

Abstract
We have previously shown that during late stages of the infectious process, serogroup B meningococci (MenB) are able to escape the phagosome of in vitro-infected human epithelial cells. They then multiply in the cytosolic environment and spread intracellularly and to surrounding cells by exploiting the microtubule cytoskeleton, as suggested by results of infections in the presence of microtubule inhibitors and evidence of nanotubes connecting neighboring cells. In this study, by using microtubule binding assays with purified microtubule asters and bundles and microtubule bundles synthesized in vitro, we demonstrate that the MenB capsule directly mediates the interaction between bacteria and microtubules. The direct interaction between the microtubules and the MenB capsular polysaccharide was confirmed by coimmunoprecipitation experiments. Unexpectedly, serogroup C meningococci (MenC), which have a capsular polysaccharide that differs from that of MenB only by its anomeric linkage, α(2→9) instead of α(2→8), were not able to interact with the microtubules, and the lack of interaction was not due to capsular polysaccharide O-acetylation that takes place in most MenC strains but not in MenB strains. Moreover, we demonstrate that the MenB capsular polysaccharide inhibits tubulin polymerization in vitro. Thus, at variance with MenC, MenB may interfere with microtubule dynamics during cell infection.
INTRODUCTION
Neisseria meningitidis (meningococcus) is a transitory colonizer of the human nasopharynx that occasionally provokes life-threatening diseases, including meningitis and sepsis. This microorganism is highly variable, and variants that are modified in their virulence and/or transmissibility are continually generated. Meningococcal surface variation serves as an adaptive mechanism to modulate tissue tropism, immune evasion, and survival in the changing host environment (1). This concept is exemplified by phase and antigenic variation of the capsular polysaccharide.
Traditionally, meningococcal strains have been classified into 13 serogroups based on the immunological specificities of their capsular polysaccharide. Of these, five serogroups (A, B, C, Y, and W135) are responsible for most of the diseases worldwide, each with various epidemiological features, including prevalence, virulence, immunogenicity, geographical, and temporal distribution (2). In particular, infection by serogroup B meningococci (MenB) is typically associated with higher morbidity and mortality rates in young infants (2–4). MenB disease can be devastating: 5 to 10% of children with the disease do not survive, and another 10 to 20% experience long-term sequelae, such as hearing loss, limb loss, and neurologic deficits. Moreover, lack of immunogenicity of the MenB polysialic acid capsule is a major problem in development of a capsule-based vaccine.
However, the meningococcal capsule plays contrasting roles depending on the host microenvironment. On one hand, the antiphagocytic capsules are essential for meningococcal growth/survival in many host environments, including the bloodstream and cerebrospinal fluid (5). Moreover, there is evidence that the MenB capsule is important for meningococcal survival within human cells, and that capsular biosynthetic genes are upregulated in the intracellular environment (6). There is also evidence that the meningococcal capsule contributes to protect the bacteria against cationic antimicrobial peptides (CAMP), including the human cathelicidin LL-37 (6, 7), while at the same time, by interacting with the capsule, LL-37 seems to inhibit the proinflammatory activity of the capsular polysaccharide (8). On the other hand, the expression of the capsular polysaccharide inhibits the colonization and invasion of the nasopharyngeal barrier by masking the meningococcal adhesins/invasins (9). For these reasons, capsular polysaccharide expression is subject to frequent phase variation via slipped-strand mispairing or reversible insertion of mobile elements (9–11) and is tightly regulated at the transcriptional level. Indeed, there is evidence that capsule biosynthesis and assembly are downregulated during the early stages of the infectious cycle to facilitate the adhesion to and invasion of the host cells (12).
In a previous study, we showed that during the late stages of the infectious process, MenB are able to escape the phagosome of in vitro-infected human epithelial cells. They then multiply at high rates in the cytosolic environment and spread to surrounding cells by exploiting the microtubule cytoskeleton, as suggested by results of infections in the presence of microtubule inhibitors (6, 13, 14). In this study, we demonstrate that the MenB capsular polysaccharide directly mediates the interaction between bacteria and microtubules and inhibits tubulin polymerization in vitro.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
N. meningitidis strains have been reported previously (11, 15–18). In particular, MenB strain B1940 (B:NT:P1.3,6,15; lipooligosaccharide [LOS] immunotype L3,7,9) and its derivatives, the B1940 cps mutant and the B1940 siaD(−C) mutant, have been described previously (9, 19). The B1940 cps mutant lacks both the capsule and the lipooligosaccharide (LOS) outer core. The B1940 siaD(−C) mutant lacks the capsule but retains the LOS outer core due to frameshift mutation, a cytosine deletion in a polycytidine repeat located in the coding region of the polysialyltransferase gene (siaD). MenB H44/76 is an isolate from a patient with invasive meningococcal disease (20) and is an international reference strain classified as B:15:P1.7,16, LOS immunotype L3,7,9 (21, 22). 205900 and XL929 are N. meningitidis serogroup A (MenA) and serogroup X (MenX) strains, respectively. Serogroup C meningococcal strain 2120 (sequence type 11 [ST-11]; OatC+) and isogenic mutant 2948 (oatC::ΩKanr) (23) were kindly provided by H. Claus (Universität Würzburg, Germany). Capsulation of meningococcal strains was checked by the latex slide agglutination test (Directigen N. meningitidis group B/Escherichia coli K1 and Directigen N. meningitidis groups A, C, Y, and W135 test kits; Becton, Dickinson and Company). Neisseria lactamica strains NL4627 and NL995 and Neisseria sicca strain NS407 have been described previously (24). All Neisseria sp. strains were cultured in gonococcus (GC) broth or agar with 1% Polyvitox at 37°C in a 5% CO2 incubator. Wild-type Escherichia coli K-12 strain FB8 (25) was grown in Luria-Bertani medium.
Cell culture and invasion assay.
For standard invasion assays, HeLa cells (ATCC CCL-2) or HEp-2 cells (ATCC CCL-23) (2 × 105) were seeded in 24-well tissue culture plates (Falcon) in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS; Gibco), 2 mM l-glutamine, and antibiotics for 24 h at 37°C in 5% CO2. Before the addition of bacteria, cells were washed three times with DMEM supplemented with 2% fetal calf serum (FCS) and 2 mM l-glutamine (without antibiotics). Meningococci grown overnight as described above were harvested from GC agar plates and resuspended in DMEM. Cells were infected at a multiplicity of infection (MOI) of 50 for 1 h. Adherence of viable bacteria was evaluated after removing nonadherent bacteria by sequential washing with phosphate-buffered saline (PBS). For invasion assays, bacteria were centrifuged (60 × g for 5 min) onto cells to start the infection. Cells then were washed twice with PBS to eliminate the majority of extracellular bacteria and then exposed to gentamicin to kill the remaining extracellular bacteria. Cells were washed extensively with PBS to remove gentamicin and dead extracellular bacteria and then lysed with saponin. Gentamicin treatment was performed at 100 μg/ml, a concentration about 10-fold above the MIC, for 30 min. We determined that this treatment was sufficient to kill all extracellular bacteria by plating the culture medium before and after gentamicin treatment onto GC agar medium. No viable bacteria were detected after treatment with either 100 or 500 μg/ml gentamicin, indicating a survival value of <10−8. When required, cells were reincubated in fresh culture medium at various time intervals (0 to 7 h) after gentamicin treatment. For quantification, bacteria present in culture medium (extracellular) or released by saponin from HeLa cells (intracellular) were plated, and CFU were counted the day after. Infected cells were also analyzed by confocal laser-scanning microscopy (CLSM) or immunofluorescence microscopy (IFM).
Immunofluorescence analysis.
