

Abstract
Diabetes affects 25.8 million people in the United States, or 8.3% of the population, and these numbers are even higher in developing countries. Diabetic patients are more susceptible to the development of chronic wounds with debilitating bacterial infections than nondiabetics. Previously, we compared the ability of the opportunistic pathogen Pseudomonas aeruginosa to cause biofilm-associated infections in chronic wounds of diabetic and nondiabetic mice (C. Watters, K. DeLeon, U. Trivedi, J. A. Griswold, M. Lyte, K. J. Hampel, M. J. Wargo, and K. P. Rumbaugh, Med. Microbiol. Immunol. 202:131–141, 2013). Unexpectedly, we observed that insulin-treated diabetic mice had significantly more biofilm in their wounds, which correlated with higher antibiotic tolerance. Here, we investigated whether insulin treatment modulates the diabetic immune system to favor P. aeruginosa biofilm formation. Utilizing a murine chronic wound model, we found that DNA protected P. aeruginosa in the wounds of insulin-treated diabetic mice from antibiotic treatment. We also observed increased numbers of neutrophils, reduced numbers of macrophages, and increased cell death in the wounds of diabetic mice on insulin therapy. Taken together, these data suggest that high levels of lysed neutrophils in the wounds of diabetic mice on insulin, combined with fewer macrophages to remove the cellular debris, contribute to increased DNA levels, which enhance P. aeruginosa biofilms.
INTRODUCTION
Diabetic chronic wound infections are considered the most significant wound care problem in the United States as well as the rest of the world. While the exact cost of care for diabetic foot ulcers is not known, it is likely to be measured in the billions of dollars (1). Wound infections with Gram-negative bacteria occur three times more often in diabetic patients than in nondiabetic patients (2), and P. aeruginosa is the most commonly isolated Gram-negative bacterium from these infections. P. aeruginosa is an opportunistic pathogen that produces a variety of virulence factors and is a notorious biofilm builder.
Biofilms are a conglomerate of bacterial cells, DNA, and proteins intercalated in a sugary slime matrix (3). The persistence of biofilms in diabetic wounds is thought to delay healing for multiple reasons. First, the continuous presentation of antigens recognized by the host's immune system leads to the induction of chronic inflammation caused by multiple host immune responses, including phagocytes, antibodies, and complement (4). For example, as phagocytes attack the biofilm by releasing granules, reactive oxygen species, and proteolytic enzymes, they stimulate the production of inflammatory cytokines, which results in collateral damage to the host tissue (5, 6). Consequently, as long as the biofilm remains, the immune system will try to remove it, resulting in prolonged inflammation and severe damage to host tissue (4). Second, the biofilm's exopolysaccharide (EPS) matrix is thought to act as a mechanical barrier to antibodies and complement, preventing penetration and protecting bacterial cells (6, 7). Lastly, the EPS may also inhibit wound healing by preventing fibroblasts, epithelial cells, and keratinocytes from migrating into the wound bed.
The clinical signs and symptoms of biofilm-associated infections, which include fever, pain, and heat, occur in response to planktonic cells that slough off the biofilm (8, 9). Although antibiotics can temporarily alleviate these symptoms by eradicating the planktonic bacteria, a recalcitrant biofilm population remains, which can be 500 to 5,000 times more tolerant of antimicrobials than planktonic bacteria (10). Multidrug tolerance observed in biofilms implies a transient, nonheritable phenotype (11), but biofilms also display a high rate of horizontal gene transfer resulting in increased antibiotic resistance. The combination of antibiotic tolerance and resistance results in a chronic biofilm infection that cannot be eradicated by either systemic or topical antimicrobials.
Previously, we developed an in vivo chronic wound, diabetic mouse model to examine the ability of the opportunistic pathogen Pseudomonas aeruginosa to cause biofilm-associated infections (12). More biofilm was observed in the wounds of diabetic mice than nondiabetic mice and correlated with higher antibiotic tolerance. Unexpectedly, we observed the highest occurrence of biofilm and highest rates of antimicrobial tolerance when diabetic mice were treated with insulin. Studies from other groups have suggested that insulin treatment affects the immune system in a way that results in P. aeruginosa transitioning from a planktonic phenotype, which results in bacteremia, to a biofilm-associated phenotype, favoring chronic infection (13, 14). Insulin treatment is also known to exert anti-inflammatory effects on both diabetic and nondiabetic human immune cells (15–18). Taken together, these previous reports and our prior findings raised the possibility that insulin treatment affects the diabetic immune response in a manner that indirectly favors P. aeruginosa infection and/or biofilm formation.
MATERIALS AND METHODS
Bacterial growth and inocula.
P. aeruginosa strain PAO1 (19), PAO1ΔrhlR (20), and Staphylococcus aureus strain SA31 (clinical isolate obtained from Abdul Hamood, TTUHSC, Lubbock, TX) were grown overnight in Luria-Bertani (LB) medium, subcultured 1:100 in fresh LB, and grown at 37°C for 3 h to an optical density of approximately 0.9 at 600 nm. Subcultures then were serially diluted, and mice were infected with approximately 104 CFU, a dose that causes a nonfatal, chronic infection in diabetic and nondiabetic mice (21). The exact inoculum of each strain was determined by plating serial dilutions of the inoculum on LB and Pseudomonas isolation agar.
Mouse chronic wound model.
Adolescent, female, Swiss Webster mice, weighing 18 to 20 g, were purchased from Charles Rivers Laboratories (Wilmington, MA). Mice were fed a standard chow diet and were acclimated for a week before wounding as previously described (12). A diabetic state was induced by the intraperitoneal injection of 250 mg/kg of body weight streptozotocin (STZ; Alexis Biochemicals, San Diego, CA). Mice were deemed diabetic if their blood glucose levels were ≥20 mmol/liter after 1 week of STZ treatment. Some groups of diabetic mice were treated daily with NPH Humulin insulin (2 U; Eli Lilly, Indianapolis, IN) given subcutaneously. Mice were anesthetized and administered 1- by 1-cm full-thickness surgical excision wounds as previously described (12). A sterile gauze pad was placed on top of the wound and then covered with an adhesive (OPSITE) dressing. A total of 104 CFU of P. aeruginosa or S. aureus was injected under the dressing and gauze bandage and on top of the wound. Mice were housed and studied according to protocols approved by the Institutional Animal Care and Use Committee in the animal facility of Texas Tech University Health Sciences Center (Lubbock, TX).
Antimicrobial tolerance assay.
