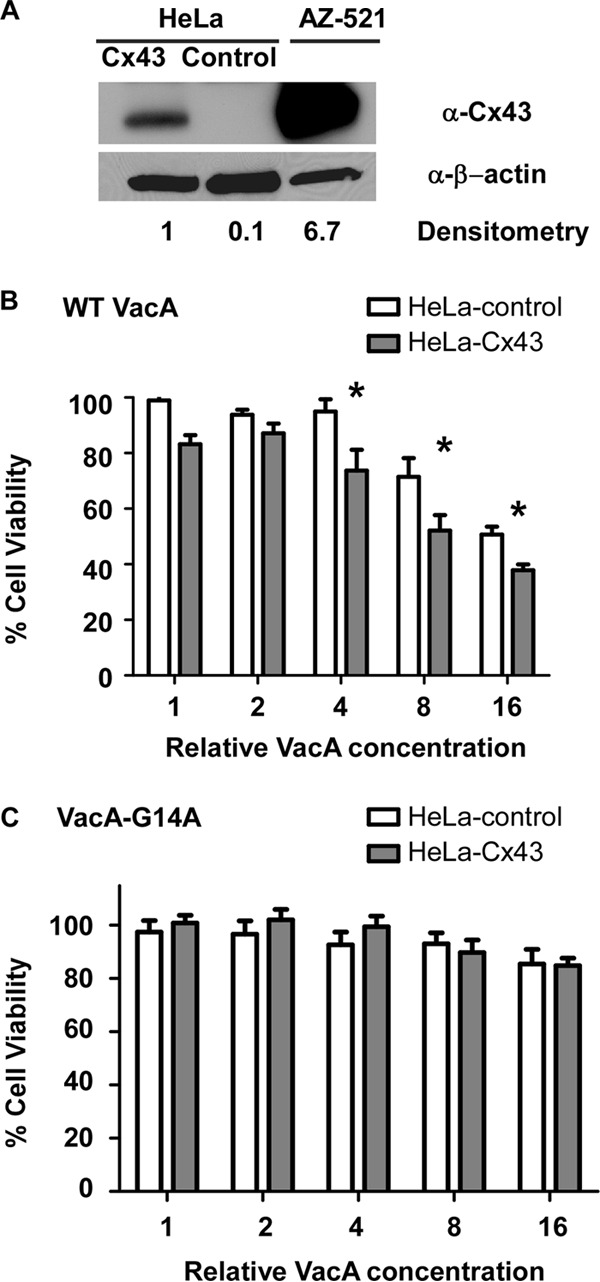
Expression of Cx43 in HeLa cells increases susceptibility to VacA. HeLa cells were transduced with lentivirus particles expressing Cx43 or a control lentivirus, and transduced cells were then selected (designated HeLa-Cx43 or HeLa-control, respectively). (A) Expression of Cx43 in AZ-521, HeLa-Cx43, and HeLa-control cells was analyzed by Western blotting using an anti-Cx43 antibody. Expression of β-actin was monitored to ensure equal loading. Levels of Cx43 were analyzed by densitometry. (B and C) HeLa-Cx43 and HeLa-control cells were incubated with serial dilutions of H. pylori broth culture supernatants containing WT VacA (B) or VacA-G14A (C) for 24 h, and cell viability was determined by the ATPlite assay. HeLa cells expressing Cx43 exhibited increased susceptibility to VacA-induced cell death compared to HeLa cells transduced with the control lentivirus. Error bars represent means plus standard errors from combined results of three independent experiments, each performed in triplicate. Asterisks indicate P < 0.05, Student's t test.
FIG 6.
