

Abstract
Salmonella enterica uses effector proteins delivered by type III secretion systems (TTSS) to colonize eukaryotic cells. Recent in vivo studies have shown that intracellular bacteria activate the TTSS encoded by Salmonella pathogenicity island-2 (SPI-2) to restrain growth inside phagocytes. Growth attenuation is also observed in vivo in bacteria colonizing nonphagocytic stromal cells of the intestinal lamina propria and in cultured fibroblasts. SPI-2 is required for survival of nongrowing bacteria persisting inside fibroblasts, but its induction mode and the effectors involved remain unknown. Here, we show that nongrowing dormant intracellular bacteria use the two-component system OmpR-EnvZ to induce SPI-2 expression and the PhoP-PhoQ system to regulate the time at which induction takes place, 2 h postentry. Dormant bacteria were shown to discriminate the usage of SPI-2 effectors. Among the effectors tested, SseF, SseG, and SseJ were required for survival, while others, such as SifA and SifB, were not. SifA and SifB dispensability correlated with the inability of intracellular bacteria to secrete these effectors even when overexpressed. Conversely, SseJ overproduction resulted in augmented secretion and exacerbated bacterial growth. Dormant bacteria produced other effectors, such as PipB and PipB2, that, unlike what was reported for epithelial cells, did not to traffic outside the phagosomal compartment. Therefore, permissiveness for secreting only a subset of SPI-2 effectors may be instrumental for dormancy. We propose that the S. enterica serovar Typhimurium nonproliferative intracellular lifestyle is sustained by selection of SPI-2 effectors that are produced in tightly defined amounts and delivered to phagosome-confined locations.
INTRODUCTION
The species Salmonella enterica comprises Gram-negative facultative intracellular bacteria that cause enteric and systemic diseases in humans and livestock (1). This pathogen uses specialized type III secretion systems (TTSS), such as those in the Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2), to inject proteins into host cells and subvert a large variety of processes, including cytoskeleton dynamics, vesicular trafficking, and nuclear responses (2–7). In vitro studies using cultured epithelial cells and macrophages differentiated the function of these two secretion systems in a temporal and spatial basis. Thus, bacterial entry and intracellular proliferation are events associated with activities of the TTSS of SPI-1 and SPI-2, respectively (2, 3, 8). These roles were also assigned based on the inability of SPI-1 mutants to cause disease in mice when administered orally (9), indicative of a defect in penetration of the intestinal barrier. In contrast, SPI-2 mutants exhibit marked attenuation of virulence following oral and systemic administration (10).
Like other virulence factors, expression of the TTSS encoded by SPI-1 and SPI-2 is tightly regulated. The SPI-1 TTSS responds to environmental signals, such as low oxygen tension and high osmolarity (11), whereas the SPI-2 TTSS is activated in response to signals encountered by intracellular Salmonella in the phagosome, including acidic pH (12–16) and limitation of phosphate and magnesium (16, 17). These signals are perceived by sensors of two-component systems that transmit the stimulus to island-encoded specific regulators, such as HilD, HilA, and HilF in SPI-1 and the two-component system SsrA-SsrB in SPI-2 (2, 5, 18–20). Cross-regulation between SPI-1 and SPI-2 has been reported (21, 22), and such functional overlap is also evident at the level of the effector proteins, some of which are recognized as substrates by both secretion systems (23, 24). These findings suggest that cross-talk between SPI-1 and SPI-2 could also occur in intracellular bacteria, although this aspect remains to be demonstrated. More recent studies have shown that intracellular bacteria express SPI-1 genes and protein effectors translocated by the TTSS encoded by this island (25–29). Representative examples are the SPI-1 effector proteins SopB and SipA. SopB is used by intracellular bacteria to modulate phagosome trafficking and host cell responses involving the prosurvival kinase Akt and the nitric oxide synthase (25, 26, 30), whereas SipA acts cooperatively with the SPI-2 effector SifA to regulate bacterial intracellular proliferation (31). Conversely, SPI-2 expression also has been shown to precede bacterial penetration of the intestine (32). This early response could facilitate the subsequent adaptation of the invading bacteria to the intracellular lifestyle.
S. enterica serovar Typhimurium behaves differently in cultured phagocytic and nonphagocytic cells, exhibiting the highest replication rates in epithelial cells (5, 33). In vivo, most of the cells colonized by S. Typhimurium in systemic sites are phagocytes, although colonization of cells distinct from macrophages is also known (34). Of interest, most in vivo studies reveal a low average number of intracellular bacteria per infected cell (35), a phenomenon also occurring in cultured fibroblasts (36). Unlike the case for other host cell types, invasion of fibroblasts by S. Typhimurium is followed by a nonproliferative state maintained by pathogen regulators, such as the two-component system PhoP-PhoQ, that prevent pathogen overgrowth. The population of intracellular bacteria behave in a homogeneous manner inside the fibroblast. Thus, most of these bacteria remain in a nongrowing state, reaching an average progeny of 3 to 4 cells per infected fibroblast at late infection times (36). In vivo, the PhoP-PhoQ system was reported to attenuate growth of intracellular bacteria located in nonphagocytic stromal cells present in the lamina propria of the mouse intestine (29). Recently, bacteria located inside phagocytes present in mouse organs have been shown to reach high numbers in the absence of SPI-2 (37). In fibroblasts, SPI-2 and the alternative sigma factor RpoE are required for survival of the nongrowing dormant bacteria (36).
Our recent transcriptomic study performed in nongrowing dormant S. Typhimurium collected from fibroblasts revealed expression of SPI-1 and SPI-2 genes (29). Of interest, enhanced SPI-2 expression was observed in the overgrowing phoP bacteria at 24 h postinfection (29). Based on these findings, we initially proposed that SPI-2 activity must be finely regulated by nongrowing dormant bacteria to establish a sustained persistence state. Here, we describe how SPI-2 is regulated by nongrowing dormant intracellular bacteria and identify which SPI-2 effectors are produced inside the fibroblast. An unsuspected discrimination of SPI-2 effectors was observed in these nongrowing intracellular bacteria, which behave competently for secreting only a subset of effectors. In addition, some of the SPI-2 effectors analyzed are secreted to subcellular locations different from those reported for other nonphagocytic cell types.
MATERIALS AND METHODS
Bacterial strains, nonphagocytic eukaryotic cell lines, and infection conditions.
The S. enterica serovar Typhimurium strains used in this study are shown in Table S1 in the supplemental material. Bacteria were grown in Luria broth (LB) at 37°C. When appropriate, kanamycin (30 μg/ml) or ampicillin (50 μg/ml) was added to the growth media. To analyze production of SPI-2-related proteins, the PCN minimal medium, adjusted to pH 5.8, was used as described previously (16). NRK-49F normal rat kidney fibroblasts (ATCC CRL-1570) and HeLa human epithelial cells (ATCC CCL-2) were used throughout the study. NRK-49F fibroblasts were propagated in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 5% (vol/vol) fetal bovine serum (FBS) and 4 mM l-glutamine. Minimum essential medium Eagle (MEM) containing 10% FBS was used to grow HeLa cells. For infection of fibroblasts or epithelial cells, bacteria were grown at 37°C in static nonaerated cultures obtained upon inoculation of 2 ml of LB medium with a bacterial colony and subsequent overnight incubation (final optical density at 600 nm [OD600] of ∼1.0). Infection was carried out for 20 min using a multiplicity of infection (MOI) of 10:1 (bacteria to eukaryotic cells) as previously described (38). After extensive washing, infected cells were incubated in fresh tissue culture medium containing 100 μg/ml gentamicin for the first 2 h postinfection and 10 μg/ml for the rest of the experiment. Infected cells were lysed at the desired postinfection times in phosphate-buffered saline (PBS), pH 7.4, 1% (vol/vol) Triton X-100. The number of viable intracellular bacteria was determined by plating.