HeLa cells were seeded onto 12-mm-diameter glass coverslips, infected as described above, and processed for immunofluorescence as described previously (10, 26). To detect intracellular bacteria, primary polyclonal antibody against whole-cell preparations of serogroup A, B, and C N. meningitidis (Omnitope, Viro Stat) or against MenB (R30167501; Remel Europe Ltd.) or serogroup C meningococci (MenC) (R30166901; Remel Europe Ltd.) capsular polysaccharides were used after permeabilization with 0.25% saponin, in combination with fluorescein isothiocyanate (FITC)-conjugated secondary antibody. To detect cellular markers, we used Alexa 568 phalloidin (Molecular Probes), anti-α-tubulin (Sigma), or anti-transferrin receptor (Roche) in combination with tetramethyl rhodamine isothiocyanate (TRITC)-conjugated secondary antibody. All antibodies used were diluted 1:500.
Interaction of meningococci with purified microtubules.
Cell-free microtubules were extracted from HeLa cells and used in an interaction assay with bacteria as described previously (27). Briefly, HeLa cells were washed with 1× PBS/protease inhibitor, scraped, and centrifuged at 20,000 × g for 20 min at 4°C; the pellet was resuspended in 5 volumes of buffer A (30 mM Tris-HCl, pH 7.5, 120 mM KCl, 5 mM Mg[OAc]2, where OAc is acetate) and centrifuged at 20,000 × g for 20 min at 4°C; the pellet then was resuspended in 2 volumes of buffer B (10 mM Tris-HCl, pH 7.5, 10 mM KCl, 1.5 mM Mg[OAc]2), incubated for 5 min on ice, and homogenized for 15 s. In the next step, 0.1 volume of buffer C (230 mM Tris-HCl, pH 7.5, 1.27 M KCl, 40 mM Mg[OAc]2) was added to the sample, and the mix was centrifuged at 20,000 × g for 20 min at 4°C. One mM paclitaxel was added to the supernatant. B1940 and the mutant bacterial cells (5 × 106 CFU) were added to purified microtubules and incubated at room temperature for 40 min. For fluorescence microscopy, meningococcal cells were either detected by using polyclonal antibody against whole-cell preparation of N. meningitidis or were stained with 4′-6-diamidino-2-phenylindole (DAPI), and the purified microtubules were labeled with tubulin tracker green reagent GT500. Each experiment was performed at least three times. Student's t test was used to analyze differences between groups. A P value below 0.05 was considered statistically significant.
In a competitive binding assay, MenB strain B1940 (5 × 106 CFU) and 3 μg of capsular polysaccharides from MenB strain B1940 or MenC strain 93/4286 were added to purified microtubules, incubated at room temperature for 40 min, and then stained with tubulin tracker green reagent GT500 and DAPI for fluorescence microscopy as described above.
Interaction of meningococci with in vitro-synthesized microtubules.
Paclitaxel-stabilized microtubules were obtained by using a >99% pure X-rhodamine-labeled tubulin preparation from porcine brain (Cytoskeleton, Inc.). The lyophilized X-rhodamine-labeled tubulin was resuspended in general tubulin buffer supplemented with 10% glycerol and 1 mM GTP to a final concentration of 3 mg/ml. Bacterial suspensions (5 × 106 CFU) from strain B1940 and from mutant strains were added to this tubulin solution and then incubated at 37°C for 30 min to polymerize the tubulin and form microtubules. In the next step, 0.7 μl of 200 μM paclitaxel stock was added to the microtubule reaction mix and incubated at 37°C for 5 min to stabilize the microtubules. The bacterial cells were stained with Syto-9, and then bacterium-microtubule interactions were visualized by fluorescence microscopy using a 100× oil immersion lens.
Preparation of extracts enriched in capsular polysaccharide.
Extracts enriched in capsular polysaccharide were prepared from MenB strain B1940 or MenC strain 93/4286 according to the protocols of Gotschlich (28), modified as follows. One liter of culture was vigorously aerated in supplemented GC broth for 16 h at 125 rpm. To precipitate the capsular polysaccharide complex, 10% hexadecyltrimethyl ammonium bromide (Cetavlon) was added to a final concentration of 0.1% (wt/vol). Samples were incubated at 4°C overnight. Hexadecyltrimethyl ammonium bromide results in bacterial lysis and release of capsular polysaccharide. The precipitate and bacterial debris were collected by centrifugation (5,000 rpm for 40 min) and then resuspended in 50 ml of distilled water. To dissociate the polysaccharide-hexadecyltrimethyl ammonium bromide complex, 1 volume of 2 M CaCl2 was added, and the mixture stirred for 2 h at 4°C. Absolute cold ethanol was added to a final concentration of 25% (vol/vol) to precipitate nucleic acids at 4°C. After 2 h, the precipitated nucleic acids and the bacterial debris were removed by centrifugation (10,000 rpm for 40 min). The ethanol concentration of the supernatant was raised to 80% (vol/vol) to precipitate polysaccharide. This polysaccharide was collected by centrifugation (10,000 rpm for 40 min), washed three times with absolute ethanol to remove CaCl2 and hexadecyltrimethyl ammonium bromide, washed three times with acetone, washed twice with diethyl ether (with centrifuging at 10,000 for 10 min between washes), and dried in a vacuum. The pellet was dissolved in 0.2 M sodium phosphate buffer, pH 7, and centrifuged at 10,000 rpm for 30 min to collect the supernatant. Contaminating proteins were then removed from the aforementioned supernatant by cold phenol extraction at neutral pH. The phenol-extracted aqueous phase was dialyzed against distilled water for 48 h. The aqueous solution was then lyophilized to yield the crude capsular polysaccharide. The lyophilized polysaccharide was treated with 1% acetic acid-hydrochloride (pH 2) for 1 h on ice to disrupt the putative phospholipid-polysaccharide bond, kept on ice for 1 h at pH 10 (by adding cold 0.1 M NaOH) to saponify lactone rings, and finally neutralized with 0.1 M acetic acid. After lyophilization, the extract was dissolved in sterile deionized water and the sialic acid content was measured by a resorcinol reaction (29) with N-acetylneuraminic acid as a standard.
Coimmunoprecipitation.
Coimmunoprecipitation assays were performed using a cross-link immunoprecipitation kit (Pierce) by following the manufacturer's instructions. Briefly, mouse anti-α-tubulin antibody (Sigma) or mouse IgG was cross-linked to a resin using disuccinimidyl suberate (DSS) and incubated with HeLa lysate that had been incubated previously with capsular extracts for 1 h at 4°C with agitation. To reduce nonspecific binding of proteins to the resin, the HeLa lysate was precleared with a control resin according to the manufacturer's instructions before coimmunoprecipitation. After washing and elution, coimmunoprecipitates were subjected to immunoblot analysis using anti-MenB or anti-MenC capsule monoclonal antibodies and anti-α-tubulin antibody (Sigma).
Immunoblot analysis.
Coimmunoprecipitates were transferred onto polyvinyl difluoride (PVDF) membranes from Millipore (Milan, Italy) under vacuum by using a slot-blot manifold (Bethesda Research Laboratories). The PVDF membranes were preventively immersed into 100% methanol for a few seconds, transferred into distilled water for 2 to 3 min, and then incubated in a transfer buffer containing 25 mM Tris, 192 mM glycine (pH 8.3), and 20% methanol. After blotting, the membranes were air dried, blocked in 5% milk in 1× PBS for 30 min at room temperature, incubated with primary antibody (anti-MenB or anti-MenC capsule-specific antibody at 1:500 dilution and anti-α tubulin antibody at 1:10,000 dilution) for 2 h, washed in 0.1% Tween in 1× PBS, and then incubated with a secondary antibody conjugated with horseradish peroxidase, diluted 1:5,000, for 1 h. Bands were visualized using enhanced chemiluminescence (GE, Milan, Italy). All incubations and washings were carried out at room temperature with gentle shaking.
In vitro tubulin polymerization assay.