The gentamicin tolerance of P. aeruginosa in wounds was examined as previously described (22). At 4 days postoperation, mice were euthanized and the gauze pads were removed from their wounds. The gauze pads were cut into equal sections, weighed, and treated with 200 μg/ml gentamicin for 5 h or placed in phosphate-buffered saline (PBS) as a nontreatment control. Following treatments, the gauze sections were neutralized in 10% Dey-Engley broth for 10 min and placed in PBS. The gauze pads were then thoroughly homogenized using the Precellys 24 tissue homogenizer/grinder (Bertin Technologies, Montigny-le-Bretonneux, France). The resulting solution was serially diluted and plated on LB agar plates and incubated at 37°C for 24 h. The colonies were counted and the CFU/g gauze was calculated. To determine the percentage of cells viable after gentamicin treatment, the CFU after treatment was divided by the CFU from the PBS-treated gauze section and multiplied by 100 (22).
DNase treatment.
Murine surgical wounds were infected with PAO1 and covered by a sterile gauze pad. At 4 days postoperation, the gauze was removed and sectioned. These sections were treated for 24 h at 37°C with 80 μl of 10× DNase reaction buffer (Promega, Madison, WI) containing either 40 U of DNase I (Promega) or sterile water. The antimicrobial tolerance to gentamicin then was examined as described above (22).
Extraction of DNA from wound biofilms.
Murine surgical wounds were infected with PAO1 and covered with a gauze pad and OPSITE dressing. Following 4 days of infection, a small portion of the gauze pad was extracted and weighed. The DNA was then extracted with the DNeasy blood and tissue kit (Qiagen, Chatsworth, CA) per the manufacturer's instructions. DNA concentrations were determined with a Take3 microvolume plate utilizing a SynergyH1 hybrid plate reader (BioTek, Winooski, VT).
MPO oxidation peroxidase assay.
Murine surgical wounds were infected with PAO1, and at 4 days postoperation the entire wound bed was extracted and weighed. The wound tissue was then homogenized and the cells were lysed in sodium phosphate with 0.5% hexadecyltrimethylammonium bromide as previously described (23). The tissue homogenates were centrifuged, and myeloperoxidase (MPO) peroxidase levels in the supernatant were measured per the manufacturer's protocol (Caymen Chemicals, Ann Arbor, MI).
TUNEL assay for apoptosis and necrosis.
Murine tissue was extracted from the wound bed-intact tissue interface and fixed in formalin, embedded in paraffin, and sectioned. Terminal deoxynucleotidyl transferase (TdT)-mediated (TUNEL) assay was performed on wound tissue 4 days postinfection to visualize apoptosis and necrosis according to the manufacturer's protocol (Promega). Sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) to stain DNA. Positive-control sections were incubated with DNase I for 5 min to induce DNA damage. Negative-control sections were incubated without TdT enzyme. Slides were visualized with epifluorescence microscopy (Eclipse 80i; Nikon, Louisville, KY), and images were captured and analyzed with the NIS Elements program. We quantified the amount of TUNEL staining utilizing ImageJ 1.46 software (NIH).
Fluorescence and immunofluorescence microscopy.
Fluorescence imaging for murine cytokine production, apoptosis, and P. aeruginosa glycocalyx was performed on formalin-fixed paraffinized tissue sections. Briefly, the tissue sections were deparaffinized with xylene, followed by rehydration in ethanol, and then they were briefly washed in 1× phosphate-buffered saline solution (PBS). For antigen retrieval and unmasking of epitopes, tissue sections were incubated in a microwave oven for 15 min in 1× Na citrate buffer (pH 6.0), rinsed in 1× PBS (for 10 min), and then treated with 10 μg/ml proteinase K for 15 min at room temperature. Tissue sections were then washed in 1× PBS (10 min). Visualization of the P. aeruginosa glycocalyx was achieved by staining tissue sections with 50 μg/ml concanavalin A (ConA) Texas red conjugate (Invitrogen, Carslbad, CA) in the dark for 5 min at room temperature, washed in 1× PBS (3 times, 5 min each), and then mounted with ProLong Gold antifade reagent (Molecular Probes, Eugene, OR) supplemented with DAPI to stain nucleic acid. For slides incubated with primary antibodies for immunofluorescence, nonspecific binding sites were blocked in 20% goat serum in 1× PBS for 45 min at room temperature. After blocking, tissue sections were incubated with primary antibodies (diluted in 2% goat serum in 1× PBS) overnight at 4°C at the following dilutions: anti-caspase 3 (active [cleaved] form) (polyclonal antibody [PAb], rabbit [Millipore, Temecula, CA, USA]), diluted 1:75, rat anti-mouse F4/80 monoclonal antibody (Abcam, Cambridge, MA), diluted 1:100, and anti-IL-6 (PAb, rabbit [Thermo Scientific, Rockford, IL], diluted 1:20). Following incubation, sections were rinsed in PBST (1× PBS plus 0.1% Tween 20) (3 times for 10 min each) and then incubated with goat anti-rabbit Alexa Fluor 488 (Life Technologies, Carlsbad, CA) diluted 1:300 in 2% goat serum in 1× PBS for 1 h at room temperature. Sections were rinsed in PBST (3 times for 10 min each) and then mounted with ProLong Gold antifade plus DAPI for visualization. Mounted slides were then visualized by fluorescence microscopy with a Nikon Eclipse 80i microscope (Nikon, Louisville, KY), and images were captured with a Nikon DS-Fi1 camera (Nikon, Louisvillek, KY) and analyzed with the NIS Elements program (version 3.00 SP7; Nikon, Japan).
RT-PCR.
Mice were euthanized, and tissue sections were harvested from the wound margin and stored in 800 μl of RNAlater solution (Ambion, Austin, TX) at −20°C. The tissue was then homogenized with a Tissue Tearor (Cole-Parmer, Vernon Hills, IL) in TRI Reagent solution (Molecular Research Center, Cincinnati, OH), and RNA was extracted from the tissue homogenate by following the manufacturer's instructions. Following extraction, RNA was treated with DNase I (Promega, Madison, WI) and purified with an RNeasy MinElute cleanup kit (Qiagen, Hilden, Germany) according to the manufacturer's directions. Total RNA from the groups of mice that were nondiabetic, diabetic, or diabetic on insulin were combined equally into separate pooled samples, and cDNA was prepared by combining 2 μg of total RNA, 400 U Superscript reverse transcriptase (Invitrogen, Carlsbad, CA), and 500 ng oligo(dT) and incubating at 37°C for 1 h. Specific primer sets for murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-A AGGTCGGAGTCAACGGATT-3′; reverse, 5′-TTGATGACAAGCTTCCCGTT-3′), tumor necrosis factor alpha (TNF-α) (forward, 5′-ACGGCATGGATCTCAAAGAC-3′; reverse, 5′-CGGACTCCGCAAAGTCTAAG-3′), and interleukin-1α (IL-1α) (forward, 5′-CGTCAGGCAGAAGTTTGTCA-3′; reverse, 5′-GTGCACCCGACTTTGTTCTT-3′) were used to amplify DNA templates in a T3 thermocycler (Biometra, Goettingen, Germany) with Taq DNA polymerase (New England BioLabs, Ipswich, MA) with 30 cycles at 94°C (0 s), 60°C (0 s), and 72°C (25 s). PCR products were then visualized under a UV light on a 1.5% agarose gel mixed with 1 μl Gelstar nucleic acid gel stain (Cambrex Bio Science Rockland Inc., Rockland, ME) per 50 ml of agarose.