Construction of chromosomal epitope-tagged genes.
The strains carrying chromosomal 3×FLAG- or hemagglutinin (HA)-epitope-tagged genes were constructed using the method described by Uzzau et al. (39). Plasmids and oligonucleotides used for this procedure are listed in Table S2 in the supplemental material. The correct insertion of the epitope at the 3′ end of the targeted gene was verified by PCR.
Large-scale infection of cultured fibroblasts to obtain protein from intracellular bacteria.
Large-scale infections were performed essentially as described previously (29). Briefly, NRK-49F normal rat fibroblasts were seeded in BioDish-XL 500-cm2 plates (reference 351040; BD Biosciences) at a density of 2 × 107 cells per dish and infected for 20 min at an MOI of 10:1 (bacteria to fibroblast). The infected cells were extensively washed with prewarmed Hank's balance salt solution (HBSS) and incubated in fresh culture medium containing 100 μg/ml of gentamicin for 1 h. The culture medium was then replaced with fresh medium containing 10 μg/ml gentamicin until the desired postinfection time (1, 2, 6, 8, or 24 h). Finally, infected fibroblasts were washed with cold PBS (pH 7.4) and lysed in a solution (30 ml per plate) containing 0.4% SDS, 1% acidic phenol, and 19% ethanol in water. After 30 min of incubation at 4°C, intracellular bacteria were collected by centrifugation (27,500 × g, 4°C, 30 min) and washed three times with 1 ml of a 1% acidic phenol, 19% ethanol solution. For protein extraction, intracellular bacteria were washed twice with cold PBS, pH 7.4, recovered by centrifugation (15,000 × g, 4°C, 10 min), and processed as described previously (29). Protein extraction in extracellular bacteria was performed by following the same treatments as those for intracellular bacteria with 1% acidic phenol, 19% ethanol, 0.4% SDS.
Subcellular fractionation of infected fibroblast and epithelial cells.
Infected NRK-49F fibroblasts or HeLa epithelial cells were either mechanically disrupted or treated with Triton X-100 as described by Knodler et al. (40). Mechanical fractionation was performed to obtain a low-speed pellet (P1 fraction) containing nuclei/cytoskeleton and intact bacteria, a high-speed pellet enriched in plasma membrane and endomembrane material (P2 fraction), and the soluble cytosolic fraction (S2 fraction). To assess secretion of the effector protein by intracellular bacteria, the infected cells were fractionated as described previously (40) in extraction buffer containing 1% (vol/vol) Triton X-100, yielding a soluble fraction (S) with cytosol and dissolved membranes and an insoluble fraction (P) containing nuclei and intact intracellular bacteria.
Protease inhibitor treatment to test stability of SPI-2 effectors in fibroblasts.
NRK-49F fibroblasts were incubated for 30 h prior to infection with protease inhibitor cocktail for tissue culture medium (catalog no. P1860; Sigma) by following the manufacturer's instructions (1:400 dilution of stock solution). These cells were infected with the respective S. Typhimurium strains tagged in SPI2 effectors and fractionated in soluble and insoluble fraction as described above. Treatment with protease inhibitors did not affect bacterial invasion or intracellular survival rates.
Antibodies, Western analysis, and immunofluorescence microscopy.
The following primary antibodies were used for Western assays and immunofluorescence microscopy studies: mouse monoclonal anti-M45, recognizing an 18-amino-acid peptide of the adenovirus protein E4-6/7 (gift from Michael Hensel, Osnabrück, Germany); mouse monoclonal 5G10, recognizing a rat membrane glycoprotein present in late endosomes and lysosomes (gift from J. Bonifacino, NIH, Bethesda, MD); mouse monoclonal anti-FLAG epitope (clone M2; Sigma); mouse monoclonal anti-HA epitope (clone 16B12; Covance); rabbit polyclonal anti-OmpA (gift of H. Schwarz, Tübingen, Germany); rabbit polyclonal anti-S. Typhimurium lipopolysaccharide (LPS), group B, factors 1:4:5:12 (Difco Laboratories); mouse monoclonal anti-DnaK from Escherichia coli (Stressgene); rabbit polyclonal anti-calnexin (Stressgen); and mouse monoclonal anti-β-tubulin (Sigma). For immunofluorescence microscopy, the following secondary antibodies were used at a 1:500 dilution: goat polyclonal anti-mouse IgG conjugated to Alexa 405 (Molecular Probes), goat polyclonal anti-rabbit IgG conjugated to Alexa 488 (Molecular Probes), and goat polyclonal anti-mouse IgG conjugated to Alexa 594. Goat polyclonal anti-mouse or anti-rabbit IgG conjugated to horseradish peroxidase (Bio-Rad) was used as the secondary antibody at a 1:5,000 dilution for Western assays. Infected NRK-49F fibroblasts were fixed and processed for immunofluorescence microscopy as previously described (38). Cells were examined in a Leica fluorescence inverted microscope (DMI6000B).
Statistical analysis.
Data were analyzed by one-way analysis of variance (ANOVA) using Prism version 5.0 with Tukey's posttest (GraphPad Software, Inc.). Differences in values with P < 0.05 were considered significant.
RESULTS
Dissimilar contribution of PhoP-PhoQ and EnvZ-OmpR to SPI-2 induction in dormant intracellular S. Typhimurium.
The transcriptome obtained in nongrowing dormant intracellular bacteria collected from fibroblasts reveals enhanced expression of SPI-1 and SPI-2 genes at a late postinfection time, 24 h (29). Of interest, at this time intracellular phoP bacteria exhibit higher expression of the SPI-2 genes than wild-type bacteria, a phenomenon not observed in SPI-1 genes (Fig. 1A) or in phoP bacteria growing extracellularly in LB broth (29). These findings suggested that the PhoP-PhoQ system could actively downregulate SPI-2 expression levels in nongrowing dormant intracellular bacteria. Such an assumption contrasts with evidence found by others reporting a requirement of PhoP-PhoQ for proper SPI-2 activation (reviewed in reference 18). Alternatively, the transcriptomic data could merely reflect an altered behavior of SPI-2 due to the lack of this regulatory system. Therefore, the exact contribution of PhoP-PhoQ and other SPI-2-positive regulators, such as the two-component system EnvZ-OmpR (14, 41–43), was assessed in nongrowing dormant bacteria. To this end, we monitored the activity of the dedicated regulatory system SsrA-SsrB (13, 43, 44). SsrB protein levels were determined in wild-type, phoP, ompR, and ompR phoP isogenic strains carrying an ssrB::3×FLAG chromosomally tagged allele. Intracellular bacteria were collected at 1, 2, 4, 8, and 24 h upon infection of NRK-49F rat fibroblasts. For comparison, extracellular bacteria grown in LB or minimal defined PCN medium, known to activate SPI-2 (16), were also examined. SsrB levels were differentially regulated by PhoP-PhoQ and EnvZ-OmpR in extracellular bacteria, with a strict requirement for OmpR in bacteria grown in LB broth (Fig. 1B). In agreement with the published transcriptomic data (29), SsrB was upregulated in nongrowing dormant intracellular wild-type bacteria and the overgrowing phoP mutant at 24 h postinfection (Fig. 1B). Nonetheless, SsrB levels differed at earlier postinfection times among strains. SsrB was detected in nongrowing intracellular wild-type bacteria from 2 h postinfection, whereas it was hardly visible in the overgrowing phoP mutant at this time (Fig. 1C). Densitometry analyses confirmed lower levels of SsrB protein in the phoP mutant at this early postinfection time (Fig. 1C). Interestingly, SsrB relative levels increased progressively during the course of infection in nongrowing wild-type bacteria, whereas in the phoP mutant SsrB levels increased at 8 h postinfection and declined at later times (Fig. 1C). The amount of SsrB protein was very low or undetectable in intracellular ompR and ompR phoP bacteria at all postinfection times tested (Fig. 1C). As expected, the expression profile of chromosomally tagged SPI-2 effectors, such as PipB and SseJ, matched that of their positive regulator, SsrB, regarding the dissimilar regulatory roles of PhoP-PhoQ and EnvZ-OmpR (Fig. 1D and E). Altogether, these observations indicate that, upon entry into the fibroblast, the PhoP-PhoQ system is primarily involved in establishing the time at which induction of SPI-2 must be initiated, ∼2 h postinfection, and that SPI-2 expression in nongrowing dormant bacteria is absolutely dependent on the OmpR regulator.
