

Abstract
Cerebral malaria (CM) is associated with excessive host proinflammatory responses and endothelial activation. The hematopoietic hormone erythropoietin (EPO) possesses neuroprotective functions in animal models of ischemic-hypoxic, traumatic, and inflammatory injuries. In the Plasmodium berghei ANKA model of experimental CM (ECM), recombinant human EPO (rhEPO) has shown evident protection against ECM. To elucidate the mechanism of EPO in this ECM model, we investigated the effect of rhEPO on host cellular immune responses. We demonstrated that improved survival of mice with ECM after rhEPO treatment was associated with reduced endothelial activation and improved integrity of the blood-brain barrier. Our results revealed that rhEPO downregulated the inflammatory responses by directly inhibiting the levels and functions of splenic dendritic cells. Conversely, rhEPO treatment led to significant expansion of regulatory T cells and increased expression of the receptor cytotoxic T lymphocyte antigen 4 (CTLA-4). The data presented here provide evidence of the direct effect of rhEPO on host cellular immunity during ECM.
INTRODUCTION
Cerebral malaria (CM), characterized by seizures and loss of consciousness, is a serious complication and a major cause of death in Plasmodium falciparum infections. Although the exact pathogenesis of CM is still not clearly defined, a current, unified view postulates that CM is associated with sequestration of parasitized red blood cells (pRBCs) in the presence or absence of leukocytes and platelets in the brain microvasculature and an overwhelming host proinflammatory response (1). Despite highly effective antimalarial drug treatments, a significant proportion of CM patients develop neurological sequelae, and therefore, it is of strategic importance to explore new adjunct therapies.
Erythropoietin (EPO), produced mainly in the kidneys, is a hematopoietic growth factor with major effects on the proliferation and survival of erythroid progenitor cells (2). EPO also possesses extrahematopoietic properties that are associated with the EPO receptor (EPOR) expressed on various nonerythroid tissues, such as brain, heart, muscles, and vascular endothelium (3, 4). For example, recombinant human EPO (rhEPO) can cross the blood-brain barrier (BBB) and is protective in animal models of ischemic-hypoxic, traumatic, and inflammatory injuries (5, 6). The erythropoietic response to EPO in RBC precursors is initiated upon EPO binding to EPOR homodimers, whereas in nonerythroid tissues, EPO utilizes a receptor complex that is composed of EPOR and CD131, the beta common receptor (βcR) shared by granulocyte-macrophage colony-stimulating factor, interleukin 3 (IL-3), and IL-5 (7, 8), thus connecting EPO with immune responses.
The immune-modulatory effects of EPO have been studied in various disease models and conditions, where EPO is frequently found to counteract the innate immune responses that are activated by proinflammatory cytokines during pathogen infections, trauma, and hypoxia (9). For example, in disease conditions linked to excessive inflammatory responses, such as experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, EPO treatment blocks clinical progression and ameliorates the disease pathology by reducing ischemia-induced production of inflammatory cytokines, such as tumor necrosis factor (TNF) and IL-6, through blockade of the proliferation of dendritic cells (DCs) and concomitant induction of regulatory T cell (Treg) expansion (10–13). Similarly, dampening host proinflammatory responses by EPO administration blunts disease severity in chemically induced colitis in mice (14). However, in multiple-myeloma patients and experimental murine models, EPO administration improves both cellular and humoral immunity, which includes CD8+ T cell activation, enhanced T cell proliferation potential, and increased antibody production (15–17). Thus, it seems that the spatial organization and temporal sequence of a plethora of mediators produced and pathways involved may dictate whether the effects are enhanced or inhibited by EPO administration. Interestingly, EPOR expression was detected in murine DCs and macrophages, and rhEPO had profound effects on these cells in culture, leading to upregulation of costimulatory molecules, cytokine secretion, and maturation of DCs (18, 19) but downregulation of NO, TNF, and IL-6 expression in macrophages (14), suggesting that EPO may affect both innate and adaptive immune responses.
The importance of EPO in malaria infection has long been recognized. High levels of EPO are found in African children suffering severe malarial anemia (20–22) and are correlated with >80% reduction in the risk of developing neurological sequelae during CM (23). In the murine experimental CM (ECM) model, administration of rhEPO significantly improves host survival without influencing parasitemia (24, 25). The neuroprotective effect of EPO in ECM includes reduced IFN-γ and TNF mRNA levels in the brain, diminished neuronal apoptosis, and activated neuronal stem cells (24–26). Furthermore, since hypoxia occurs in brain tissues of ECM and hypoxia induces EPO expression (27), systemic EPO administration substantially reduces the degree of neural hypoxia (28). In this report, we aimed to elucidate the mediators of EPO's neuroprotection during ECM. We focus on the roles of EPO in cell-mediated immunity using a well-established ECM model, C57BL/6 mice infected with Plasmodium berghei ANKA.
MATERIALS AND METHODS
Mice, parasite, and EPO treatment.
Female C57BL/6 mice, aged 6 to 8 weeks, were purchased from Benxi Animal Institute, Liaoning, China. Infections were initiated by intraperitoneal injection of 106 P. berghei ANKA pRBCs into C57BL/6 mice. In the EPO treatment group (P. berghei ANKA plus EPO), mice were treated daily by intravenous injection of 50 U rhEPO/mouse (Roche, Basel, Switzerland) dissolved in phosphate-buffered saline (PBS) at 2 to 4 days postinfection (p.i.). The control received the same volume of PBS at the same time points. For the mortality and parasitemia experiment, 10 mice were used in each group. Parasitemia was monitored by counting the pRBCs per 1,000 RBCs by light microscopy of Giemsa-stained blood smears, and mortality was monitored daily. Hematological parameters (RBCs and platelets) were determined in five mice in each group by using an automated analyzer with blood collected from the retro-orbital venous plexus and anticoagulated by EDTA. For the study of immune responses, nine mice were divided into three replicates at each time point for each treatment. All experiments were performed in compliance with animal ethics committee requirements in China.
