

Abstract
Candida lusitaniae is an emerging opportunistic yeast and an attractive model to discover new virulence factors in Candida species by reverse genetics. Our goal was to create a dpp3Δ knockout mutant and to characterize the effects of this gene inactivation on yeast in vitro and in vivo interaction with the host. The secretion of two signaling molecules in Candida species, phenethyl alcohol (PEA) and tyrosol, but not of farnesol was surprisingly altered in the dpp3Δ knockout mutant. NO and reactive oxygen species (ROS) production as well as tumor necrosis factor alpha (TNF-α) and interleukin 10 (IL-10) secretion were also modified in macrophages infected with this mutant. Interestingly, we found that the wild-type (WT) strain induced an increase in IL-10 secretion by zymosan-activated macrophages without the need for physical contact, whereas the dpp3Δ knockout mutant lost this ability. We further showed a striking role of PEA and tyrosol in this modulation. Last, the DPP3 gene was found to be an essential contributor to virulence in mice models, leading to an increase in TNF-α secretion and brain colonization. Although reinsertion of a WT DPP3 copy in the dpp3Δ knockout mutant was not sufficient to restore the WT phenotypes in vitro, it allowed a restoration of those observed in vivo. These data support the hypothesis that some of the phenotypes observed following DPP3 gene inactivation may be directly dependent on DPP3, while others may be the indirect consequence of another genetic modification that systematically arises when the DPP3 gene is inactivated.
INTRODUCTION
Candida species are the fourth leading cause of nosocomial infections in the United States, with a mortality rate reaching 50% (1–3). Even though Candida albicans is the most common and the most studied species (4), non-albicans species represent 50% of the reported episodes of candidemia (3). Candida lusitaniae is an emerging environmental opportunistic pathogen (5–7) frequently associated with antifungal resistance (5, 6, 8). Candida lusitaniae is a dimorphic microorganism due to its ability to make pseudohyphae. It has a haploid genome, and easily employed molecular tools for rapid gene deletion have already been developed (9). These features make this yeast a convenient and attractive model to discover new virulence factors and potential antifungal targets.
The anti-Candida immune response seems to be mainly mediated by innate immunity (10), with macrophages being among the first immune cells to intervene. As professional phagocytes and antigen-presenting cells, they kill pathogens and stimulate a protective immune response using a combination of factors, including the production of nitric oxide (NO) and reactive oxygen species (ROS), and the secretion of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) (11–14). They also have a role in downregulating and controlling the proinflammatory response by secreting cytokines such as transforming growth factor beta (TGF-β) and interleukin 10 (IL-10) (15).
Recently, it has been reported that several signaling molecules produced by C. albicans modulate the host-pathogen interaction (16–19). Among them, farnesol, the first quorum-sensing molecule discovered in eukaryotes (20), is now considered a contributor to virulence (21) with multiple signaling roles (20–25). Farnesol is derived from the ergosterol biosynthesis pathway. In C. albicans, farnesyl pyrophosphate is converted to farnesol by a pyrophosphate phosphatase encoded by the DPP3 gene (16, 21). A dpp3Δ knockout mutant produces less farnesol and is less pathogenic to mice than the wild-type (WT) strain (21). The role of this gene in other Candida species has not been investigated. Other signaling molecules such as phenethyl alcohol (PEA) and tyrosol have also been identified (26, 27), but they have not been as extensively studied as farnesol and their role in the host-pathogen interaction is still unclear.
The purpose of this work was to create a dpp3Δ knockout mutant in C. lusitaniae and to determine how the DPP3 inactivation affects yeast interaction with macrophages in vitro and virulence in mice.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were carried out in strict accordance with the French legislation (Rural Code articles L 214-1 to L 214-122 and associated penal consequences) and the European Economic Community (86-6091 EEC) guidelines for the care of laboratory animals and were approved by the Ethical Committee (CEEA50) of the Centre National de la Recherche Scientifique, Région Aquitaine, and by the University of Bordeaux 2 animal care and use committee. All efforts were made to minimize animal suffering.
Strains, media, and growth conditions.
Candida lusitaniae CBS 6936 MATa (ATCC 38533) was used as the wild-type (WT) strain in all of the experiments. The uracil auxotrophic ura3Δ MATa and the uracil leucine auxotrophic ura3Δ leu2Δ MATa strains used for genetic transformations (9) were derived from this wild-type strain. Strain 5094 MATα (Centraalbureau voor Schimmelcultures, Baarn, The Netherlands) was crossed with the ura3Δ leu2Δ MATa strain, and a ura3Δ leu2Δ MATα descendant was selected for genetic transformations. All strains used or constructed during this work are listed in Table S1 in the supplemental material. Yeasts were grown in YPD medium (1% yeast extract, 2% bactopeptone, 2% glucose). In some experiments, yeast cells were labeled with 10 μg/ml of calcofluor white (CFW) (Sigma). The minimum medium YNB (yeast nitrogen base) without amino acid (Difco) was used to select prototrophic strains.
Escherichia coli JM109 (Stratagene) was used for cloning assays. Bacteria were cultured at 37°C in LB medium (tryptone, 10 g/liter; yeast extract, 5 g/liter; NaCl, 5 g/liter) eventually supplemented with ampicillin antibiotic at 50 μg/ml. Plasmid pGURA3, derived from a pGEMT (Promega) harboring the C. lusitaniae URA3 gene, was used for genetic constructions (9).
Murine macrophage cell line J774A.1 (ATCC TIB-67) was cultured as previously described (28).
Growth rate tests.
Growth rates were tested for all strains in every medium used for experimentation either at 35°C in liquid media or at 30°C on solid media. In liquid media, the optical density at 600 nm (OD600) was measured at between 1 and 24 h for each strain. On solid media, drop tests were performed on agar plates using different yeast concentrations (from 10 yeasts/droplet to 108 yeasts/droplet).
Macrophage infection with yeasts.
Macrophages were infected as previously described (28) in RPMI medium. A multiplicity of infection (MOI) of 1 macrophage to 5 yeasts (1M:5Y) was used in all macrophage infection assays. An MOI of 10M:1Y was used only when determining the internalized yeast survival rate since it allowed a complete phagocytosis of all yeasts present in the infection medium after 24 h. For the complementation assays, phenethyl alcohol (Sigma) (PEA) and tyrosol (Aldrich Chemistry) were used at concentrations ranging from 0.1 μM to 10 mM. PEA and tyrosol were added either directly to the macrophages immediately after yeast infection or to yeast cultures in the stationary phase (from overnight cultures in YPD) for a 2-h treatment. Following this treatment, yeasts were washed and resuspended in RPMI medium prior to macrophage infection.
Fluorimetry and flow cytometry assays.