FIG 1.

Distinct contribution of PhoP-PhoQ and OmpR-EnvZ to SPI-2 regulation in dormant intracellular S. Typhimurium. (A) Heat map showing the expression prolife of S. Typhimurium pathogenicity island genes (SP-1, SPI-2, SPI-3, SPI-4, and SPI-5) in nongrowing (wild-type) and overgrowing bacteria (phoP mutant) at 24 h postinfection of NRK-49F fibroblasts. As reference, the expression profile of extracellular wild-type bacteria grown to stationary phase (Extra-WT-ST) is shown. The Venn diagram indicates genes that are expressed at higher levels in intracellular phoP bacteria than in nongrowing dormant wild-type bacteria, including genes of the SPI-2 regulon. (B) Levels of the SPI-2 regulator SsrB in isogenic strains with an ssrB::3×FLAG chromosomal allele and differences in the functionality of the PhoP-PhoQ and OmpR-EnvZ two-component systems. The analysis was performed in extracellular bacteria grown in rich (LB) or chemically defined (PCN) nutrient media, and intracellular bacteria were collected at 24 h postinfection. (C) Kinetics revealing the postinfection time (∼2 h) at which intracellular dormant bacteria initiate the production of SsrB. Note the delay and reduction in the production of SsrB levels displayed by phoP and ompR mutants, respectively. Such delay was also observed upon quantification by densitometry of the relative levels of SsrB with respect to the internal control, OmpA. (D) Expression pattern of the SPI-2 effector protein PipB in extracellular bacteria grown in LB and PCN media and in intracellular bacteria at 24 h postinfection of NRK-49F fibroblasts. Note the correspondence between these data and those obtained for the SPI-2 dedicated regulator SsrB. Kinetics analysis performed in parallel also showed such similarities. (E) Relative levels of the SPI-2 effector SseJ in intracellular bacteria collected at 24 h postinfection of NRK-49F fibroblasts. The expression prolife matches those of SsrB and PipB. OmpA and calnexin were used as loading controls.
Nongrowing dormant intracellular S. Typhimurium uses a defined set of SPI-2 effectors to survive inside fibroblasts.
More than 20 effectors are predicted to be secreted by the SPI-2 TTSS (2, 20, 23, 24, 45). Some of these effectors engage diverse host cell processes, including cytoskeleton dynamics, vesicular trafficking, host cell death, and inflammatory signals (46). The levels at which these effectors are produced or secreted in different host cell types remain poorly understood. Redundancy in function for some SPI-2 effectors, especially in epithelial cells, has been claimed in some studies due to the absence of a phenotype for mutants lacking individual SPI-2 effectors (47). Exceptions are SseF, SseG, and SseJ, for which different studies have reported contrasting data relative to their requirement for S. Typhimurium proliferation in HeLa epithelial cells at distinct postinfection times (47–51). In macrophages, SPI-2 mutants defective in secretion display impaired proliferation and survival (13, 52–54). In fibroblasts, the role of SPI-2 in survival is inferred from the rapid loss of viability experienced by a mutant defective in SpiA, a structural protein of the SPI-2 TTSS apparatus (36). Inactivation of SPI-2 in a phoP mutant (phoP spiA double mutant) reduces the enhanced growth rate associated with the loss of the PhoP-PhoQ regulatory system (36). These data indicate that upon entry into fibroblasts, SPI-2 plays an important role in ensuring survival of nongrowing dormant bacteria and that in the artificial nonphysiological case of a mutational loss of the PhoP-PhoQ system, it can also promote pathogen intracellular proliferation. We then focused on how SPI-2 effectors could contribute to survival of dormant bacteria and analyzed those effectors known that subvert host vesicular trafficking, such as SifA, SifB, SseF, SseG, SseJ, PipB, and PipB2 (40, 55–58). Isogenic mutants defective in each of these effectors were tested in fibroblasts together with a mutant lacking the SPI-2 structural protein SsaV, used as a negative control of secretion. Among these strains, the ssaV mutant and those lacking the effectors SseF, SseG, or SseJ lost viability at 24 h postinfection (Fig. 2). These data supported the idea of nongrowing dormant intracellular S. Typhimurium using exclusively a subset of SPI-2 effectors to establish a persistence state inside fibroblasts.
FIG 2.
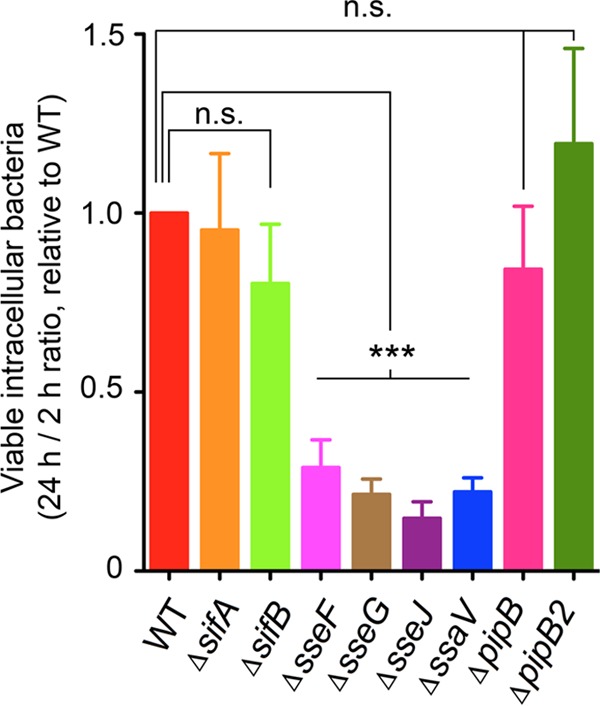
Involvement of a subset of SPI-2 effectors in maintenance of S. Typhimurium viability inside fibroblasts. Shown is the ratio between numbers of viable intracellular bacteria counted at 24 h versus those at 2 h postinfection of NRK-49F fibroblasts for each of the indicated strains relative to the ratio of wild-type bacteria, which had an average of 5.3 in three independent experiments. Note the overgrowing phenotype displayed by the phoP mutant and the requirement of SseF, SseG, and SseJ for viability maintenance. The ssaV mutant, lacking a structural protein of the secretion apparatus, was used as the negative control for secretion via the SPI-2 TTSS. Data represent the means and standard deviations from three independent experiments. ***, P < 0.001; n.s., not significant by a one-way ANOVA with Tukey's posttest.