CLSM.
After 4 days of infection, mice were euthanized and wound tissue sections were extracted and stained for 20 min with 10 μM propidium iodide (PI; Invitrogen, Carlsbad, CA) and 2.5 μM SYTO 9 (Invitrogen) as previously described (24). PI was used to visualize extracellular DNA and SYTO 9 for visualizing live cells. The tissue then was gently rinsed in water and placed in an imaging chamber (Molecular Probes) that contained antifade reagent (Molecular Probes). The tissue was placed in a CoverWell perfusion chamber (Grace Bio-Labs, Bend, OR) and visualized with an Olympus IX71 Fluoview 300 confocal laser-scanning microscope (CLSM; Olympus America, Melville, NY). All images were obtained through a 203×, 0.40 numeric aperture, phase 1 objective utilizing a green helium neon laser (propidium iodide) or argon laser (Alexa Fluor 488). All CLSM instrument settings were consistent for each individual experimental parameter tested. In order to obtain quantitative structural analysis of live and dead cells within the wound, three image stacks each (of various sizes) were acquired from PAO1- and PAO1ΔrhlR-infected tissue sections and analyzed using the COMSTAT program (25). Biovolume, mean thickness, and roughness coefficient were determined. Values represent the means ± standard errors of the means (SEM), and each experiment was performed in triplicate.
RESULTS
eDNA in the wounds of diabetic mice on insulin therapy contributes to the increased antibiotic tolerance of P. aeruginosa.
We previously showed that the diabetic environment promoted P. aeruginosa biofilm development in mouse chronic wounds, as significantly more biofilm was visualized in wound sections from diabetic than nondiabetic mice (12). Insulin treatment appeared to potentiate this effect, evidenced by the increased presence of biofilm and significantly higher antibiotic tolerance in the wounds of diabetic mice treated with insulin. Extracellular DNA (eDNA), originating from lysed bacteria and host cells, is an integral component of the biofilm EPS matrix (26, 27), and it has been shown in vitro to promote protection from antibiotics, especially aminoglycosides (28–30). Recently, DNase was shown to increase P. aeruginosa susceptibility to gentamicin treatment in vitro (31). Thus, we first determined if DNA in the EPS of biofilms from diabetic murine wounds protected P. aeruginosa from gentamicin. To accomplish this, we examined two groups of mice: diabetic mice treated with insulin and their nondiabetic littermates. These mice were given a full-thickness surgical excision wound, which was covered with a gauze pad and infected with PAO1, as previously described (22). After 4 days, the gauze pads were removed, sectioned, and treated for 24 h with a mixture containing either DNase buffer combined with active DNase enzyme or DNase buffer combined with sterile water (as a negative control). We then performed an antibiotic tolerance assay on the samples as described in Materials and Methods.
As we have previously published (12), we observed marked colocalization of ConA, which binds to mannose residues present in the glycocalyx, in areas with bacterial aggregates (i.e., the wound beds) (Fig. 1A and B). Tolerance to gentamicin was greatly enhanced in diabetic mice on insulin therapy compared to that in nondiabetic mice (Fig. 1D, compare column 1 to column 3). Degrading DNA decreased tolerance to gentamicin in both nondiabetic and diabetic mice; however, we observed a significant difference only in the diabetic mice on insulin therapy. We also quantified the concentration of DNA from the gauze pads and observed significantly more DNA in samples from the diabetic mice on insulin therapy (Fig. 1E). It should be noted that the concentration of DNA from the wound beds of diabetic mice not on insulin therapy was also significantly increased (data not shown). Taken together, these data suggest that the increased tolerance we observe in biofilms from diabetic mice treated with insulin depends on the increased presence of DNA in the EPS.
FIG 1.

DNase treatment reduced the antimicrobial tolerance of P. aeruginosa from wounds of diabetic mice treated with insulin. P. aeruginosa establishes biofilms in the chronic wounds of mice, as demonstrated by the increased concentration of ConA staining within the infected wound beds of diabetic mice on insulin treatment at 3 (A) and 14 (B) days postinfection. (C) A section from an area of wound tissue lacking bacterial aggregates is shown to demonstrate the level of ConA background staining. (D) Tolerance to gentamicin, with and without DNase pretreatment, was assessed in wounds after 4 days of infection, as described in Materials and Methods (nondiabetic, light gray; diabetic mice treated with insulin, dark gray). One-way ANOVA on the trimmed means was followed by Bonferroni's multiple-comparison test to determine significant differences between groups. ***, P < 0.001 (n = 10 mice/group). (E) DNA concentrations from wound samples of nondiabetic (ND) and diabetic mice on insulin treatment (D+I) were quantified as described in Materials and Methods. An unpaired, two-tailed t test was performed to determine significant differences between groups. *, P < 0.05 (n = 12 [4 mice/3 samples per mouse]).
Wounds from diabetic mice on insulin therapy displayed increased neutrophil infiltration and increased apoptosis.
A prolonged and abundant infiltration of neutrophils into the wound bed is a characteristic of human chronic wounds (32) and a phenomenon we have consistently seen in wound sections from our mice. However, neutrophils from both diabetic humans and rodents display impaired functions (33–36). Since the incorporation of genomic DNA from lysed neutrophils has been shown to enhance biofilms in vitro, we next sought to determine the relative abundance and viability of neutrophils present in the wounds of nondiabetic, diabetic, and diabetic mice on insulin. We estimated the numbers of neutrophils indirectly by measuring the peroxidase activity of MPO, a lysosomal protein abundantly found in neutrophils. MPO is stored within the azurophilic granules of leukocytes and is responsible for producing antimicrobial oxidizing compounds in these cells (37). The peroxidase activity of MPO can be used to approximate the tissue infiltration of neutrophils in rodent models even in the presence of a mixture of inflammatory cells (23). We measured the peroxidase activity in the extracts of 4-day-old wounds and observed 1.8- and 3.4-fold increases in the activity from wound extracts of untreated diabetic mice and diabetic mice on insulin therapy compared to nondiabetic wound extracts (nondiabetic, 139 ± 17.6 relative fluorescence units [RFUs]/min; untreated diabetic, 257 ± 52 RFUs/min; diabetic on insulin, 468 ± 141 RFUs/min; n = 9 mice/group). While these data suggested that there were more neutrophils in the wounds of diabetic mice when they were on insulin therapy, it did not provide information about their viability or ability to neutralize invading bacteria.