Histopathology, immunohistochemistry, and BBB permeability.
Three mice from each of the three groups (uninfected control, P. berghei ANKA, and P. berghei ANKA plus EPO) were euthanized at 5 days p.i. The brains were immediately removed, fixed in 4% paraformaldehyde for 24 h, and embedded in paraffin. Serial 4-μm-thick horizontal sections were made and processed for hematoxylin and eosin staining and immunohistochemical studies. To detect ICAM-1 and VCAM-1 along the endothelial lining, immunohistochemical staining was performed with specific polyclonal antibodies against ICAM-1 (Abcam, USA) and VCAM-1 (Santa Cruz Biotechnology, USA) using a previously described method (29), which was followed by biotin-conjugated goat anti-rabbit IgG antibody. Finally, streptavidin-conjugated peroxidase was added, and color development was done using 3-amino-9-ethylcarbazole as the substrate. In each experiment, ICAM-1- or VCAM-1-positive vessels were visualized by microscopy at ×400 magnification for each section. The vessels in 20 fields were counted in each mouse. The integrity of the BBB was examined using Evans blue extravasation as previously described (30). At 5 days p.i., mice were injected intravenously with 200 μl of 2% Evans blue solution (Sigma, St. Louis, MO, USA). The mice were euthanized 1 h later, and the brains were isolated and incubated in 2 ml of formamide for 48 h at 37°C. The amount of Evans blue in 100 μl of the brain tissue extracts was determined by measuring absorbance at 620 nm (31).
Splenocyte culture and quantification of cytokines and nitrite.
Splenocytes were harvested and cultured essentially as previously described (32). Aliquots (5 × 106 cells/well) of the cell suspensions were seeded in 24-well flat-bottom tissue culture plates (Falcon) in triplicate in a humidified 5% CO2 incubator. For testing the in vitro effect of rhEPO on splenycytes, 5 × 106 cells were incubated with 5 U/ml of rhEPO for 48 h at 37°C. Levels of gamma interferon (IFN-γ), TNF, and IL-10 in culture supernatants and plasma samples were measured by enzyme-linked immunosorbent assays (ELISA) (R&D Systems, Minneapolis, MN, USA). Concentrations of NO2−, the stable oxidation product of NO, in cell supernatants were measured by the Griess reaction (33).
Flow cytometry.
Splenocytes from a portion of each group were collected to differentiate Th1-type cells, Tregs, the subsets of splenic DCs (myeloid DCs [mDCs] and plasmacytoid DCs [pDCs]), and the expression of major histocompatibility complex class II (MHC-II), CD86, Toll-like receptor 4 (TLR4), and TLR9 on CD11c+ DCs. Unless otherwise indicated, antibodies were purchased from BD Biosciences. To measure Th1-type cells (CD4+ T-bet+ IFN-γ+), 107 fresh splenocytes were stimulated in 12-well plates with phorbol myristate acetate (50 ng/ml) and ionomycin (0.5 μg/ml). After 2 h, 2 μM Golgi stop reagent (BD Pharmingen) containing brefeldin A was added to block cytokine export. After 4 additional hours of culture, the cells were stained with anti-CD4–fluorescein isothiocyanate (FITC) (clone H1.2F3). The cells were then fixed, permeabilized, and stained with anti-T-bet–phycoerythrin (PE) (clone eBio4B10), and anti-IFN-γ–allophycocyanin (APC) (XMG1.2). To assess Tregs, splenocytes were incubated with FITC–anti-CD4 (clone H1.2F3), peridinin chlorophyll protein (PerCP)–anti-CD25 (clone PC61), and PE–anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) (clone UC10-4F10-11) for surface staining in 100 μl of PBS supplemented with 3% fetal calf serum (FCS). Then, the cells were fixed and permeabilized, and intracytoplasmic staining was performed using APC–anti-Foxp3 antibody (clone FJK16s) (34).
For DCs, cells were double stained with FITC-conjugated CD11c monoclonal antibody (MAb) (clone HL3) and PE-conjugated anti-CD11b (clone M1/70), CD86 (clone GL1), MHC-II MAb (clone M5/114.15.2), PerCP-conjugated CD45R/B220 (clone RA3-6B2), or TLR4 (clone MTS510). To assess the expression of TLR9 in CD11c+ DCs, spleen cells were stained first with FITC-conjugated CD11c MAb. After fixation and permeabilization, the cells were incubated with biotinylated anti-TLR9 MAb (clone 5G5; Hycult Biotech), followed by PE-conjugated streptavidin.
Brain mononuclear cells were isolated from brains on day 5 p.i. A single-cell suspension was obtained by grinding the tissue and resuspending the resulting cells in 5 ml of RPMI 1640, which contained 100 U/ml type IV collagenase (Invitrogen, USA), and incubated at 42°C for 45 min. The cells were pelleted at 300 × g for 10 min, resuspended in 30% Percoll in PBS (Sigma), layered on 70% Percoll, and centrifuged at 515 × g for 30 min at room temperature. The cells at the interface were isolated, washed twice, resuspended in PBS, and labeled with the following antibodies: FITC–anti-CD4, PerCP–anti-CD8 (clone 53-6.7), and APC–anti-CD3 (clone 145-2C11). The cells were incubated at 4°C for 30 min and washed twice with PBS.
Flow cytometric analysis was performed using a FACSCalibur, and data were analyzed with the FlowJo software.