SseJ is an SPI-2 effector with cholesterol acyltransferase activity that promotes localization of the Salmonella-containing vacuole (SCV) to the Golgi area and the formation of Salmonella-induced filaments (Sifs) (59, 60). Neither of these phenomena are as prominent in Salmonella-infected fibroblasts as in epithelial cells (61). Since the sseJ mutant exhibited decreased survival in fibroblasts (Fig. 2), we reasoned that the amount and/or localization of this effector could differ in infected NRK-49F fibroblasts with respect to HeLa epithelial cells. Twenty-four hours after infection of fibroblasts, the chromosomally tagged SseJ-3×FLAG protein accumulated in phagosome-confined regions with scarce amounts of the effector detected outside the phagosomal compartment (Fig. 3). No signal was detected in fibroblasts infected with wild-type bacteria with untagged SseJ protein or with sseJ::3×FLAG-tagged strains having no functional SPI-2 TTSS (data not shown). The distribution of SseJ in fibroblasts contrasted with that observed in HeLa-infected cells, in which elaborate filamentous structures containing the effector were observed in cells having similar bacterial loads (Fig. 3). These findings, obtained with physiological levels of the effector produced in two distinct nonphagocytic host cell types, indicated that translocated SseJ traffics differently inside fibroblasts and epithelial cells.
FIG 3.

SPI-2 effector SseJ is distributed along the membranous system of the infected cell to a lesser extent in fibroblasts than in epithelial cells. Shown are immunofluorescence microscopy images of NRK-49F fibroblasts and HeLa epithelial cells infected with the S. Typhimurium chromosomally tagged strain sseJ::3×FLAG for 24 and 6 h, respectively. The distribution of the endogenous tagged SseJ-3×FLAG protein was examined with anti-FLAG antibodies. Note the distinct pattern between host cell types even with similar numbers of intracellular bacteria. Arrows point to intracellular bacteria located inside the fibroblasts, and the arrowheads point to SseJ present in filamentous membranous structures of HeLa epithelial cells.
Secretion of the SPI-2 effector PipB by nongrowing intracellular S. Typhimurium is confined to the phagosomal compartment.
pipB and pipB2 genes are among the most highly upregulated genes in nongrowing dormant intracellular bacteria located in fibroblasts (29). In epithelial cells, PipB and PipB2 are enriched in lipid rafts upon translocation, and PipB2 is required for Sif formation (40, 62). Moreover, PipB2 targets the motor protein kinesin-1 (63). Since no prominent Sifs are observed in S. Typhimurium-infected fibroblasts, we wondered whether the localization of these two effectors could differ in this host cell type. Their distribution was monitored in NRK-49F fibroblasts using pipB::3×FLAG and pipB2::HA chromosomally tagged isogenic strains. Unexpectedly, PipB was visualized exclusively in defined locations confined to the phagosome-containing nongrowing bacteria (Fig. 4A and B). No signal was detected in fibroblasts infected with wild-type bacteria encoding untagged PipB protein or with pipB::3×FLAG-tagged strains mutants having no functional SPI-2 TTSS (data not shown). The apparent exclusive presence of PipB in phagosome-confined areas was confirmed by mechanical fractionation experiments, which demonstrated that PipB was absent from membranous material collected as postnuclear pellets (Fig. 4C, P2 fraction). Fractionation experiments based on nonionic detergent (1% Triton X-100)-mediated cell lysis led to the visualization of part of the PipB molecules in the soluble fraction, demonstrating that part of the PipB molecules was secreted, whereas the rest remained in the bacteria (Fig. 4D). Additional experiments, in which the subcellular distribution of PipB was monitored together with that of a rat membrane glycoprotein present in late endosomes and lysosomes recognized by mouse monoclonal 5G10 (64), unequivocally demonstrated that PipB translocation is confined to the phagosome-containing nongrowing dormant bacteria (Fig. 4E). Single-copy tagged alleles of genes encoding SPI-2 proteins were used to analyze effector production and subcellular location. Thus, we could not rule out that the different subcellular distribution observed for PipB in fibroblasts compared to that in epithelial cells resulted from the fact than in epithelial cells, this effector has been analyzed only upon ectopic overexpression (62). To test this possibility, the pipB::3×FLAG-tagged strain was used to infect HeLa epithelial cells. In contrast to fibroblasts, the tagged PipB-3×FLAG protein was visualized in phagosomal and extraphagosomal locations as well as in filamentous structures of HeLa-infected cells (Fig. 4F). Of interest, PipB-3×FLAG was also observed in extraphagosomal locations in overgrowing phoP mutant colonizing fibroblasts, although in this case no filamentous structures containing the effector were visible (data not shown). Taken together, these observations demonstrated that the confinement of the PipB effector to the phagosomal region upon translocation is a phenomenon occurring in nongrowing dormant bacteria.
FIG 4.

SPI-2 effector PipB is translocated by nongrowing dormant intracellular S. Typhimurium to phagosome-confined regions. (A) Distribution of endogenous chromosomally tagged PipB-3×FLAG in NRK-49F fibroblasts at 24 h postinfection. Arrows indicate four fields containing intracellular bacteria that are magnified in panel B. (B) Magnified images of fields selected from panel A showing the distribution of PipB-3×FLAG in dormant intracellular bacteria. Note the lack of effector labeling in some bacteria. (C) Mechanical fractionation denotes that endogenous PipB-3×FLAG is not released to the endomembrane system. P1, nuclear/bacterial fraction; P2, high-speed particulate material, membranous fraction; S2, cytosol, soluble fraction (see Materials and Methods for details). (D) Translocation of PipB-3×FLAG by nongrowing dormant intracellular bacteria shown by the detection of the effector in a subcellular fraction containing material solubilized with 1% (vol/vol) Triton X-100. P, pellet, material insoluble in detergent; S, material solubilized with the detergent. (E) Triple labeling showing that the distribution of PipB-3×FLAG in NRK-49F fibroblasts is observed inside that of a membrane marker positioned in the phagosomal membrane, a glycoprotein recognized by the mouse monoclonal 5G10 (64). (F) Distribution of endogenous PipB-3×FLAG in HeLa epithelial cells at 6 h postinfection. Note that PipB shows a distribution confined to the phagosomal region exclusively in nongrowing dormant bacteria located inside fibroblasts.
Similar experiments were performed with a pipB2::HA chromosomally tagged strain. In this case, much smaller amounts of the PipB2-HA-tagged protein were detected in nongrowing dormant wild-type bacteria, and no obvious subcellular location could be assigned for this effector upon fibroblast infection (Fig. 5A). Fractionation experiments suggested that, similar to PipB, PipB2 secretion was limited to the phagosomal compartment, since no PipB2 was detected in the P2 fraction (membranous material) or the cytosol upon mechanical rupture of infected fibroblasts (Fig. 5B). A small amount of the effector could be visualized upon 1% Triton X-100 treatment (Fig. 5C), suggesting that part of the PipB2-HA molecules produced by dormant bacteria could be secreted. PipB2 was visualized in large amounts in phagosomal and extraphagosomal locations in fibroblasts infected with the phoP pipB2::HA mutant (data not shown). It was noteworthy that, unlike the response exhibited in fibroblasts, wild-type bacteria secreted large amounts of chromosomally tagged PipB2::HA into membranous structures of HeLa epithelial cells (Fig. 5D). Altogether, these findings indicated that PipB2 is an effector produced and translocated by dormant nongrowing intracellular S. Typhimurium in small amounts inside fibroblasts, and that it does not traffic to extraphagosomal or filamentous structures of the infected cell.
FIG 5.