The effects of MenB capsular polysaccharide on tubulin polymerization in vitro were determined by a turbidity assay developed by Cytoskeleton, Inc. (HTS-tubulin polymerization assay kit; CytoDYNAMIX Screen 01). Lyophilized porcine tubulin (HTS03; Cytoskeleton, Inc.) was resuspended in G-PEM buffer (80 mM 1,4-piperazinediethane sulfonic acid, pH 6.9, 0.5 mM EGTA, 2 mM MgCl2, 1 mM GTP) to a final concentration of 3 mg/ml and kept at 4°C. The extracts enriched in capsular polysaccharide from strain B1940 and the control extract from strain B1940 siaD(−C) were dotted onto prewarmed 96-well plates (product no. 3696; Corning Costar, Lowell, MA). Cold tubulin was added to the wells, plate contents were mixed by shaking, and the plates were immediately transferred to a 37°C plate reader (Victor X5 multilabel plate reader; Perkin-Elmer). The absorbance at 340 nm was read every minute for 200 min, and the percentages of tubulin polymerization were calculated by following the manufacturer's instructions.
RESULTS
Adherence of serogroup B meningococci to purified microtubules.
Consistent with previous findings, CLSM images of MenB infecting HeLa cells show that, at late infection time points, the internalized meningococci were mostly found at the periphery of the cells, close to the cell membrane (Fig. 1a to d). They were detected rarely in the perinuclear region, while they were often observed in cell protrusions joining adjacent cells resembling nanotubes that contained microtubules and actin filaments. CLSM images also demonstrate extensive rearrangements of the actin cytoskeleton of infected cells with an apparent decrease in cortical actin and formation of many actin-containing intercellular bridges (Fig. 1a and b). Similar images were obtained by immunofluorescence microscopy (IFM) with MenB infecting human HEp-2 epithelial cells (see Fig. S1 in the supplemental material).
FIG 1.
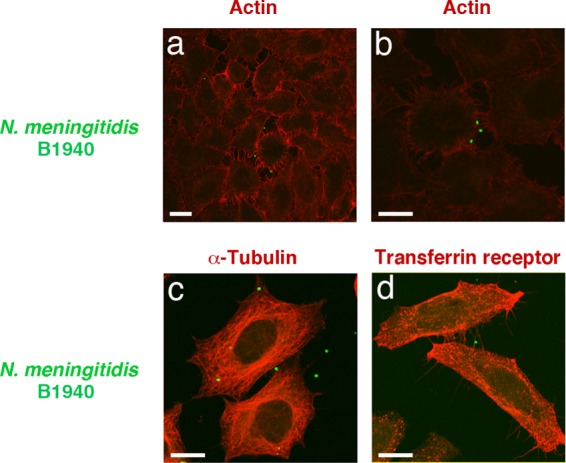
CLSM analysis of HeLa cells infected by N. meningitidis strains. HeLa cells were infected with N. meningitidis serogroup B strain B1940. After application of gentamicin treatment to selectively kill extracellular bacteria, infected cells were analyzed by CLSM. Images were taken 7 h after infection. To detect intracellular bacteria, polyclonal antibody against a whole-cell preparation of N. meningitidis was used after permeabilization with saponin, in combination with FITC-conjugated secondary antibody. To detect cellular markers (actin, α-tubulin, and transferring receptor), we used Alexa 568 phalloidin (a and b), anti-α-tubulin (c), or anti-transferrin receptor (d) antibody in combination with TRITC-conjugated secondary antibody. Merged images of the different channels are shown. Bars, 10 μm. Panels a and b are different views of the same bacteria infecting phalloidin-stained HeLa cells.
This finding prompted us to examine the possibility that a microtubule network provides a track by which N. meningitidis can move and spread. Therefore, we tested the ability of N. meningitidis to bind paclitaxel-induced microtubule asters and bundles compared to that of Escherichia coli K-12 or other nonpathogenic Neisseria species, such as Neisseria lactamica and Neisseria sicca. E. coli, N. lactamica, or N. sicca bound to microtubules only occasionally, and often asters on coverslips were completely devoid of bacteria (Fig. 2e and f and unpublished data). In contrast, MenB strains H44/76 and B1940 specifically bound to asters and bundles (Fig. 2a to d). In some cases, binding was prominent at the plus end of the filaments, but often the entire microtubules were covered with bacteria (Fig. 2c and d; also see File S1 in the supplemental material).
FIG 2.
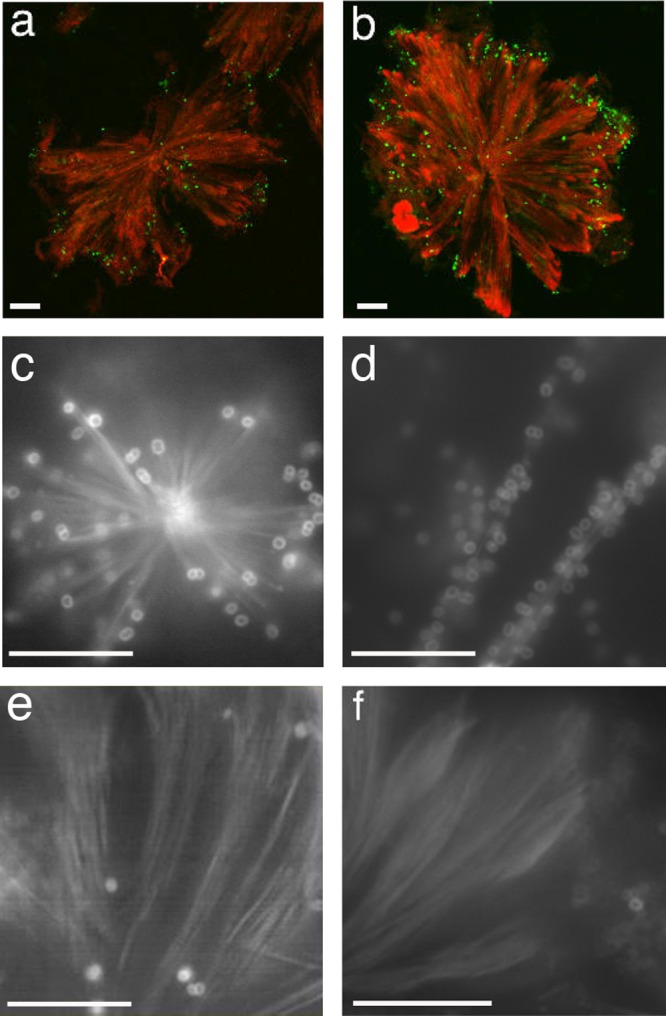
Interaction between N. meningitidis strains and purified microtubules. Paclitaxel-induced microtubule asters and bundles were purified from HeLa cells and incubated with MenB strain B1940 (a) or H44/76 (b to d) or with the N. lactamica NL995 (e) or NL4627 (f) strain. After incubation, samples were analyzed by IFM. Microtubules were detected using an anti-α-tubulin antibody followed by a secondary antibody labeled with TRITC. Bacteria were detected by using a primary polyclonal antibody against a whole-cell preparation of N. meningitidis followed by a secondary antibody labeled with FITC (a and b) or labeled with FM4-64 dye (c to f). Bars, 10 μm.
Involvement of serogroup B capsular polysaccharide in the adherence to purified microtubules.
To gain insight into the meningococcal surface structure responsible for adherence to microtubules, we used two isogenic derivative strains of B1940: the B1940 cps mutant (lacking both the capsule and the LOS outer core) and the B1940 siaD(−C) mutant (lacking the capsule but possessing the LOS outer core). DAPI-stained mutant or wild-type bacteria were incubated with purified microtubules labeled with tubulin tracker green reagent GT500. The IFM images shown in Fig. 3 confirm tight adhesion of encapsulated bacteria to microtubule asters, with most bacteria localizing at microtubule tips (Fig. 3a and b). Interestingly, adherence was dramatically reduced in bacteria lacking capsule or both capsule and LOS outer core (Fig. 3c to e), suggesting a prominent role of the capsular polysaccharide in microtubule binding.