Statistical analysis.
Data are presented as the mean and standard error of the mean (SEM). Survival analysis was performed using the Kaplan-Meier log rank test. The statistical significance of the differences was analyzed by the t test or one-way analysis of variance (ANOVA) (SPSS 17.0). A P value of <0.05 was considered significant.
RESULTS
EPO treatment protects mice from ECM and ameliorates ECM pathology.
Through a pilot study of rhEPO treatment of P. berghei ANKA-infected mice at three daily dosages (10, 50, or 200 U/mouse) for 3 days (2 to 4 days p.i.), we selected the 50-U/mouse dosage in subsequent experiments (see Fig. S1 in the supplemental material). P. berghei ANKA-infected C57BL/6 mice typically displayed neurological symptoms around day 7 p.i., and most succumbed to death between days 7 and 12 (Fig. 1A). Consistent with earlier reports (24, 25), rhEPO treatment offered significant protection of mice against ECM compared with the untreated group (Kaplan-Meier analysis; P < 0.05; log rank test), and 80% of the mice in the P. berghei ANKA-plus-EPO group survived longer than 15 days (Fig. 1A). No significant differences in the levels of daily parasitemia were observed between the treatment groups (Fig. 1B). Since the protective effect of rhEPO may be associated with increased erythropoiesis from rhEPO treatments, we monitored the RBC and platelet densities. Compared with the baseline levels, daily RBC and platelet counts at 3 and 5 days p.i. did not differ significantly between the untreated and rhEPO-treated groups (Fig. 1C and D).
FIG 1.

Treatment with rhEPO increases survival of mice with P. berghei ANKA-induced ECM. P. berghei ANKA-plus-EPO group mice (n = 10) were infected with P. berghei ANKA and received 50 U/mouse rhEPO at 2 to 4 days p.i. The control group (P. berghei ANKA) was treated with PBS (n = 10). (A) Cumulative survival analysis. P. berghei ANKA (PbA)-infected mice were monitored daily for survival. (B) Parasitemia was monitored by microscopic evaluation of thin blood films with Giemsa staining. The values represent the means ± SEM. (C and D) Effects of rhEPO treatment (50 U) on total numbers of RBCs (C) and platelets (PLT) (D). The values represent the means ± SEM (n = 5 mice per group).
A common feature of CM pathology is sequestration of pRBCs and inflammatory cells in the brain microvasculature and alteration of BBB integrity (35, 36). In addition, endothelial activation and increased expression of adhesive molecules in cerebral microvessels are responsible for sequestration of pRBCs and inflammatory cells during CM (37, 38). At 5 days p.i., when ECM symptoms became evident, P. berghei ANKA-infected mice contained intense intravascular leukocyte aggregates and vascular obstruction in various brain regions (Fig. 2). In contrast, sequestration of inflammatory cells in the cerebral microvasculature was much less in rhEPO-treated mice. Immunohistochemistry confirmed dramatically increased expression of the adhesive molecules ICAM-1 and VCAM-1 in the brain microvasculature of infected mice, whereas rhEPO administration significantly decreased their expression (P < 0.001, t test), indicating reduced endothelial activation (Fig. 2). Further, brain vascular leakage was evident in infected mice, whereas rhEPO treatment significantly reduced the amount of dye in the brains of infected mice (Fig. 2), indicating better-preserved BBB integrity in rhEPO-treated mice.
FIG 2.

Treatment with rhEPO decreases endothelium activation and improves BBB integrity. (A) At 5 days p.i., three mice from each of the three groups (uninfected, P. berghei ANKA, and P. berghei ANKA plus EPO) were processed for histology with hematoxylin and eosin (H&E) staining and immunohistochemical analysis with anti-ICAM-1 and -VCAM-1 antibodies. The arrows point to the cerebral microvessels. (B) Quantification of ICAM-1- and VCAM-1-positive vessels. ICAM-1- or VCAM-1-positive microvessels per microscopic field were quantified in 20 fields per mouse. The values are means and SEM from three mice in each group. (C) Representative brain images for qualitative evaluation of vascular leakage using Evans blue extravasation. (D). Quantitative assessment of BBB leakage shown as the optical density (OD) readings of brain extracts at 620 nm. The values are means and SEM (n = 3). **, significant difference at a P value of <0.05 (t test) between treatment groups.
EPO treatment decreases plasma IFN-γ and TNF levels.
Both TNF and IFN-γ are implicated in the pathogenesis of CM (39). rhEPO treatment has been shown to downregulate the expression of IFN-γ and TNF mRNAs in the brains of infected mice on day 6 p.i. (24). Analysis of the plasma IFN-γ, TNF, and IL-10 levels found that P. berghei ANKA infection drastically increased the levels of these cytokines in plasma at 5 days p.i. (Fig. 3). However, rhEPO treatment significantly reduced the plasma IFN-γ and TNF levels (P < 0.05; t test). Conversely, the level of the anti-inflammatory cytokine IL-10 in plasma was significantly increased in rhEPO-treated mice (P < 0.05; t test).
FIG 3.
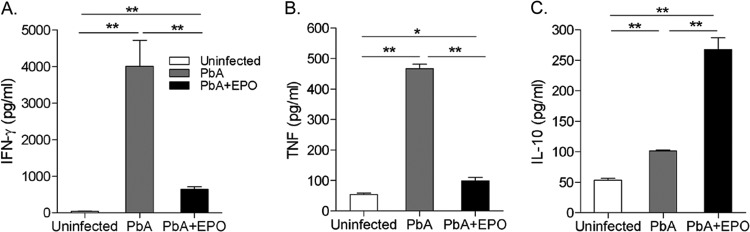
Effects of rhEPO treatment on plasma IFN-γ (A), TNF (B), and IL-10 (C) levels. For each experiment, three mice were used per group. Each experiment was repeated three times. The values represent the means and SEM. * and **, significant differences at P values of <0.05 and <0.01, respectively.