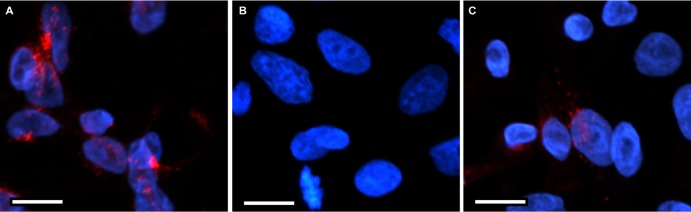
Expression and localization of Cx43 in AZ-521 and HeLa cells. HeLa cells were transduced with lentiviral particles expressing Cx43 or a control lentivirus, and transduced cells were then selected (designated HeLa-Cx43 or HeLa-control, respectively). AZ-521, HeLa-control, and HeLa-Cx43 cells were examined for expression and localization of Cx43. AZ-521, HeLa-control, and HeLa-Cx43 cells were fixed, permeabilized, and stained to detect Cx43, as described in Materials and Methods. The nucleus is shown in blue, and Cx43 is shown in red. Bars, 10 μm. Cx43 was detected in AZ-521 cells (A) and HeLa-Cx43 cells (C) but not in HeLa-control cells (B).
DISCUSSION
One of the important virulence factors produced by H. pylori is the vacuolating toxin VacA (7–14). Several host cell factors are known to be required for VacA-induced cellular alterations. These include cell surface receptors (e.g., sphingomyelin, RPTPα, RPTPβ, low-density lipoprotein [LDL] receptor-related protein 1 [LRP-1], and β2 integrin) (21–26), host cell factors required for VacA internalization and trafficking (dynamin and Rab7), and host factors involved in processes that are perturbed by VacA (e.g., Bax, Bak, Drp1, and the vacuolar ATPase) (7–14, 27–29, 38, 39, 42, 44). Nearly all of the previously identified host cell factors required for VacA action were discovered either by testing specific hypotheses about whether a known factor contributed to VacA action or by using biochemical approaches to identify proteins that interact with VacA. In the current study, we used insertional mutagenesis using gene entrapment (gene trap) and shRNA libraries as systematic unbiased approaches to identify host cell factors that are required for VacA-induced cell death.
Several previous studies have suggested that the use of hypodiploid cell lines facilitates gene trap selections (68, 69). However, the findings in the current study and several other studies indicate that gene trap selections can be used successfully in diploid or hyperdiploid cell lines (70–73). We identified VacA-resistant clones with disrupted expression of Cx43 in both gene trap library and shRNA library experiments, and therefore, we analyzed the role of this host cell protein in further depth. In addition to identifying VacA-resistant clones with disrupted expression of Cx43, we also identified VacA-resistant gene trap clones with disruptions of 13 other genes (Table 1). Further studies will be required to validate a role of these other genes in the cellular actions of VacA and to determine by what mechanisms they contribute to VacA activity.
Connexins are components of gap junctions, which are important for many physiological processes (74, 75). Gap junctions are comprised of two hexameric structures, known as hemichannels or connexons, with one hemichannel originating from each of two adjacent cells. Each connexon is comprised of six individual connexin subunits (54, 76–78). Direct intercellular communication by gap junctions is mediated primarily by passive diffusion of small hydrophilic molecules, such as glucose, glutamate, glutathione, cAMP, ATP, inositol triphosphate (IP3), calcium, potassium, and sodium (48, 54, 74). Gap junction proteins (and Cx43 in particular) can regulate cell death and survival, cell growth, and cell differentiation through several mechanisms, which are either dependent or independent of channel activity (79–83). Cx43 has been localized not only to gap junctions but also to intracellular sites, including mitochondria (48, 76, 79, 80, 84).
In the current study, we demonstrated by multiple approaches that Cx43 contributes to VacA-induced cell death in AZ-521 cells. Specifically, analysis of a gene trap library yielded multiple independent VacA-resistant clones that all harbored gene trap vector insertions in Cx43, and analysis of an shRNA library also yielded a VacA-resistant clone in which expression of Cx43 was targeted. Subsequent shRNA experiments validated the results of the library analyses, and the production of Cx43 in HeLa cells (which ordinarily fail to produce Cx43) conferred increased susceptibility to VacA-induced cell death. AZ-521 cells in which Cx43 expression was inhibited remained susceptible to high concentrations of VacA; this may be attributable to incomplete knockdown of Cx43 expression. We did not detect colocalization of VacA with Cx43, which suggests that VacA-induced cell death does not depend on a direct interaction between VacA and Cx43.