FIG 3.
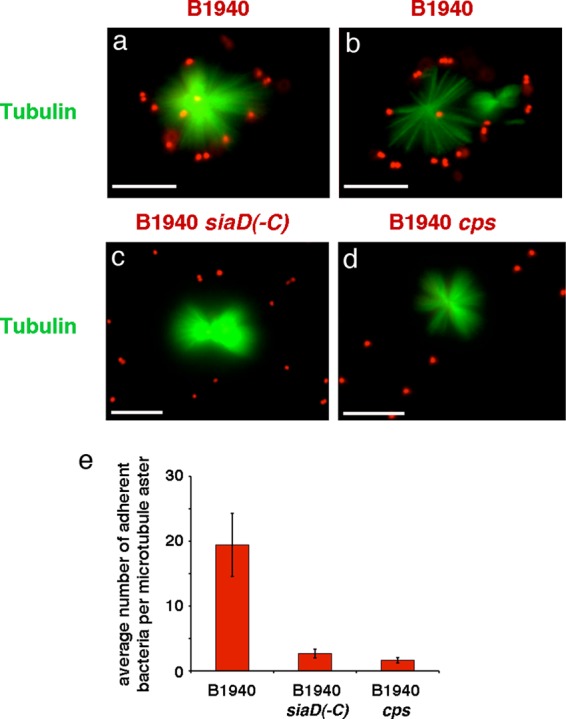
Interaction between MenB encapsulated and unencapsulated strains and purified microtubules. Paclitaxel-induced microtubule asters and bundles were purified from HeLa cells, labeled with tubulin tracker green reagent GT500, and incubated in the presence of DAPI-stained MenB strain B1940 (a and b) or the B1940 siaD(−C) (c) or B1940 cps (d) mutant as indicated. After incubation, samples were analyzed by IFM. The two channels for tubulin tracker green reagent GT500 (green) and DAPI (blue) staining were merged using Adobe Photoshop, version 6. The blue channel for DAPI staining was replaced with the red channel to improve visualization of DAPI-stained bacteria. Bars, 10 μm. Panels a and b are different views of B1940 interacting with microtubules. The average numbers ± standard deviations (SD) of adherent bacteria per microtubule aster are reported in panel e. For statistical analysis, at least 100 asters were counted for each strain. Asterisks indicate statistically significant differences (P < 0.05).
Specificity of serogroup B capsular polysaccharide in the adherence to purified microtubules.
We tested the specificity of the interaction of the capsular polysaccharide with purified microtubules by using N. meningitidis strains belonging to different serogroups. MenB capsular polysaccharide is composed of a linear homopolymer of α(2→8) N-acetyl-neuraminic acid (polysialic acid). MenC have a capsular polysaccharide that differs from that of MenB only by its anomeric linkage, α(2→9) instead of α(2→8). In spite of this small difference, MenC of strain 93/4286 were not able to bind to purified microtubules with specificity. Only occasionally were these bacteria binding to microtubule asters (Fig. 4a, b, and g). In contrast, both MenA (strain 205900) (Fig. 4c, d, and g) and MenX (strain XL929) (Fig. 4e to g), which have capsular polysaccharides composed of homopolymers of α(1→6) N-acetyl-d-mannosamine-1-phosphate and α(1→4) N-acetyl-d-glucosamine-1-phosphate, respectively, were able to interact with purified microtubules in our assay. These results demonstrated that the interaction was, at the same time, specific and extended to other serogroups.
FIG 4.

Interaction between MenC, MenA, and MenX strains and purified microtubules. Paclitaxel-induced microtubule asters and bundles were purified from HeLa cells, labeled with tubulin tracker green reagent GT500, and incubated in the presence of DAPI-stained meningococcal strain 93/4286 (MenC) (a and b), 205900 (MenA) (c and d), XL929 (MenX) (e and f), 2120 (MenC OatC+) (h and i), or 2948 (MenC OatC−) (j and k) as indicated. After incubation, samples were analyzed by IFM. The two channels for tubulin tracker green reagent GT500 (green) and DAPI (blue) staining were merged using Adobe Photoshop, version 6. The blue channel for DAPI staining was replaced with the red channel to improve visualization of DAPI-stained bacteria. Bars, 10 μm. Average numbers ± SD of adherent bacteria per microtubule aster are reported in panels g and l. For statistical analysis, at least 100 asters were counted for each strain. Asterisks indicate statistically significant differences (P < 0.05).
The specificity of MenB interaction with purified microtubules was further tested using a bacterial cell extract enriched in capsular polysaccharide from MenB strain B1940 as the inhibitor of the interaction (see Fig. S2 in the supplemental material). Results demonstrated a physical interaction between microtubules and capsular polysaccharide (which was stained by DAPI molecules carrying a divalent positive charge), preventing binding of bacteria to microtubule asters (see Fig. S2a). Very rarely, bacteria were seen adhering to microtubule tips (see Fig. S2b). In contrast, the capsular polysaccharide from MenC strain 93/4286 did not inhibit the interaction between strain B1940 and asters (see Fig. S2c and d).
The different behaviors of MenB and MenC in our assay were rather surprising, because these bacteria have similar polysialic acid capsules. However, at variance with MenB, MenC capsule can be O-acetylated at C-7 or C-8 of the sialic acid residue (30) by the capsular polysaccharide O-acetyltransferase encoded by the phase-variable gene oatC (23). Therefore, we decided to test the possible effect of O-acetylation on meningococcal interaction with microtubule asters. For this purpose, the serogroup C strain 2120 (OatC+) and its isogenic mutant 2948 (OatC−) were used in the microtubule-binding assay (Fig. 4h to l). The results clearly showed that the behavior of these two strains was similar to that of the previously tested 93/4286, indicating that the interaction with microtubules was not prevented by capsular polysaccharide O-acetylation.
Adherence of serogroup B meningococci to microtubules synthesized in vitro.
To dissect at the molecular level the nature of the interaction between encapsulated meningococci and the microtubule cytoskeleton, the adhesion process was studied by using microtubules synthesized in vitro. Paclitaxel-stabilized microtubule bundles were obtained by using a >99% pure X-rhodamine-labeled tubulin preparation from porcine brain. Microtubule bundles were then challenged with Syto-9-labeled N. meningitidis strain B1940, B1940 cps, or B1940 siaD(−C) and analyzed by IFM. The images shown in Fig. 5 demonstrate the ability of the encapsulated strain to adhere to the in vitro-synthesized microtubules. Also in this assay, adherence was greatly reduced in bacteria lacking capsule or both capsule and LOS outer core. Noticeably, encapsulated bacteria formed chains along the microtubule bundles, suggesting cooperative binding with polymerized tubulin.
FIG 5.
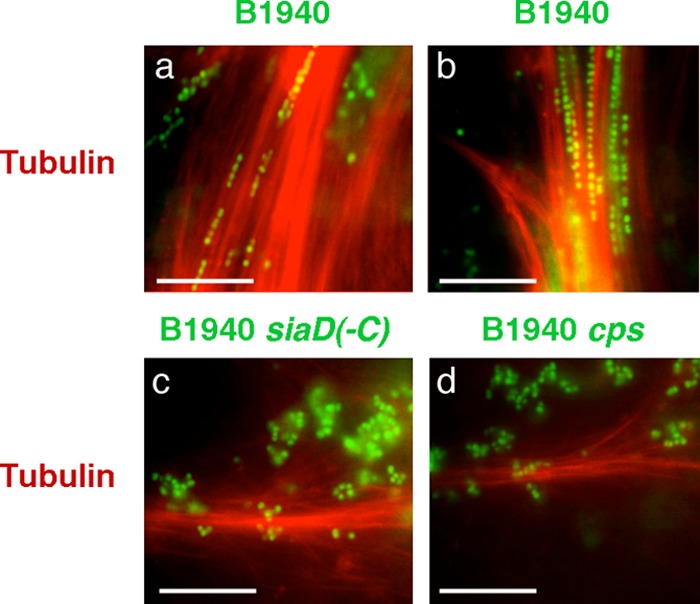
Interaction between N. meningitidis strains and microtubule bundles synthesized in vitro. Paclitaxel-stabilized microtubule bundles were obtained by using a >99% pure X-rhodamine-labeled tubulin preparation from porcine brain. Microtubule bundles were then challenged with Syto-9-labeled N. meningitidis strain B1940 (a and b), B1940 siaD(−C) (c), or B1940 cps (d) as indicated. After incubation, samples were analyzed by IFM. Bars, 10 μm.