Limited translocation of the SPI-2 effector PipB2 in nongrowing intracellular S. Typhimurium. (A) Distribution of endogenous chromosomally tagged PipB2-HA in NRK-49F fibroblasts at 24 h postinfection. (B) Mechanical fractionation denotes that endogenous PipB2-HA is not released to the endomembrane system. P1, nuclear/bacterial fraction; P2, high-speed particulate material, membranous fraction; S2, cytosol, soluble fraction (see Materials and Methods for details). (C) PipB2 is translocated by dormant intracellular bacteria, as denoted by its detection in a subcellular fraction containing material solubilized with 1% (vol/vol) Triton X-100. (D) Distribution of endogenous PipB2-HA in HeLa epithelial cells at 6 h postinfection. Arrows indicate the presence of the effector in endomembranous filamentous structures.
Nongrowing S. Typhimurium discriminates among SPI-2 effectors for production and translocation inside fibroblasts.
The data described above evidenced two major differences concerning the biology of SPI-2 effectors when S. Typhimurium colonizes fibroblasts or epithelial cells. First, at least two of the three SPI-2 effectors analyzed (PipB and PipB2) were distributed in the phagosomal area to a greater extent in fibroblasts than in epithelial cells (Fig. 4 and 5). Second, unlike the case for epithelial cells, the lack of individual SPI-2 effectors compromises the viability of intracellular bacteria (Fig. 2). Based on these observations, we sought to determine whether S. Typhimurium could discriminate SPI-2 effectors to adapt to a nongrowing intracellular lifestyle. For this purpose, different SPI-2 effectors tagged with the M45 epitope of the adenovirus protein E4-6/7 (65) were expressed in trans. These effectors included SifA-M45, SifB-M45, SseJ-M45, SseF-45, and SseG-M45 (24, 45), which are produced by bacteria growing in LB (Fig. 6A). Under this growth condition, it has been shown that bacteria do not secrete any of these SPI-2 effectors (66). Secretion of these M45-tagged effectors was reported to occur in extracellular bacteria growing in acidified minimal medium (66). In fibroblasts, nongrowing dormant bacteria produced all of these SPI-2-tagged effectors (Fig. 6B). However, only SseF-M45 and SseJ-M45 were observed in subcellular fractions containing material solubilized with a low concentration of a nonionic detergent, 1% (vol/vol) Triton X-100 (Fig. 6B). Such translocation was dependent on an active SPI-2 TTSS, since it was not observed in the ssaV mutant (Fig. 6C). Control experiments with bacteria grown extracellularly discarded that the Triton X-100 treatment extracted SseF and SseJ from intact bacteria (Fig. 6D). Thus, we concluded that SseF and SseJ are efficiently translocated by intracellular S. Typhimurium inside fibroblasts when overexpressed in trans. Strikingly, no translocation was observed for SifA, SifB, or SseG (Fig. 6B). This difference could be due to a distinct stability of the translocated effector dictated by the host cell type or to a relatively low level of production, as seems to be case for SseG. Experiments in which a protease inhibitor was used ruled out an action of proteases on translocated SifA-M45 or SifB-M45 (Fig. 6E). When tested in HeLa epithelial cells, SifA-M45, SifB-M45, and SseG-M45 were efficiently translocated when overexpressed on an individual basis (Fig. 6F). Similar to the data obtained in fibroblasts, SseJ-M45 and SseF-M45 are efficiently translocated by bacteria proliferating inside HeLa cells (Fig. 6F). Thus, with the exception of SseG-M45, which was barely detected in nongrowing dormant bacteria-colonizing fibroblasts (Fig. 6B), these results demonstrated that bacteria persisting inside fibroblasts are not competent for secreting SifA or SifB even when these two effectors are artificially overproduced in trans. These observations led us to examine whether overproduction of SPI-2 effectors that were efficiently translocated in fibroblasts, such as SseJ, could have different consequences for the bacterial fate compared to those effectors remaining inside the bacteria (SifB). Microscopy analyses performed in NRK-49F fibroblasts revealed that intracellular bacteria overexpressing SseJ-M45 were in much larger numbers, and that this effector localized in phagosomal and extraphagosomal locations, including filamentous structures resembling Sifs (Fig. 7A), defined by some authors as globular membranous compartments (56). Importantly, increased proliferation was not observed in bacteria overexpressing an SPI-2 effector not efficiently translocated, such as SifB (Fig. 7A). Quantification of bacterial loads in fibroblasts infected with these strains also denotes a significant increase in bacterial proliferation upon SseJ translocation (Fig. 7B). Altogether, these data supported the idea of nongrowing dormant intracellular S. Typhimurium translocating only a discrete subset of SPI-2 effectors upon entry into fibroblasts. Moreover, the data suggested that the amount of effector secreted must be finely adjusted to ensure control of the intracellular growth rate.
FIG 6.

Nongrowing dormant intracellular S. Typhimurium is competent for translocating only defined SPI-2 effectors inside fibroblasts. (A) Expression of M45-tagged SPI-2 effectors in extracellular S. Typhimurium upon overnight growth at 37°C in LB medium at neutral pH under nonshaking conditions. Indicated are the respective SPI-2 effectors analyzed. (B) Selective translocation of tagged SseF-M45 and SseJ-M45 effectors occurs in nongrowing dormant intracellular fibroblasts. Note that there is no translocation of SifA-M45 or SifB-M45, even when there is prominent production of both effectors. (C) Control assays demonstrating that SseF-M45 and SseJ-M45 translocation depends on a functional SPI-2 TTSS. (D) Extraction of M45-tagged SPI-2 effectors in extracellular S. Typhimurium in buffer containing 1% (vol/vol) Triton X-100. Note the negligible amount of the bacterium-associated effector extracted by the detergent, which contrasts with the amount of effector detected outside the bacteria in infected fibroblasts (Fig. 4B and 5B). (E) Translocation rate of the effectors SifA, SseJ, and SifB in intracellular bacteria infecting NRK-49F fibroblasts pretreated for 30 h with a protease inhibitor cocktail or left untreated. (F) Efficient translocation of M45-tagged SseF, SseG, SifA, SifB, and SseJ by intracellular bacteria proliferating inside HeLa epithelial cells.
FIG 7.

Increased translocation of SseJ results in augmented proliferation of S. Typhimurium inside NRK-49F fibroblasts. (A) Immunofluorescence microscope images denoting the increased proliferation of intracellular bacteria overexpressing the SseJ-M45-tagged effector. Note that the phenomenon is accompanied by the formation of globular filamentous structures containing the effector that resemble Sifs (arrows). None of these effects were observed in intracellular wild-type overexpression of the SPI-2 effector SifB-M45. (B) Quantification of these phenotypes was performed by enumeration of intracellular bacteria in each cell, counting a minimum of 100 infected fibroblasts. Shown are the combined data from two independent experiments. ∗, P = 0.01 to 0.05; ∗∗, P = 0.001 to 0.01 (by a one-way ANOVA with Tukey's posttest). Note the significant increase in bacterial proliferation upon SseJ overexpression.