Cx43 can potentially contribute to VacA-induced cell death through multiple mechanisms, including leakage of cellular constituents through Cx43 hemichannels, intracellular actions of Cx43, or Cx43-dependent paracrine signaling between cells (48, 76, 79, 80, 84). Previous publications have provided evidence indicating that Cx43 can cause cell death through each of these mechanisms (79, 80, 85–87). We have shown previously that incubation of Cx43-expressing gastric epithelial cells with VacA results in a reduction of cellular ATP content (35), and this reduction in ATP content could occur at least in part by release of ATP through connexin 43 channels (88). Another possibility is that VacA alters Cx43-dependent signaling cascades. Cx43 plays a major role in regulating intracellular Ca2+ levels and cell volume (76), which are both relevant determinants of cell survival and cell death (89) and previously were shown to be altered in VacA-treated cells (35, 90, 91). Furthermore, Cx43-dependent intercellular communication through gap junctions can have a role in the spread of cell death signals between neighboring cells (80, 92, 93), involving the use of Ca2+, cAMP, cGMP, and ATP as candidate messengers (80, 94). Further experiments will be necessary to determine which of these molecular actions of Cx43 are most relevant in VacA-induced cell death.
We examined several cultured cell lines for expression of Cx43 and observed that among the cell lines tested, AZ-521 cells were the only cells that expressed detectable levels of the Cx43 protein. These findings are consistent with the results of previous studies, which reported that HeLa cells (64, 65), as well as many other cultured human cell lines and human cancers, lack Cx43 protein expression (66, 67). Similarly, gap junction-mediated intercellular communication is often impaired in cultured human cell lines compared to that in corresponding noncancerous cells (49–51, 65–67, 95). Interestingly, HeLa cells (which fail to express Cx43) are susceptible to VacA-induced cell vacuolation. This observation suggests that although Cx43 contributes to VacA-induced cell death, it is not required for VacA-induced cell vacuolation.
In summary, this study provides strong evidence that Cx43 has an important role in VacA-induced death of gastric epithelial cells. These results are consistent with a growing body of evidence indicating that Cx43 can contribute to cell death in many different cell types and tissues (48, 76, 79, 80, 84). VacA-mediated death of gastric epithelial cells through a Cx43-dependent pathway may be particularly important in the pathogenesis of H. pylori-associated peptic ulceration. We speculate that the reestablishment of epithelial integrity in the setting of gastric ulceration and persistent H. pylori infection might favor proliferation of cells that exhibit decreased expression of Cx43, and such cells may have increased potential for malignant transformation.
ACKNOWLEDGMENTS
This work was supported by NIH R01AI039657, P01 CA116087, T32 AI007281, and F32 AI102568 and the Bob and Red Buisson Foundation. Experiments in the Vanderbilt Cell Imaging Shared Resource were supported by Vanderbilt University Digestive Disease Research Center (NIH grant P30DK058404) and the Vanderbilt University Ingram Cancer Center (NIH grant P30 CA068485).
The content is solely our responsibility and does not necessarily represent the official views of the funding agencies. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 4 November 2013
REFERENCES
- 1.Cover TL, Blaser MJ. 2009. Helicobacter pylori in health and disease. Gastroenterology 136:1863–1873. 10.1053/j.gastro.2009.01.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atherton JC, Blaser MJ. 2009. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest. 119:2475–2487. 10.1172/JCI38605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suerbaum S, Michetti P. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175–1186. 10.1056/NEJMra020542 [DOI] [PubMed] [Google Scholar]