Interaction of MenB capsular polysaccharides with microtubules in coimmunoprecipitation experiments.
To prove directly that the interaction with microtubules was mediated by the meningococcal capsule, the bacterial cell extracts enriched in capsular polysaccharide from MenB strain B1940 or MenC strain 93/4286 were challenged with HeLa cell lysate in coimmunoprecipitation assays by using mouse anti-α-tubulin antibody cross-linked to a resin. Coimmunoprecipitates and control extracts were then analyzed by immunoblotting with anti-α-tubulin and anti-MenB or anti-MenC capsule antibodies (Fig. 6). The results clearly confirmed that binding of MenB to microtubule cytoskeleton was mediated by the capsular polysaccharide and that binding was capsule specific, because the anti-α-tubulin antibody did not immunoprecipitate the MenC capsular polysaccharide.
FIG 6.
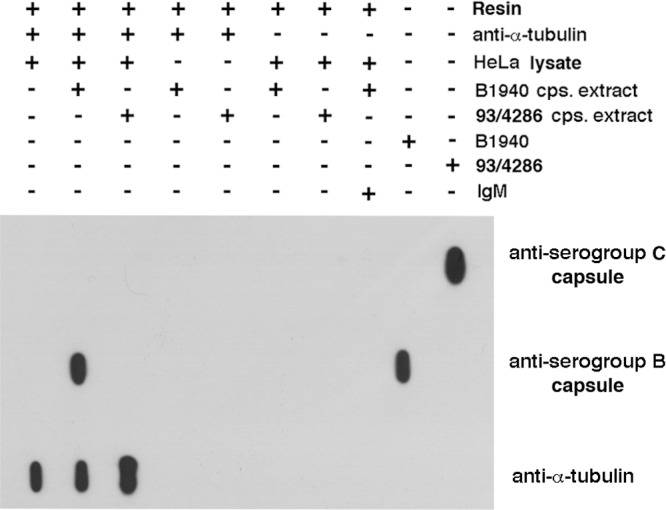
Interaction between MenB capsular polysaccharide and microtubules in coimmunoprecipitation experiments. Bacterial cell extracts enriched in capsular polysaccharide (cps) from MenB strain B1940 or MenC strain 93/4286 were challenged with precleared HeLa cell lysate in coimmunoprecipitation assays by using mouse anti-α-tubulin antibody cross-linked to a resin. Coimmunoprecipitates and control extracts were then analyzed by immunoblotting with anti-α-tubulin and anti-MenB or anti-MenC capsule antibodies.
Effects of purified capsular polysaccharides on microtubule polymerization in vitro.
Considering its ability to bind α-tubulin, we wondered whether the capsular polysaccharide could interfere with microtubule polymerization. To this purpose, the microtubule polymerization assay in vitro was carried out in the presence of the extract enriched in capsular polysaccharide from B1940 and the control extract from B1940 siaD(−C). The assay is based on an adaptation of the original method of Shelanski et al. (31) and Lee and Timasheff (32), which demonstrated that light is scattered by microtubules to an extent that is proportional to the concentration of microtubule polymer. Polymerization was started at 37°C and monitored spectrophotometrically in real time by monitoring the change in optical density (OD) at 340 nm (Fig. 7). Under the assay conditions, polymerization reached a maximal OD variation (about 0.06, approximately corresponding to 0.6 mg/ml of polymer mass and 20% of the tubulin polymerized) within 200 min in both the untreated sample and in the sample containing the control extract. In contrast, the extract from B1940 slowed down microtubule polymerization in a dose-response manner (Fig. 8).
FIG 7.
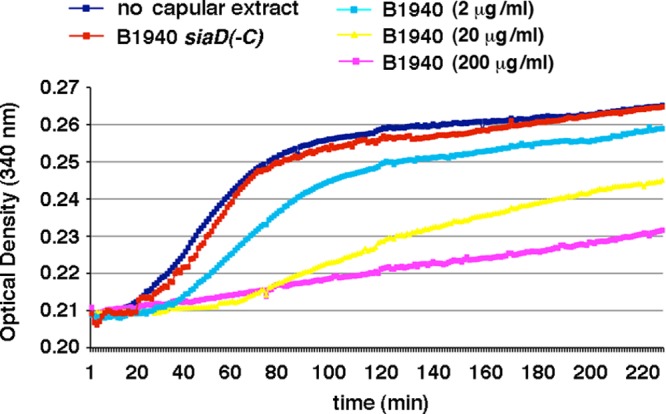
Effects of purified capsular polysaccharides on microtubule polymerization in vitro. The microtubule polymerization assay in vitro was carried out in the presence of different amounts (0.01 to 1 μl; corresponding to a final sialic acid content of 2 to 200 μg/ml) of a bacterial cell extract enriched in capsular polysaccharide from strain B1940 or a control extract (1 μl) from strain B1940 siaD(−C). Polymerization was started at 37°C and was monitored spectrophotometrically in real time by monitoring the change in optical density (OD) at 340 nm.
FIG 8.

Adhesion, invasion, intracellular replication, and exit from infected HeLa cells of MenB and MenC strains. HeLa cells were infected at a multiplicity of infection (MOI) of 50 for 1 h. (a) Adherence of viable bacteria was evaluated by the CFU method after removing nonadherent bacteria by sequential PBS washes. Values are relative to B1940 adherence, and the data are expressed as the means ± SD from at least three independent experiments with triplicate samples. (b) Relative invasion rates (determined as the normalized CFU ratio between intracellular and adherent bacteria) of strains B1940 (MenB) and 93/4286 (MenC) are reported. The number of viable intracellular bacteria was determined after 1 h of infection, followed by 30 min of gentamicin treatment to kill extracellular/adherent bacteria and lysis with 0.1% saponin to release intracellular bacteria. Lysates were serially plated on GC agar to determine the CFU. Values are means ± SD from at least three independent experiments with triplicate samples and are relative to B1940 with a ratio between intracellular and adherent B1940 bacteria equal to about 1.59%. (c) To monitor bacterial intracellular replication, HeLa cells were infected for 1 h, treated with gentamicin for 30 min, washed to remove gentamicin, reincubated for different time intervals (0 to 7 h) in DMEM, and lysed with 0.1% saponin. Lysates were serially plated on GC agar to determine the CFU. (d) To count the number of extracellular bacteria, HeLa cells were infected as described for panel c. Medium was collected at different time intervals after infection and serially plated onto GC agar. In panels c and d, values are relative to B1940 at time zero (0 h). The data are expressed as the means ± SD from at least three independent experiments with triplicate samples. (e) Ratios between extracellular and intracellular bacteria. (f) The numbers of infected cells at two time intervals (0 and 7 h) after 1 h of infection and 30 min of gentamicin treatment. Data were obtained by IFM by analyzing at least 100 microscope fields for each strain in a single experiment. Values are expressed as the means ± SD. In panels a to f, asterisks indicate statistically significant differences (P < 0.05). (g) HeLa cells were infected with strain 93/4286. After application of gentamicin treatment to selectively kill extracellular bacteria, infected cells were analyzed by CLSM. Images were taken 7 h after infection. To detect intracellular bacteria, antibody against MenC capsule was used after permeabilization with saponin, in combination with FITC-conjugated secondary antibody. Actin was stained by Alexa 568 phalloidin. Merged images of the different channels are shown. Bars, 10 μm.