DISCUSSION
In this study, we analyzed the contribution of SPI-2 to the persistence state that S. Typhimurium mounts upon entry into fibroblasts. Unlike the case in other host cell types, in fibroblasts this pathogen utilizes distinct regulators to attenuate intracellular growth. One of these regulators, the two-component system PhoP-PhoQ, is linked to the fine tuning of SPI-2 gene expression (reviewed in reference 18). Our former studies in fibroblasts revealed that the lack of SPI-2 causes loss of viability of nongrowing dormant bacteria, whereas a nonfunctional PhoP-PhoQ system led to exacerbated intracellular bacterial growth (36). Based on these findings, we examined the relationship between SPI-2 and PhoP-PhoQ in the fibroblast infection model and also implicated the OmpR-EnvZ system, known to play an important role in SPI-2 induction (14, 41, 67). Usage of chromosomally tagged strains proved to be a convenient strategy to monitor in a timely fashion SPI-2 induction based on the detection of physiological levels of the SPI-2 regulator SsrB. In a previous study, Xu and Hensel analyzed the kinetics of induction of the SPI-2 regulon using luciferase-based transcriptional fusions in bacteria grown in PCN, a minimal defined medium that mimics SPI-2-inducing conditions (68). Consistent with the protein data reported here for nongrowing dormant intracellular bacteria, these authors noted a delayed expression of the SPI-2 regulon in the phoP mutant, a phenomenon not observed in other mutant backgrounds, such as ompR-envZ or slyA (68). This trend was reproduced in different luc fusions, such as sseJ::luc, sifA::luc, and sseA::luc, but the basis of this temporal regulation was not investigated further. In our work, the apparent delayed expression of the SPI-2 regulon exhibited by the phoP mutant correlates with exacerbated bacterial growth. Importantly, we were able to detect differences at the protein level between nongrowing dormant wild-type bacteria and overgrowing phoP bacteria, as was the case for SseJ, which is produced at slightly higher levels by the phoP mutant at late postinfection times (Fig. 1). Since we did not investigate all predicted and known SPI-2 effectors, it cannot be discarded that the exacerbated growth exhibited by the phoP mutant results from an altered production and/or secretion of SPI-2 effectors at defined postinfection times. For the ompR mutant, the major defect observed was the inability of the bacteria to produce normal amounts of SsrB and SPI-2 effectors (Fig. 1). This erroneous manner of inducing the SPI-2 regulon could explain the mild increase in bacterial growth rate observed for the ompR mutant inside fibroblasts (36).
An unexpected result of our study was the apparent discrimination of SPI-2 effectors relative to the role played by SPI-2 in survival of nongrowing dormant intracellular bacteria (36). These differences are not obvious in other nonphagocytic cells, such as epithelial cells, in which it has not yet been determined whether all or only a subset of SPI-2 effectors are produced and translocated upon bacterial entry. In the epithelial cell infection model, some studies showed that null mutants with global defects in the SPI-2 regulon, such as the ssaR mutant, do not exhibit any phenotype (47). In other cases, a role of SPI-2 effectors, such as SseF and SseG, in promoting bacterial growth inside epithelial cells was claimed (48, 49). Remarkably, the same study that investigated in a comprehensive manner the role of individual SPI-2 effectors in epithelial cells monitored the phenotype of the respective mutants in macrophages, as well as in organs and in the presence of bile (47). In RAW264.7 macrophages, the data implicated SPI-2 effectors such as SifA and SifB in bacterial replication/survival but did not reveal any change for mutants lacking SseF or SseG. More recently, the lack of SPI-2 effectors, such as SifA, SseJ, SopD2, SseG, SseF, and SrfH, has been reported to affect the capacity of S. Typhimurium to proliferate within mouse bone marrow-derived macrophages (69). Of these effectors, SseJ, SseG, and SseF play an important role in survival of nongrowing dormant bacteria located in fibroblasts. Despite this apparent similarity, differences between fibroblasts and cultured macrophages are likely to exist, since in the former cells SPI-2 is used by intracellular S. Typhimurium mainly for survival rather than for proliferation (36, 61). Our study also shows that SifA is dispensable for intracellular survival, in marked contrast to the key role of this effector in bacterial growth inside macrophages (47, 69). Interestingly, a recent study by Grant et al. reported a role of SPI-2 in negatively modulating the growth rate in vivo in phagocytic cells (37). Therefore, the analysis of phenotypes shown by S. Typhimurium in fibroblasts may provide new insights into how this pathogen establishes a nonproliferative intracellular state and the extent to which the lifestyles that the pathogen adopts in fibroblasts and phagocytes are comparable.
To our knowledge, this study uncovered a fact not previously reported for any SPI-2 effector: the possibility of reaching different final destinations depending on the type of host cell that is colonized. The limited distribution observed for all SPI-2 effectors tested in persistently infected fibroblasts compared to that of epithelial cells may result from different dynamics in the vesicular traffic directed to or emanating from the SCV. Thus, the proximity of the SCV to the microtubule organizing center and the Golgi apparatus reported to occur in epithelial cells (48, 49) is a rare phenomenon in fibroblasts (our unpublished data). Such proximity to the Golgi apparatus following the subversion of key motor proteins, such as kinesin and dynein, by dedicated SPI-2 effectors (70–72) is fundamental for S. Typhimurium proliferation. This phenomenon is not prominent in bacteria located in fibroblasts, with the SCV maintaining a scattered distribution in the infected cell long after bacterial entry (our unpublished data). Since the data reported here sustain an important role of SPI-2 effectors previously linked to SCV positioning in survival of dormant bacteria located in fibroblasts, it is tempting to speculate that the effectors SseJ, SseF, and SseG play different roles depending on the host cell type that is colonized. On the other hand, SifA, an SPI-2 effector that plays a master role in subversion of kinesin activity and SCV positioning in epithelial cells (72), is dispensable in fibroblast cells for intracellular survival. Overall, these data support the idea that differences exist in the trafficking and biogenesis of the SCV in distinct nonphagocytic cell types.
Lastly, the data reported here allow us to tentatively hypothesize why S. Typhimurium, as well as many other intracellular pathogens, has such a vast repertoire of effector proteins that evolved to subvert host cell functions. The fact that in fibroblasts certain effectors, such as SifA and SifB, are not translocated by nongrowing intracellular bacteria even upon their artificial production from plasmids suggests that the SPI-2 secretion apparatus displays permissiveness for only certain protein effectors depending on the host cell type that is colonized. Such permissiveness could be dictated by signals coming from the host cell type. The molecular details for such restriction in protein secretion is challenging, considering that the available transcriptomic data infer activity of the SPI-1 secretion apparatus in S. Typhimurium persisting inside fibroblasts (29). Cross-talk between the two type III secretion systems is also inferred by the existence of effectors that can be recognized in vitro as substrates by both apparatuses (23) and by the interconnected role of SPI-1 regulators acting on SPI-2 (21, 22). Future analyses should address how communication between the infected host cell and intracellular S. Typhimurium can lead to utilization of a defined subset of SPI-2 effectors for ensuring the survival of nongrowing dormant intracellular bacteria.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Holden, Michael Hensel, and Víctor H. Bustamante for providing plasmids and mutants used in this study. We also thank Diana Barroso and Pablo García for technical assistance.
A.A. and C.N-H. held fellowships from the Consejería de Educación de la Comunidad de Madrid. This work was supported by grants BIO2010-18885 (to F.G-P.), CSD2008-00013-INTERMODS (to F.G.-P. and J.C.), and BIO2010-15023 (to J.C.) from the Spanish Ministry of Economy and Competitiveness and by PIE-201320E020 (to F.G-P.) from the Agencia Estatal CSIC.
Footnotes
Published ahead of print 21 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01304-13.