We hypothesized that if the large number of neutrophils in the wounds of diabetic mice on insulin were dead or dying, their DNA debris could be incorporated into P. aeruginosa EPS. To determine if there was an increased presence of dead or dying cells, we utilized TUNEL staining, which fluorescently labels fragmented or damaged DNA (38). Additionally, we used activated caspase-3 immunolabeling to specifically identify apoptotic regions in the tissue. We identified apoptotic/necrotic cells in the wound tissue from the fourth day of infection from nondiabetic, untreated diabetic, and diabetic mice treated with insulin (Fig. 2A). TUNEL identifies apoptotic, necrotic, or damaged tissue because it nonspecifically labels damaged DNA (39), while activated caspase-3 is a specific marker of apoptosis (40). We utilized DAPI, which stains all nucleic acids, to distinguish cell morphology. We observed increased apoptosis/necrosis in the diabetic wound sections, especially in those from diabetic mice treated with insulin (Fig. 2A). Prolific TUNEL and caspase-3 staining was confined to the edge of the wound margin in all groups, indicating the high number of damaged cells in or near the wound bed and site of infection. We quantitated the relative amount of TUNEL and activated caspase-3 immunofluorescent staining using Image J (Fig. 2B), and we observed a significantly higher fluorescent signal, corresponding to increased staining, in the wound sections of the diabetic mice on insulin therapy. These data indicate that there is a large influx of neutrophils into the wounds of the diabetic mice treated with insulin which undergo apoptosis and/or necrosis.
FIG 2.

Increased host cell death was detected in the wound tissue from diabetic mice on insulin therapy. Tissue was harvested from the wound margin on the fourth day of infection from nondiabetic (ND), diabetic (D), and diabetic mice treated with insulin (D+I). (A) TUNEL and caspase-3 immunofluorescent staining was performed on sections of formalin-fixed wound tissue. Images were taken at the same exposure (200 ms), and fluorescent intensity, corresponding to TUNEL and caspase-3, was measured in two sections from 3 mice per group. Blue color, DAPI-stained nuclei; green, TUNEL or caspase-3. (B) ImageJ analysis of green fluorescent intensity indicative of TUNEL and caspase-3 staining. One-way ANOVA, followed by a Tukey-Kramer multiple-comparison test, was used to determine the differences between groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (n = 6 images).
Immunofluorescence revealed decreased numbers of macrophages in the wounds of diabetic mice on insulin therapy.
We observed a large number of apoptotic/necrotic cells in images from wound sections of diabetic mice treated with insulin, suggesting deficient clearing of cellular debris, a process known as efferocytosis (41). Macrophages are the primary cell population responsible for efferocytosis; thus, we sought to quantify macrophage numbers in wound tissue sections. Immunofluorescence, using an antibody to F4/80, was performed on wound sections from nondiabetic, untreated diabetic, and diabetic mice treated with insulin (Fig. 3). F4/80 is a mouse homologue of Emr1, a transmembrane protein present on the cell surface of human macrophages (42). The F4/80 antigen is also found on Langerhans cells in the skin, but their morphology is easily distinguishable from that of macrophages. To quantify the number of macrophages in tissue sections, positively stained cells were counted from 10 random images taken at 100× magnification from 3 wound sections/group of mice. We observed a decreased number of macrophages in sections from diabetic mouse wounds, with the fewest macrophages in the wounds of insulin-treated diabetic mice. Fewer macrophages at the wound site could result in insufficient efferocytosis and could help explain the abundance of apoptotic cells we detected in wound sections from the same group of mice.
FIG 3.
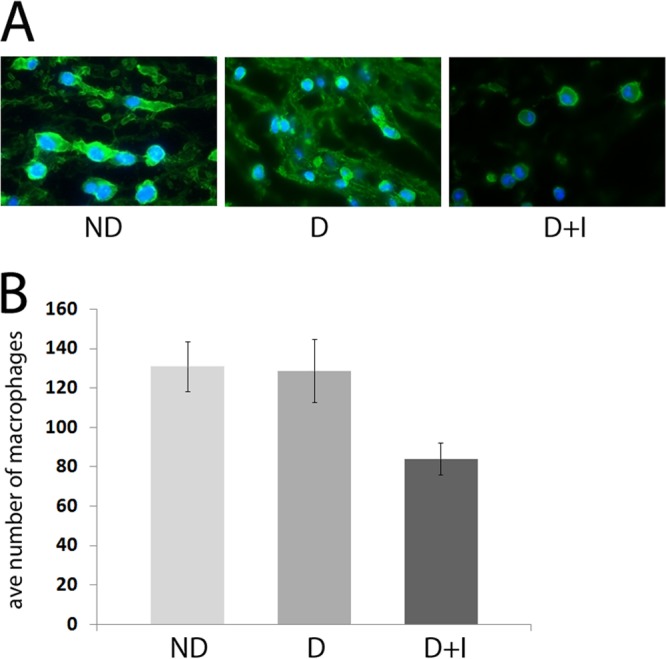
Fewer macrophages were visualized in wound sections from diabetic mice on insulin therapy. Tissue was harvested from the wound margin of nondiabetic (ND), untreated diabetic (D), and diabetic mice treated with insulin (D+I) on day 4 postinfection. Fluorescent IHC with antibodies to F4/80 was performed on formalin-fixed tissue sections to visualize macrophages (n = 3 mice/group). Ten representative images were taken and scored for positive F4/80 fluorescence. Blue color, DAPI-stained nuclei; green, F4/80. One-way ANOVA was used to determine the difference between groups (n = 3 mice/group).
Diabetic chronic wound infections are associated with a reduced early inflammatory response.
Having observed more neutrophils and fewer macrophages in the infected wounds of diabetic mice on insulin, we sought to determine how this affected the expression of proinflammatory cytokines in the wound. We measured the local mRNA levels of TNF-α and IL-1α in the wounds of mice at 6 and 14 days postinfection. RT-PCR analysis revealed lower levels of both cytokines in the diabetic groups on day 6, but by day 14 the levels appeared similar in all groups (Fig. 4A). Immunohistochemistry (IHC) with an antibody to murine IL-6 revealed that it too appeared to be present at lower levels in the wounds of diabetic mice on insulin therapy (Fig. 4B), which is consistent with previous reports describing the anti-inflammatory effect insulin treatment has on P. aeruginosa-infected burn wounds (43–46). TNF-α, IL-1α, and IL-6 are major proinflammatory cytokines produced by both murine neutrophils and macrophages in response to infectious agents, and their reduction in the wounds of diabetic mice could be due to the lower numbers of macrophages and/or nonviable neutrophils.