Dynamics of infection of HeLa cells with MenB and MenC strains.
The lack of interaction of MenC with microtubules led us to suspect some differences between MenB and MenC in cell infection dynamics. To clarify this matter, HeLa cells were infected with strain B1940 (MenB) or 93/4286 (MenC), and a number of parameters were evaluated (Fig. 8). The two strains exhibited similar adherence to HeLa cells (Fig. 8a), while the relative invasion rate (determined as the normalized CFU ratio between intracellular and adherent bacteria) of 93/4286 was slightly lower (about 60%) than that of B1940 (Fig. 8b). In these assays, the number of adherent bacteria was evaluated by CFU counts after 1 h of infection, while that of intracellular bacteria was determined after 1 h of infection followed by 30 min of gentamicin treatment to kill extracellular/adherent bacteria. Under these experimental conditions, the ratio between intracellular and adherent B1940 bacteria was equal to about 1.59%.
To monitor intracellular replication, exit from infected cells, and extracellular replication, HeLa cells were infected for 1 h with the bacteria, treated with gentamicin for 30 min, and then reincubated for various times (0 to 7 h) (Fig. 8c to e). Bacteria were then recovered from saponin-lysed HeLa cells or from the culture medium. In these assays, the two strains behaved similarly. Indeed, the number of recoverable CFU from cells decreased during the first 3 h after infection and then started to increase from 5 up to 7 h (Fig. 8c). A parallel increase in the number of recoverable bacteria from the culture medium was observed between 5 and 7 h after infection (Fig. 8d). Even though the dynamics of the infection appeared similar, some quantitative differences were observed between the two strains. In particular, at later time points (7 h), the number of recoverable intracellular bacteria was about 1.9-fold higher for 93/4286 than for B1940 (Fig. 8c), and a parallel increase of percentages of infected cells was observed by IFM (Fig. 8f). The number of recoverable extracellular bacteria was also significantly higher for 93/4286 (about 3.3-fold) than for B1940 (Fig. 8d). By assuming similar growth rates in the extracellular environment (B1940 and 93/4286 exhibited similar growth rates in DMEM cell culture medium [data not shown]) and negligible reinfection (due to low invasion rate), the ratio between extracellular and intracellular bacteria was taken as a measure of bacteria egression from infected cells (Fig. 8e). The results suggested egression rates that are significantly higher for 93/4286 than for B1940.
All observed quantitative differences could be unrelated (or only partially related) to the nature of the capsular polysaccharide, as the two strains analyzed were not isogenic, and other bacterial determinants might be involved. More interestingly, the extensive rearrangements of the actin cytoskeleton, which were often observed with MenB strains with a decrease in cortical actin and formation of intercellular bridges (Fig. 1a and b), were never detected in HeLa cells infected with MenC 93/4286 (Fig. 8g).
DISCUSSION
A crucial factor in meningococcal pathogenesis is the ability of N. meningitidis to traverse cellular barriers in humans. This pathogen, after crossing the nasopharyngeal mucosa, occasionally spreads into the bloodstream before moving across the blood-brain barrier, causing fatal sepsis and meningitis in otherwise healthy individuals. In spite of numerous efforts, the mechanism(s) by which N. meningitidis crosses these cellular barriers remains one of the most poorly elucidated aspects of meningococcal pathogenesis. There are conflicting data regarding the route of traversal (paracellular or transcellular) and the intracellular location (cytosolic or vacuolar) of meningococci in experiments with organ cultures or cell lines (14, 33–39). Although we are aware of the inherent limitations of in vitro systems in their ability to model processes that occur in vivo, we have previously shown that during the late stages of the infectious process MenB have a tendency to localize primarily to the cytosol in in vitro-infected human epithelial cells (6, 13, 14). They were mostly found at the periphery of the cells, close to the cell membrane, and were often observed in cell protrusions joining adjacent cells resembling nanotubes, raising the possibility that they could move and spread to surrounding cells by using the microtubule and actin cytoskeleton (Fig. 1; also see Fig. S1 in the supplemental material).
By using a number of complementary approaches, in this study we provide evidence that MenB have the ability to interact with the microtubule cytoskeleton of human epithelial cells. Encapsulated bacteria bind to paclitaxel-induced microtubule asters and bundles purified from HeLa cells (Fig. 2 and 3) and to microtubule bundles synthesized in vitro (Fig. 5). Binding was dramatically reduced in isogenic unencapsulated bacteria (Fig. 3), leading us to hypothesize a direct role of capsule in binding. This hypothesis was confirmed by competition and coimmunoprecipitation experiments with partially purified MenB or MenC capsules (Fig. 6; also see Fig. S2 in the supplemental material). Interestingly, MenC were not able to bind the microtubules in spite of a small difference in the capsular polysaccharide with respect to MenB supporting specificity in binding, and the lack of interaction was not due to capsular polysaccharide O-acetylation that takes place in most of the MenC strains but not in MenB strains (Fig. 4). Together these findings raise questions concerning the biological significance of the observed interaction and its effect on the infectious cycle.
During the infectious process, many pathogenic bacteria modulate the dynamics of microtubules in the host cells (40). The role of microtubule dynamics in bacterial infection varies between different species, and different processes may be affected by bacterial manipulation of the microtubule network, ranging from invasion to intracellular movement to cell-to-cell spreading (40–42). In some ways the behavior of serogroup B N. meningitidis within infected cells is similar to that of the oral periodontopathogen Actinobacillus actinomycetemcomitans (43, 44). Similar to MenB, after entering epithelial cells, this bacterium escapes from the host cell vacuole and spreads intracellularly and to adjacent cells via intercellular protrusion. Drugs that inhibit microtubule polymerization and those which stabilize polymerized microtubules prevent both spreading and egression from host cells, indicating a role in microtubule dynamics. Interestingly, strains that are both invasive and egressive interact specifically with the plus end of the filaments of microtubule asters in a KB cell extract, suggesting that the surfaces of these microorganisms have a kinesin- or dynein-like protein that mediates the bacterium-microtubule interaction (44).
Here, we show that the capsular polysaccharide directly mediates the interaction of MenB with the microtubule network and inhibits microtubule polymerization in vitro (Fig. 7). The video microscope sequences (see Movie S1 in the supplemental material) give a visual demonstration of how the MenB-microtubule interaction was strong, with the bacteria firmly attached to the microtubules withstanding the forces produced by fluid flow. Under these conditions, the tight attachment of bacteria to microtubules may represent an obstacle to their intracellular spreading, and capsule shedding (45) might be required for detachment of bacteria from microtubules and their movement along the microtubule axis. This hypothesis seems to be supported by reduced intracellular replication of B1940 (MenB) with respect to 93/4286 (MenC) in the infection assay with HeLa cells (Fig. 8), although these data should be confirmed in an isogenic background. Indeed, MenC, which seem to be unable to interact with microtubules, should move and replicate faster than MenB in the host cell. In this context, it is noteworthy that an epidemiological study suggested that the acquisition of the serogroup C polysaccharide capsule results in increased virulence and a higher case fatality rate (CFR) in strains belonging to the hypervirulent ST-32 complex (46).
Whether there is any biological significance of the observed inhibition of microtubule polymerization in vitro by MenB capsule is another question that should be addressed.