EPO counteracts Th1 cell subsets in the spleen during ECM.
To determine the in vivo effect of rhEPO on the differentiation of Th1-type cells, we quantified the Th1 cell CD4+ T-bet+ IFN-γ+ subtype. rhEPO treatment had no effect on the numbers of this cell subtype in uninfected mice. However, rhEPO treatment significantly reduced the CD4+ T-bet+ IFN-γ+ Th1 cells in the P. berghei ANKA-plus-EPO group at both 3 and 5 days p.i. (P < 0.05) (Fig. 4A). Murine macrophages, as important effector cells, express considerable amounts of EPOR and CD131 and are therefore targets of EPO (14, 40). EPO has been shown to downregulate the expression of inflammatory cytokines by macrophages in vitro. Compared with the baseline levels before inoculation, P. berghei ANKA infection increased the numbers of splenic macrophages (F4/80+ cells) at 3 days p.i., and a significant increase could be detected at 5 days p.i. In comparison, rhEPO treatment reduced the numbers of splenic macrophages in P. berghei ANKA mice on both days, though the differences between the P. berghei ANKA and P. berghei ANKA-plus-EPO groups did not achieve statistical significance (Fig. 4B).
FIG 4.

Effects of rhEPO treatment on splenic Th1 cells, macrophages, and secretion of cytokines. (A) Absolute numbers (top) and proportion (bottom; representative dot plots) of CD4+ T-bet+ IFN-γ+ Th1 cells in spleens were quantified by flow cytometry. d, day. (B) Absolute numbers (top) and proportion (bottom; representative histograms) of macrophages (F4/80+ cells) in spleens were quantified by flow cytometry. (C to E) Splenocytes were cultured, and the supernatants were analyzed for concentrations of nitrite (C) and cytokines IFN-γ (D) and TNF (E). For each experiment, three mice were used per group. Each experiment was repeated three times. The values represent the means and SEM. * and **, significant differences compared with baseline levels on day 0 at P values of <0.05 and <0.01, respectively. # and ##, significant differences between P. berghei ANKA-infected and rhEPO-treated groups at P values of < 0.05 and <0.01, respectively.
We then evaluated the effects of in vitro rhEPO treatment on the production of proinflammatory molecules by splenic cells. Primary splenic cells from P. berghei ANKA-plus-EPO mice produced significantly smaller amounts of nitrite and IFN-γ than those from P. berghei ANKA mice at both 3 and 5 days p.i. (P < 0.05) (Fig. 4C and D). rhEPO treatment markedly reduced the expression of TNF from the splenocytes on both days, although the difference between the P. berghei ANKA and P. berghei ANKA-plus-EPO groups was significant only at 5 days p.i. (P < 0.05) (Fig. 4E). Similarly, treatment of the in vitro-cultured splenocytes with 5 U/ml of rhEPO resulted in reduced production of TNF and IFN-γ (see Fig. S2 in the supplemental material).
EPO treatment reduces trafficking of CD4+ and CD8+ T cells to the brain.
Increased T cell trafficking to the brain is involved in the pathogenesis of ECM (41). To determine whether EPO treatment affects T cell trafficking to the brain, we isolated brain mononuclear cells from control mice and untreated and rhEPO-treated P. berghei ANKA-infected mice at 5 days p.i. As expected, CD4+ and CD8+ T cells were present in the brains of uninfected mice at baseline levels, whereas P. berghei ANKA infection resulted in a significant increase of the CD4+ T cells (P < 0.05; t test), as well as an elevation of the CD8+ T cell population in the brains of infected mice (Fig. 5). However, rhEPO treatment lowered the numbers of CD4+ (P < 0.05; t test) and CD8+ T cells in the brains of P. berghei ANKA-infected mice (Fig. 5), indicating reduced trafficking of these T cells to the brain.
FIG 5.

rhEPO inhibits migration of CD4+ and CD8+ T cells to the brains of P. berghei ANKA-infected mice. At 5 days p.i., three mice from each of the three groups (uninfected, P. berghei ANKA, and P. berghei ANKA plus EPO) were processed for the quantification of CD4+ and CD8+ T cells in the brains. The values are means and SEM (n = 3). * and **, significant differences at P values of <0.05 and <0.01 (t test) between treatment groups, respectively.
EPO treatment causes Tregs to proliferate and express higher levels of CTLA-4.
Tregs are specialized T-cell subgroups with potent immune-regulatory functions to prevent ECM via CTLA-4 (42, 43). Compared with the P. berghei ANKA group, we detected a highly significant elevation of CD4+ CD25+ FoxP3+ Tregs in rhEPO-treated mice at 3 and 5 days p.i. (P < 0.05) (Fig. 6A). Concomitantly, significantly increased expression of CTLA-4 was detected in CD4+ CD25+ FoxP3+ Tregs from P. berghei ANKA-plus-EPO mice at 5 days p.i. compared to the P. berghei ANKA group (Fig. 6B). These results indicated that rhEPO treatment triggered the expansion of Tregs, which expressed higher levels of CTLA-4.
FIG 6.

rhEPO stimulates expansion of Tregs and increases CTLA-4 expression by splenocytes. Absolute numbers (left) and proportion (right; representative dot plots) of Tregs (CD4+ CD25+ Foxp3+) in the spleens (A) and mean fluorescence intensity (MFI) of CTLA-4 (B) on Tregs were quantified by flow cytometry. For each experiment, three mice were used per group. Each experiment was repeated three times. The values represent the means and SEM. * and **, significant differences compared with baseline levels on day 0 at P values of <0.05 and <0.01, respectively. #, significant difference between P. berghei ANKA and P. berghei ANKA-plus-EPO groups (P < 0.05).