REFERENCES
- 1.Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6:53–66. 10.1038/nrmicro1788 [DOI] [PubMed] [Google Scholar]
- 2.Figueira R, Holden DW. 2012. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158:1147–1161. 10.1099/mic.0.058115-0 [DOI] [PubMed] [Google Scholar]
- 3.Kuhle V, Hensel M. 2004. Cellular microbiology of intracellular Salmonella enterica: functions of the type III secretion system encoded by Salmonella pathogenicity island 2. Cell. Mol. Life Sci. 61:2812–2826. 10.1007/s00018-004-4248-z [DOI] [PubMed] [Google Scholar]
- 4.Galan JE, Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573. 10.1038/nature05272 [DOI] [PubMed] [Google Scholar]
- 5.Malik-Kale P, Jolly CE, Lathrop S, Winfree S, Luterbach C, Steele-Mortimer O. 2011. Salmonella–at home in the host cell. Front. Microbiol. 2:125. 10.3389/fmicb.2011.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coburn B, Sekirov I, Finlay BB. 2007. Type III secretion systems and disease. Clin. Microbiol. Rev. 20:535–549. 10.1128/CMR.00013-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agbor TA, McCormick BA. 2011. Salmonella effectors: important players modulating host cell function during infection. Cell Microbiol. 13:1858–1869. 10.1111/j.1462-5822.2011.01701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, Heikenwalder M, Stallmach T, Hensel M, Pfeffer K, Akira S, Hardt WD. 2005. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 174:1675–1685 [DOI] [PubMed] [Google Scholar]
- 9.Galan JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387. 10.1073/pnas.86.16.6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shea JE, Hensel M, Gleeson C, Holden DW. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 93:2593–2597. 10.1073/pnas.93.6.2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bajaj V, Lucas RL, Hwang C, Lee CA. 1996. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol. Microbiol. 22:703–714. 10.1046/j.1365-2958.1996.d01-1718.x [DOI] [PubMed] [Google Scholar]
- 12.Odendall C, Rolhion N, Forster A, Poh J, Lamont DJ, Liu M, Freemont PS, Catling AD, Holden DW. 2012. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe 12:657–668. 10.1016/j.chom.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188. 10.1046/j.1365-2958.1998.01048.x [DOI] [PubMed] [Google Scholar]
- 14.Lee AK, Detweiler CS, Falkow S. 2000. OmpR regulates the two-component system SsrA-ssrB in Salmonella pathogenicity island 2. J. Bacteriol. 182:771–781. 10.1128/JB.182.3.771-781.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beuzon CR, Banks G, Deiwick J, Hensel M, Holden DW. 1999. pH-dependent secretion of SseB, a product of the SPI-2 type III secretion system of Salmonella typhimurium. Mol. Microbiol. 33:806–816. 10.1046/j.1365-2958.1999.01527.x [DOI] [PubMed] [Google Scholar]
- 16.Lober S, Jackel D, Kaiser N, Hensel M. 2006. Regulation of Salmonella pathogenicity island 2 genes by independent environmental signals. Int. J. Med. Microbiol. 296:435–447. 10.1016/j.ijmm.2006.05.001 [DOI] [PubMed] [Google Scholar]
- 17.Deiwick J, Nikolaus T, Erdogan S, Hensel M. 1999. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol. Microbiol. 31:1759–1773. 10.1046/j.1365-2958.1999.01312.x [DOI] [PubMed] [Google Scholar]
- 18.Fass E, Groisman EA. 2009. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12:199–204. 10.1016/j.mib.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen-Wester I, Hensel M. 2001. Salmonella pathogenicity islands encoding type III secretion systems. Microbes Infect. 3:549–559. 10.1016/S1286-4579(01)01411-3 [DOI] [PubMed] [Google Scholar]
- 20.Srikanth CV, Mercado-Lubo R, Hallstrom K, McCormick BA. 2011. Salmonella effector proteins and host-cell responses. Cell. Mol. Life Sci. 68:3687–3697. 10.1007/s00018-011-0841-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH. 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol. Microbiol. 80:1637–1656. 10.1111/j.1365-2958.2011.07674.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bustamante VH, Martinez LC, Santana FJ, Knodler LA, Steele-Mortimer O, Puente JL. 2008. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. U. S. A. 105:14591–14596. 10.1073/pnas.0801205105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos-Morales F. 2012. Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. 2012:1–36. 10.5402/2012/787934 [DOI] [Google Scholar]
- 24.van der Heijden J, Finlay BB. 2012. Type III effector-mediated processes in Salmonella infection. Future Microbiol. 7:685–703. 10.2217/fmb.12.49 [DOI] [PubMed] [Google Scholar]
- 25.Drecktrah D, Knodler LA, Galbraith K, Steele-Mortimer O. 2005. The Salmonella SPI1 effector SopB stimulates nitric oxide production long after invasion. Cell Microbiol. 7:105–113. 10.1111/j.1462-5822.2004.00436.x [DOI] [PubMed] [Google Scholar]
- 26.Knodler LA, Finlay BB, Steele-Mortimer O. 2005. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J. Biol. Chem. 280:9058–9064. 10.1074/jbc.M412588200 [DOI] [PubMed] [Google Scholar]
- 27.Hautefort I, Thompson A, Eriksson-Ygberg S, Parker ML, Lucchini S, Danino V, Bongaerts RJ, Ahmad N, Rhen M, Hinton JC. 2008. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell Microbiol. 10:958–984. 10.1111/j.1462-5822.2007.01099.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372. 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 29.Nunez-Hernandez C, Tierrez A, Ortega AD, Pucciarelli MG, Godoy M, Eisman B, Casadesus J, Garcia-Del Portillo F. 2013. Genome expression analysis of nonproliferating intracellular Salmonella enterica serovar Typhimurium unravels an acid pH-dependent PhoP-PhoQ response essential for dormancy. Infect. Immun. 81:154–165. 10.1128/IAI.01080-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hernandez LD, Hueffer K, Wenk MR, Galan JE. 2004. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805–1807. 10.1126/science.1098188 [DOI] [PubMed] [Google Scholar]
- 31.Brawn LC, Hayward RD, Koronakis V. 2007. Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 1:63–75. 10.1016/j.chom.2007.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown NF, Vallance BA, Coombes BK, Valdez Y, Coburn BA, Finlay BB. 2005. Salmonella pathogenicity island 2 is expressed prior to penetrating the intestine. PLoS Pathog. 1:e32. 10.1371/journal.ppat.0010032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brumell JH, Perrin AJ, Goosney DL, Finlay BB. 2002. Microbial pathogenesis: new niches for salmonella. Curr. Biol. 12:R15–R17. 10.1016/S0960-9822(01)00640-6 [DOI] [PubMed] [Google Scholar]
- 34.Monack DM, Bouley DM, Falkow S. 2004. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J. Exp. Med. 199:231–241. 10.1084/jem.20031319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mastroeni P, Grant A, Restif O, Maskell D. 2009. A dynamic view of the spread and intracellular distribution of Salmonella enterica. Nat. Rev. Microbiol. 7:73–80. 10.1038/nrmicro2034 [DOI] [PubMed] [Google Scholar]
- 36.Cano DA, Martinez-Moya M, Pucciarelli MG, Groisman EA, Casadesus J, Garcia-Del Portillo F. 2001. Salmonella enterica serovar Typhimurium response involved in attenuation of pathogen intracellular proliferation. Infect. Immun. 69:6463–6474. 10.1128/IAI.69.10.6463-6474.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grant AJ, Morgan FJ, McKinley TJ, Foster GL, Maskell DJ, Mastroeni P. 2012. Attenuated Salmonella Typhimurium lacking the pathogenicity island-2 type 3 secretion system grow to high bacterial numbers inside phagocytes in mice. PLoS Pathog. 8:e1003070. 10.1371/journal.ppat.1003070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aiastui A, Pucciarelli MG, Garcia-del Portillo F. 2010. Salmonella enterica serovar typhimurium invades fibroblasts by multiple routes differing from the entry into epithelial cells. Infect. Immun. 78:2700–2713. 10.1128/IAI.01389-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U. S. A. 98:15264–15269. 10.1073/pnas.261348198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knodler LA, Vallance BA, Hensel M, Jackel D, Finlay BB, Steele-Mortimer O. 2003. Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol. Microbiol. 49:685–704. 10.1046/j.1365-2958.2003.03598.x [DOI] [PubMed] [Google Scholar]