- 4.Atherton JC. 2006. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 1:63–96. 10.1146/annurev.pathol.1.110304.100125 [DOI] [PubMed] [Google Scholar]
- 5.Parkin DM, Bray F, Ferlay J, Pisani P. 2005. Global cancer statistics, 2002. CA Cancer J. Clin. 55:74–108. 10.3322/canjclin.55.2.74 [DOI] [PubMed] [Google Scholar]
- 6.Fox JG, Wang TC. 2007. Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 117:60–69. 10.1172/JCI30111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cover TL, Blanke SR. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320–332. 10.1038/nrmicro1095 [DOI] [PubMed] [Google Scholar]
- 8.Palframan SL, Kwok T, Gabriel K. 2012. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell Infect. Microbiol. 2:92. 10.3389/fcimb.2012.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boquet P, Ricci V. 2012. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 20:165–174. 10.1016/j.tim.2012.01.008 [DOI] [PubMed] [Google Scholar]
- 10.Kim IJ, Blanke SR. 2012. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front. Cell Infect. Microbiol. 2:37. 10.3389/fcimb.2012.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rassow J, Meinecke M. 2012. Helicobacter pylori VacA: a new perspective on an invasive chloride channel. Microbes Infect. 14:1026–1033. 10.1016/j.micinf.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 12.de Bernard M, Cappon A, Del Giudice G, Rappuoli R, Montecucco C. 2004. The multiple cellular activities of the VacA cytotoxin of Helicobacter pylori. Int. J. Med. Microbiol. 293:589–597. 10.1078/1438-4221-00299 [DOI] [PubMed] [Google Scholar]
- 13.Fischer W, Prassl S, Haas R. 2009. Virulence mechanisms and persistence strategies of the human gastric pathogen Helicobacter pylori. Curr. Top. Microbiol. Immunol. 337:129–171. 10.1007/978-3-642-01846-6_5 [DOI] [PubMed] [Google Scholar]
- 14.Rassow J. 2011. Helicobacter pylori vacuolating toxin A and apoptosis. Cell Commun. Signal 9:26. 10.1186/1478-811X-9-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Telford JL, Ghiara P, Dell'Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce MF, Censini S, Covacci A, Xiang Z, Papini E, Montecucco C, Parent L, Rappuoli R. 1994. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179:1653–1658. 10.1084/jem.179.5.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitt W, Haas R. 1994. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Mol. Microbiol. 12:307–319. 10.1111/j.1365-2958.1994.tb01019.x [DOI] [PubMed] [Google Scholar]
- 17.Cover TL, Tummuru MK, Cao P, Thompson SA, Blaser MJ. 1994. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 269:10566–10573 [PubMed] [Google Scholar]
- 18.Cover TL, Blaser MJ. 1992. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 267:10570–10575 [PubMed] [Google Scholar]
- 19.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB. 2007. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. U. S. A. 104:16293–16298. 10.1073/pnas.0707447104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cover TL, Krishna US, Israel DA, Peek RM., Jr 2003. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 63:951–957 [PubMed] [Google Scholar]
- 21.Gupta VR, Patel HK, Kostolansky SS, Ballivian RA, Eichberg J, Blanke SR. 2008. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 4:e1000073. 10.1371/journal.ppat.1000073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta VR, Wilson BA, Blanke SR. 2010. Sphingomyelin is important for the cellular entry and intracellular localization of Helicobacter pylori VacA. Cell Microbiol. 12:1517–1533. 10.1111/j.1462-5822.2010.01487.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yahiro K, Niidome T, Kimura M, Hatakeyama T, Aoyagi H, Kurazono H, Imagawa K, Wada A, Moss J, Hirayama T. 1999. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase beta. J. Biol. Chem. 274:36693–36699. 10.1074/jbc.274.51.36693 [DOI] [PubMed] [Google Scholar]
- 24.Yahiro K, Wada A, Nakayama M, Kimura T, Ogushi K, Niidome T, Aoyagi H, Yoshino K, Yonezawa K, Moss J, Hirayama T. 2003. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 278:19183–19189. 10.1074/jbc.M300117200 [DOI] [PubMed] [Google Scholar]