FIG 4.
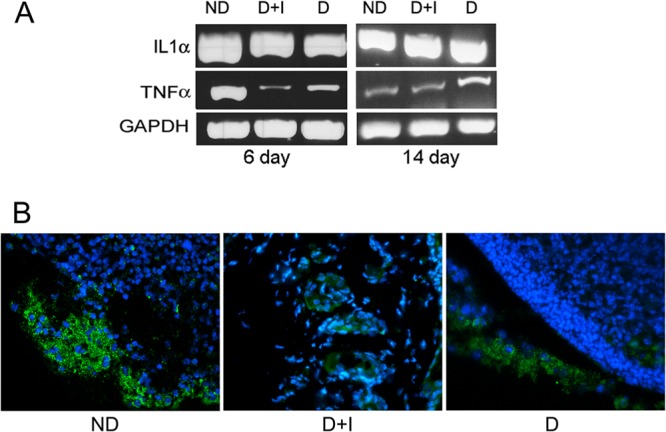
Lower levels of proinflammatory cytokines were detected in the P. aeruginosa-infected wounds of diabetic mice on insulin therapy. Tissue was harvested from the wound margin of PAO1-infected nondiabetic (ND), untreated diabetic (D), and diabetic mice treated with insulin (D+I) at the indicated time point and was either fixed in formalin or stored in RNAlater. (A) The mRNA levels of genes encoding the inflammatory mediators IL-1α and TNF-α were assessed by RT-PCR. GAPDH was used as a loading control. RNA from 3 to 5 mice per treatment was pooled and used for RT-PCR. (B) Fluorescent IHC with antibodies to IL-6 was performed on day 4 postinfection with formalin-fixed tissue sections (n = 3 mice/group). Blue color, DAPI-stained nuclei; green, IL-6. Representative images from each group are shown.
Rhamnolipid is not responsible for host cell death or increased tolerance in P. aeruginosa-infected wounds.
In the chronic wound environment, bacterial necrosis of the short-lived neutrophil occurs consistently and rapidly, and clearance of cellular debris is ineffective (47, 48). P. aeruginosa has been shown to lyse neutrophils very quickly in vitro and in vivo through the production of rhamnolipids (24, 49). To determine if rhamnolipid-dependent lysis of neutrophils enhanced biofilm formation and subsequent tolerance to gentamicin in the diabetic wound environment, we compared gentamicin tolerance in the wounds of mice infected with wild-type P. aeruginosa or a rhamnolipid mutant (PAO1ΔrhlR) (20). The rhamnolipid mutant displayed the same tolerance to gentamicin as wild-type P. aeruginosa in the wounds of diabetic mice treated with insulin (Fig. 5B). In addition, we did not observe a difference in the number of dead cells in tissue infected with wild-type P. aeruginosa or PAO1ΔrhlR (Fig. 5A and Table 1). These data suggest that enhanced tolerance to gentamicin is rhamnolipid independent in our chronic wound model. Many studies suggest that other virulence factors can compensate for the loss of a single virulence factor (50). In support of this, P. aeruginosa has been shown to lyse up to 80% of neutrophils in 6 h in vitro via the ExoU toxin independent of rhamnolipid (51). However, an ExoU mutant still lysed 33% of neutrophils in 6 h, suggesting a redundant role for rhamnolipid. Thus, it is likely that ExoU and/or some other P. aeruginosa virulence factors are responsible for host cell death in our model.
FIG 5.

Loss of rhamnolipid did not affect host cell damage or gentamicin tolerance. (A) Diabetic mice on insulin therapy were given surgical wounds and infected with PAO1 or PAO1ΔrhlR. On day 4, tissue was harvested from the wound bed and stained with PI (red) and SYTO 9 (green) to indicate dead or live cells, respectively. The wounds were then visualized with CLSM. (B) Diabetic mice on insulin therapy were given surgical wounds, which were infected with PAO1 or PAO1ΔrhlR and covered by a sterile gauze pad. At 4 days postoperation, the gauze was removed and the tolerance of the bacterial biofilms to gentamicin was determined. An unpaired, two-tailed t test was performed to determine significant differences between groups (n = 8 for the PAO1 group and n = 10 for the PAO1ΔrhlR group). txt, treatment.
TABLE 1.
COMSTAT analysis of live/dead stained wound tissue
| Cell type | Image stacka (total no. of slices) | Total biovolumeb (μm3/μm2) | Mean thicknessc (μm) | Roughness coefficientd |
|---|---|---|---|---|
| PAO1-infected dead | 237–369 | 0.7367 ± 0.06 | 0.8033 ± 0.05 | 0.3233 ± 0.07 |
| PAO1-infected live | 186–316 | 0.2633 ± 0.06 | 0.1967 ± 0.05 | 0.6767 ± 0.07 |
| PAO1ΔrhlR-infected dead | 179–300 | 0.8033 ± 0.09 | 0.7367 ± 0.13 | 0.4100 ± 0.04 |
| PAO1ΔrhlR-infected live | 194–291 | 0.2033 ± 0.09 | 0.2633 ± 0.13 | 0.5900 ± 0.04 |
Each experiment was done in triplicate. The number of slices per image stack is indicated.
Estimates the biomass of positively stained cells within the tissue section (P = 0.5805).
Measures the spatial size of the positively stained cells within the tissue section (P = 0.6568).
Assesses the variations of thickness among positively stained cells (P = 0.3465).
Lastly, we wanted to determine if insulin-induced antimicrobial tolerance was specific to P. aeruginosa infection. Nondiabetic, untreated diabetic, and diabetic mice on insulin therapy were wounded as described above and infected with 104 CFU S. aureus. At 4 days postinfection, mice were euthanized and gentamicin tolerance assays were performed. While 14% (±3.7%) of S. aureus organisms in the wounds of nondiabetic mice (n = 7) were tolerant to gentamicin, 26.9% (±5%) were tolerant to gentamicin in the wounds of diabetic mice (n = 7), and that tolerance increased to 33.9% (±6.2%) when the diabetic mice (n = 6) were on insulin therapy (P < 0.034 by analysis of variance [ANOVA] with Tukey-Kramer multiple-comparison test). Thus, similar to our observations with P. aeruginosa infection, S. aureus tolerance to gentamicin was also significantly increased in diabetic mice on insulin therapy. However, while we observed a 2.4-fold increase in the number of S. aureus organisms that were viable after gentamicin treatment from the wounds of diabetic mice on insulin compared to that of nondiabetic mice, we previously reported a 9-fold increase in the number of P. aeruginosa organisms that remained viable in the insulin-treated diabetic mice compared to that of nondiabetic mice (12). These data indicate that insulin-induced antimicrobial tolerance is not unique to P. aeruginosa infection and support the hypothesis of a host-related mechanism.