EPO inhibits DC expansion.
DCs have been found to express EPOR and are potentially targeted by EPO (18, 19). To determine the potential impacts of EPO on DCs, we measured the levels of DC differentiation, maturation, and activation. rhEPO significantly inhibited differentiation of DCs; the numbers of mDCs and pDCs were significantly lower in the P. berghei ANKA-plus-EPO group than in the P. berghei ANKA group at both 3 and 5 days p.i. (P < 0.01) (Fig. 7A, a, B, and b). We further compared the expression of the maturation markers MHC-II and CD86 on splenic CD11c+ DCs, which are essential for induction of T cell responses. The expression of both MHC-II and CD86 was decreased in the rhEPO-treated group at 5 days p.i., though the difference between the rhEPO-treated and untreated P. berghei ANKA groups was not significant (Fig. 7C, c, D, and d). Finally, the expression of TLR4 and TLR9 on DCs was decreased by rhEPO treatment, although the difference between treated and untreated groups was significant only at 3 days p.i. (P < 0.05) (Fig. 7E, e, F, and f). Together, these results indicate that rhEPO administration inhibits the expansion and functions of DCs.
FIG 7.

rhEPO inhibits expansion of DC subsets and DC maturation and function. All DC subpopulations and markers were analyzed by flow cytometry. Shown are the absolute numbers (A) and proportion (a) of mDCs, absolute numbers (B) and proportion (b) of pDCs, absolute numbers (C) and proportion (c) of DCs expressing MHC-II, absolute numbers (D) and proportion (d) of DCs expressing costimulatory marker CD86, absolute numbers (E) and proportion (e) of DCs expressing intracellular TLR-9, and absolute numbers (F) and proportion (f) of DCs expressing TLR-4. The data are presented as means and SEM (n = 3). The results are representative of three independent experiments. * and **, significant differences compared with baseline levels on day 0 at P values of <0.05 and <0.01, respectively. # and ##, significant differences between P. berghei ANKA-infected and EPO-treated groups at P values of <0.05 and <0.01, respectively.
DISCUSSION
Although the early Th1 immune responses triggered by malaria parasites are crucial for preventing rapid proliferation of the parasites, excessive effects mediated by Th1 cells and related inflammatory cytokines, such as TNF, IFN-γ, and IL-1β, promote the occurrence of CM (1, 44). Consistent with the anti-inflammatory effect of EPO, we found that rhEPO treatment significantly reduced the splenic CD4+ T-bet+ IFN-γ+ T cell and macrophage populations in P. berghei ANKA-infected mice. Since IFN-γ-producing CD4+ T cells play an essential regulatory role in the cerebral accumulation of CD8+ T cells (45), the effector cells responsible for ECM mortality (46), rhEPO-mediated reduction of the CD4+ IFN-γ+ population may result in lower levels of accumulation of the CD8+ T cells in the brain (46–49). In addition, rhEPO may directly inhibit expression of NO by macrophages through interaction with the EPOR, since mouse macrophages express a relatively high level of EPOR mRNA (14).
Tregs play an essential role in balancing protective immune responses and immune-mediated pathology (42). Tregs can suppress the development of P. berghei ANKA-specific Th1 responses (50). Accordingly, increasing the number of Tregs in vivo provides protection of mice against ECM (43). The CTLA-4 receptor is expressed on activated T cells, including Tregs. Recently, the protective function of Tregs against ECM has been shown to be highly dependent on CTLA-4 expression but modestly affected by IL-10 blockade (43). Furthermore, Tregs impair antigen-specific CD4+ T cell and CD8+ T cell responses that would normally promote parasite tissue sequestration in this model (43). Our data are consistent with the role of CTLA-4 in pathogenic T cell responses, demonstrating that elevated Tregs and upregulation of CTLA-4 as a result of rhEPO treatment could protect mice against ECM (51).
DCs are central to T lymphocyte activation and differentiation and play a key role in initiating adaptive immune response (52). Murine studies show that DCs can phagocytize Plasmodium chabaudi pRBCs (53) and present related antigens to CD4+ T cells to initiate the development of Th1 immune responses (54, 55). Among the DC populations, the balance of mDC and pDC subpopulations regulates the types of T cell immune responses, and Th1 responses are primarily mediated by mDCs (48, 56). During vivax malaria infection, mDCs and pDCs exhibit distinct proliferative activities under the same stimuli (57). In this study, rhEPO suppressed the differentiation of both mDCs and pDCs and downregulated the expression of MHC class II and the costimulatory molecule CD86 on DCs. The functions of DCs are regulated through the pathogen recognition receptors (58), and activation of TLRs induces maturation of DCs (59). During malaria infections, parasite-derived glycosylphosphatidylinositol induces signaling in host cells via TLR-2 and -4 (60), while hemozoin-induced and/or parasite nucleosome-induced immune activation involves TLR-9 (61, 62). This study showed downregulation of TLR4 and TLR9 expression in CD11c+ DCs in rhEPO-treated mice, implying impaired activation and maturation of DCs during ECM. These results indicate that the abilities of DCs to mount an effective innate immune response and initiate T cell responses are diminished by rhEPO treatment.