- 25.Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, Suzuki H, Nomura F, Noda M, Moss J, Hirayama T. 2012. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J. Biol. Chem. 287:31104–31115. 10.1074/jbc.M112.387498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, Osicka R, Schiemann M, Busch DH, Semmrich M, Holzmann B, Sebo P, Haas R. 2008. Integrin subunit CD18 is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3:20–29. 10.1016/j.chom.2007.11.003 [DOI] [PubMed] [Google Scholar]
- 27.Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. 2005. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol. Biol. Cell 16:4852–4866. 10.1091/mbc.E05-05-0398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gauthier NC, Monzo P, Gonzalez T, Doye A, Oldani A, Gounon P, Ricci V, Cormont M, Boquet P. 2007. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 177:343–354. 10.1083/jcb.200609061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Wandinger-Ness A, Goldenring JR, Cover TL. 2004. Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol. Biol. Cell 15:1946–1959. 10.1091/mbc.E03-08-0618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willhite DC, Blanke SR. 2004. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol. 6:143–154. 10.1046/j.1462-5822.2003.00347.x [DOI] [PubMed] [Google Scholar]
- 31.Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD, Scorrano L, de Bernard M. 2010. Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 17:1707–1716. 10.1038/cdd.2010.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. 2009. Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5:370–379. 10.4161/auto.5.3.7663 [DOI] [PubMed] [Google Scholar]
- 33.Schraw W, Li Y, McClain MS, van der Goot FG, Cover TL. 2002. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 277:34642–34650. 10.1074/jbc.M203466200 [DOI] [PubMed] [Google Scholar]
- 34.Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. 1999. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18:5517–5527. 10.1093/emboj/18.20.5517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radin JN, Gonzalez-Rivera C, Ivie SE, McClain MS, Cover TL. 2011. Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect. Immun. 79:2535–2543. 10.1128/IAI.01370-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho SJ, Kang NS, Park SY, Kim BO, Rhee DK, Pyo S. 2003. Induction of apoptosis and expression of apoptosis related genes in human epithelial carcinoma cells by Helicobacter pylori VacA toxin. Toxicon 42:601–611. 10.1016/j.toxicon.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 37.Kuck D, Kolmerer B, Iking-Konert C, Krammer PH, Stremmel W, Rudi J. 2001. Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect. Immun. 69:5080–5087. 10.1128/IAI.69.8.5080-5087.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, Van Obberghen E, Boquet P. 2000. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 19:6361–6370. 10.1093/emboj/19.23.6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Domanska G, Motz C, Meinecke M, Harsman A, Papatheodorou P, Reljic B, Dian-Lothrop EA, Galmiche A, Kepp O, Becker L, Gunnewig K, Wagner R, Rassow J. 2010. Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog. 6:e1000878. 10.1371/journal.ppat.1000878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foo JH, Culvenor JG, Ferrero RL, Kwok T, Lithgow T, Gabriel K. 2010. Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J. Mol. Biol. 401:792–798. 10.1016/j.jmb.2010.06.065 [DOI] [PubMed] [Google Scholar]
- 41.Manente L, Perna A, Buommino E, Altucci L, Lucariello A, Citro G, Baldi A, Iaquinto G, Tufano MA, De Luca A. 2008. The Helicobacter pylori's protein VacA has direct effects on the regulation of cell cycle and apoptosis in gastric epithelial cells. J. Cell. Physiol. 214:582–587. 10.1002/jcp.21242 [DOI] [PubMed] [Google Scholar]
- 42.Jain P, Luo ZQ, Blanke SR. 2011. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. U. S. A. 108:16032–16037. 10.1073/pnas.1105175108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanke SR. 2005. Micro-managing the executioner: pathogen targeting of mitochondria. Trends Microbiol. 13:64–71. 10.1016/j.tim.2004.12.007 [DOI] [PubMed] [Google Scholar]