Destabilization of host microtubules is a strategy adopted by many bacterial pathogens, such as Shigella flexneri, enteropathogenic E. coli, Citrobacter rodentium, Chlamydia, Edwardsiella tarda, and Listeria monocytogenes (40). Microtubule destabilization benefits these pathogens in many ways, including the free movement of pathogens through the cytoplasm and modulation of actin cytoskeleton through the activation of small GTPases, leading to formation of membrane ruffles and pseudopodia that promote bacterial invasion and intercellular spreading (40). By inhibiting tubulin polymerization, the capsular polysaccharide might induce microtubule destabilization locally. Several lines of evidence indicate that microtubule dynamics and microfilament reorganization are linked. Actin is regulated by small GTPases of the Rho family, in particular Rac1, Rho, and Cdc42, and there is evidence that microtubule dynamics locally modulate the activity of Rho GTPases and influence the actin cytoskeleton (47, 48). In this regard, it may be noteworthy that CLSM images show extensive rearrangements of the actin cytoskeleton of HeLa cells infected by MenB with destruction of cortical actin cytoskeleton and formation of many actin-containing intercellular bridges (Fig. 1a to c). Intriguingly, these rearrangements of the actin cytoskeleton were never observed in HeLa cells infected with MenC, although several aspects of cell infection dynamics, including the ability to survive/replicate inside and egress from infected cells were similar in MenB and MenC strains (Fig. 8).
Thus, at variance with MenC, by exploiting microtubule dynamics, MenB might modulate the actin cytoskeleton. It is interesting that tunneling nanotubes are now recognized as structures for intercellular communication and organelle transfer (49–51). There are at least two types of nanotubes: thin nanotubes contain actin, while nanotubes with diameters of at least 0.7 μm contain both actin and microtubules (49–51). These nanotubes are also exploited by pathogens to spread among cells. Indeed, bacterial, viral, and prion infective agents spread intracellularly and extracellularly using these structures (50). Therefore, it is tempting to speculate that MenB induces the formation of nanotubes by acting on microtubule and actin cytoskeleton. Experiments with isogenic MenB and MenC strains and actin cytoskeleton inhibitors will be useful to test this hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Claus for generously providing us with meningococcal strains.
This work was partially supported by grants from the Italian MIUR to P.A. (PRIN 2008) and C.B. (PRIN 2010-2011).
Footnotes
Published ahead of print 28 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00501-13.
REFERENCES
- 1.Taha MK, Deghmane AE, Antignac A, Zarantonelli ML, Larribe M, Alonso JM. 2002. The duality of virulence and transmissibility in Neisseria meningitidis. Trends Microbiol. 10:376–382. 10.1016/S0966-842X(02)02402-2 [DOI] [PubMed] [Google Scholar]
- 2.Harrison LH, Trotter CL, Ramsay ME. 2009. Global epidemiology of meningococcal disease. Vaccine 27(Suppl 2):B51–B63. 10.1016/j.vaccine.2009.04.063 [DOI] [PubMed] [Google Scholar]
- 3.Cohn AC, MacNeil JR, Harrison LH, Hatcher C, Theodore J, Schmidt M, Pondo TT, Arnold KE, Baumbach J, Bennett N, Craig AS, Farley M, Gershman K, Petit S, Lynfield R, Reingold A, Schaffner W, Shutt KA, Zell ER, Mayer LW, Clark T, Stephens D, Messonnier NE. 2010. Changes in Neisseria meningitidis disease epidemiology in the United States, 1998–2007: implications for prevention of meningococcal disease. Clin. Infect. Dis. 50:184–191. 10.1086/649209 [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization 2011. Meningococcal vaccines: WHO position paper, November 2011. Wkly. Epidemiol. Rec. 86:521–539 [PubMed] [Google Scholar]
- 5.Jarvis GA, Vedros NA. 1987. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect. Immun. 55:174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spinosa MR, Progida C, Tala A, Cogli L, Alifano P, Bucci C. 2007. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect. Immun. 75:3594–3603. 10.1128/IAI.01945-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones A, Geörg M, Maudsdotter L, Jonsson AB. 2009. Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J. Bacteriol. 191:3861–3868. 10.1128/JB.01313-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zughaier SM, Svoboda P, Pohl J, Stephens DS, Shafer WM. 2010. The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 5:e13627. 10.1371/journal.pone.0013627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammerschmidt S, Muller A, Sillmann H, Muhlenhoff M, Borrow R, Fox A, van Putten J, Zollinger WD, Gerardy-Schahn R, Frosch M. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 20:1211–1220. 10.1111/j.1365-2958.1996.tb02641.x [DOI] [PubMed] [Google Scholar]
- 10.Hammerschmidt S, Hilse R, van Putten JP, Gerardy-Schahn R, Unkmeir A, Frosch M. 1996. Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J. 15:192–198 [PMC free article] [PubMed] [Google Scholar]
- 11.Bucci C, Lavitola A, Salvatore P, Del Giudice L, Massardo DR, Bruni CB, Alifano P. 1999. Hypermutation in pathogenic bacteria: frequent phase variation in meningococci is a phenotypic trait of a specialized mutator biotype. Mol. Cell 3:435–445. 10.1016/S1097-2765(00)80471-2 [DOI] [PubMed] [Google Scholar]
- 12.Deghmane AE, Giorgini D, Larribe M, Alonso JM, Taha MK. 2002. Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol. Microbiol. 43:1555–1564. 10.1046/j.1365-2958.2002.02838.x [DOI] [PubMed] [Google Scholar]
- 13.Talà A, De Stefano M, Bucci C, Alifano P. 2008. Reverse transcriptase-PCR differential display analysis of meningococcal transcripts during infection of human cells: up-regulation of priA and its role in intracellular replication. BMC Microbiol. 8:131. 10.1186/1471-2180-8-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talà A, Progida C, De Stefano M, Cogli L, Spinosa MR, Bucci C, Alifano P. 2008. The HrpB-HrpA two-partner secretion system is essential for intracellular survival of Neisseria meningitidis. Cell. Microbiol. 10:2461–2482. 10.1111/j.1462-5822.2008.01222.x [DOI] [PubMed] [Google Scholar]
- 15.Lavitola A, Bucci C, Salvatore P, Maresca G, Bruni CB, Alifano P. 1999. Intracistronic transcription termination in polysialyltransferase gene (siaD) affects phase variation in Neisseria meningitidis. Mol. Microbiol. 33:119–127. 10.1046/j.1365-;2958.1999.01454.x [DOI] [PubMed] [Google Scholar]
- 16.Pagliarulo C, Salvatore P, De Vitis LR, Colicchio R, Monaco C, Tredici M, Tala A, Bardaro M, Lavitola A, Bruni CB, Alifano P. 2004. Regulation and differential expression of gdhA encoding NADP-specific glutamate dehydrogenase in Neisseria meningitidis clinical isolates. Mol. Microbiol. 51:1757–1772. 10.1111/j.1365-2958.2003.03947.x [DOI] [PubMed] [Google Scholar]
- 17.Salvatore P, Pagliarulo C, Colicchio R, Zecca P, Cantalupo G, Tredici M, Lavitola A, Bucci C, Bruni CB, Alifano P. 2001. Identification, characterization, and variable expression of a naturally occurring inhibitor protein of IS1106 transposase in clinical isolates of Neisseria meningitidis. Infect. Immun. 69:7425–7436. 10.1128/IAI.69.12.7425-7436.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvatore P, Bucci C, Pagliarulo C, Tredici M, Colicchio R, Cantalupo G, Bardaro M, Del Giudice L, Massardo DR, Lavitola A, Bruni CB, Alifano P. 2002. Phenotypes of a naturally defective recB allele in Neisseria meningitidis clinical isolates. Infect. Immun. 70:4185–4195. 10.1128/IAI.70.8.4185-4195.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frosch M, Schultz E, Glenn-Calvo E, Meyer TF. 1990. Generation of capsule-deficient Neisseria meningitidis strains by homologous recombination. Mol. Microbiol. 4:1215–1218. 10.1111/j.1365-2958.1990.tb00697.x [DOI] [PubMed] [Google Scholar]