DISCUSSION
Diabetics are more susceptible than nondiabetics to debilitating wound infections that can remain unhealed for months or years. While most of these patients are on some form of insulin to control their disease, these treatments do not appear to significantly improve wound healing. P. aeruginosa is one of the most common causes of bacterial infections in chronic wounds, and it is thought to persist in the wound by establishing biofilms which confer antibiotic tolerance and impair wound healing.
Previously, we examined how the diabetic environment promotes the chronicity and severity of P. aeruginosa biofilm-associated wound infections. Our most surprising finding was that biofilms were more prevalent in the wounds of diabetic mice on insulin therapy and displayed significantly higher tolerance to gentamicin than did bacteria in the wounds of nondiabetic mice or diabetic mice that were not treated with insulin. This phenomenon was explained in part by the finding that insulin could directly enhance P. aeruginosa biofilm formation in vitro (12). However, we also speculated that the immune system, influenced by insulin, could promote biofilm formation.
In this study, we explored the possibility that insulin treatment could alter the immune response to favor bacterial biofilm development. We utilized a diabetic chronic wound mouse model to compare the antimicrobial tolerance and numbers of neutrophils, macrophages, and dead/damaged cells in nondiabetic and diabetic wounds. We used STZ to induce diabetes in otherwise healthy, outbred Swiss Webster mice. STZ destroys the beta islet cells of the pancreas, and STZ-treated mice are unable to produce insulin. STZ-induced diabetes mimics type I diabetes, causing the mice to become severely hyperglycemic and unable to gain weight (52). STZ-treated diabetic rodents display a diminished immune response, attributed to both hyperglycemia and STZ directly. Specifically, neutrophils and macrophages from STZ diabetic rodents display diminished phagocytosis, which has also been frequently observed in diabetic patients (33–36). Hyperglycemia is thought to impair the function of these cells (53), and poorly controlled blood glucose levels is a risk factor for bacterial infection (54). However, even diabetic patients on insulin therapy display defective bactericidal activity against P. aeruginosa (55), demonstrating that insulin therapy does not completely alleviate the negative effects of diabetes in regard to immune function. In accordance with this, we have previously observed that insulin treatment does not improve bacterial clearance or wound closure in diabetic mice (12).
Neutrophils act as a first line of defense against bacterial pathogens and can phagocytize and kill bacteria or capture and kill bacterial cells through the release of neutrophil extracellular traps (NETs) composed predominantly of DNA and histones (known as NETosis [56]). In vitro, NETs have been shown to kill planktonic P. aeruginosa cells, but clinical isolates from cystic fibrosis patients demonstrated increased resistance to NET killing (57). In fact, human neutrophils (both intact and lysed) actually enhanced initial P. aeruginosa biofilm development through the formation of polymers comprised of actin and DNA (58, 59). Neutrophil-enhanced P. aeruginosa biofilms were more tolerant to antipseudomonal antibiotics, but when treated with DNase, these biofilms were readily disrupted (59). Similarly, we observed significantly enhanced efficacy of gentamicin when sections from diabetic mice on insulin therapy were pretreated with DNase. While DNase was shown to increase P. aeruginosa susceptibility to gentamicin treatment in vitro (31), to our knowledge we are the first to demonstrate the efficacy of DNase to enhance antibiotic clearance of biofilms grown in vivo. Our data also demonstrated that there was a greater amount of DNA in wound sections from diabetic mice on insulin, suggesting that eDNA plays an important role in antibiotic tolerance in vivo.
One potential source of this eDNA is from host cells, and we observed enhanced MPO peroxidase activity in the wounds of diabetic mice treated with insulin, which is indicative of higher numbers of neutrophils. We also observed significantly increased cell death in these same tissues. Efferocytosis is key to resolving inflammation by removing cellular debris; however, diabetes is known to impair macrophage function (41), and we detected fewer macrophages in the wounds of diabetic mice on insulin. We also observed lower expression of several major proinflammatory cytokines early in the infection. Inflammation is a key early step to resolving wound infections and initiating healing (32). Ideally, an early and robust inflammatory response will efficiently resolve or prevent infection, and neutrophils and macrophages are essential in this response, because they are the primary cells responsible for removing foreign materials, bacteria, and damaged tissue from the wound (32). During the normal healing cascade, the inflammatory phase ends once the wound site is cleared and a proliferative phase is initiated, during which new cell growth and migration begins in the wound bed. A hallmark of chronic wounds is a prolonged inflammatory response that perturbs healing. Insulin treatment has been shown to reduce the levels of proinflammatory cytokines in burned mice and rats treated with insulin (43–46); therefore, diabetes- and/or insulin-related inhibition of this response could provide invading bacteria the opportunity to gain a foothold by establishing a biofilm within the wound. Taken together, our data suggest that the high levels of lysed neutrophils in the wounds of diabetic mice on insulin, combined with fewer macrophages to clear away the cellular detritus, contribute to enhanced eDNA levels of P. aeruginosa biofilms, making them more tolerant to gentamicin.