Fluorimetry and flow cytometry assays were conducted as previously described (28). Fluorimetry was used to determine the ratio of fungal phagocytosis, and flow cytometry helped to determine the ratio of macrophages engaged in phagocytosis, as well as the macrophage and yeast mortality rates. To evaluate yeast mortality inside macrophages, the macrophages were infected with CFW-labeled yeasts at an MOI of 10M:1Y in a 96-well plate. After 24 h, the wells were washed, the macrophages were detached and then lysed with 0.1% Triton X-100 (Acros Organics), and the collected yeasts were stained with 1 μg/ml propidium iodide (PI) (Sigma) for flow cytometry analysis. PI enters only dead cells. Heat-killed yeasts were used as a positive control and live yeasts were used as a negative control for PI staining. For analysis, the CFW population was selected and the ratio of the PI population was then measured in order to determine yeast mortality.
Nitric oxide (NO) and reactive oxygen species (ROS) measurement.
Macrophages were infected with yeasts at an MOI of 1M:5Y. After 24 h, wells were washed in phosphate-buffered saline (PBS). Then, a 5 μM concentration of either 5,6-diaminofluorescein-diacetate (DAF-2DA; Sigma) or 2,7-dichlorofluorescein-diacetate (DCFH-DA; Sigma) was added for 40 min at 37°C in the dark, in order to measure the generation of intracellular nitrite or reactive oxygen intermediates, respectively. Cells were washed twice and kept in PBS for 15 min. The fluorescence intensity was measured with an Optima plate reader (BMG Labtech, Germany) (excitation/emission, 485/520 nm).
Cytokine assays.
Tumor necrosis factor alpha (TNF-α) and interleukin 10 (IL-10) were measured in the supernatants of macrophages infected with yeasts for 6 h at an MOI of 1M:5Y or from mouse sera. Assays were performed using enzyme-linked immunosorbent assay (ELISA) Ready-Set-Go kits (eBioscience) according to the manufacturer's instructions. Cytokines were also measured when yeasts were physically separated from macrophages using transwells. In 1 ml of RPMI medium containing fetal bovine serum (FBS), 106 macrophages were left to adhere overnight in 24-well plates. The next day, the medium was replaced with RPMI medium without FBS. Zymosan A (Sigma) was prepared following the manufacturer's instructions and was added to stimulate the macrophages at 1.2 mg/ml (1M:5 zymosan particles). Transwells were placed, and 5 × 106 yeasts, with or without a 2-h pretreatment with PEA or tyrosol, were carefully added inside.
Experimental infection of mice.
Adult female DBA/2J and BALB/cByJ mice were purchased at 6 weeks of age from the Jackson Laboratory (Bar Harbor, ME), and female NOG mice (NOD.Cg-Prkdescid Il2rgtm1 Wjl/Szj) were bred locally under specific-pathogen-free conditions and used for experiments at 7 weeks of age. BALB/cByJ mice were immunocompromised by two injections of 200 μg/g cyclophosphamide (Endoxan) at days 1 and 4 prior to yeast infection.
Yeasts were grown overnight in YPD medium and then inoculated into fresh medium until the early exponential phase was reached (OD600, 1 to 3). Yeasts were then washed and prepared in PBS at a concentration of 3 × 108/ml for inoculation. Mice were anesthetized with isoflurane and injected intravenously via the retro-orbital sinus (29) with 100 μl of yeast suspension or PBS alone as a negative control. All mice were evaluated on a daily basis for 21 days. Blood samples were collected after 4 days by eye bleed in 100-μl capillary tubes coated with Na-heparin, which were then sealed and centrifuged, and the blood was allowed to clot in order to get sera. Mice underwent euthanasia after either 4 days for brain collection or 21 days. Once collected in PBS, brains were pulverized by roughly mixing and vortexing in the presence of glass beads. The suspension was then diluted and plated on YPD agar plates for CFU counting.
Microscopy.
A Nikon Eclipse 50i microscope with digital camera and an EVOS f1 AMG inverted microscope were used to capture images of yeast cells after pulverization of the mouse brains or after infection of the macrophages.
dpp3 knockout and reconstituted wild-type DPP3 strains.
PCR amplification, Southern hybridization (Roche Molecular Biochemicals), and yeast transformation were conducted as previously described (9, 30) and according to the manufacturer's instructions. The total RNA was purified from C. lusitaniae cells cultured in YPD, using selective precipitations with LiCl as previously described (31). Extracted RNA were treated with DNase (Ambion TURBO DNA free) following the manufacturer's recommendations. Then, 100 ng of RNA was used for reverse transcriptase PCR (RT-PCR; Invitrogen Superscript II) following the manufacturer's recommendations. DNA was sequenced using a BigDye Terminator v1.1 kit (Applied Biosystems), at the Genotyping-Sequencing Pole of the Functional Genomic Platform of Bordeaux, Bordeaux, France. All primers used in this study were synthesized by Eurofins MWG Operon. All constructed strains were verified by PCR, Southern blot analysis, and sequencing and are presented in Table S1 in the supplemental material. All of the details regarding the constructions of the different strains are provided in the supplemental material.
Using the URA3 marker, three different DPP3 gene knockouts were generated by gene replacement and gene disruption strategies in two different recipient strains, ura3Δ MATa and ura3Δ leu2Δ MATα. Thus, the ura3::URA3 dpp3Δ::0 MATa (knockout [KO]), the ura3Δ leu2Δ dpp3Δ::URA3 MATα (KOα), and the ura3Δ dpp3Δ:: pGURA3-DPP3(130) MATa (KOdis) mutants were obtained.
Four different methods were used in order to reinsert a wild-type (WT) copy of the DPP3 gene into the different knockout mutants. A WT copy of DPP3 was inserted either at the inactivated DPP3 locus by single (RECpGURA3-DPP3) or double (REC and RECα) crossing over or elsewhere in the genome after ectopic integration (RECect). One last reconstituted strain was obtained without any transformation step, by forcing the KOdis to autoexcise the inactivation cassette (RECexc).
A dpp2Δ knockout mutant inactivated for the closest DPP3 paralog (CLUG-02164) and a double dpp3Δ dpp2Δ knockout (dbKO) mutant were created after replacing the DPP2 gene with an inactivation cassette containing the URA3 marker.
Farnesol, phenethyl alcohol, and tyrosol extraction and measurement.