- 44.Yamasaki E, Wada A, Kumatori A, Nakagawa I, Funao J, Nakayama M, Hisatsune J, Kimura M, Moss J, Hirayama T. 2006. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 281:11250–11259. 10.1074/jbc.M509404200 [DOI] [PubMed] [Google Scholar]
- 45.Willhite DC, Cover TL, Blanke SR. 2003. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J. Biol. Chem. 278:48204–48209. 10.1074/jbc.M304131200 [DOI] [PubMed] [Google Scholar]
- 46.Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A, Sciullo A, Sommi P, Fabbri A, Ricci V, Boquet P. 2009. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 5:e1000603. 10.1371/journal.ppat.1000603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura M, Goto S, Wada A, Yahiro K, Niidome T, Hatakeyama T, Aoyagi H, Hirayama T, Kondo T. 1999. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb. Pathog. 26:45–52. 10.1006/mpat.1998.0241 [DOI] [PubMed] [Google Scholar]
- 48.Vinken M, Decrock E, Leybaert L, Bultynck G, Himpens B, Vanhaecke T, Rogiers V. 2012. Non-channel functions of connexins in cell growth and cell death. Biochim. Biophys. Acta 1818:2002–2008. 10.1016/j.bbamem.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 49.Tang B, Peng ZH, Yu PW, Yu G, Qian F, Zeng DZ, Zhao YL, Shi Y, Hao YX, Luo HX. 2013. Aberrant expression of cx43 is associated with the peritoneal metastasis of gastric cancer and cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PLoS One 8:e74527. 10.1371/journal.pone.0074527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang B, Peng ZH, Yu PW, Yu G, Qian F. 2011. Expression and significance of Cx43 and E-cadherin in gastric cancer and metastatic lymph nodes. Med. Oncol. 28:502–508. 10.1007/s12032-010-9492-5 [DOI] [PubMed] [Google Scholar]
- 51.Kadle R, Zhang JT, Nicholson BJ. 1991. Tissue-specific distribution of differentially phosphorylated forms of Cx43. Mol. Cell. Biol. 11:363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spray DC, Chanson M, Moreno AP, Dermietzel R, Meda P. 1991. Distinctive gap junction channel types connect WB cells, a clonal cell line derived from rat liver. Am. J. Physiol. 260:C513–C527 [DOI] [PubMed] [Google Scholar]
- 53.Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G. 2002. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 383:725–737. 10.1515/BC.2002.076 [DOI] [PubMed] [Google Scholar]
- 54.Saez JC, Contreras JE, Bukauskas FF, Retamal MA, Bennett MV. 2003. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol. Scand. 179:9–22. 10.1046/j.1365-201X.2003.01196.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oviedo-Orta E, Howard Evans W. 2004. Gap junctions and connexin-mediated communication in the immune system. Biochim. Biophys. Acta 1662:102–112. 10.1016/j.bbamem.2003.10.021 [DOI] [PubMed] [Google Scholar]
- 56.Neijssen J, Pang B, Neefjes J. 2007. Gap junction-mediated intercellular communication in the immune system. Prog. Biophys. Mol. Biol. 94:207–218. 10.1016/j.pbiomolbio.2007.03.008 [DOI] [PubMed] [Google Scholar]
- 57.Mendoza-Naranjo A, Bouma G, Pereda C, Ramirez M, Webb KF, Tittarelli A, Lopez MN, Kalergis AM, Thrasher AJ, Becker DL, Salazar-Onfray F. 2011. Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J. Immunol. 187:3121–3132. 10.4049/jimmunol.1100378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuczma M, Lee JR, Kraj P. 2011. Connexin 43 signaling enhances the generation of Foxp3+ regulatory T cells. J. Immunol. 187:248–257. 10.4049/jimmunol.1003785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen TD, Taffet SM. 2009. A model system to study Connexin 43 in the immune system. Mol. Immunol. 46:2938–2946. 10.1016/j.molimm.2009.06.022 [DOI] [PubMed] [Google Scholar]
- 60.Machtaler S, Dang-Lawson M, Choi K, Jang C, Naus CC, Matsuuchi L. 2011. The gap junction protein Cx43 regulates B-lymphocyte spreading and adhesion. J. Cell Sci. 124:2611–2621. 10.1242/jcs.089532 [DOI] [PubMed] [Google Scholar]