This study confirmed the neuroprotective role of rhEPO during ECM and suggested that an immune-regulatory function of EPO was an important mechanism. The anti-inflammatory effects of EPO in ECM are highly compatible with that in disease conditions such as EAE, which involves excessive or dysregulated inflammatory responses (10–12, 14). The immunosuppressive action of EPO might be exerted on DCs and macrophages, since these cells express remarkable amounts of EPOR-βcR (14, 40). In contrast, the expression of heterodimeric EPOR complexes is relatively low in CD4+ T cells (14). The inhibitory effect of EPO on the expression of proinflammatory genes, such as TNF and iNOS, in activated macrophages has been attributed to reduced NF-κB p65 activation (14). This mechanism is compatible with the observed reduction of DC differentiation, downregulation of MHC-II and costimulatory factors, and lower expression of TLRs in DCs during ECM, which indicate a reduced ability of DCs to mount innate immune responses and to prime adaptive immune responses. As a result, reduction of the mDC level and its activation by rhEPO lead to the taming of Th1 responses and elevation of Tregs and anti-inflammatory cytokines, such as IL-10 and transforming growth factor beta (TGF-β), which prevent the occurrence of ECM in mice.
The extrahematopoietic functions of EPO have been widely acknowledged, especially for their neurotropic and neuroprotective effects in the brain (63). In this study, we confirmed the protective role of rhEPO in ECM (24, 25) and showed that EPO may downregulate the inflammatory response by directly inhibiting the differentiation and function of splenic DCs during ECM. In addition, EPO, as a major hematopoietic hormone, may also ameliorate anemia induced by malaria. Thus, these data provide a further theoretical basis for clinical adjunct treatment of human CM patients with EPO.
Supplementary Material
ACKNOWLEDGMENT
This study was funded by the National Institute of Allergy and Infectious Diseases, NIH (R01AI099611 to Y.C. and U19AI089672 to L.C.).
Footnotes
Published ahead of print 14 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00929-13.
REFERENCES
- 1.van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. 2006. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22:503–508. 10.1016/j.pt.2006.09.002 [DOI] [PubMed] [Google Scholar]
- 2.Krantz SB. 1991. Erythropoietin. Blood 77:419–434 [PubMed] [Google Scholar]
- 3.Brines M, Cerami A. 2005. Emerging biological roles for erythropoietin in the nervous system. Nat. Rev. Neurosci. 6:484–494. 10.1038/nrn1687 [DOI] [PubMed] [Google Scholar]
- 4.Arcasoy MO. 2008. The non-haematopoietic biological effects of erythropoietin. Br. J. Haematol. 141:14–31. 10.1111/j.1365-2141.2008.07014.x [DOI] [PubMed] [Google Scholar]
- 5.Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R. 1998. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. U. S. A. 95:4635–4640. 10.1073/pnas.95.8.4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A. 2000. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 97:10526–10531. 10.1073/pnas.97.19.10526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su-Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A. 2004. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc. Natl. Acad. Sci. U. S. A. 101:14907–14912. 10.1073/pnas.0406491101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Andrea RJ, Gonda TJ. 2000. A model for assembly and activation of the GM-CSF, IL-3 and IL-5 receptors: insights from activated mutants of the common beta subunit. Exp. Hematol. 28:231–243. 10.1016/S0301-472X(99)00159-9 [DOI] [PubMed] [Google Scholar]
- 9.Brines M, Cerami A. 2006. Discovering erythropoietin's extra-hematopoietic functions: biology and clinical promise. Kidney Int. 70:246–250. 10.1038/sj.ki.5001546 [DOI] [PubMed] [Google Scholar]
- 10.Yuan R, Maeda Y, Li W, Lu W, Cook S, Dowling P. 2008. Erythropoietin: a potent inducer of peripheral immuno/inflammatory modulation in autoimmune EAE. PLoS One 3:e1924. 10.1371/journal.pone.0001924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agnello D, Bigini P, Villa P, Mennini T, Cerami A, Brines ML, Ghezzi P. 2002. Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res. 952:128–134. 10.1016/S0006-8993(02)03239-0 [DOI] [PubMed] [Google Scholar]
- 12.Li W, Maeda Y, Yuan RR, Elkabes S, Cook S, Dowling P. 2004. Beneficial effect of erythropoietin on experimental allergic encephalomyelitis. Ann. Neurol. 56:767–777. 10.1002/ana.20274 [DOI] [PubMed] [Google Scholar]
- 13.Chen SJ, Wang YL, Lo WT, Wu CC, Hsieh CW, Huang CF, Lan YH, Wang CC, Chang DM, Sytwu HK. 2010. Erythropoietin enhances endogenous haem oxygenase-1 and represses immune responses to ameliorate experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 162:210–223. 10.1111/j.1365-2249.2010.04238.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nairz M, Schroll A, Moschen AR, Sonnweber T, Theurl M, Theurl I, Taub N, Jamnig C, Neurauter D, Huber LA, Tilg H, Moser PL, Weiss G. 2011. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-kappaB-inducible immune pathways. Immunity 34:61–74. 10.1016/j.immuni.2011.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittelman M, Neumann D, Peled A, Kanter P, Haran-Ghera N. 2001. Erythropoietin induces tumor regression and antitumor immune responses in murine myeloma models. Proc. Natl. Acad. Sci. U. S. A. 98:5181–5186. 10.1073/pnas.081275298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prutchi-Sagiv S, Golishevsky N, Oster HS, Katz O, Cohen A, Naparstek E, Neumann D, Mittelman M. 2006. Erythropoietin treatment in advanced multiple myeloma is associated with improved immunological functions: could it be beneficial in early disease? Br. J. Haematol. 135:660–672. 10.1111/j.1365-2141.2006.06366.x [DOI] [PubMed] [Google Scholar]