For the farnesol measurement, 30 ml of RPMI medium (buffered to pH 7.0 with 0.165 M morpholinepropanesulfonic acid and supplemented with 2% glucose) was inoculated to a final concentration of 107 yeasts/ml at 35°C and 215 rpm for 24 h. For the PEA and tyrosol measurement, 5 ml of YPD was inoculated for 16 h. Cultures were centrifuged at 4,000 × g for 15 min. Supernatants were extracted with a one-fourth volume of ethyl acetate. Gas chromatography (GC) and GC-mass spectrometry (GC/MS) were used to measure PEA and tyrosol levels. For the farnesol measurement, the ethyl acetate was removed under conditions of nitrogen evaporation and the residue was suspended in 200 μl of acetonitrile for high-performance liquid chromatography–tandem MS (HPLC-MS/MS) analysis. Details of GC, GC/MS, and HPLC-MS/MS are provided in the supplemental material.
PPi measurement assay.
In 30 ml of RPMI medium, 107 yeasts/ml were cultured for 24 h at 35°C and 215 rpm. Then, 17 × 108 yeasts were collected and resuspended in 17 ml of phosphate buffer (NaHPO4) supplemented with 1 M sorbitol (pH 7.5). A volume of 170 μl of a 10 mg/ml Zymolyase 20T (Seikagaku Biobusiness) freshly prepared solution and 34 μl of β-mercaptoethanol were added, and the suspension was left for 1 h at 37°C for spheroplast formation. Cells were then centrifuged at 4,000 × g for 10 min and washed twice with the phosphate buffer. A last wash was performed in phosphate buffer without sorbitol, and cells were pelleted and resuspended in 500 μl of HEPES buffer (pH 7). For lysis, the suspension was left for 1 h on ice while being subjected to a vigorous vortex procedure every 10 min. The protein concentration was determined by Bradford's method (32) using bovine serum albumin as the standard. For the total pyrosphosphate (PPi) measurement assay, either equivalent volumes of 50 μl of the cell lysate preparation or equivalent protein quantities were added to a 96-well flat-bottom black plate (Greiner Bio-one). In order to measure substrate-specific pyrophosphatase activity by detecting a potential increase in PPi levels, farnesyl pyrophosphate (Santa Cruz), geranyl pyrophosphate (Sigma), geranylgeranyl pyrophosphate (Sigma), diacylglycerol pyrophosphate (Avanti), or isopentyl pyrophosphate (Sigma) was added in the plates for 5 to 60 min at final concentrations of 10 μM to 500 μM. PPi was measured using a PhosphoWorks fluorimetric pyrophosphate assay kit (AAT Bioquest) following the manufacturer's protocol.
Crude extract preparation.
In order to prepare yeast crude extract for Western blotting, 24 × 109 cells of an overnight culture in 30 ml YPD were resuspended in 50 ml of phosphate buffer (50 mM NaHPO4, 1 M sorbitol, pH 7.5). A 500-μl volume of a freshly prepared Zymolyase 20T (Seikagaku Biobusiness) solution at 30 mg/ml and 500 μl of β-mercaptoethanol were added, and the suspension was incubated for 2 h at 37°C for spheroplast formation. Cells were then centrifuged at 4,000 × g for 10 min and washed twice in the phosphate buffer. A last wash was made with the phosphate buffer without sorbitol, and the cells were resuspended in a 2-ml volume containing 1 ml of HEPES buffer (50 mM, pH 7) and 1 ml of lysis buffer (100 mM Tris-HCl, 2 mM dithiothreitol [DTT], 0.2% SDS, 0.2% Triton X-100 [pH 8], protease inhibitor [Roche] [1 tablet/10 ml]). The suspension was left for 3 h on ice and shaken vigorously every 15 min for spheroplast lysis.
Western blot analysis.
Rabbit polyclonal antibodies were produced and purified by ProteoGenix against the C-terminal (cys-EEIAPESGYSPVEEV) and N-terminal (TRSTFGLENIKGGR-cys) peptides of the C. lusitaniae Dpp3 protein. Mouse anti-α-tubulin antibody was kindly provided by Derrick Robinson and was used as a positive control.
Proteins from identical volumes of yeast crude extracts were used in a 12% SDS-polyacrylamide gel. After gel migration, proteins were transferred to a polyvinylidene difluoride (PVDF) (Amersham Hybond-P, General Electric) membrane following the manufacturer's instructions. Then, the membrane was incubated in PBS–Tween-Milk (0.05% [vol/vol] Tween 20, 5% [wt/vol] milk) for 20 min. After the blocking step, the solution was discarded and the primary antibody diluted 1:100 in PBS–Tween-Milk was added. The membrane was incubated overnight at 4°C on a rotary shaker. The next day, the membrane was washed with PBS–Tween-Milk four times for 10 min each time. Then, the secondary antibody, goat anti-rabbit or rabbit anti-mouse (Sigma) coupled to peroxidase and diluted 1/10,000 or 1/5,000, respectively, in PBS–Tween-Milk, was added. After 1 h of incubation, the membrane was washed twice in PBS–Tween-Milk and once in PBS. Finally, the immunodetection and the revelation were made using a Clarity Western ECL kit (Bio-Rad) and “ImageQuant LAS 4000” (General Electric) following the manufacturer's recommendations.
Statistical analysis.
The differences presented in Results were all tested and confirmed for significance using analysis of variance (ANOVA) and t tests. Differences were significant at P values < 0.05.
RESULTS
The dpp3Δ mutant is altered in PEA and tyrosol secretion.
We performed a tBLASTn analysis to identify the ortholog of C. albicans DPP3 in C. lusitaniae and found CLUG-05720 to be the most closely related gene, with 54% identity and 70% similarity (E-value 0). The two genes share a conserved catalytic domain of the phosphatidic acid phosphatase (PAP2) family (Pfam 01569). A phylogenetic analysis of the DPP3 gene in the Candida clade is shown in Fig. S1 in the supplemental material. To investigate the effects of this gene inactivation in C. lusitaniae, we constructed three dpp3Δ knockout mutants in two different genetic backgrounds, either by the gene replacement or the gene disruption technique (see Supplemental Materials and Methods and Table S1 in the supplemental material). All mutants shared identical phenotypes; thus, we show the data for a representative mutant obtained by gene replacement that we named “KO.” The KO and the wild-type (WT) strains had similar growth rates in all media used in experimentation. In YPD rich medium at 35°C under conditions of agitation, the KO strain had a division time of 74 ± 4 min whereas the WT strain had a division time of 80 ± 6 min.
To assess the importance of Dpp3 as a pyrophosphatase in C. lusitaniae, intracellular pyrophosphate (PPi) was measured for the KO and the WT strains. Interestingly, we found that the KO strain had 8.6 ± 0.7 μM intracellular PPi and that the WT strain had 10.7 ± 0.8 μM (Fig. 1). Thus, the inactivation of the DPP3 gene resulted in a 20% decrease in the total intracellular PPi. To further characterize the pyrophosphatase activity of Dpp3, we tested several potential substrates. Among all of the substrates tested, we observed only a slight (7% ± 1%) increase in the PPi levels when farnesyl pyrophosphate and geranylgeranyl pyrophosphate were added at 500 μM to yeast lysates. However, this increase was the same in the WT and the KO strains.