- 61.McClain MS, Iwamoto H, Cao P, Vinion-Dubiel AD, Li Y, Szabo G, Shao Z, Cover TL. 2003. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 278:12101–12108. 10.1074/jbc.M212595200 [DOI] [PubMed] [Google Scholar]
- 62.Cover TL, Hanson PI, Heuser JE. 1997. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 138:759–769. 10.1083/jcb.138.4.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osipovich AB, White-Grindley EK, Hicks GG, Roshon MJ, Shaffer C, Moore JH, Ruley HE. 2004. Activation of cryptic 3′ splice sites within introns of cellular genes following gene entrapment. Nucleic Acids Res. 32:2912–2924. 10.1093/nar/gkh604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mesnil M, Krutovskikh V, Piccoli C, Elfgang C, Traub O, Willecke K, Yamasaki H. 1995. Negative growth control of HeLa cells by connexin genes: connexin species specificity. Cancer Res. 55:629–639 [PubMed] [Google Scholar]
- 65.Eckert R, Dunina-Barkovskaya A, Hulser DF. 1993. Biophysical characterization of gap-junction channels in HeLa cells. Pflugers Arch. 424:335–342. 10.1007/BF00384361 [DOI] [PubMed] [Google Scholar]
- 66.Linnainmaa K, Pelin K, Vanhala E, Tuomi T, Piccoli C, Fitzgerald DJ, Yamasaki H. 1993. Gap junctional intercellular communication of primary and asbestos-associated malignant human mesothelial cells. Carcinogenesis 14:1597–1602. 10.1093/carcin/14.8.1597 [DOI] [PubMed] [Google Scholar]
- 67.Zhang ZQ, Zhang W, Wang NQ, Bani-Yaghoub M, Lin ZX, Naus CC. 1998. Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin43. Carcinogenesis 19:1889–1894. 10.1093/carcin/19.11.1889 [DOI] [PubMed] [Google Scholar]
- 68.Banks DJ, Bradley KA. 2007. SILENCE: a new forward genetic technology. Nat. Methods 4:51–53. 10.1038/nmeth991 [DOI] [PubMed] [Google Scholar]
- 69.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. 2009. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326:1231–1235. 10.1126/science.1178955 [DOI] [PubMed] [Google Scholar]
- 70.Fennessey CM, Sheng J, Rubin DH, McClain MS. 2012. Oligomerization of Clostridium perfringens epsilon toxin is dependent upon caveolins 1 and 2. PLoS One 7:e46866. 10.1371/journal.pone.0046866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ivie SE, Fennessey CM, Sheng J, Rubin DH, McClain MS. 2011. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens epsilon-toxin. PLoS One 6:e17787. 10.1371/journal.pone.0017787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sheng J, Organ EL, Hao C, Wells KS, Ruley HE, Rubin DH. 2004. Mutations in the IGF-II pathway that confer resistance to lytic reovirus infection. BMC Cell Biol. 5:32. 10.1186/1471-2121-5-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murray JL, Mavrakis M, McDonald NJ, Yilla M, Sheng J, Bellini WJ, Zhao L, Le Doux JM, Shaw MW, Luo CC, Lippincott-Schwartz J, Sanchez A, Rubin DH, Hodge TW. 2005. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J. Virol. 79:11742–11751. 10.1128/JVI.79.18.11742-11751.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexander DB, Goldberg GS. 2003. Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 10:2045–2058. 10.2174/0929867033456927 [DOI] [PubMed] [Google Scholar]
- 75.Lecanda F, Warlow PM, Sheikh S, Furlan F, Steinberg TH, Civitelli R. 2000. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 151:931–944. 10.1083/jcb.151.4.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodriguez-Sinovas A, Cabestrero A, Lopez D, Torre I, Morente M, Abellan A, Miro E, Ruiz-Meana M, Garcia-Dorado D. 2007. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog. Biophys. Mol. Biol. 94:219–232. 10.1016/j.pbiomolbio.2007.03.003 [DOI] [PubMed] [Google Scholar]
- 77.Evans WH, De Vuyst E, Leybaert L. 2006. The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem. J. 397:1–14. 10.1042/BJ20060175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dbouk HA, Mroue RM, El-Sabban ME, Talhouk RS. 2009. Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Commun. Signal 7:4. 10.1186/1478-811X-7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Decrock E, Vinken M, De Vuyst E, Krysko DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. 2009. Connexin-related signaling in cell death: to live or let die? Cell Death Differ 16:524–536. 10.1038/cdd.2008.196 [DOI] [PubMed] [Google Scholar]