- 17.Katz O, Gil L, Lifshitz L, Prutchi-Sagiv S, Gassmann M, Mittelman M, Neumann D. 2007. Erythropoietin enhances immune responses in mice. Eur. J. Immunol. 37:1584–1593. 10.1002/eji.200637025 [DOI] [PubMed] [Google Scholar]
- 18.Prutchi Sagiv S, Lifshitz L, Orkin R, Mittelman M, Neumann D. 2008. Erythropoietin effects on dendritic cells: potential mediators in its function as an immunomodulator? Exp. Hematol. 36:1682–1690. 10.1016/j.exphem.2008.07.010 [DOI] [PubMed] [Google Scholar]
- 19.Lifshitz L, Prutchi-Sagiv S, Avneon M, Gassmann M, Mittelman M, Neumann D. 2009. Non-erythroid activities of erythropoietin: Functional effects on murine dendritic cells. Mol. Immunol. 46:713–721. 10.1016/j.molimm.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 20.Newton CR, Warn PA, Winstanley PA, Peshu N, Snow RW, Pasvol G, Marsh K. 1997. Severe anaemia in children living in a malaria endemic area of Kenya. Trop. Med. Int. Health 2:165–178. 10.1046/j.1365-3156.1997.d01-238.x [DOI] [PubMed] [Google Scholar]
- 21.Kurtzhals JA, Rodrigues O, Addae M, Commey JO, Nkrumah FK, Hviid L. 1997. Reversible suppression of bone marrow response to erythropoietin in Plasmodium falciparum malaria. Br. J. Haematol. 97:169–174. 10.1046/j.1365-2141.1997.82654.x [DOI] [PubMed] [Google Scholar]
- 22.Casals-Pascual C, Kai O, Cheung JO, Williams S, Lowe B, Nyanoti M, Williams TN, Maitland K, Molyneux M, Newton CR, Peshu N, Watt SM, Roberts DJ. 2006. Suppression of erythropoiesis in malarial anemia is associated with hemozoin in vitro and in vivo. Blood 108:2569–2577. 10.1182/blood-2006-05-018697 [DOI] [PubMed] [Google Scholar]
- 23.Casals-Pascual C, Idro R, Gicheru N, Gwer S, Kitsao B, Gitau E, Mwakesi R, Roberts DJ, Newton CR. 2008. High levels of erythropoietin are associated with protection against neurological sequelae in African children with cerebral malaria. Proc. Natl. Acad. Sci. U. S. A. 105:2634–2639. 10.1073/pnas.0709715105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaiser K, Texier A, Ferrandiz J, Buguet A, Meiller A, Latour C, Peyron F, Cespuglio R, Picot S. 2006. Recombinant human erythropoietin prevents the death of mice during cerebral malaria. J. Infect. Dis. 193:987–995. 10.1086/500844 [DOI] [PubMed] [Google Scholar]
- 25.Wiese L, Hempel C, Penkowa M, Kirkby N, Kurtzhals JA. 2008. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar. J. 7:3. 10.1186/1475-2875-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Core A, Hempel C, Kurtzhals JA, Penkowa M. 2011. Plasmodium berghei ANKA: erythropoietin activates neural stem cells in an experimental cerebral malaria model. Exp. Parasitol. 127:500–505. 10.1016/j.exppara.2010.09.010 [DOI] [PubMed] [Google Scholar]
- 27.Marti HH. 2004. Erythropoietin and the hypoxic brain. J. Exp. Biol. 207:3233–3242. 10.1242/jeb.01049 [DOI] [PubMed] [Google Scholar]
- 28.Hempel C, Combes V, Hunt NH, Kurtzhals JA, Grau GE. 2011. CNS hypoxia is more pronounced in murine cerebral than noncerebral malaria and is reversed by erythropoietin. Am. J. Pathol. 179:1939–1950. 10.1016/j.ajpath.2011.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, III, Pober JS, Lowenstein CJ. 2007. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc. Natl. Acad. Sci. U. S. A. 104:1301–1306. 10.1073/pnas.0602035104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belayev L, Busto R, Zhao W, Ginsberg MD. 1996. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 739:88–96. 10.1016/S0006-8993(96)00815-3 [DOI] [PubMed] [Google Scholar]
- 31.Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, Intaglietta M, van der Heyde HC. 2006. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat. Med. 12:1417–1422. 10.1038/nm1499 [DOI] [PubMed] [Google Scholar]
- 32.Chen G, Liu J, Wang QH, Wu Y, Feng H, Zheng W, Guo SY, Li DM, Wang JC, Cao YM. 2009. Effects of CD4(+)CD25(+)Foxp3(+)regulatory T cells on early Plasmodium yoelii 17XL infection in BALB/c mice. Parasitology 136:1107–1120. 10.1017/S0031182009990370 [DOI] [PubMed] [Google Scholar]
- 33.Cao Y, Tsuboi T, Torii M. 1998. Nitric oxide inhibits the development of Plasmodium yoelii gametocytes into gametes. Parasitol. Int. 47:157–166. 10.1016/S1383-5769(98)00014-2 [DOI] [Google Scholar]
- 34.Zheng W, Wang QH, Feng H, Liu J, Meng HR, Cao YM. 2009. CD4+CD25+Foxp3+ regulatory T cells prevent the development of Th1 immune response by inhibition of dendritic cell function during the early stage of Plasmodium yoelii infection in susceptible BALB/c mice. Folia Parasitol. 56:242–250 [DOI] [PubMed] [Google Scholar]
- 35.Renia L, Wu HS, Claser C, Charlotte Gruner A, Suwanarusk R, Hui Teo T, Russell B, Ng LF. 2012. Cerebral malaria: mysteries at the blood-brain barrier. Virulence 3:193–201. 10.4161/viru.19013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medana IM, Turner GD. 2006. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 36:555–568. 10.1016/j.ijpara.2006.02.004 [DOI] [PubMed] [Google Scholar]
- 37.Favre N, Da Laperousaz C, Ryffel B, Weiss NA, Imhof BA, Rudin W, Lucas R, Piguet PF. 1999. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. 1:961–968. 10.1016/S1286-4579(99)80513-9 [DOI] [PubMed] [Google Scholar]