FIG 1.

Measurement of intracellular pyrophosphate. Intracellular PPi was measured in WT, KO, dbKO, and REC yeast lysates by fluorimetry assay. Results are expressed as mean values ± standard deviations (SD) of data from three independent experiments. Means with different letters are significantly different (P < 0.05).
Then, we measured yeast farnesol secretion and we found that the WT strain secreted low (0.1 μM) levels of farnesol, but surprisingly, the dpp3Δ mutant was not affected with regard to its farnesol secretion. To rule out the possibility of a functional redundancy, we knocked out the closest DPP3 paralog in C. lusitaniae (CLUG-02164), and we named the resulting construct DPP2. Neither the dpp2Δ knockout nor the dpp3Δ dpp2Δ double-knockout (dbKO) strain showed reduced farnesol production. The dbKO strain also had a level of intracellular PPi similar to that seen with the KO strain (Fig. 1).
On the other hand, two aromatic alcohols, phenethyl alcohol (PEA) and tyrosol, were the only compounds found to be secreted in concentrations that differed between the WT and the KO strains in the YPD rich medium. The WT strain produced 116 μM PEA and 80 μM tyrosol, whereas the KO strain produced approximately 62 μM PEA and 121 μM tyrosol (Fig. 2). The reconstituted (REC) DPP3 strain showed no restoration of the wild-type phenotypes (Fig. 1 and 2). Taken together, these results showed that in C. lusitaniae, the KO mutant is affected with regard to pyrophosphatase activity as well as with regard to PEA and tyrosol secretion.
FIG 2.
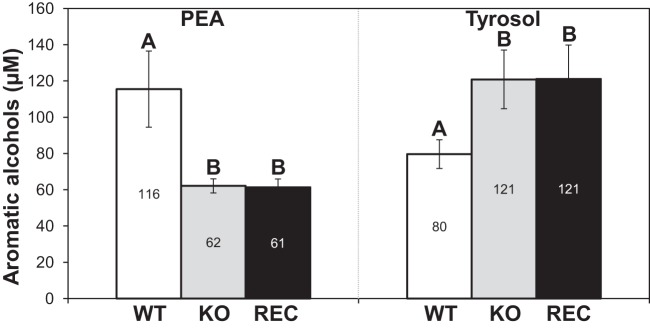
Production of aromatic alcohols. PEA and tyrosol produced in WT, KO, and REC yeast cell cultures were quantified by GC/MS. Results are expressed as mean values ± SD of data from eight independent experiments. Means with different letters are significantly different (P < 0.05).
The dpp3Δ mutant shows decreased mortality and a decreased capacity to stimulate NO and ROS production in macrophages in vitro.
We previously developed an experimental model to infect J774 cell line macrophages with yeasts in RPMI medium (28). This model was used for in vitro study of the different parameters of the interaction between yeast strains and macrophages. During the infection, the KO and the WT strains showed similar morphologies: both were able to produce pseudohyphae (see Fig. S2 in the supplemental material). No significant differences were noted in the uptake of fungal cells, in the amount of macrophages engaged in yeast phagocytosis, or in macrophage mortality between the KO and the WT strains (data not shown). In contrast, the KO mutant showed higher resistance to macrophage killing after 24 h of infection (Fig. 3A).
FIG 3.

Macrophage killing of intracellular yeasts and ROS production. (A) Infected macrophages were lysed after 24 h, and WT, KO, and REC yeast mortality was assessed. (B) Fluorimetric measurement of intracellular ROS production by macrophages infected with the WT, the WT plus 10 μM tyrosol (Tyr), the KO, the KO plus 10 μM PEA, or the REC strain. Noninfected macrophages produced basal levels of ROS production that were designated “0.” Results are expressed as mean values ± SD of data from three independent experiments. Means with different letters are significantly different (P < 0.05). A.F.U., arbitrary fluorescence units.
Because ROS and NO production greatly contributes to the killing of intracellular pathogens (11), the levels of intracellular ROS and NO were measured in the macrophages during the infection with the KO or the WT strain. The results were similar between ROS and NO; hence, only the ROS levels are presented (Fig. 3B). After 24 h of infection, we found that macrophages infected with the KO mutant produced half the amount of ROS and NO compared to macrophages infected with the WT strain. The reconstituted DPP3 strain (REC) again showed no restoration of the wild-type phenotypes (Fig. 3).
Adding PEA or tyrosol to uninfected macrophages (see Fig. S3 in the supplemental material) or to macrophages immediately after yeast infection did not induce ROS and NO production (data not shown). In contrast, when yeasts in the stationary phase were incubated with PEA or tyrosol at 10 μM or 100 μM for 2 h prior to infection, ROS production and NO production were dramatically affected. Interestingly, pretreating the KO mutant with PEA restored ROS and NO production to the WT level, and inversely, pretreating the WT strain with tyrosol reduced ROS and NO production (Fig. 3B). In contrast, pretreating the KO mutant with tyrosol and pretreating the WT strain with PEA had no effect on ROS and NO production (see Fig. S3). Even though ROS and NO production was modulated, the mortality of phagocytosed yeasts was not affected when yeasts were treated before infection (data not shown). This result was not surprising since yeast killing relies on multiple factors during the interaction with macrophages (33, 34).
The dpp3Δ mutant stimulates a different cytokine response by macrophages in vitro.
To further explore macrophage response to yeasts, TNF-α and IL-10 levels were measured in the infection media. At 6 h postinfection, macrophages secreted 46% more TNF-α and 41% less IL-10 when infected with the KO strain compared to the WT strain (Fig. 4A).
FIG 4.

Cytokine secretion by macrophages. (A) TNF-α and IL-10 measurement by ELISA at 6 h postinfection. (B) IL-10 secretion by macrophages after 6 h of coincubation with yeasts in transwells. Zymosan was added to stimulate macrophages under every set of conditions. Uninfected macrophages (Mac+Z) were used as a control. Basal levels of cytokine secretion corresponding to noninfected and nonstimulated macrophages were designated “0.” Results are expressed as mean values ± SD of data from three independent experiments. Means with different letters are significantly different (P < 0.05).