- 20.Holten E. 1979. Serotypes of Neisseria meningitidis isolated from patients in Norway during the first six months of 1978. J. Clin. Microbiol. 9:186–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Høiby EA, Rosenqvist E, Frøholm LO, Bjune G, Feiring B, Nøkleby H, Rønnild E. 1991. Bactericidal antibodies after vaccination with the Norwegian meningococcal serogroup B outer membrane vesicle vaccine: a brief survey. NIPH Ann. 14:147–156 [PubMed] [Google Scholar]
- 22.Fredriksen JH, Rosenqvist E, Wedege E, Bryn K, Bjune G, Frøholm LO, Lindbak AK, Møgster B, Namork E, Rye U, Stabbetorp G, Winsnes R, Aase B, Closs O. 1991. Production, characterization and control of MenB-Vaccine “Folkehelsa”: an outer membrane vesicle vaccine against group B meningococcal disease. NIPH Ann. 14:67–80 [PubMed] [Google Scholar]
- 23.Claus H, Borrow R, Achtman M, Morelli G, Kantelberg C, Longworth E, Frosch M, Vogel U. 2004. Genetics of capsule O-acetylation in serogroup C, W-135 and Y meningococci. Mol. Microbiol. 51:227–239 [DOI] [PubMed] [Google Scholar]
- 24.Cantalupo G, Bucci C, Salvatore P, Pagliarulo C, Roberti V, Lavitola A, Bruni CB, Alifano P. 2001. Evolution and function of the neisserial dam-replacing gene. FEBS Lett. 495:178–183. 10.1016/S0014-5793(01)02388-2 [DOI] [PubMed] [Google Scholar]
- 25.Kasai T. 1974. Regulation of the expression of the histidine operon in Salmonella typhimurium. Nature 249:523–527. 10.1038/249523a0 [DOI] [PubMed] [Google Scholar]
- 26.Makino S, van Putten JPM, Meyer TF. 1991. Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J. 10:1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rose JE, Meyer H, Fives-Taylor PM. 1998. Detection of bacteria-microtubule interactions in a cell-free extract. Methods Cell Sci. 19:325–330. 10.1023/A:1009733318063 [DOI] [Google Scholar]
- 28.Gotschlich EC. 1975. Development of polysaccharide vaccines for the prevention of meningococcal disease. Monogr. Allergy 9:245–251 [PubMed] [Google Scholar]
- 29.Miettinen TA, Takki-Luukkainen I-T. 1959. Use of butylacetate in determination of sialic acid. Acta Chem. Scand. 13:856–858. 10.3891/acta.chem.scand.13-0856 [DOI] [Google Scholar]
- 30.Jennings HJ, Bhattacharjee AK, Bundle DR, Kenny CP, Martin A, Smith IC. 1977. Structures of the capsular polysaccharides of Neisseria meningitidis as determined by 13C-nuclear magnetic resonance spectroscopy. J. Infect. Dis. 136:S78–S83. 10.1093/infdis/136.Supplement.S78 [DOI] [PubMed] [Google Scholar]
- 31.Shelanski ML, Gaskin F, Cantor CR. 1973. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. U. S. A. 70:765–768. 10.1073/pnas.70.3.765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JC, Timasheff SN. 1977. In vitro reconstitution of calf brain microtubules: effects of solution variable. Biochemistry 16:1754–1762. 10.1021/bi00627a037 [DOI] [PubMed] [Google Scholar]
- 33.Stephens DS. 1989. Gonococcal and meningococcal pathogenesis as defined by human cell, cell culture, and organ culture assays. Clin. Microbiol. Rev. 2(Suppl):S104–S111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pujol C, Eugene E, de Saint Martin L, Nassif X. 1997. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect. Immun. 65:4836–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Birkness KA, Swisher BL, White EH, Long EG, Ewing EP, Jr, Quinn FD. 1995. A tissue culture bilayer model to study the passage of Neisseria meningitidis. Infect. Immun. 63:402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merz AJ, So M. 2000. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16:423–457. 10.1146/annurev.cellbio.16.1.423 [DOI] [PubMed] [Google Scholar]
- 37.De Vries FP, Van det Ende A, Van Putten JPM, Dankert J. 1996. Invasion of primary nasopharyngeal epithelial cells by Neisseria meningitidis is controlled by phase variation of multiple surface antigens. Infect. Immun. 64:2998–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutherland TC, Quattroni P, Exley RM, Tang CM. 2010. Transcellular passage of Neisseria meningitidis across a polarized respiratory epithelium. Infect. Immun. 78:3832–3847. 10.1128/IAI.01377-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens DS, Hoffman LH, McGee ZA. 1983. Interaction of Neisseria meningitidis with human nasopharyngeal mucosa: attachment and entry into columnar epithelial cells. J. Infect. Dis. 148:369–376. 10.1093/infdis/148.3.369 [DOI] [PubMed] [Google Scholar]
- 40.Radhakrishnan GK, Splitter GA. 2012. Modulation of host microtubule dynamics by pathogenic bacteria. Concepts 3:571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henry T, Gorvel JP, Méresse S. 2006. Molecular motors hijacking by intracellular pathogens. Cell Microbiol. 8:23–32. 10.1111/j.1462-5822.2005.00649.x [DOI] [PubMed] [Google Scholar]
- 42.Yoshida S, Sasakawa C. 2003. Exploiting host microtubule dynamics: a new aspect of bacterial invasion. Trends Microbiol. 11:139–143. 10.1016/S0966-842X(03)00023-4 [DOI] [PubMed] [Google Scholar]
- 43.Meyer DH, Mintz KP, Fives-Taylor PM. 1997. Models of invasion of enteric and periodontal pathogens into epithelial cells: a comparative analysis. Crit. Rev. Oral Biol. Med. 8:389–409. 10.1177/10454411970080040301 [DOI] [PubMed] [Google Scholar]
- 44.Meyer DH, Rose JE, Lippmann JE, Fives-Taylor PM. 1999. Microtubules are associated with intracellular movement and spread of the periodontopathogen Actinobacillus actinomycetemcomitans. Infect. Immun. 67:6518–6525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uria MJ, Zhang Q, Li Y, Chan A, Exley R, Gollan B, Chan H, Feavers I, Yarwood A, Abad R, Borrow R, Fleck RA, Mulloy B, Vazquez JA, Tang CM. 2008. A generic mechanism in Neisseria meningitidis for enhanced resistance against bactericidal antibodies. J. Exp. Med. 205:1423–1434. 10.1084/jem.20072577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith I, Caugant DA, Høiby EA, Wentzel-Larsen T, Halstensen A. 2006. High case-fatality rates of meningococcal disease in western Norway caused by serogroup C strains belonging to both sequence type (ST)-32 and ST-11 complexes, 1985–2002. Epidemiol. Infect. 134:1195–1202. 10.1017/S0950268806006248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wittmann T, Waterman-Storer CM. 2001. Cell motility: can Rho GTPases and microtubules point the way? J. Cell Sci. 114:3795–3803 [DOI] [PubMed] [Google Scholar]
- 48.Etienne-Manneville S. 2004. Actin and microtubules in cell motility: which one is in control? Traffic 5:470–477. 10.1111/j.1600-0854.2004.00196.x [DOI] [PubMed] [Google Scholar]
- 49.Gerdes HH, Carvalho RN. 2008. Intracellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 20:470–475. 10.1016/j.ceb.2008.03.005 [DOI] [PubMed] [Google Scholar]
- 50.Kimura S, Hase K, Ohno H. 2013. The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue Res. 352:67–76. 10.1007/s00441-012-1518-1 [DOI] [PubMed] [Google Scholar]
- 51.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. 2004. Nanotubular highways for intercellular organelle transport. Science 303:1007–1010. 10.1126/science.1093133 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.