ACKNOWLEDGMENTS
This study was supported by American Diabetes Association research grant 1-08-RA-165 and a grant from The CH Foundation to K.P.R.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1.Cunningham AB. 2006. Biofilms: the hypertextbook. Montana State University, Bozeman, MT [Google Scholar]
- 2.Stamm WE, Martin SM, Bennett JV. 1977. Epidemiology of nosocomial infection due to Gram-negative bacilli: aspects relevant to development and use of vaccines. J. Infect. Dis. 136(Suppl):S151–S160. 10.1093/infdis/136.Supplement.S151 [DOI] [PubMed] [Google Scholar]
- 3.Fuxman Bass JI, Russo DM, Gabelloni ML, Geffner JR, Giordano M, Catalano M, Zorreguieta A, Trevani AS. 2010. Extracellular DNA: a major proinflammatory component of Pseudomonas aeruginosa biofilms. J. Immunol. 184:6386–6395. 10.4049/jimmunol.0901640 [DOI] [PubMed] [Google Scholar]
- 4.Wolcott RD, Rhoads DD, Dowd SE. 2008. Biofilms and chronic wound inflammation. J. Wound Care 17:333–341 [DOI] [PubMed] [Google Scholar]
- 5.Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR, Otto M. 2004. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell. Microbiol. 6:269–275. 10.1046/j.1462-5822.2004.00367.x [DOI] [PubMed] [Google Scholar]
- 6.Thomson CH. 2011. Biofilms: do they affect wound healing? Int. Wound J. 8:63–67. 10.1111/j.1742-481X.2010.00749.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobsen JN, Andersen AS, Sonnested MK, Laursen I, Jorgensen B, Krogfelt KA. 2011. Investigating the humoral immune response in chronic venous leg ulcer patients colonised with Pseudomonas aeruginosa. Int. Wound J. 8:33–43. 10.1111/j.1742-481X.2010.00741.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolcott RD, Ehrlich GD. 2008. Biofilms and chronic infections. JAMA 299:2682–2684. 10.1001/jama.299.22.2682 [DOI] [PubMed] [Google Scholar]
- 9.Percival SL, Hill KE, Malic S, Thomas DW, Williams DW. 2011. Antimicrobial tolerance and the significance of persister cells in recalcitrant chronic wound biofilms. Wound Repair Regen. 19:1–9. 10.1111/j.1524-475X.2010.00651.x [DOI] [PubMed] [Google Scholar]
- 10.Anwar H, Dasgupta MK, Costerton JW. 1990. Testing the susceptibility of bacteria in biofilms to antibacterial agents. Antimicrob. Agents Chemother. 34:2043–2046. 10.1128/AAC.34.11.2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gefen O, Balaban NQ. 2009. The importance of being persistent: heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol. Rev. 33:704–717. 10.1111/j.1574-6976.2008.00156.x [DOI] [PubMed] [Google Scholar]
- 12.Watters C, DeLeon K, Trivedi U, Griswold JA, Lyte M, Hampel KJ, Wargo MJ, Rumbaugh KP. 2013. Pseudomonas aeruginosa biofilms perturb wound resolution and antibiotic tolerance in diabetic mice. Med. Microbiol. Immunol. 202:131–141. 10.1007/s00430-012-0277-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gauglitz GG, Toliver-Kinsky TE, Williams FN, Song JQ, Cui WH, Herndon DN, Jeschke MG. 2010. Insulin increases resistance to burn wound infection-associated sepsis. Crit. Care Med. 38:202–208. 10.1097/CCM.0b013e3181b43236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeschke MG, Kulp GA, Kraft R, Finnerty CC, Mlcak R, Lee JO, Herndon DN. 2010. Intensive insulin therapy in severely burned pediatric patients: a prospective randomized trial. Am. J. Resp. Crit. Care Med. 182:351–359. 10.1164/rccm.201002-0190OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dandona P, Chaudhuri A, Mohanty P, Ghanim H. 2007. Anti-inflammatory effects of insulin. Curr. Opin. Clin. Nutr. Metabolic Care 10:511–517. 10.1097/MCO.0b013e3281e38774 [DOI] [PubMed] [Google Scholar]
- 16.Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, Ahmad S. 2001. Insulin inhibits intranuclear nuclear factor kappa B and stimulates I kappa B in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J. Clin. Endocrinol. Metabolism 86:3257–3265. 10.1210/jc.86.7.3257 [DOI] [PubMed] [Google Scholar]
- 17.Martins JO, Ferracini M, Ravanelli N, Landgraf RG, Jancar S. 2008. Insulin suppresses LPS-induced iNOS and COX-2 expression and NF-kappa B activation in alveolar macrophages. Cell. Physiol. Biochem. 22:279–286. 10.1159/000149806 [DOI] [PubMed] [Google Scholar]
- 18.Kidd LB, Schabbauer GA, Luyendyk JP, Holscher TD, Tilley RE, Tencati M, Mackman N. 2008. Insulin activation of the phosphatidylinositol 3-kinase/protein kinase B (Akt) pathway reduces lipopolysaccharide-induced inflammation in mice. J. Pharmacol. Exp. Ther. 326:348–353. 10.1124/jpet.108.138891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 43:73–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brint JM, Ohman DE. 1995. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J. Bacteriol. 177:7155–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown RL, Greenhalgh DG. 1997. Mouse models to study wound closure and topical treatment of infected wounds in healing-impaired and normal healing hosts. Wound Repair Regen. 5:198–204. 10.1046/j.1524-475X.1997.50213.x [DOI] [PubMed] [Google Scholar]
- 22.Wolcott RD, Rumbaugh KP, James G, Schultz G, Phillips P, Yang Q, Watters C, Stewart PS, Dowd SE. 2010. Biofilm maturity studies indicate sharp debridement opens a time-dependent therapeutic window. J. Wound Care 19:320–328 [DOI] [PubMed] [Google Scholar]
- 23.Schneider T, Issekutz AC. 1996. Quantitation of eosinophil and neutrophil infiltration into rat lung by specific assays for eosinophil peroxidase and myeloperoxidase. Application in a Brown Norway rat model of allergic pulmonary inflammation. J. Immunol. Methods 198:1–14 [DOI] [PubMed] [Google Scholar]
- 24.van Gennip M, Christensen LD, Alhede M, Qvortrup K, Jensen PO, Hoiby N, Givskov M, Bjarnsholt T. 2012. Interactions between polymorphonuclear leukocytes and Pseudomonas aeruginosa biofilms on silicone implants in vivo. Infect. Immun. 80:2601–2607. 10.1128/IAI.06215-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146(Part 10):2395–2407 [DOI] [PubMed] [Google Scholar]
- 26.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. 10.1126/science.295.5559.1487 [DOI] [PubMed] [Google Scholar]
- 27.Spoering AL, Gilmore MS. 2006. Quorum sensing and DNA release in bacterial biofilms. Curr. Opin. Microbiol. 9:133–137. 10.1016/j.mib.2006.02.004 [DOI] [PubMed] [Google Scholar]