To inspect a possible role of signaling molecules in the differences observed in cytokine secretion, transwells were used to physically separate yeasts from macrophages in the infection medium. Without physical contact with yeasts, phagocytes did not secrete TNF-α or IL-10 above their basal levels (data not shown). In order to activate the macrophages, zymosan was added. It stimulated the secretion of both TNF-α and IL-10, but yeasts added in the transwells had no effect on the TNF-α level (data not shown). In contrast, the C. lusitaniae WT strain was remarkably able to increase macrophage IL-10 secretion through the transwells. However, the KO mutant was unable to do so (Fig. 4B). Once again, the reconstituted DPP3 strain (REC) showed no restoration of the wild-type phenotypes. These data suggest that the KO mutant lost its capacity to increase IL-10 secretion by J774 macrophages via one or more diffusible molecules. Using GC/MS or HPLC-MS/MS, we attempted to identify those molecules without success. Adding PEA or tyrosol to uninfected macrophages or to macrophages immediately after yeast infection did not affect the cytokine response (data not shown). In contrast, even though yeasts were physically separated from the phagocytes, pretreating the KO strain with 10 μM or 100 μM PEA strikingly restored the capacity of yeasts to induce IL-10 secretion, and pretreating the WT strain with 10 μM or 100 μM tyrosol disabled it (Fig. 4B). To our knowledge, this is the first report where PEA and tyrosol are shown to be implicated in the modulation of the macrophagic response (IL-10, ROS, NO) during yeast-phagocyte interaction.
DPP3 is essential for C. lusitaniae virulence in mice.
To investigate whether the dpp3Δ mutant also exhibited defects in vivo, we performed virulence assays in our murine model of disseminated candidemia where DBA2/J mice were infected with the C. lusitaniae WT, KO, or REC strain (KO with a reconstituted WT DPP3 locus; see Table S1 in the supplemental material). When infected with the WT strain, mice showed a low survival rate of 5%. In contrast, the dpp3Δ mutant appeared remarkably avirulent, with a mouse survival rate of 95% (Fig. 5A). Unlike the WT strain, which was highly virulent, the KO strain was also avirulent in NOG and in immunocompromised BALB/cByJ mice (data not shown). These data indicate that the C. lusitaniae DPP3 gene played an essential role in virulence in a manner independent of the mouse strain infected.
FIG 5.

Effects of yeast infection on DBA2/J mouse survival rates, TNF-α levels, and brain colonization. (A) Mouse survival rates after infection with the WT, the KO, and the REC strains. For each strain, 17 to 19 mice were used. (B) TNF-α secretion in mouse sera after 4 days of infection with the WT, the KO, and the REC strains. Mice injected with PBS served as controls. Six to eight mice per strain were used for serum collection. (C) Brain colonization by the WT, the KO, and the REC strains. After 4 days, the brains of six mice from each infection group were collected and pulverized and the suspension was plated on YPD agar for CFU counting. Means with different letters are significantly different (P < 0.05).
Four days after infection, we measured the levels of TNF-α and IL-10 in sera of DBA2/J-infected mice. No difference in IL-10 levels was observed between mice infected with the different yeast strains and control mice injected with PBS (data not shown). In contrast, mice infected with the KO mutant had TNF-α levels similar to those of control mice, whereas the infection with the WT strain multiplied those levels by two (Fig. 5B).
Loss of balance in mice led us to assess yeast cell colonization of the brain. The numbers of C. lusitaniae CFU recovered from the brains of infected mice were compared 4 days after infection. Mice infected with the WT strain had approximately three times more CFU in their brains than those infected with the KO mutant (Fig. 5C). When brain pulverizations were observed under the microscope, the WT, the KO, and the REC strains were all found in yeast form (data not shown). Reconstructing a WT DPP3 locus in the KO strain (REC) restored to a great extent the WT phenotypes observed in vivo (Fig. 5). Taken together, these data show that the DPP3 gene played a major role in C. lusitaniae virulence in mice, by promoting TNF-α secretion and brain colonization.
Reinserting a WT DPP3 copy in the dpp3Δ mutant is sufficient to restore the WT phenotypes in vivo but not in vitro.
In order to bypass any issue related to a specific molecular technique, four different methods were used to reinsert a WT copy of the DPP3 gene in the different knockout mutants (see Supplemental Materials and Methods and Table S1 in the supplemental material). RT-PCR and Western blot analysis confirmed that the DPP3 gene was transcribed and translated in the reconstituted strains (Fig. 6). Even though the WT phenotypes were greatly restored in vivo, unfortunately, the in vitro WT phenotypes were not restored regardless of the molecular strategy and the genetic background used.
FIG 6.
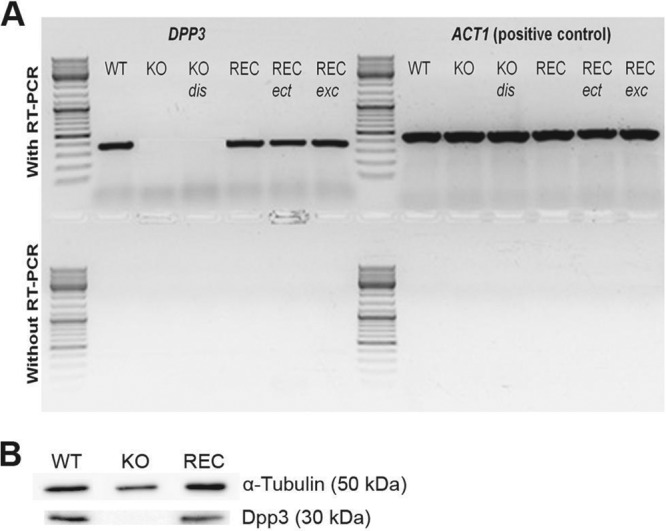
RT-PCR and Western blot analyses showing the transcription and translation of the DPP3 gene. (A) DPP3 mRNA presence was detected by RT-PCR. The ACT1 housekeeping gene was used as a positive control. DNA contamination of RNA preparations was verified by PCR without the prior use of a reverse transcriptase (RT). (B) Dpp3 protein (30-kDa) presence was detected by Western blot analysis. α-Tubulin protein (50-kDa) detection was used as a positive control.
DISCUSSION
Signaling molecules implicated in the interaction of C. lusitaniae with the host have not yet been explored. In order to get insights into these signaling molecules, we chose to study the effect of inactivation of the DPP3 gene which was shown in C. albicans to be involved in the biosynthesis of farnesol (21), the most studied signaling molecule in Candida species. Candida lusitaniae and C. albicans are two different yeast species; thus, what is already known for one might not necessarily apply to the other. In fact, from our previous work (28) and our unpublished data in the laboratory, we know that these two species have many differences in their physiobiologies and their metabolisms. Therefore, the aim of this work was to create a dpp3Δ knockout mutant in C. lusitaniae and to characterize its different phenotypes from signaling molecule production to interaction with macrophages in vitro and virulence in vivo.