- 38.Bauer PR, Van Der Heyde HC, Sun G, Specian RD, Granger DN. 2002. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation 9:463–470. 10.1038/sj.mn.7800159 [DOI] [PubMed] [Google Scholar]
- 39.de Kossodo S, Grau GE. 1993. Role of cytokines and adhesion molecules in malaria immunopathology. Stem Cells 11:41–48. 10.1002/stem.5530110108 [DOI] [PubMed] [Google Scholar]
- 40.Lifshitz L, Tabak G, Gassmann M, Mittelman M, Neumann D. 2010. Macrophages as novel target cells for erythropoietin. Haematologica 95:1823–1831. 10.3324/haematol.2010.025015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC. 1996. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 157:1620–1624 [PubMed] [Google Scholar]
- 42.Hansen DS, Schofield L. 2010. Natural regulatory T cells in malaria: host or parasite allies? PLoS Pathog. 6:e1000771. 10.1371/journal.ppat.1000771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haque A, Best SE, Amante FH, Mustafah S, Desbarrieres L, de Labastida F, Sparwasser T, Hill GR, Engwerda CR. 2010. CD4+ natural regulatory T cells prevent experimental cerebral malaria via CTLA-4 when expanded in vivo. PLoS Pathog. 6:e1001221. 10.1371/journal.ppat.1001221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dodoo D, Omer FM, Todd J, Akanmori BD, Koram KA, Riley EM. 2002. Absolute levels and ratios of proinflammatory and anti-inflammatory cytokine production in vitro predict clinical immunity to Plasmodium falciparum malaria. J. Infect. Dis. 185:971–979. 10.1086/339408 [DOI] [PubMed] [Google Scholar]
- 45.Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, Hafalla JC, Blount DG, Hunter CA, Riley EM, Couper KN. 2012. IFN-gamma-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 189:968–979. 10.4049/jimmunol.1200688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Renia L. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 169:6369–6375 [DOI] [PubMed] [Google Scholar]
- 47.McQuillan JA, Mitchell AJ, Ho YF, Combes V, Ball HJ, Golenser J, Grau GE, Hunt NH. 2011. Coincident parasite and CD8 T cell sequestration is required for development of experimental cerebral malaria. Int. J. Parasitol. 41:155–163. 10.1016/j.ijpara.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 48.deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KP, Hill GR, Engwerda CR. 2007. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J. Immunol. 178:6033–6037 [DOI] [PubMed] [Google Scholar]
- 49.Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, Villadangos JA, Crabb BS, Heath WR. 2008. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 105:14509–14514. 10.1073/pnas.0806727105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nie CQ, Bernard NJ, Schofield L, Hansen DS. 2007. CD4+ CD25+ regulatory T cells suppress CD4+ T-cell function and inhibit the development of Plasmodium berghei-specific TH1 responses involved in cerebral malaria pathogenesis. Infect. Immun. 75:2275–2282. 10.1128/IAI.01783-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freitas do Rosario AP, Langhorne J. 2012. T cell-derived IL-10 and its impact on the regulation of host responses during malaria. Int. J. Parasitol. 42:549–555. 10.1016/j.ijpara.2012.03.010 [DOI] [PubMed] [Google Scholar]
- 52.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. 10.1038/32588 [DOI] [PubMed] [Google Scholar]
- 53.Ing R, Segura M, Thawani N, Tam M, Stevenson MM. 2006. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J. Immunol. 176:441–450 [DOI] [PubMed] [Google Scholar]
- 54.Pouniotis DS, Proudfoot O, Bogdanoska V, Scalzo K, Kovacevic S, Coppel RL, Plebanski M. 2005. Selectively impaired CD8+ but not CD4+ T cell cycle arrest during priming as a consequence of dendritic cell interaction with plasmodium-infected red cells. J. Immunol. 175:3525–3533 [DOI] [PubMed] [Google Scholar]
- 55.Shortman K, Liu YJ. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. 10.1038/nri746 [DOI] [PubMed] [Google Scholar]
- 56.Liu YJ. 2001. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 106:259–262. 10.1016/S0092-8674(01)00456-1 [DOI] [PubMed] [Google Scholar]
- 57.Jangpatarapongsa K, Chootong P, Sattabongkot J, Chotivanich K, Sirichaisinthop J, Tungpradabkul S, Hisaeda H, Troye-Blomberg M, Cui L, Udomsangpetch R. 2008. Plasmodium vivax parasites alter the balance of myeloid and plasmacytoid dendritic cells and the induction of regulatory T cells. Eur. J. Immunol. 38:2697–2705. 10.1002/eji.200838186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trinchieri G, Sher A. 2007. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7:179–190. 10.1038/nri2038 [DOI] [PubMed] [Google Scholar]
- 59.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 20:621–667. 10.1146/annurev.immunol.20.100301.064828 [DOI] [PubMed] [Google Scholar]
- 60.Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. 2005. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 280:8606–8616. 10.1074/jbc.M413541200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201:19–25. 10.1084/jem.20041836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu X, Gowda NM, Kumar S, Gowda DC. 2010. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 184:4338–4348. 10.4049/jimmunol.0903824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jelkmann W. 2005. Effects of erythropoietin on brain function. Curr. Pharm. Biotechnol. 6:65–79. 10.2174/1389201053167257 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.