- 80.Decrock E, De Vuyst E, Vinken M, Van Moorhem M, Vranckx K, Wang N, Van Laeken L, De Bock M, D'Herde K, Lai CP, Rogiers V, Evans WH, Naus CC, Leybaert L. 2009. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death Differ. 16:151–163. 10.1038/cdd.2008.138 [DOI] [PubMed] [Google Scholar]
- 81.Fujimoto E, Satoh H, Negishi E, Ueno K, Nagashima Y, Hagiwara K, Yamasaki H, Yano T. 2004. Negative growth control of renal cell carcinoma cell by connexin 32: possible involvement of Her-2. Mol. Carcinog. 40:135–142. 10.1002/mc.20025 [DOI] [PubMed] [Google Scholar]
- 82.Jiang JX, Gu S. 2005. Gap junction- and hemichannel-independent actions of connexins. Biochim. Biophys. Acta 1711:208–214. 10.1016/j.bbamem.2004.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krysko DV, Mussche S, Leybaert L, D'Herde K. 2004. Gap junctional communication and connexin43 expression in relation to apoptotic cell death and survival of granulosa cells. J. Histochem. Cytochem. 52:1199–1207. 10.1369/jhc.3A6227.2004 [DOI] [PubMed] [Google Scholar]
- 84.Decrock E, Vinken M, Bol M, D'Herde K, Rogiers V, Vandenabeele P, Krysko DV, Bultynck G, Leybaert L. 2011. Calcium and connexin-based intercellular communication, a deadly catch? Cell Calcium 50:310–321. 10.1016/j.ceca.2011.05.007 [DOI] [PubMed] [Google Scholar]
- 85.Frank DK, Szymkowiak B, Josifovska-Chopra O, Nakashima T, Kinnally KW. 2005. Single-cell microinjection of cytochrome c can result in gap junction-mediated apoptotic cell death of bystander cells in head and neck cancer. Head Neck 27:794–800. 10.1002/hed.20235 [DOI] [PubMed] [Google Scholar]
- 86.Huang R, Liu YG, Lin Y, Fan Y, Boynton A, Yang D, Huang RP. 2001. Enhanced apoptosis under low serum conditions in human glioblastoma cells by connexin 43 (Cx43). Mol. Carcinog. 32:128–138. 10.1002/mc.1072 [DOI] [PubMed] [Google Scholar]
- 87.Huang RP, Hossain MZ, Huang R, Gano J, Fan Y, Boynton AL. 2001. Connexin 43 (cx43) enhances chemotherapy-induced apoptosis in human glioblastoma cells. Int. J. Cancer 92:130–138. 10.1002/1097-0215(200102)9999:9999 [DOI] [PubMed] [Google Scholar]
- 88.Ali SR, Timmer AM, Bilgrami S, Park EJ, Eckmann L, Nizet V, Karin M. 2011. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 35:34–44. 10.1016/j.immuni.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Piper HM, Garcia-Dorado D, Ovize M. 1998. A fresh look at reperfusion injury. Cardiovasc. Res. 38:291–300. 10.1016/S0008-6363(98)00033-9 [DOI] [PubMed] [Google Scholar]
- 90.Wang F, Xia P, Wu F, Wang D, Wang W, Ward T, Liu Y, Aikhionbare F, Guo Z, Powell M, Liu B, Bi F, Shaw A, Zhu Z, Elmoselhi A, Fan D, Cover TL, Ding X, Yao X. 2008. Helicobacter pylori VacA disrupts apical membrane-cytoskeletal interactions in gastric parietal cells. J. Biol. Chem. 283:26714–26725. 10.1074/jbc.M800527200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Bernard M, Cappon A, Pancotto L, Ruggiero P, Rivera J, Del Giudice G, Montecucco C. 2005. The Helicobacter pylori VacA cytotoxin activates RBL-2H3 cells by inducing cytosolic calcium oscillations. Cell Microbiol. 7:191–198. 10.1111/j.1462-5822.2004.00446.x [DOI] [PubMed] [Google Scholar]
- 92.Cusato K, Zakevicius J, Ripps H. 2003. An experimental approach to the study of gap-junction-mediated cell death. Biol. Bull. 205:197–199. 10.2307/1543250 [DOI] [PubMed] [Google Scholar]
- 93.Krutovskikh VA, Piccoli C, Yamasaki H. 2002. Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene 21:1989–1999. 10.1038/sj.onc.1205187 [DOI] [PubMed] [Google Scholar]
- 94.Kalvelyte A, Imbrasaite A, Bukauskiene A, Verselis VK, Bukauskas FF. 2003. Connexins and apoptotic transformation. Biochem. Pharmacol. 66:1661–1672. 10.1016/S0006-2952(03)00540-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML. 2005. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 1719:125–145. 10.1016/j.bbamem.2005.11.004 [DOI] [PubMed] [Google Scholar]