Three different dpp3Δ mutants were constructed by gene replacement and gene disruption strategies in two recipient strains. This ensured that the phenotypes observed in vitro were the result of DPP3 gene inactivation and were not related to the molecular method employed or the genetic background of the recipient strain. All of the mutants displayed the same in vitro phenotypes. DPP3 encodes a putative pyrophosphatase with a catalytic domain of the phosphatidic acid phosphatase (PAP2) family (Pfam 01569). Here, we found lower intracellular pyrophosphate (PPi) levels in the dpp3Δ knockout (KO) strain than in the WT strain, revealing a missing pyrophosphatase activity in the mutant. However, unlike in C. albicans, the KO mutant in C. lusitaniae was not affected for farnesol synthesis. In addition, neither the dpp2Δ knockout nor the dpp3Δ dpp2Δ double-knockout strain had reduced farnesol production, thus eliminating a paralogous functional redundancy hypothesis. From these data, we conclude that these genes were not implicated in farnesol production in C. lusitaniae. In previous studies on C. albicans, authors did not rule out the implication of Dpp3 in cell activities other than farnesol production (21). Furthermore, in Saccharomyces cerevisiae, the Dpp3 ortholog Dpp1 is shown to be a pyrophosphatase with many potential substrates (35). Here, the substrate-specific enzyme activity assay that we carried out was not conclusive. On the other hand, we showed that the KO mutant in C. lusitaniae secreted less PEA but more tyrosol than the WT strain. Interestingly, the sums of the PEA and the tyrosol secreted in the two strains were very similar, suggesting that the difference observed was a potential shift of a PEA/tyrosol ratio. PEA and tyrosol are aromatic alcohols that derive from phenylalanine and from tyrosine or tryptophan, respectively, but it is still not completely clear how they are synthesized in C. albicans (36). These two aromatic alcohols differ only by an additional hydroxyl group present in tyrosol, but a direct link connecting their biosynthesis pathways is not yet known. Assuming that it can exist, it would be altered in the KO mutant. Taking these data altogether, we show that the KO mutant was not affected in terms of farnesol synthesis but lost significant pyrophosphatase activity and had altered PEA and tyrosol secretion.
The host response to a Candida infection relies essentially on innate immunity, with macrophages representing an effective first line of defense. Here we found that after 24 h of infection, the KO mutant had enhanced resistance to macrophage killing compared to the WT strain. Furthermore, the infection with the KO mutant resulted in lower NO and ROS production by the phagocytes. Moreover, at 6 h postinfection, macrophages produced less IL-10 and more TNF-α when infected with the KO mutant. Additionally, we showed that when transwells were used to physically separate the two cell types, the WT strain of C. lusitaniae was remarkably able to increase IL-10 secretion by zymosan-treated macrophages. However, the KO mutant was unable to do so. Since we knew that the KO mutant produced less PEA than the WT strain, PEA was directly added in the infection medium, but the addition did not restore the WT phenotypes. Interestingly, when KO cells were incubated with PEA 2 h prior to infection, not only NO and ROS production but also IL-10 secretion by macrophages in the transwell assay was restored to WT levels. In contrast, when tyrosol was added to the WT cells, the macrophagic responses observed were similar to those triggered by the KO mutant. The PEA/tyrosol balance is one of possibly many factors that differ between the WT and the KO strains, but here we showed that these two aromatic alcohols had direct effects on yeast cells, possibly by modulating their secretome, which ultimately led to different macrophagic responses. All efforts will now focus on revealing these secretome modulations.
Our in vivo studies gave new insights into the importance of DPP3 in virulence in mice. Here we showed that, unlike the WT strain, which was severely virulent, the KO mutant was almost completely avirulent, independently of the mouse strain infected. After 4 days of infection, mice seemed completely disoriented and had a loss of balance. At this time point, brain colonization and cytokine expression were assessed. Although TNF-α confers protection against a number of pathogens (12–14), excessive levels are often associated with organ failure, septic shock, and tissue damage (37, 38). In this study, mortality was correlated with high TNF-α levels in mouse sera. These data could also be connected to higher colonization of mouse brains. The KO mutant showed no altered morphology compared to the WT strain; both were able to make pseudohyphae when infecting macrophages in vitro, and both were in yeast form inside mouse brains in vivo. Thus, we believe that morphology played no obvious role in the avirulent phenotype of the KO mutant, and even though we do not know if the WT and the KO strains equally multiply inside the mice, the role of the DPP3 gene as an essential contributor to virulence for C. lusitaniae is clear.
When a WT copy of the DPP3 gene was reinserted in the KO mutant (REC strain), a restoration of all the WT phenotypes analyzed in vivo, including virulence and TNF-α and CFU levels in the brains, was observed. This restoration did not reach 100% of the WT levels. This phenomenon is not completely uncommon, since it was observed in many cases studying microorganism interaction with the host (39, 40), including C. albicans (41, 42). However, no restoration of the WT phenotypes was achieved in vitro despite several techniques being used to reconstitute a WT copy of the gene in the dpp3Δ mutants, including a reconstitution of the DPP3 locus by autoexcision of the inactivation cassette (RECexc; see Supplemental Materials and Methods and Table S1 in the supplemental material). RT-PCR and Western blot analysis confirmed that DPP3 mRNA and protein were absent in the different knockout mutants and present in the strains carrying a WT copy of the DPP3 gene. We believe that a seemingly adaptive and irreversible response might have occurred in yeasts after the DPP3 gene was inactivated. The difficulties encountered in attempts to restore the WT phenotype by reinserting a WT copy of the inactivated gene have been described for the ASH1 gene, implicated in the morphology of C. albicans, for which only 1 of 48 independent reintegrant strains had a restored phenotype (43). Taking these data altogether, we believe there are two relevant hypotheses that might explain this lack of complementation in vitro. The first is based on the possible existence of posttranscriptional or posttranslational modifications that would be required for a fully active Dpp3 protein. Some of these modifications would be missing in the reconstituted strains for unknown reasons. This hypothesis could be supported by the fact that even though the REC strain expressed the mRNA and protein of DPP3, it showed no restoration of a pyrophosphatase activity. The second hypothesis is based on the possible existence of compensatory mutations that might have occurred once the DPP3 gene was inactivated and which were not restored when a WT copy of the gene was reinserted or reconstituted. The acquirement of a compensatory mutation elsewhere in the genome was described in a fis1Δ knockout mutant in S. cerevisiae, where every time the gene was inactivated, the same compensatory mutation was selected (44). If this hypothesis is valid in the case of this study, then the compensatory mutations would more likely be the ones responsible for the phenotypes observed in vitro and could also slightly contribute to the in vivo phenotypes, since no complete restoration of these phenotypes was achieved after the reconstitution of a WT DPP3 locus.