- 41.Garmendia J, Beuzon CR, Ruiz-Albert J, Holden DW. 2003. The roles of SsrA-SsrB and OmpR-EnvZ in the regulation of genes encoding the Salmonella typhimurium SPI-2 type III secretion system. Microbiology 149:2385–2396. 10.1099/mic.0.26397-0 [DOI] [PubMed] [Google Scholar]
- 42.Cameron AD, Dorman CJ. 2012. A fundamental regulatory mechanism operating through OmpR and DNA topology controls expression of Salmonella pathogenicity islands SPI-1 and SPI-2. PLoS Genet. 8:e1002615. 10.1371/journal.pgen.1002615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim CC, Falkow S. 2004. Delineation of upstream signaling events in the salmonella pathogenicity island 2 transcriptional activation pathway. J. Bacteriol. 186:4694–4704. 10.1128/JB.186.14.4694-4704.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hensel M. 2000. Salmonella pathogenicity island 2. Mol. Microbiol. 36:1015–1023. 10.1046/j.1365-2958.2000.01935.x [DOI] [PubMed] [Google Scholar]
- 45.Steele-Mortimer O. 2008. The Salmonella-containing vacuole: moving with the times. Curr. Opin. Microbiol. 11:38–45. 10.1016/j.mib.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auweter SD, Bhavsar AP, de Hoog CL, Li Y, Chan YA, van der Heijden J, Lowden MJ, Coombes BK, Rogers LD, Stoynov N, Foster LJ, Finlay BB. 2011. Quantitative mass spectrometry catalogues Salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J. Biol. Chem. 286:24023–24035. 10.1074/jbc.M111.224600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buckner MMC, Croxen M, Arena ET, Finlay BB. 2011. A comprehensive study of the contribution of Salmonella enterica serovar Typhimurium SPI2 effectors to bacterial colonization, survival, and replication in typhoid fever, macrophage, and epithelial cell infection models. Virulence 2:208–216. 10.4161/viru.2.3.15894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salcedo SP, Holden DW. 2003. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 22:5003–5014. 10.1093/emboj/cdg517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deiwick J, Salcedo SP, Boucrot E, Gilliland SM, Henry T, Petermann N, Waterman SR, Gorvel JP, Holden DW, Meresse S. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 74:6965–6972. 10.1128/IAI.00648-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abrahams GL, Hensel M. 2006. Manipulating cellular transport and immune responses: dynamic interactions between intracellular Salmonella enterica and its host cells. Cell Microbiol. 8:728–737. 10.1111/j.1462-5822.2006.00706.x [DOI] [PubMed] [Google Scholar]
- 51.Malik-Kale P, Winfree S, Steele-Mortimer O. 2012. The bimodal lifestyle of intracellular Salmonella in epithelial cells: replication in the cytosol obscures defects in vacuolar replication. PLoS One 7:e38732. 10.1371/journal.pone.0038732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174. 10.1046/j.1365-2958.1998.01047.x [DOI] [PubMed] [Google Scholar]
- 53.Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804. 10.1073/pnas.93.15.7800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salcedo SP, Noursadeghi M, Cohen J, Holden DW. 2001. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in vivo. Cell Microbiol. 3:587–597. 10.1046/j.1462-5822.2001.00137.x [DOI] [PubMed] [Google Scholar]
- 55.Kuhle V, Hensel M. 2002. SseF and SseG are translocated effectors of the type III secretion system of Salmonella pathogenicity island 2 that modulate aggregation of endosomal compartments. Cell Microbiol. 4:813–824. 10.1046/j.1462-5822.2002.00234.x [DOI] [PubMed] [Google Scholar]
- 56.Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. 2002. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 44:645–661. 10.1046/j.1365-2958.2002.02912.x [DOI] [PubMed] [Google Scholar]
- 57.Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249. 10.1093/emboj/19.13.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Freeman JA, Ohl ME, Miller SI. 2003. The Salmonella enterica serovar typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella-containing vacuole. Infect. Immun. 71:418–427. 10.1128/IAI.71.1.418-427.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohlson MB, Fluhr K, Birmingham CL, Brumell JH, Miller SI. 2005. SseJ deacylase activity by Salmonella enterica serovar Typhimurium promotes virulence in mice. Infect. Immun. 73:6249–6259. 10.1128/IAI.73.10.6249-6259.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nawabi P, Catron DM, Haldar K. 2008. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol. 68:173–185. 10.1111/j.1365-2958.2008.06142.x [DOI] [PubMed] [Google Scholar]
- 61.Martinez-Moya M, de Pedro MA, Schwarz H, Garcia-del Portillo F. 1998. Inhibition of Salmonella intracellular proliferation by non-phagocytic eucaryotic cells. Res. Microbiol. 149:309–318. 10.1016/S0923-2508(98)80436-1 [DOI] [PubMed] [Google Scholar]
- 62.Knodler LA, Steele-Mortimer O. 2005. The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell 16:4108–4123. 10.1091/mbc.E05-04-0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Henry T, Couillault C, Rockenfeller P, Boucrot E, Dumont A, Schroeder N, Hermant A, Knodler LA, Lecine P, Steele-Mortimer O, Borg JP, Gorvel JP, Meresse S. 2006. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. U. S. A. 103:13497–13502. 10.1073/pnas.0605443103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonifacino JS, Perez P, Klausner RD, Sandoval IV. 1986. Study of the transit of an integral membrane protein from secretory granules through the plasma membrane of secreting rat basophilic leukemia cells using a specific monoclonal antibody. J. Cell Biol. 102:516–522. 10.1083/jcb.102.2.516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obert S, O'Connor RJ, Schmid S, Hearing P. 1994. The adenovirus E4-6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol. Cell. Biol. 14:1333–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansen-Wester I, Stecher B, Hensel M. 2002. Type III secretion of Salmonella enterica serovar Typhimurium translocated effectors and SseFG. Infect. Immun. 70:1403–1409. 10.1128/IAI.70.3.1403-1409.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feng X, Oropeza R, Kenney LJ. 2003. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol. Microbiol. 48:1131–1143. 10.1046/j.1365-2958.2003.03502.x [DOI] [PubMed] [Google Scholar]
- 68.Xu X, Hensel M. 2010. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 78:49–58. 10.1128/IAI.00931-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Figueira R, Watson KG, Holden DW, Helaine S. 2013. Identification of Salmonella pathogenicity island-2 type III secretion system effectors involved in intramacrophage replication of S. enterica serovar Typhimurium: implications for rational vaccine design. mBio 4:e00065. 10.1128/mBio.00065-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramsden AE, Holden DW, Mota LJ. 2007. Membrane dynamics and spatial distribution of Salmonella-containing vacuoles. Trends Microbiol. 15:516–524. 10.1016/j.tim.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 71.Henry T, Gorvel JP, Meresse S. 2006. Molecular motors hijacking by intracellular pathogens. Cell Microbiol. 8:23–32. 10.1111/j.1462-5822.2005.00649.x [DOI] [PubMed] [Google Scholar]
- 72.Leone P, Meresse S. 2011. Kinesin regulation by Salmonella. Virulence 2:63–66. 10.4161/viru.2.1.14603 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.