- 28.Tetz GV, Artemenko NK, Tetz VV. 2009. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 53:1204–1209. 10.1128/AAC.00471-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulcahy H, Charron-Mazenod L, Lewenza S. 2008. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4:e1000213. 10.1371/journal.ppat.1000213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiang WC, Nilsson M, Jensen PO, Hoiby N, Nielsen TE, Givskov M, Tolker-Nielsen T. 2013. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 57:2352–2361. 10.1128/AAC.00001-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aspe M, Jensen L, Melegrito J, Sun M. 2012. The role of alginate and extracellular DNA in biofilm-meditated Pseudomonas aeruginosa gentamicin. J. Exp. Microbiol. Immunol. 16:42–48 [Google Scholar]
- 32.Diegelmann RF, Evans MC. 2004. Wound healing: an overview of acute, fibrotic and delayed healing. Front. Biosci. 9:283–289. 10.2741/1184 [DOI] [PubMed] [Google Scholar]
- 33.Nabi AH, Islam LN, Rahman MM, Biswas KB. 2005. Polymorphonuclear neutrophil dysfunctions in streptozotocin-induced type 1 diabetic rats. J. Biochem. Mol. Biol. 38:661–667. 10.5483/BMBRep.2005.38.6.661 [DOI] [PubMed] [Google Scholar]
- 34.Saiki O, Negoro S, Tsuyuguchi I, Yamamura Y. 1980. Depressed immunological defence mechanisms in mice with experimentally induced diabetes. Infect. Immun. 28:127–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass EJ, Stewart J, Matthews DM, Collier A, Clarke BF, Weir DM. 1987. Impairment of monocyte “lectin-like” receptor activity in type 1 (insulin-dependent) diabetic patients. Diabetologia 30:228–231. 10.1007/BF00270420 [DOI] [PubMed] [Google Scholar]
- 36.Wilson RM, Reeves WG. 1986. Neutrophil phagocytosis and killing in insulin-dependent diabetes. Clin. Exp. Immunol. 63:478–484 [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison JE, Schultz J. 1976. Studies on the chlorinating activity of myeloperoxidase. J. Biol. Chem. 251:1371–1374 [PubMed] [Google Scholar]
- 38.Loo DT. 2002. TUNEL assay. An overview of techniques. Methods Mol. Biol. 203:21–30. 10.1385/1-59259-179-5:21 [DOI] [PubMed] [Google Scholar]
- 39.de Torres C, Munell F, Ferrer I, Reventos J, Macaya A. 1997. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci. Lett. 230:1–4. 10.1016/S0304-3940(97)00445-X [DOI] [PubMed] [Google Scholar]
- 40.Jeruc J, Vizjak A, Rozman B, Ferluga D. 2006. Immunohistochemical expression of activated caspase-3 as a marker of apoptosis in glomeruli of human lupus nephritis. Am. J. Kidney Dis. 48:410–418. 10.1053/j.ajkd.2006.05.019 [DOI] [PubMed] [Google Scholar]
- 41.Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. 2010. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 5:e9539. 10.1371/journal.pone.0009539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Austyn JM, Gordon S. 1981. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 11:805–815. 10.1002/eji.1830111013 [DOI] [PubMed] [Google Scholar]
- 43.Klein D, Schubert T, Horch RE, Jauch KW, Jeschke MG. 2004. Insulin treatment improves hepatic morphology and function through modulation of hepatic signals after severe trauma. Ann. Surg. 240:340–349. 10.1097/01.sla.0000133353.57674.cd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeschke MG, Klein D, Bolder U, Einspanier R. 2004. Insulin attenuates the systemic inflammatory response in endotoxemic rats. Endocrinology 145:4084–4093. 10.1210/en.2004-0592 [DOI] [PubMed] [Google Scholar]
- 45.Jeschke MG, Klein D, Herndon DN. 2004. Insulin treatment improves the systemic inflammatory reaction to severe trauma. Ann. Surg. 239:553–560. 10.1097/01.sla.0000118569.10289.ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Przkora R, Herndon DN, Finnerty CC, Jeschke MG. 2007. Insulin attenuates the cytokine response in a burn wound infection model. Shock 27:205–208. 10.1097/01.shk.0000238069.84826.1b [DOI] [PubMed] [Google Scholar]
- 47.Ramos AN, Cabral ME, Noseda D, Bosch A, Yantorno OM, Valdez JC. 2012. Antipathogenic properties of Lactobacillus plantarum on Pseudomonas aeruginosa: the potential use of its supernatants in the treatment of infected chronic wounds. Wound Repair Regen. 20:552–562. 10.1111/j.1524-475X.2012.00798.x [DOI] [PubMed] [Google Scholar]
- 48.Bjarnsholt T, Kirketerp-Moller K, Jensen PO, Madsen KG, Phipps R, Krogfelt K, Hoiby N, Givskov M. 2008. Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen. 16:2–10. 10.1111/j.1524-475X.2007.00283.x [DOI] [PubMed] [Google Scholar]
- 49.Jensen PO, Bjarnsholt T, Phipps R, Rasmussen TB, Calum H, Christoffersen L, Moser C, Williams P, Pressler T, Givskov M, Hoiby N. 2007. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 153:1329–1338. 10.1099/mic.0.2006/003863-0 [DOI] [PubMed] [Google Scholar]
- 50.Schaber JA, Carty NL, McDonald NA, Graham ED, Cheluvappa R, Griswold JA, Hamood AN. 2004. Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 53:841–853. 10.1099/jmm.0.45617-0 [DOI] [PubMed] [Google Scholar]
- 51.Diaz MH, Shaver CM, King JD, Musunuri S, Kazzaz JA, Hauser AR. 2008. Pseudomonas aeruginosa induces localized immunosuppression during pneumonia. Infect. Immun. 76:4414–4421. 10.1128/IAI.00012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu KK, Huan Y. 2008. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. Chapter 5:Unit 5.47. 10.1002/0471141755.ph0547s40 [DOI] [PubMed] [Google Scholar]
- 53.Turina M, Fry DE, Polk HC., Jr 2005. Acute hyperglycemia and the innate immune system: clinical, cellular, and molecular aspects. Crit. Care Med. 33:1624–1633. 10.1097/01.CCM.0000170106.61978.D8 [DOI] [PubMed] [Google Scholar]
- 54.Wheat LJ. 1980. Infection and diabetes mellitus. Diabetes Care 3:187–197. 10.2337/diacare.3.1.187 [DOI] [PubMed] [Google Scholar]
- 55.Naghibi M, Smith RP, Baltch AL, Gates SA, Wu DH, Hammer MC, Michelsen PB. 1987. The effect of diabetes mellitus on chemotactic and bactericidal activity of human polymorphonuclear leukocytes. Diabetes Res. Clin. Pract. 4:27–35. 10.1016/S0168-8227(87)80030-X [DOI] [PubMed] [Google Scholar]
- 56.Kaplan MJ, Radic M. 2012. Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 189:2689–2695. 10.4049/jimmunol.1201719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, Taylor-Cousar JL, Saavedra MT, Randell SH, Vasil ML, Burns JL, Moskowitz SM, Nick JA. 2011. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One 6:e23637. 10.1371/journal.pone.0023637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, Fessler MB, Malcolm KC, Vasil ML, Nick JA. 2005. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect. Immun. 73:3693–3701. 10.1128/IAI.73.6.3693-3701.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parks QM, Young RL, Poch KR, Malcolm KC, Vasil ML, Nick JA. 2009. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: human F-actin and DNA as targets for therapy. J. Med. Microbiol. 58:492–502. 10.1099/jmm.0.005728-0 [DOI] [PMC free article] [PubMed] [Google Scholar]