In conclusion, our work characterized the interaction of the C. lusitaniae WT strain with the host and determined to which extent this interaction was affected in a dpp3Δ mutant. Compared to the WT strain, this mutant showed lower PPi levels, had altered PEA and tyrosol secretion, stimulated different ROS and cytokine responses by macrophages in vitro, and displayed severely diminished virulence in vivo. Some of these phenotypes were directly dependent on DPP3; others might have been the result of other genetic modifications due to DPP3 inactivation. On the other hand, this study also revealed the interesting implication of PEA and tyrosol in the interaction of yeasts with macrophages. Besides decrypting the detailed mechanisms of action connecting PEA and tyrosol to the yeast-macrophage interaction, the role of these two aromatic alcohols in virulence in mice remains to be explored. Last, since the WT strain was remarkably able to modulate macrophage IL-10 secretion without physical contact, our next effort will consist of unraveling the chemical nature of the diffusible molecules involved and determining their subsequent effect on T helper cell differentiation. Characterizing the potentially implicated receptors and the underlying pathways will also be of great importance.
Supplementary Material
ACKNOWLEDGMENTS
A.S. is supported by the French Ministère de l'Enseignement Supérieur et de la Recherche.
We thank Derrick Robinson (Laboratoire de Microbiologie Fondamentale et Pathogénicité UMR-CNRS 5234, Bordeaux, France) for kindly providing the primary anti-α-tubulin antibody used in the Western blot assay. We thank Loïc Rivière for critical reading of the manuscript. We are grateful to the Flow Cytometry platform of the Federative Research Institute “Infectious Diseases and Cancers” (SFR TransBiomed) of Bordeaux and to the Animalerie A2 of the University of Bordeaux. We acknowledge the Genotyping and Sequencing facility of Bordeaux for participating in the sequencing of the DPP3 gene and the platforms Métabolome-Lipidome-Fluxome of Bordeaux for contributing to the GC-FID and the GC-MS analysis.
Footnotes
Published ahead of print 4 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01263-13.
REFERENCES
- 1.Bustamante CI. 2005. Treatment of Candida infection: a view from the trenches! Curr. Opin. Infect. Dis. 18:490–495. 10.1097/01.qco.0000191516.43792.61 [DOI] [PubMed] [Google Scholar]
- 2.Kullberg BJ, Filler SG. 2002. Candidemia, p 327–340 In Calderone RA. (ed), Candida and candidiasis. ASM Press, Washington, DC [Google Scholar]
- 3.Pfaller MA, Jones RN, Messer SA, Edmond MB, Wenzel RP. 1998. National surveillance of nosocomial blood stream infection due to Candida albicans: frequency of occurrence and antifungal susceptibility in the SCOPE Program. Diagn. Microbiol. Infect. Dis. 31:327–332. 10.1016/S0732-8893(97)00240-X [DOI] [PubMed] [Google Scholar]
- 4.Calderone RA. (ed). 2002. Candida and candidiasis. ASM Press, Washington [Google Scholar]
- 5.Pappagianis D, Collins MS, Hector R, Remington J. 1979. Development of resistance to amphotericin B in Candida lusitaniae infecting a human. Antimicrob. Agents Chemother. 16:123–126. 10.1128/AAC.16.2.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawkins JL, Baddour LM. 2003. Candida lusitaniae infections in the era of fluconazole availability. Clin. Infect. Dis. 36:e14–e18. 10.1086/344651 [DOI] [PubMed] [Google Scholar]
- 7.Hazen KC. 1995. New and emerging yeast pathogens. Clin. Microbiol. Rev. 8:462–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Favel A, Michel-Nguyen A, Peyron F, Martin C, Thomachot L, Datry A, Bouchara J-P, Challier S, Noël T, Chastin C, Regli P. 2003. Colony morphology switching of Candida lusitaniae and acquisition of multidrug resistance during treatment of a renal infection in a newborn: case report and review of the literature. Diagn. Microbiol. Infect. Dis. 47:331–339. 10.1016/S0732-8893(03)00094-4 [DOI] [PubMed] [Google Scholar]
- 9.El-Kirat-Chatel S, Dementhon K, Noël T. 2011. A two-step cloning-free PCR-based method for the deletion of genes in the opportunistic pathogenic yeast Candida lusitaniae. Yeast 28:321–330. 10.1002/yea.1836 [DOI] [PubMed] [Google Scholar]
- 10.Romani L. 2000. Innate and adaptive immunity in Candida albicans infections and saprophytism. J. Leukoc. Biol. 68:175–179 [PubMed] [Google Scholar]
- 11.Wink DA, Hines HB, Cheng RYS, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. 13 January 2011. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 10.1189/jlb.1010550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufmann SH. 1993. Immunity to intracellular bacteria. Annu. Rev. Immunol. 11:129–163. 10.1146/annurev.iy.11.040193.001021 [DOI] [PubMed] [Google Scholar]
- 13.Salgame P. 2005. Host innate and Th1 responses and the bacterial factors that control Mycobacterium tuberculosis infection. Curr. Opin. Immunol. 17:374–380. 10.1016/j.coi.2005.06.006 [DOI] [PubMed] [Google Scholar]
- 14.Westendorp RG, Langermans JA, Huizinga TW, Elouali AH, Verweij CL, Boomsma DI, Vandenbrouke JP. 1997. Genetic influence on cytokine production and fatal meningococcal disease. Lancet 349:170–173. 10.1016/S0140-6736(96)06413-6 [DOI] [PubMed] [Google Scholar]
- 15.Netea MG, Brown GD, Kullberg BJ, Gow NAR. 2008. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6:67–78. 10.1038/nrmicro1815 [DOI] [PubMed] [Google Scholar]
- 16.Shea JM, Del Poeta M. 2006. Lipid signaling in pathogenic fungi. Curr. Opin. Microbiol. 9:352–358. 10.1016/j.mib.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 17.Erb-Downward JR, Noverr MC. 2007. Characterization of prostaglandin E2 production by Candida albicans. Infect. Immun. 75:3498–3505. 10.1128/IAI.00232-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins de Lima T, Gorjão R, Hatanaka E, Cury-Boaventura MF, Portioli Silva EP, Procopio J, Curi R. 13 June 2007. Mechanisms by which fatty acids regulate leucocyte function. Clin. Sci. (Lond) 10.1042/CS20070006 [DOI] [PubMed] [Google Scholar]
- 19.Haas-Stapleton EJ, Lu Y, Hong S, Arita M, Favoreto S, Nigam S, Serhan CN, Agabian N. 2007. Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS One 2:e1316. 10.1371/journal.pone.0001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornby JM, Jensen EC, Lisec AD, Tasto JJ, Jahnke B, Shoemaker R, Dussault P, Nickerson KW. 2001. Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl. Environ. Microbiol. 67:2982–2992. 10.1128/AEM.67.7.2982-2992.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navarathna DH, Hornby JM, Krishnan N, Parkhurst A, Duhamel GE, Nickerson KW. 2007. Effect of farnesol on a mouse model of systemic candidiasis, determined by use of a DPP3 knockout mutant of Candida albicans. Infect. Immun. 75:1609–1618. 10.1128/IAI.01182-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarathna DHMLP, Nickerson KW, Duhamel GE, Jerrels TR, Petro TM. 2007. Exogenous farnesol interferes with the normal progression of cytokine expression during candidiasis in a mouse model. Infect. Immun. 75:4006–4011. 10.1128/IAI.00397-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramage G, Saville SP, Wickes BL, Lopez-Ribot JL. 2002. Inhibition of Candida albicans biofilm formation by farnesol, a quorum-sensing molecule. Appl. Environ. Microbiol. 68:5459–5463. 10.1128/AEM.68.11.5459-5463.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semighini CP, Hornby JM, Dumitru R, Nickerson KW, Harris SD. 2006. Farnesol-induced apoptosis in Aspergillus nidulans reveals a possible mechanism for antagonistic interactions between fungi. Mol. Microbiol. 59:753–764. 10.1111/j.1365-2958.2005.04976.x [DOI] [PubMed] [Google Scholar]
- 25.Deveau A, Piispanen AE, Jackson AA, Hogan DA. 2010. Farnesol induces hydrogen peroxide resistance in Candida albicans yeast by inhibiting the Ras-cyclic AMP signaling pathway. Eukaryot. Cell 9:569–577. 10.1128/EC.00321-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H. 2006. Feedback control of morphogenesis in fungi by aromatic alcohols. Genes Dev. 20:1150–1161. 10.1101/gad.1411806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Fujita M, Feng Q, Clardy J, Fink GR. 2004. Tyrosol is a quorum-sensing molecule in Candida albicans. Proc. Natl. Acad. Sci. U. S. A. 101:5048–5052. 10.1073/pnas.0401416101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dementhon K, El-Kirat-Chatel S, Noël T. 2012. Development of an in vitro model for the multi-parametric quantification of the cellular interactions between Candida yeasts and phagocytes. PLoS One 7:e32621. 10.1371/journal.pone.0032621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trammell RA, Cox L, Pikora J, Murphy LL, Toth LA. 2012. Evaluation of an extract of North American ginseng (Panax quinquefolius L.) in Candida albicans-infected complement-deficient mice. J. Ethnopharmacol. 139:414–421. 10.1016/j.jep.2011.11.026 [DOI] [PubMed] [Google Scholar]
- 30.François F, Chapeland-Leclerc F, Villard J, Noël T. 2004. Development of an integrative transformation system for the opportunistic pathogenic yeast Candida lusitaniae using URA3 as a selection marker. Yeast 21:95–106 [DOI] [PubMed] [Google Scholar]
- 31.Ramon A, Gil R, Burgal M, Sentandreu R, Valentin E. 1996. A novel cell wall protein specific to the mycelial form of Yarrowia lipolytica. Yeast 12:1535–1548 [DOI] [PubMed] [Google Scholar]
- 32.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 7:248–254 [DOI] [PubMed] [Google Scholar]
- 33.Frohner IE, Bourgeois C, Yatsyk K, Majer O, Kuchler K. 2009. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol. Microbiol. 71:240–252. 10.1111/j.1365-2958.2008.06528.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jamieson DJ, Stephen DW, Terrière EC. 1996. Analysis of the adaptive oxidative stress response of Candida albicans. FEMS Microbiol. Lett. 138:83–88. 10.1111/j.1574-6968.1996.tb08139.x [DOI] [PubMed] [Google Scholar]
- 35.Faulkner A, Chen X, Rush J, Horazdovsky B, Waechter CJ, Carman GM, Sternweis PC. 1999. The LPP1 and DPP1 gene products account for most of the isoprenoid phosphate phosphatase activities in Saccharomyces cerevisiae. J. Biol. Chem. 274:14831–14837. 10.1074/jbc.274.21.14831 [DOI] [PubMed] [Google Scholar]
- 36.Han T-L, Cannon RD, Villas-Bôas SG. 2011. The metabolic basis of Candida albicans morphogenesis and quorum sensing. Fungal Genet. Biol. 48:747–763. 10.1016/j.fgb.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 37.Mencacci A, Cenci E, Del Sero G, d' Ostiani CF, Mosci P, Montagnoli C, Bacci A, Bistoni F, Quesniaux VF, Ryffel B. 1998. Defective co-stimulation and impaired Th1 development in tumor necrosis factor/lymphotoxin-alpha double-deficient mice infected with Candida albicans. Int. Immunol. 10:37–48. 10.1093/intimm/10.1.37 [DOI] [PubMed] [Google Scholar]
- 38.Mootoo A, Stylianou E, Arias MA, Reljic R. 2009. TNF-alpha in tuberculosis: a cytokine with a split personality. Inflamm. Allergy Drug Targets. 8:53–62. 10.2174/187152809787582543 [DOI] [PubMed] [Google Scholar]
- 39.Converse PJ, Karakousis PC, Klinkenberg LG, Kesavan AK, Ly LH, Allen SS, Grosset JH, Jain SK, Lamichhane G, Manabe YC, McMurray DN, Nuermberger EL, Bishai WR. 2009. Role of the dosR-dosS two-component regulatory system in Mycobacterium tuberculosis virulence in three animal models. Infect. Immun. 77:1230–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russell-Goldman E, Xu J, Wang X, Chan J, Tufariello JM. 2008. A Mycobacterium tuberculosis Rpf double-knockout strain exhibits profound defects in reactivation from chronic tuberculosis and innate immunity phenotypes. Infect. Immun. 76:4269–4281. 10.1128/IAI.01735-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chamilos G, Nobile CJ, Bruno VM, Lewis RE, Mitchell AP, Kontoyiannis DP. 2009. Candida albicans Cas5, a regulator of cell wall integrity, is required for virulence in murine and Toll mutant fly models. J. Infect. Dis. 200:152–157. 10.1086/599363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarathna DHMLP, Lionakis MS, Lizak MJ, Munasinghe J, Nickerson KW, Roberts DD. 2012. Urea amidolyase (DUR1,2) contributes to virulence and kidney pathogenesis of Candida albicans. PLoS One 7:e48475. 10.1371/journal.pone.0048475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braun BR, Head WS, Wang MX, Johnson AD. 2000. Identification and characterization of TUP1-regulated genes in Candida albicans. Genetics 156:31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng W-C, Teng X, Park HK, Tucker CM, Dunham MJ, Hardwick JM. 2008. Fis1 deficiency selects for compensatory mutations responsible for cell death and growth control defects. Cell Death Differ. 15:1838–1846. 10.1038/cdd.2008.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.